

Abstract
Background
Lipopolysaccharide (LPS), also referred to as endotoxin, is the major constituent of the outer leaflet of the outer membrane of virtually all Gram-negative bacteria. The lipid A moiety, which anchors the LPS molecule to the outer membrane, acts as a potent agonist for Toll-like receptor 4/myeloid differentiation factor 2-mediated pro-inflammatory activity in mammals and, thus, represents the endotoxic principle of LPS. Recombinant proteins, commonly manufactured in Escherichia coli, are generally contaminated with endotoxin. Removal of bacterial endotoxin from recombinant therapeutic proteins is a challenging and expensive process that has been necessary to ensure the safety of the final product.
Results
As an alternative strategy for common endotoxin removal methods, we have developed a series of E. coli strains that are able to grow and express recombinant proteins with the endotoxin precursor lipid IVA as the only LPS-related molecule in their outer membranes. Lipid IVA does not trigger an endotoxic response in humans typical of bacterial LPS chemotypes. Hence the engineered cells themselves, and the purified proteins expressed within these cells display extremely low endotoxin levels.
Conclusions
This paper describes the preparation and characterization of endotoxin-free E. coli strains, and demonstrates the direct production of recombinant proteins with negligible endotoxin contamination.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-015-0241-5) contains supplementary material, which is available to authorized users.
Keywords: Escherichia coli, Lipopolysaccharide, Lipid A, Endotoxic activity, Recombinant protein, TLR4/MD-2 activation
Background
In the last thirty years, the biopharmaceutical industry has brought more than 220 biologics to market, giving rise to sales of approximately $70 - $80 billion per year [1]. One third of the unique recombinant protein therapeutics [2] and approximately one half of all products approved [3] are produced using an Escherichia coli based expression platform. However, the outer membrane of E. coli, like that of most Gram-negative bacteria, contains the potent immunostimulatory molecule lipopolysaccharide (LPS). In mammalian hosts, LPS (also known as endotoxin) can induce a pyrogenic response and ultimately trigger septic shock. Contaminating LPS must therefore be removed from recombinant therapeutic proteins expressed in E. coli before they can be safely administered to human patients. The removal of endotoxin from recombinant therapeutics and the testing to demonstrate endotoxin levels below a minimal threshold requires considerable effort, adding significant developmental and manufacturing costs. To date, no post-expression methodologies have been described that can remove endotoxin entirely [4]. Common endotoxin removal methods, such as ultrafiltration, Triton X phase separation, anion-exchange chromatography, adsorption on activated carbon, or treatment with polymyxin B- or histamine-immobilized affinity resins, are plagued by low efficiency and unsatisfactory selectivity [5]. In this context, it is important to note that commercially available recombinant proteins manufactured in E. coli may contain residual endotoxin in low but still sufficient quantities to activate human immune cells [6].
Cells of the innate immune system mediate the endotoxic response in mammals. LPS-mediated activation of a cell-surface receptor, consisting of Toll-like receptor 4 (TLR4) complexed with myeloid differentiation factor 2 (MD-2), results in the production of pro-inflammatory cytokines and type-1 interferons that are the chief effectors of the endotoxic response [7]. Research relating the structure of LPS to the activation of TLR4/MD-2 has demonstrated that lipid A is the component of LPS that is responsible for its TLR4/MD-2-dependent endotoxic activity [8]. Alterations in the structure of lipid A, chiefly modifications to the structure, number, and attachment sites of the hydrophobic acyl chains to the di-glucosamine backbone, significantly impacts endotoxic activity through altering TLR4/MD-2 mediated signaling [9,10]. When all the secondary acyl chains are removed, the under-acylated lipid A precursor lipid IVA not only lacks endotoxic activity in human immune cells, but also becomes a hTLR4/MD-2 receptor antagonist [8].
Until recently, it was believed that the structural features of LPS required to maintain the integrity of the Gram-negative outer membrane were essentially the same as the structural features of LPS required to elicit an endotoxic immune response in mammalian cells. The minimal structure essential for survival of typical laboratory strains of E. coli K-12 was thought to consist of a molecule of lipid A glycosylated at the 6′ position with two 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) sugar residues. This view changed when we reported the construction and characterization of KPM22, a derivative of the E. coli K-12 wild-type strain BW30270 that is unable to synthesize Kdo and yet retains viability with lipid IVA as the predominant outer membrane LPS component [11]. Subsequent research identified gain of function suppressor mutations in the LPS transport apparatus that apparently promote flipping of lipid IVA across the inner membrane [12]. These mutations both remove toxic side effects normally associated with lipid IVA accumulation in the inner membrane as well as provide a sufficient concentration of lipid IVA to support outer membrane biogenesis. The discovery of KPM22 presented us with the opportunity to construct recombinant protein expression strains of E. coli with low intrinsic endotoxic potential by rationally reprogramming the outer membrane biosynthesis pathway to elaborate solely lipid IVA.
This report describes the construction of stable E. coli strains, including derivatives of the popular expression strain BL21 (DE3), capable of efficiently expressing recombinant proteins which are essentially free of endogenous endotoxin contamination under standard laboratory conditions. To evaluate the utility of this expression platform, we applied it to the production of two different human proteins: apolipoprotein A-1 (ApoA-1) and heat shock protein 70 (Hsp70), both known to avidly bind endotoxin, and demonstrate marked reduction in endotoxin activity from minimally purified recombinant proteins.
Results and discussion
Engineering of LPS biosynthesis in E. coli
We began our construction of LPS-free E. coli K-12 using the Kdo-depleted strain KPM22 L11 [12]. This strain contains deletions of kdsD and gutQ, which encode d-arabinose 5-phosphate isomerases essential for the biosynthesis of Kdo [13,14], and a C:G to T:A transition at position 52 of msbA, which acts as a suppressor of the normally lethal ∆Kdo phenotype [12]. To produce a strain that contains lipid IVA as the only LPS outer membrane component and that cannot revert to synthesize endotoxic derivatives, we sequentially generated mutant strains with unmarked deletions of the genes lpxL, lpxM, pagP, lpxP and eptA. These genes encode enzymes acting downstream of Kdo incorporation into lipid IVA, and are either part of the constitutive pathway of Kdo2-hexaacyl lipid A biosynthesis (the Kdo2-lipid IVA lauroyl-ACP acyltransferase LpxL and the Kdo2-lauroyl-lipid IVA myristoyl-ACP acyltransferase LpxM), modify lipid A with additional acyl chains (the phospholipid:lipid A palmitoyl transferase PagP and the Kdo2-lipid IVA palmitoleoyl-ACP acyltransferase LpxP), or append phosphoethanolamine (P-EtN) under certain conditions (the lipid A P-EtN transferase EptA) [15] (Figure 1). Analysis of the LPS isolated from this strain, designated KPM318, using electrospray-ionization Fourier-transformed ion cyclotron mass spectrometry (ESI FT-ICR) revealed a single primary peak at 1404.85 u, consistent with the structure of the unmodified tetraacyl-1,4′-bisphosphate LPS precursor lipid IVA (Figure 2A).
Figure 1.
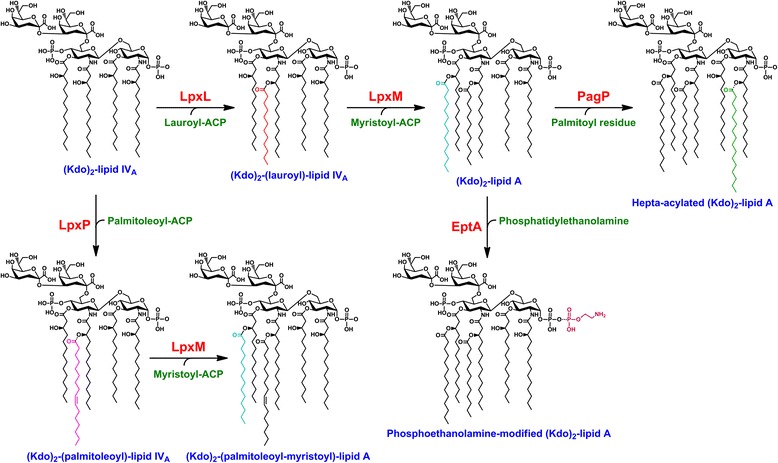
Biosynthetic reactions targeted during the construction of E. coli strains KPM318 and KPM404. The late acyltransferases LpxL and LpxM of the constitutive pathway transfer laurate and myristate, respectively, to Kdo2-lipid IVA to form the characteristic acyloxyacyl units of hexaacylated Kdo2-lipid A. In contrast, LpxP, PagP and EptA are regulated in response to certain stimuli such as incorporation of palmitoleate in place of laurate by LpxP at low growth temperatures or PagP-catalyzed palmitoylation of lipid A upon phospholipid translocation to the outer leaflet of the outer membrane, for example, in strains defective in LPS biosynthesis. The acyl-acyl carrier protein (ACP) serves as the preferred acyl donor for various lipid A-acyltransferases.
Figure 2.
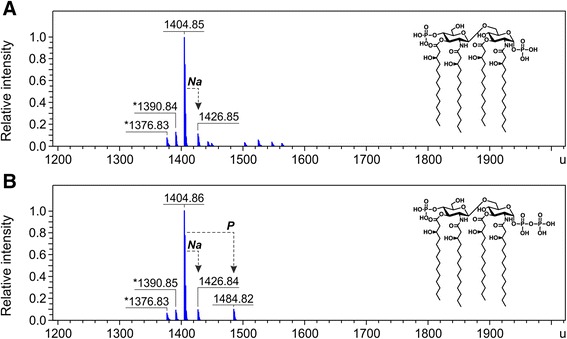
Charge deconvoluted ESI FT-ICR mass spectra in negative ion mode of lipid IVA isolated from BW30270-derived mutants. The lipid IVA (calculated mass 1404.854 u) was extracted from E. coli (K-12) strains KPM318 (A) and KPM335 (B). Mass numbers given refer to the monoisotopic masses of neutral molecules. Peaks representing molecules with variations in acyl chain length are labeled with asterisks (∆m = 14.02 u). The molecular ion at 1484.82 u in panel B indicates the presence of a minor fraction of 1-diphosphate lipid IVA. The structures of lipid IVA and 1-diphosphate lipid IVA are shown as insets in panels A and B, respectively.
The successful construction of KPM318 demonstrated the viability of E. coli containing only lipid IVA. However, like other E. coli K-12 strains with partially defective outer membranes such as the ∆Kdo prototype strain KPM22 [11], KPM318 displayed growth defects at temperatures above 40°C. To overcome this, we isolated a series of stable temperature-resistant derivatives of KPM318 capable of growing exponentially at 42°C. KPM335 was the most robust of the spontaneous mutants. Whole-genome sequencing of KPM335 identified a single de novo mutation in comparison to the temperature sensitive KPM318 parent strain, a G:C to T:A transversion at base number 181 of the frr gene (Additional file 1: Table S1). The frr gene encodes an essential ribosome recycling factor which has been described to play multiple cellular roles, such as in disassembly of the post-termination complex [16], prevention of translation errors [17], promoting translational coupling [18], and increasing cell viability through polyamine stimulation of stationary-phase E. coli cultures [19]. The derivation of KPM335 argues strongly for a direct correlation between the development of the frr181 allele and the ability of the strain to tolerate the ∆Kdo phenotype at 42°C. However, elucidation of the underlying mechanism must await further investigations.
ESI FT-ICR analysis of the LPS isolated from KPM335 revealed no significant changes in the LPS composition, with lipid IVA remaining the predominant LPS-related molecule as in the parent KPM318 strain (Figure 2B). However, in contrast to KPM318, spectra of KPM335 displayed a minor peak with a molecular mass of 1484.82 u, consistent with the structure of 1-diphosphate lipid IVA. Touzé and coworkers have shown previously that transfer of a second phosphate group to the 1 position of lipid A is catalyzed by LpxT, an inner membrane protein of the undecaprenyl-pyrophosphate phosphatase family, which is able to phosphorylate Kdo2-lipid IVAin vitro, but not lipid acceptors lacking Kdo [20]. Thus, the presence of a minor tris-phosphorylated lipid IVA fraction in KPM335 argues against an absolute requirement of LpxT for Kdo-glycosylated lipid A acceptors under in vivo conditions. It remains unclear, however, why phosphorylation of lipid IVA, albeit with a very low efficiency, can occur in KPM335, but apparently not in its KPM318 parent strain.
Apart from the frr181 mutation in KPM335, a total of 12 mutations were specific to both KPM318 and KPM335, including ∆kdsD, ∆gutQ, ∆lpxL, ∆lpxM, ∆pagP, ∆lpxP, ∆eptA and msbA52 as a prerequisite for synthesis of lipid IVA as the predominant LPS-related outer membrane component. For another four mutations, we believe that they arose spontaneously during the generation of mutant strains, namely a silent mutation located in yqiI, two missense mutations in gor and waaY, and a point mutation in the non-coding region upstream of the deleted eptA gene. The latter mutation is most likely a result of KPM274 construction as a donor strain of the ΔeptA::kan cassette. The mutation is located within the sequence of the homology region of primer ECOeptAH1 (Additional file 2: Table S2) and suggests an error in PCR amplification of the kanamycin resistance cassette targeting the eptA gene and ultimately integration into the genome of KPM318. Compared to the reference genome sequence of the common E. coli MG1655 progenitor, the strains BW30270, KPM318 and KPM335 share sequence variations at six locations. Of these, both a silent nucleotide substitution at position 114 and a single nucleotide insertion at base number 253 of ylbE, and deletion of nucleotide 151 within the glpR gene have been described recently as genetic variations in common E. coli MG1655 stock cultures [21]. Furthermore, while E. coli MG1655 has been shown to express a defective ribonuclease PH due to a frameshift by deletion of the first nucleotide of codon 223 of the pseudogene rph-1 [22], insertion of a single nucleotide as the first base of codon 224 of rph-1 predicts reconstitution of RNase PH function and elimination of the polar effect of the rph-1 mutation on the downstream pyrE gene in BW30270, KPM318 and KPM335.
Next, we decided to replicate the unique set of unmarked genomic deletions in the genetic background of the popular E. coli expression strain BL21 (DE3). As a first step towards the construction of an LPS-free E. coli BL21 (DE3) derivative, we replaced the wild-type msbA gene with the msbA148 suppressor allele of strain KPM22 L1 [12]. This allowed the resulting strain, MWB03, to tolerate null mutations in otherwise essential genes of the LPS biosynthesis pathway. We then sequentially deleted the same genes that had been deleted during the creation of KPM318. Whole-genome sequencing of the final strain, KPM404, confirmed the presence of the msbA148 suppressor mutation (Additional file 3: Table S3), and verified the absence of the kdsD, gutQ, lpxL, lpxM, pagP, lpxP and eptA genes. In comparison to the genome sequence of E. coli BL21 (DE3), we further identified a silent mutation in yceJ, three missense changes within the coding sequences of YadG, ECD_00513 and RstA, and a point mutation in the intergenic region between nudC and hemE. Finally, a total of five single nucleotide substitutions were counted in the region between nucleotides 46 and 222 of the basR gene downstream of ∆eptA. These substitutions perfectly matched the sites of basR sequence variations in E. coli B and K-12, indicating that about one third of the BasR-encoding gene of KPM404 was replaced by the corresponding basR sequence of E. coli K-12. Just as for the construction of KPM318, the E. coli K-12 strain KPM274 served as the donor for transfer of the ΔeptA::kan cassette via P1vir transduction to yield strain KPM403, which well explains the generation of a basR hybrid sequence by co-transduction of the ΔeptA::kan cassette and adjacent basR sequences. In fact, as described above, the use of KPM274 as a ΔeptA::kan donor strain also explains why KPM404 carries a point mutation at the same position upstream of the deleted eptA gene as in KPM318.
Mass spectrometric analyses of the LPS profiles from mutants established in the BL21 (DE3) genetic background underscored the need for radical modification of LPS biosynthesis to accomplish synthesis of only lipid IVA in KPM404 (Figures 3 and 4). While disruption of the gutQ, kdsD and lpxL genes in the intermediate mutant strain KPM396 resulted in synthesis of non-glycosylated lipid IVA precursors lacking the secondary lauroyl and myristoyl chains, mass spectrometry revealed a rather heterogeneous mixture of differently modified lipid IVA species. The spectra displayed four prominent peaks with molecular masses of lipid IVA substituted with one P-EtN group (1527.86 u), lipid IVA modified with two P-EtN moieties (1650.87 u), and palmitoylated lipid IVA molecules carrying either one (1766.09 u) or two (1889.10 u) P-EtN residues. Since lipid A palmitoylation seems to be an indication for an adaptive response to aberrant translocation of phospholipids to the outer leaflet of the outer membrane [23], we suspect that PagP-mediated transfer of palmitate to lipid IVA is triggered by perturbations of outer membrane lipid asymmetry in Kdo-depleted strains of the KPM series. As shown for the LPS sample of KPM400, complete loss of the palmitoylated lipid IVA fraction was achieved by deletion of the pagP gene, leaving lipid IVA molecules modified with either one or two P-EtN groups unaffected.
Figure 3.
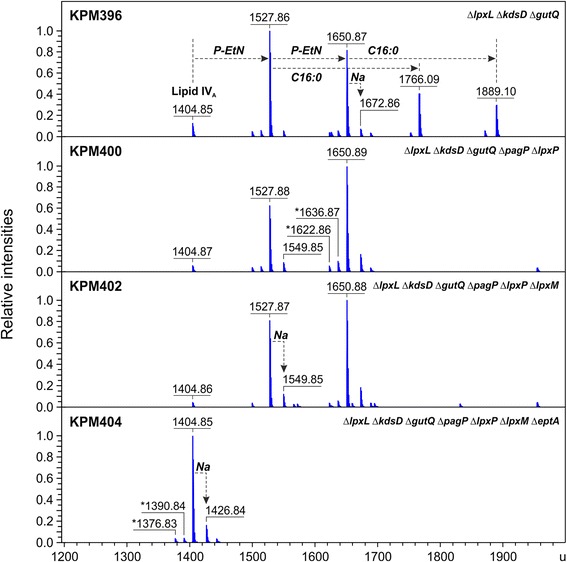
Charge deconvoluted ESI FT-ICR mass spectra in negative ion mode of LPS isolated from BL21 (DE3)-derived mutants. Mass numbers given refer to the monoisotopic masses of neutral molecules. Peaks representing molecules with variations in acyl chain length are labeled with asterisks (∆m = 14.02 u). The mass spectra depict the progress in elimination of lipid IVA heterogeneity by sequential deletion of genes encoding the addition of acyl chains and P-EtN to the lipid A precursor.
Figure 4.
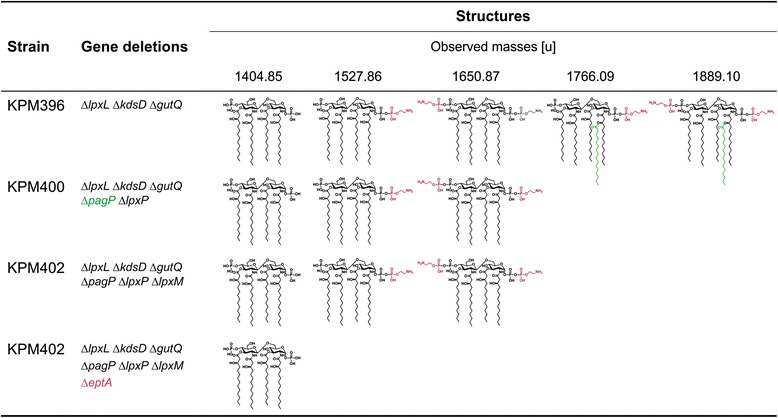
Structures and molecular masses of lipid IVA molecules identified by ESI FT-ICR mass spectrometry in BL21 (DE3)-derived KPM mutants. The ESI FT-ICR mass spectra are shown in Figure 3. Modifications of lipid IVA with palmitate (green) and P-EtN (magenta) are catalyzed by PagP and EptA, respectively.
Due to the block in Kdo biosynthesis and the lack of LpxL, Kdo2-lipid IVA and Kdo2-(lauroyl)-lipid IVA, the preferred substrates for LpxP and LpxM, respectively [24-26], cannot be synthesized in strains derived from KPM396. In addition, previous work has shown that expression of LpxP is induced under conditions of cold shock (12°C) to incorporate an unsaturated C16:1 acyl chain at the expense of a laurate (C12:0), perhaps reflecting the demand to adjust membrane fluidity in the cold [25,27]. It was therefore not surprising that deletion of the lpxP and lpxM genes did not exhibit an obvious effect on lipid IVA composition of KPM400 and KPM402, respectively. There is no data indicating that LpxP and LpxM are capable of using lipid IVA as an acceptor substrate. It seems to be quite possible, however, that both enzymes show low levels of activity under specific conditions. Contrary to the proposed physiological role of LpxP in adaptation of E. coli cells to low growth temperatures, limited induction of LpxP expression has been demonstrated as a potential compensatory mechanism even at 30°C in lpxL and lpxL lpxM mutants of E. coli W3110 [28]. Likewise, LpxM was able to transfer a myristoyl chain directly to Kdo2-lipid IVA in E. coli W3110 strains lacking lpxL and lpxL lpxP [28].
Like PagP and LpxP, EptA-dependent modification of lipid A with P-EtN is part of the complex regulatory network associated with the structural redesign of LPS upon exposure to changing environmental conditions or envelope stress factors. As shown for E. coli K-12, modification of lipid A with P-EtN occurs under certain conditions such as in response to external stimuli like ammonium metavanadate [29] or mild acid pH [30]. Although P-EtN appears to be transferred predominantly to the 1-phosphate group of lipid A [31], double P-EtN substitutions at the 1- und 4′-phosphate positions were evident in lipid A of E. coli K-12 lacking LpxT activity [32] and lipid IVA of a mutant strain defective in MsbA-dependent translocation of LPS across the inner membrane [12]. Based upon ESI FT-ICR analysis presented here, deletion of the eptA gene was clearly necessary as well as sufficient to prevent lipid IVA of KPM404 from being substituted with one or two P-EtN residues. Thus, our data not only corroborate previous findings on the ability of EptA to transfer P-EtN to both the 1- and the 4′-phosphate group of lipid A [32], but also provide experimental evidence for its ability to use lipid IVA as a substrate for single and double P-EtN modifications.
In contrast to the E. coli K-12 strain KPM318, integration of lipid IVA into the outer membrane of KPM404 did not result in a temperature-sensitive phenotype of the BL21 (DE3)-derived mutant (data not shown). Although closely related at the genomic level [33], combined analysis of the genomes, transcriptomes, proteomes and phenomes of E. coli K-12 and BL21 (DE3) revealed significant differences in their cellular physiology and metabolism [34], which may explain the differences in the ability of KPM318 and KPM404 to maintain outer membrane integrity in the presence of lipid IVA at temperatures above 40°C.
Biological activity of engineered E. coli cells and LPS
To test the endotoxic potential of the engineered E. coli K-12 and B strains, we performed TLR4/MD-2 activation assays using HEK-Blue hTLR4 cells. Stimulation of these cells, which express human TLR4, MD-2 and CD14 on their surfaces, induces the production of the NF-κB- and activator protein-1 (AP-1)-dependent reporter secreted embryonic alkaline phosphatase (SEAP). The phosphatase levels can be determined by reading the absorbance at 655 nm using a colorimetric substrate. In order to address the question of whether NF-κB is specifically induced via the hTLR4/MD-2 signaling pathway, HEK-Blue Null2 cells, the parental cell line of HEK-Blue hTLR4 cells lacking the hTLR4/MD-2 receptor complex, were used as a control in all hTLR4/MD-2 activation assays. The strains KPM318, KPM335 and KPM404 as analyzed by challenging HEK-Blue hTLR4 cells with an increasing number of colony forming units (cfu) of up to 106 cfu/ml were virtually free of hTLR4/MD-2-stimulating activity, whereas their parental strains BW30270 and BL21 (DE3) elicited a substantial hTLR4/MD-2 activation already at 103 cfu/ml (Figures 5A, B, 6A and B). When the extracted LPS of the strains was subjected to the TLR4-specific assay, we could confirm the lack of endotoxic activity of the samples isolated from KPM318, KPM335 and KPM404 (Figures 5C, D, 6C and D). The data also demonstrated that palmitoylation of lipid IVA (when PagP was expressed) and/or modification of lipid IVA with one or two P-EtN groups (when EptA was expressed) in KPM396, KPM400 and KPM402 are capable of conferring certain hTLR4/MD-2 stimulatory activity on the otherwise endotoxically inactive tetraacylated lipid A precursor (Figure 6). Our results allow us to draw the major conclusion that inactivation of regulated lipid IVA modifications as demonstrated herein to be present in BL21 (DE3)-based intermediate mutant strains is a crucial prerequisite to yield consistently endotoxin-free E. coli strains.
Figure 5.
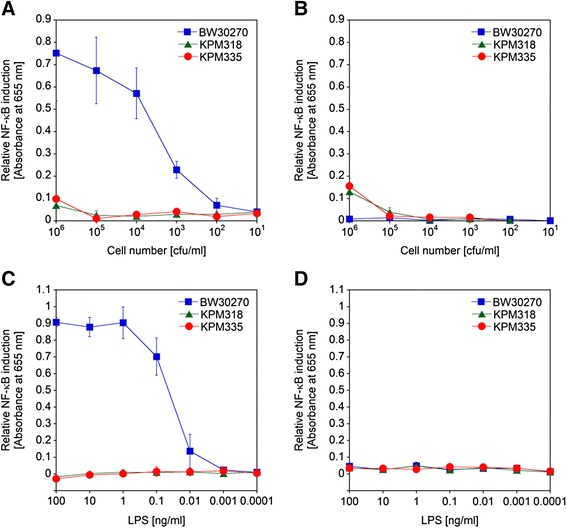
Dose–response curves of NF-κB induction by whole bacterial cells and LPS of E. coli K-12 strains. The samples were assayed with HEK-Blue hTLR4 cells for hTLR4/MD-2-mediated NF-κB induction by colorimetric determination of NF-κB-dependent SEAP activity (A and C). HEK-Blue Null2 cells, the parental cell line of HEK-Blue hTLR4 cells lacking the hTLR4/MD-2 receptor complex, were used as a control (B and D). HEK-Blue hTLR4 and Null2 cells were stimulated with tenfold serial dilutions of whole bacterial cells (A and B) and LPS extracts (C and D) of KPM318 and KPM335 in comparison to their parental strain BW30270, respectively. The values represent the means and standard deviations from three individual experiments. In all experiments, assayed samples showed low level stimulation of HEK-Blue Null2 cells, indicating that NF-κB-dependent SEAP expression was specifically induced via the hTLR4/MD-2 signaling pathway in HEK-Blue hTLR4 cells.
Figure 6.
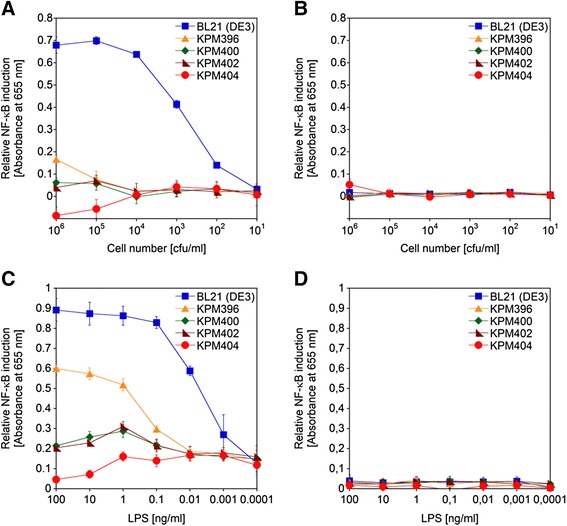
Dose–response curves of NF-κB induction by whole bacterial cells and LPS of E. coli BL21 (DE3) strains. The samples were assayed with HEK-Blue hTLR4 (A and C) and Null2 (B and D) cells for relative NF-κB induction by colorimetric determination of NF-κB-dependent SEAP activity. Relative NF-κB induction was measured following stimulation of HEK-Blue hTLR4 and Null2 cells with tenfold serial dilutions of whole bacterial cells (A and B) and LPS extracts (C and D) of E. coli BL21 (DE3) and BL21 (DE3)-derived KPM mutants, respectively. The values represent the means and standard deviations from three individual experiments. In all experiments, assayed samples displayed negligible activation of parental HEK-Blue Null2 cells, suggesting specific induction of NF-κB-dependent SEAP expression via the hTLR4/MD-2 signaling pathway in HEK-Blue hTLR4 cells.
As one of the critical mediators induced in response to endotoxin, we assayed the release of TNF-α upon stimulation of human macrophages by lipid IVA samples of KPM318, KPM335 and KPM404. The samples exhibited very low biological activity as demonstrated by their low capacity to provoke TNF-α production in human macrophages even at concentrations of 0.1-1 μg/ml (Figure 7A). Compared to the LPS from the parental strains, which induced maximal TNF-α release at 0.01 μg/ml, TNF-α induction was reduced by about 80-95% even at 100-fold higher levels of mutant extracts. The well-documented ability of lipid IVA to act as an antagonist for the hTLR4/MD-2 signaling pathway [35,36] prompted us to examine the inhibition of the agonistic activity of S-form LPS from Salmonella enterica subspecies enterica serovar Abortusequi (S. Abortusequi) by lipid IVA samples from KPM318, KPM335 and KPM404. As shown in Figure 7B, pre-exposure of macrophages to 0.1 μg/ml and 1 μg/ml of the lipid IVA extracts resulted in 72.0 ± 11.2% and 75.9 ± 2.0% (mean percent inhibition ± SD) inhibition of TNF-α production induced by LPS from S. Abortusequi, respectively. Thus, lipid IVA from KPM318, KPM335 and KPM404 displayed potent antagonistic activity against biologically active wild-type LPS.
Figure 7.
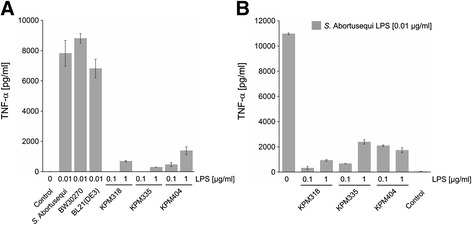
Biological activity of LPS from KPM mutants in human macrophages. Macrophages were differentiated from human blood monocytes of healthy donors. On day seven of differentiation, macrophages were seeded at 1 × 105 cells/well and stimulated with LPS at the indicated amounts (LPS from S. Abortusequi, BW30270 and BL21 (DE3) at 0.01 μg/ml; LPS from strains KPM318, KPM335 and KPM404 at 0.1 μg/ml and 1 μg/ml, respectively) for 4 h at 37°C (A). To determine the antagonistic activity of LPS from KPM strains, macrophages were incubated with LPS samples from KPM318, KPM335 or KPM404 at 0.1 μg/ml or 1 μg/ml for 30 min at 37°C, followed by stimulation of the cells with 0.01 μg/ml of LPS from S. Abortusequi for 4 h (B). Cell-free supernatants were analyzed for TNF-α content by ELISA. The values represent the means and standard deviations from three independent experiments using cells from different donors.
Endotoxin-free expression of ApoA-1 and Hsp70
Previous studies have shown that a BL21 (DE3) ∆lpxM::cat mutant synthesizing a non-myristoylated LPS can be used to express heterologous proteins with reduced stimulatory activity in human LPS-responsive cells [37]. To test the ability of KPM318, KPM335 and KPM404 to serve as hosts for the production of endotoxin-free recombinant proteins, we selected the heterologously expressed human proteins ApoA-1 and Hsp70 as model systems. ApoA-1, a major component of high-density lipoprotein and important mediator in maintaining cholesterol homeostasis [38], is particularly challenging because the 28-kDa protein is known to be directly involved in neutralization of LPS toxicity and, therefore, difficult to separate from endotoxic activity [39,40]. Yet another challenging protein able to associate with LPS is Hsp70 [41]. Moreover, the 70-kDa molecular chaperone has been suggested to function as an endogenous damage-associated molecular pattern protein for activation of the TLR4 signaling pathway upon tissue injury [42]. As Hsp70-induced stimulation of innate immune system cells in many respects resembles the effects of LPS, removal of endotoxin contamination, as frequently present in recombinant Hsp preparations [43], remains a key issue to discriminate between LPS- and Hsp-induced effects.
For production of ApoA-1, we used the T5 promoter-based plasmid pApo404 in E. coli strains BW30270, KPM318 and KPM335, whereas Hsp70 was expressed from pHsp70His under the control of a T7 promoter in BL21 (DE3) and KPM404. SDS-PAGE analysis of the protein samples following minimal purification of each soluble protein extract, by immobilized metal affinity chromatography (IMAC), revealed that the endotoxin-free strains KPM318/pApo404, KPM335/pApo404 and KPM404/pHsp70His produced recombinant ApoA-1 and Hsp70 in approximately equal quantities and had similar impurity profiles as their parental strains, respectively (Figure 8). Since ApoA-1 has been described to also associate with proteins of the host cell [44], it was not surprising to detect relatively high levels of protein contaminants in the IMAC-purified ApoA-1 samples. Regardless, we did not perform any further protein purification or endotoxin removal steps to investigate the biological activity of the ApoA-1 and Hsp70 samples.
Figure 8.
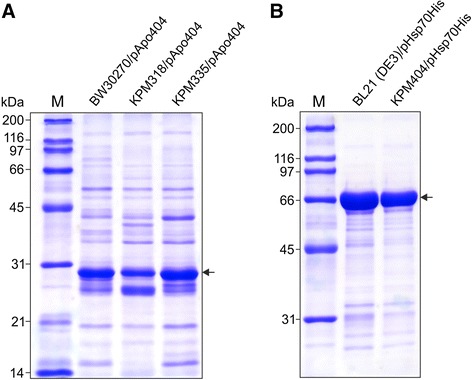
SDS-PAGE gels of ApoA-1 and Hsp70. The proteins were expressed in endotoxin-free derivatives of E. coli strains BW30270 (A) and BL21 (DE3) (B), respectively, and minimally purified using IMAC on HisTrap HP (1 ml) columns. The recombinant ApoA-1 and Hsp70 samples (6 μg each) were resolved under denaturing conditions using 12% and 10% polyacrylamide gels, respectively. Molecular mass protein markers (kDa) were run in lanes M. Arrows indicate the positions of ApoA-1 and Hsp70.
The first test of biological activity employed was the Limulus amebocyte lysate (LAL) assay, an FDA-approved method that is based on activation of a coagulation cascade in the LAL by trace amounts of endotoxin [45]. In this assay, the endotoxin unit equivalents determined in ApoA-1 samples from KPM318/pApo404 and KPM335/pApo404 were significantly reduced by 92.7 ± 1.3% and 82.2 ± 3.9% (mean percent inhibition ± SD) compared to those found in the ApoA-1 sample from BW30270/pApo404, respectively (Figure 9). Furthermore, when KPM404/pHsp70His was used as a host for production of Hsp70, the LAL response to the protein decreased by 97.2 ± 0.5% in comparison to the response elicited by Hsp70 obtained from BL21 (DE3)/pHsp70His. The LAL assay, though widely used for the detection and quantitation of endotoxins, is an inappropriate method to discriminate between endotoxically active hexaacylated LPS and endotoxically inactive tetraacylated lipid IVA due to the presence of the LAL-activating 4′-monophosphoryl-diglucosamine backbone in both lipid structures [46,47]. As such, the LAL clotting cascade is activated by a wider spectrum of LPS/lipid A variants than LPS-responsive cells of the human immune system. The residual LAL reactivity of the proteins from the lipid IVA host strains most likely reflects the non-specific nature of the assay, giving rise to false positive endotoxic results.
Figure 9.
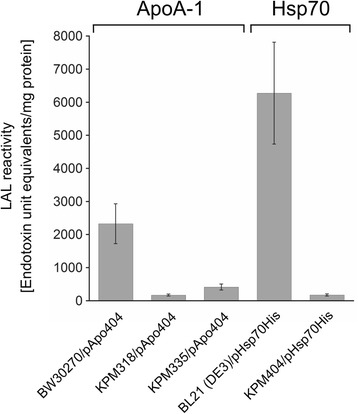
Reactivity of ApoA-1 and Hsp70 in the Limulus amebocyte lysate (LAL) assay. ApoA-1 was produced in E. coli strain BW30270/pApo404 and its endotoxin-free derivatives KPM318/pApo404 and KPM335/pApo404, whereas Hsp70 was obtained from KPM404/pHsp70His and its parental strain BL21 (DE3)/pHsp70His. The proteins were minimally purified by IMAC and assayed with the LAL test. The measurements represent the means and standard deviations from three individual experiments.
In order to specifically address the endotoxic activity of the ApoA-1 and Hsp70 samples, we utilized the HEK-Blue hTLR4/MD-2 cell activation assay. The ApoA-1 and Hsp70 samples derived from the endotoxin-free strains did not trigger an endotoxic response in HEK-Blue hTLR4 cells, even when present in the assay at 10 μg/ml, whereas the proteins produced in the parental strains showed substantial NF-κB activation already at concentrations in the range between 0.1 μg/ml and 1 μg/ml (Figure 10). These results were in excellent agreement with the inability of KPM318, KPM335 and KPM404 cells and LPS to stimulate the hTLR4/MD-2 signaling pathway (Figures 5 and 6).
Figure 10.
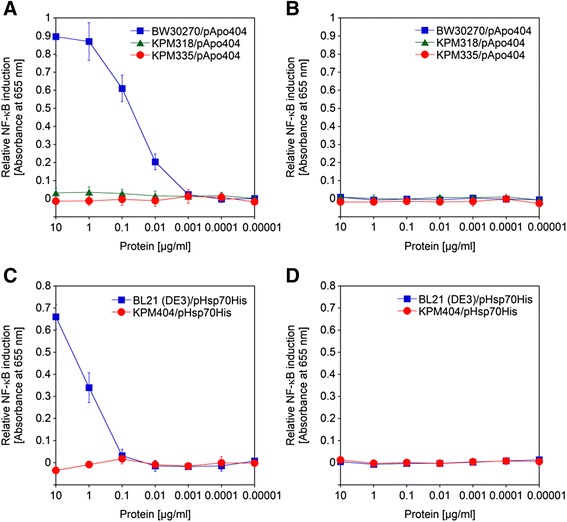
Stimulation of hTLR4/MD-2 by ApoA-1 and Hsp70 produced in endotoxin-free E. coli strains. The proteins were minimally purified by IMAC and assayed with HEK-Blue hTLR4 cells for their ability to activate NF-κB-dependent SEAP expression (A and C). HEK-Blue Null2 cells served as a control (B and D). Relative NF-κB induction was measured following stimulation of HEK-Blue hTLR4 and Null2 cells with tenfold serial dilutions of ApoA-1 (A and B) and Hsp70 (C and D) samples obtained by heterologous expression in BW30270/pApo404, KPM318/pApo404 and KPM335/pApo404, and BL21 (DE3)/pHsp70His and KPM404/pHsp70His, respectively. The values represent the means and standard deviations from three individual experiments. In all experiments, the ApoA-1 and Hsp70 samples did not activate NF-κB-dependent SEAP expression in HEK-Blue Null2 cells, suggesting that NF-κB-dependent SEAP expression was due to specific activation of the hTLR4/MD-2 signaling pathway in HEK-Blue hTLR4 cells.
As exemplified herein by heterologous expression of ApoA-1 and Hsp70, proteins prepared from engineered E. coli strains are innately free of endotoxic activity in human LPS-responsive cells, consistent with earlier observations that KPM335 is also a suitable host for production of functional endotoxin-free inclusion bodies of the aggregation-prone fluorescent fusion protein VP1GFP [48]. However, it must be taken into account that lipid IVA may act agonistically in other mammalian hosts such as mouse [49], Chinese hamster [50] or equine [36] cells, which reflects species-specific lipid IVA sensing by the TLR4/MD-2 complex [51,52]. The application of proteins in cells other than human cells may therefore also require lipid IVA depletion. Due to the lack of specificity, the LAL assay cannot be used to assess endotoxic activity but is ideally suited to detect residual lipid IVA in recombinant proteins prepared from LPS-free E. coli strains. We are aware that we currently cannot respond to the question of whether further purification of ApoA1 and Hsp70 would result in complete loss of lipid IVA. However, our data on significantly decreased LAL reactivities of minimally purified ApoA1 and Hsp70 proteins from endotoxin-free bacteria give reason to assume that lipid IVA, being homogeneous, is much easier to remove from downstream products than mature LPS. Despite a common backbone structure, LPS is synthesized as a heterogeneous mixture of structurally related molecules that are decorated with various substituents, usually present in non-stoichiometric amounts [53]. These substitutions, which may vary significantly depending on growth conditions, contribute to considerable physico-chemical heterogeneity of LPS molecules, posing a major challenge to the development of a generally applicable method for endotoxin removal from proteins produced in common E. coli expression strains [5].
Conclusions
Recombinant proteins manufactured in E. coli are inherently contaminated with endotoxin. In light of the enormous diversity of recombinant proteins expressed in E. coli, none of the purification methods are universally applicable to significantly reduce the endotoxin content. Instead of removing endotoxin from the protein samples, the work presented here demonstrates that it is now possible to eliminate endotoxin at the source by producing proteins in an endotoxin-free environment using LPS-free derivatives of E. coli K-12 and BL21 (DE3) strains. These non-conditional mutants clearly lack hTLR4/MD-2 signaling pathway agonists yet can retain viability through predominantly synthesizing the tetraacylated, endotoxically inactive lipid A precursor lipid IVA. The design of the strains also prohibits them from easily regaining the potential to synthesize normal LPS or endotoxically active lipid IVA derivatives through acquiring mutations. This has been accomplished by incorporating a total of seven non-reverting genetic deletions that disrupt Kdo biosynthesis and prevent lipid IVA from being modified with enzymes of both the constitutive and the regulated LPS pathway, while the compensating mutations msbA52 and msbA148 enable the E. coli K-12- and BL21 (DE3)-derived cells to maintain viability, respectively. The derivation of E. coli strains with dramatically modified LPS affords the unique opportunity to produce endotoxin-free recombinant proteins suitable for downstream experiments with human cells. These strains allow researchers to save time-consuming cleanup steps that may affect yield and functionality of the end product.
Methods
Bacterial strains, plasmids and growth conditions
All strains and plasmids used in the present study are described in Additional file 4: Table S4. The bacteria were grown aerobically with shaking (220 rpm) at 37°C in standard LB-Miller medium containing 10 g/l of NaCl to maintain a non-mucoid phenotype in ∆Kdo strains [54]. To induce LPS biosynthesis and restore susceptibility of Kdo-depleted KPM strains to phage P1vir, the media was supplemented with 15 μM d-arabinose 5-phosphate and 10 μM d-glucose 6-phosphate [11,12]. Ampicillin (100 μg/ml), carbenicillin (200 μg/ml), kanamycin (30 μg/ml) or chloramphenicol (30 μg/ml) was added to the media as required.
DNA manipulations
Chromosomal deletions were constructed with the phage λ Red recombinase system [55] using the plasmids pKD4 and pKD3 as templates for amplification of the kanamycin and chloramphenicol resistance cassettes, respectively (for targeting cassette primers see Additional file 2: Table S2). To prevent insertion of the targeting cassettes into scar sequences of previously deleted genes, the insert cassettes targeting the lpxM, pagP, lpxP and eptA genes were generated first in separate donor strains and then transferred successively to the KPM strains by P1vir transduction according to standard protocols [56]. The P1vir donor strains were constructed by the phage λ Red recombinase procedure, except strain MWB06 was generated using the “Gene doctoring” method [57]. The kanamycin resistance cassette targeting the lpxM gene of BL21 (DE3) was amplified from pDOC-K with targeting cassette primers DOCBL21lpxMH1 and DOCBL21lpxMH2, digested with EcoRI/SpeI and ligated into the EcoRI/SpeI sites of pDOC-C to yield pDOClpxM::kan. The lpxM gene was then replaced with lpxM::kan by co-transformation of BL21 (DE3) with pDOClpxM::kan and the recombineering plasmid pACBSCE as described [57]. Unmarked deletion mutants were obtained by excision of the antibiotic resistance markers in the presence of the FLP recombinase encoded either by pCP20 [55] or pFLP2 [58], followed by curing of the helper plasmids at 37°C or sucrose selection, respectively. All strains were tested for loss of the relevant genes by PCR (for control primers see Additional file 2: Table S2). The nonconditional suppressor strain KPM22 L11 maintaining the normally lethal ∆gutQ and ∆kdsD mutations [12] was used as the parent for construction of KPM strains with the E. coli K-12 genetic background.
Strain KPM318, showing temperature-sensitive growth defects at temperatures above 40°C, was adapted to elevated temperatures by continued propagation of the bacterial cells at 42°C, which yielded strain KPM335 capable of sustained growth at 42°C.
To convert E. coli BL21 (DE3) into a nonconditional suppressor strain able to tolerate null mutations in essential Kdo pathway genes, the wild-type msbA gene of the strain was replaced by the msbA148 allele, which was previously identified as a suppressor of the lethal phenotype associated with Kdo depletion in E. coli KPM22 L1 [12]. Using the primers 5ECycaI and 3ECycaQ (Additional file 2: Table S2), msbA148 was amplified from the E. coli KPM22 L1 suppressor strain. A kanamycin resistance cassette targeting the essential msbA wild-type gene was generated as reported [12] and inserted into the chromosome of BL21 (DE3) containing the temperature-sensitive helper plasmids pKD46 [55] and pMAK705-ECmsbA. The latter plasmid was constructed by amplification of the msbA gene of E. coli BW30270 with primer pair 5HindIIIECmsbA/3BamHIECmsbA (Additional file 2: Table S2), digestion of the PCR product with HindIII/BamHI and cloning into the HindIII/BamHI sites of pMAK705 [59]. After transformation of E. coli BL21 (DE3) ∆msbA::kan/pKD46/pMAK705-ECmsbA with msbA148, screening for replacement of the ∆msbA::kan cassette by the suppressor allele was performed at 37°C to select for transformants that retained viability upon loss of the functional copy of the msbA wild-type gene encoded on the temperature-sensitive plasmid pMAK705-ECmsbA. The successful insertion of msbA148 was then verified by PCR amplification and sequence analysis. As a result, the strain E. coli MWB03 was obtained, which subsequently served as the host for deletion of Kdo/lipid A genes.
The plasmid pApo404 carrying the synthetic gene with optimized E. coli codon usage for expression of human ApoA-1 as a C-terminally histidine-tagged fusion protein was manufactured by DNA2.0, Menlo Park, CA. The codon-optimized gene for human Hsp70 was synthesized by GenScript, Piscataway, NJ, digested with NdeI and HindIII, and ligated into the NdeI/HindIII sites of pET-22b to yield plasmid pHsp70His.
Whole-genome sequencing and data analysis
The genomes of strains BW30270, KPM318 und KPM335 were sequenced at the Scripps Research Institute on the Illumina HiSeq instrument after library preparation with a customized protocol. Genomic DNA samples were sheared to a size range between 200 and 300 bp using an S2 Covaris ultrasonicator. The fragments were then end repaired, A-tailed with Taq polymerase, phosphorylated and ligated to standard Illumina TruSeq barcoded adapters following Illumina recommended protocols. The libraries were then PCR amplified for 12 cycles, followed by gel purification of the amplified libraries to select insert sizes between 200 and 250 bp.
For strains BL21 (DE3) and KPM404, the genomic DNAs were sequenced at the Research Center Borstel using the Illumina MiSeq sequencer, Nextera XT library preparation kits, and 500 cycle v2 sequencing kits as instructed by the manufacturer. Resulting reads were mapped to the genome of either E.coli K-12 MG1655 [GenBank:NC_000913.2] for strains BW30270, KPM318, and KPM335, or E. coli BL21 (DE3) [GenBank:CP001509.3] for strains BL21 (DE3) and KPM404, using the exact alignment program SARUMAN [60]. Single nucleotide polymorphisms and Indels were extracted from mapped reads by customized Perl scripts using a minimum coverage of ten reads and a minimum allele frequency of 75% as thresholds for detection.
The whole-genome sequence data of this study have been submitted to the NCBI Sequence Read Archive [SRA:PRJNA212553].
LPS and lipid IVA isolation and characterization
The dried biomasses of stationary phase 2-liter cultures were used to isolate either LPS by the original phenol/chloroform/light petroleum (PCP) procedure [61] or lipid IVA by a modified PCP protocol [12]. For ESI FT-ICR, the samples were prepared as described previously [62]. The mass spectra were recorded in the negative ion mode using a 7-Tesla hybrid Apex Qe Instrument (Bruker Daltonics).
Overexpression and purification of human ApoA-1 and Hsp70
Each strain was grown to an OD600 of 0.6 - 0.7 before the heterologous protein was expressed in the presence of 0.4 mM isopropyl-β-d-thiogalactoside for 3 h. All subsequent steps were performed at 4°C. The cells were harvested by centrifugation at 9,000 × g for 20 min, washed with phosphate-buffered saline, re-suspended in 45 ml of 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl, and incubated with lysozyme (0.2 mg/ml) for 30 min and continuous stirring. Prior to disintegration of the cells by three passages through a French pressure cell at 20,000 psi, the suspension was supplemented with DNase I (50 μg/ml), RNase A (50 μg/ml) and Complete Protease Inhibitor Cocktail (EDTA-free) according to the recommendations of the manufacturer (Roche), followed by centrifugation of the lysate at 10,000 × g for 30 min to remove cellular debris. Imidazole was added to a final concentration of 10 mM before the cleared lysate was loaded onto a HisTrap HP (1 ml) column (GE Healthcare) pre-equilibrated with 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl, 10 mM imidazole. The column was extensively washed with a stepwise gradient of 20 mM, 80 mM and 100 mM imidazole in 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl, and the heterologously expressed protein was eluted using 500 mM imidazole in the same buffer. Fractions containing the protein were pooled and dialyzed against 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl.
Protein samples were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) [63] and visualized by staining with Coomassie Brilliant Blue R-250.
Toll-like receptor activation assays
The stimulation assays using whole bacterial cells, extracted LPS samples and heterologously expressed human proteins were performed with HEK-Blue™ hTLR4 and HEK-Blue™ Null2 cells in accordance with the specifications of the supplier of the cell lines (InvivoGen). The HEK-Blue hTLR4 cells were grown at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/l glucose (PAA Laboratories), 2 mM l-glutamine (PAA Laboratories), 10% fetal bovine serum, PAA Clone (PAA Laboratories), 100 μg/ml Normocin™ (InvivoGen), 1 × HEK-Blue™ Selection solution (InvivoGen), and 1 × penicillin-streptomycin (Pen-Strep) solution (PAA Laboratories). The parental cell line of HEK-Blue hTLR4, HEK-Blue Null2, was grown under the same conditions in DMEM supplemented with 4.5 g/l glucose, 2 mM l-glutamine, 10% fetal bovine serum, PAA Clone, 100 μg/ml Normocin, 100 μg/ml Zeocin™ (InvivoGen), and 1 × Pen-Strep solution. When a 60-80% confluency was reached, the cells were detached in the presence of Dulbecco’s phosphate-buffered saline (DPBS) (PAA Laboratories), washed with DPBS and resuspended at a cell density of 1.4 × 105 cells/ml in HEK-Blue test medium consisting of DMEM with 4.5 g/l glucose, 2 mM l-glutamine, 10% fetal bovine serum, PAA Clone, 100 μg/ml Normocin, and 1 × Pen-Strep solution. For stimulation of HEK-Blue cells, each sample (20 μl) was mixed with 180 μl of the cell suspension (25,000 cells) in one well of a 96-well plate (COS96ft - Corning 96 Flat Bottom Transparent Polystyrol) and incubated at 37°C for 20 h in a humidified atmosphere with 5% CO2. The supernatant of each HEK-Blue cell suspension (20 μl) was then added to 180 μl of reconstituted QUANTI-Blue™ substrate (InvivoGen) per well, followed by incubation of the samples at 37°C for 3 h. NF-κB-dependent SEAP activity was determined by reading the absorbance at 655 nm using a Tecan Infinite M200 NanoQuant Microplate Reader. The positive and negative control reactions were prepared with 25,000 HEK-Blue cells per well and assayed under the same conditions as described above. For the positive control experiments, tenfold serial dilutions of LPS from E. coli K-12 (InvivoGen) and recombinant human TNF-α (200 ng/well) (InvivoGen) were used. To determine the basal levels of SEAP activity, pyrogen-free water, 10 × concentrated Pen-Strep in DPBS, and 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl, served as negative controls to assay LPS samples, whole bacterial cells and heterologously expressed proteins, respectively. The basal level of SEAP activity in untreated HEK-Blue cells was subtracted from the relative SEAP activity measured for each treated sample.
Whole bacterial cells were prepared for the assays from aliquots of overnight cultures. The cells were sedimented by centrifugation, washed with DPBS, re-suspended in a 10 × concentrated Pen-Strep solution in DPBS, and incubated first at 22°C for 3 h and then at 4°C overnight. On the basis of CFU counts obtained from platings of the initial overnight cultures, the Pen-Strep-treated cells were serially diluted appropriately in 10 × concentrated Pen-Strep in DPBS. Serial dilutions of LPS and protein samples were performed in pyrogen-free water and 25 mM Tris–HCl, pH 7.5, 0.1 M NaCl, respectively.
Activation of human macrophages
Biological activity of LPS was tested on human macrophages derived from blood of healthy donors. The Ethics Committee of the University of Lübeck, Germany, approved the procedures. Monocytes were isolated, differentiated to macrophages and stimulated with LPS samples as described [64]. Briefly, MNCs were cultivated in teflon bags in RPMI medium supplemented with 200 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 4% heat-inactivated human serum type AB and 2 ng/ml macrophage colony-stimulating factor (R&D Systems, Wiesbaden, Germany) at 37°C for 7 days in a humidified atmosphere with 5% CO2. Macrophages were harvested on day 7, washed twice in serum-free RPMI, and seeded in serum-free RPMI containing 200 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin for experiments. After 4 h of stimulation with LPS, TNF-α was determined in cell-free supernatants using the human TNF-α ELISA set according to the recommendations of the manufacturer (BD Biosciences). Lyophilized LPS samples were re-suspended in pyrogen-free water at 1 mg/ml stock by sonication for 30 min and temperature-cycled twice between 4°C and 56°C. The samples were stored at least for 12 h at 4°C before biological experiments.
Limulus amebocyte lysate assay
The Limulus amebocyte lysate (LAL) assay was performed using the Endosafe® Portable Test System (PTS) with single-use Endosafe®-PTS cartridges as specified by the supplier (Charles River Laboratories).
Acknowledgements
We thank Michael Weinkauf, Brigitte Kunz, Sabrina Groth, Irina von Cube, Michaela Ramhold, Julia Zallet and Tanja Ubben for technical assistance. Plasmid pFLP2 was kindly provided by Herbert P. Schweizer (Department of Microbiology, Colorado State University, Fort Collins, Colorado, USA), and plasmids pDOC-K, pDOC-C and pACBSCE by David J. Lee (School of Biosciences, University of Birmingham, Birmingham, UK). The work was financially supported by Research Corporation Technologies, Inc., in-house funding of the Research Center Borstel, the National Institutes of Health Grant U19 A1063603 to SRH, and the European Union PathoNgenTrace (FP7- 278864-2) project to TAK. The authors thank Tod Holler at the College of Pharmacy of the University of Michigan, Ann Arbor, for his input and helpful discussions on the manuscript.
Abbreviations
- ApoA-1
Apolipoprotein A-1
- ESI FT-ICR
Electrospray-ionization Fourier-transformed ion cyclotron mass spectrometry
- Hsp70
Heat shock protein 70
- Kdo
3-deoxy-d-manno-oct-2-ulosonic acid
- LAL
Limulus amebocyte lysate
- MD-2
Myeloid differentiation factor 2
- P-EtN
Phosphoethanolamine
- SEAP
Secreted embryonic alkaline phosphatase
- TLR4
Toll-like receptor 4
- u
Unified atomic mass unit
Additional files
Summary of genome sequence data of E. coli strains BW30270, KPM318 and KPM335.
Primers used in this study.
Summary of genome sequence data of E. coli strains BL21 (DE3) and KPM404.
Bacterial strains and plasmids used in this study.
Footnotes
Competing interests
CS is employed by Research Corporation Technologies, Inc. (RCT). ClearColi® is the registered trademark of endotoxin-free E. coli strains owned by RCT. The ClearColi® technology is covered by U.S. Patent 8,303,964 and other U.S. and foreign pending patents. E. coli KPM404 is offered commercially as ClearColi® BL21 (DE3) by RCT and Lucigen Corp., Middleton, WI 53562. All other authors declare no competing interests.
Authors’ contributions
UM, RWW, DB, TCM and CS conceived the project and designed experiments. KW, UM, ABS, BL, TAK, JLC and LS performed experiments. UM, DB, ABS, BL, TAK, AV, SRH and RWW analyzed data. UM and RWW wrote the paper, with input from all other authors. All authors read and approved the manuscript.
Contributor Information
Uwe Mamat, Email: umamat@fz-borstel.de.
Kathleen Wilke, Email: kathleen_wilke@gmx.de.
David Bramhill, Email: dbramhill@gmail.com.
Andra Beate Schromm, Email: aschromm@fz-borstel.de.
Buko Lindner, Email: blindner@fz-borstel.de.
Thomas Andreas Kohl, Email: tkohl@fz-borstel.de.
José Luis Corchero, Email: jlcorchero1967@gmail.com.
Antonio Villaverde, Email: antoni.villaverde@uab.es.
Lana Schaffer, Email: schaffer@scripps.edu.
Steven Robert Head, Email: shead@scripps.edu.
Chad Souvignier, Email: csouvignier@rctech.com.
Timothy Charles Meredith, Email: tcmered@gmail.com.
Ronald Wesley Woodard, Email: rww@med.umich.edu.
References
- 1.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–24. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 2.Huang CJ, Lin H, Yang X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J Ind Microbiol Biotechnol. 2012;39:383–99. doi: 10.1007/s10295-011-1082-9. [DOI] [PubMed] [Google Scholar]
- 3.Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discov Today. 2010;15:773–80. doi: 10.1016/j.drudis.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Wakelin SJ, Sabroe I, Gregory CD, Poxton IR, Forsythe JL, Garden OJ, et al. “Dirty little secrets” - endotoxin contamination of recombinant proteins. Immunol Lett. 2006;106:1–7. doi: 10.1016/j.imlet.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Petsch D, Anspach FB. Endotoxin removal from protein solutions. J Biotechnol. 2000;76:97–119. doi: 10.1016/S0168-1656(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz H, Schmittner M, Duschl A, Horejs-Hoeck J. Residual endotoxin contaminations in recombinant proteins are sufficient to activate human CD1c+ dendritic cells. PLoS One. 2014;9:e113840. doi: 10.1371/journal.pone.0113840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol. 2010;8:8–14. doi: 10.1038/nrmicro2266. [DOI] [PubMed] [Google Scholar]
- 8.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Wang Z, Chen J, Ernst RK, Wang X. Influence of lipid a acylation pattern on membrane permeability and innate immune stimulation. Mar Drugs. 2013;11:3197–208. doi: 10.3390/md11093197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc Natl Acad Sci U S A. 2013;110:1464–9. doi: 10.1073/pnas.1218080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meredith TC, Aggarwal P, Mamat U, Lindner B, Woodard RW. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem Biol. 2006;1:33–42. doi: 10.1021/cb0500015. [DOI] [PubMed] [Google Scholar]
- 12.Mamat U, Meredith TC, Aggarwal P, Kühl A, Kirchhoff P, Lindner B, et al. Single amino acid substitutions in either YhjD or MsbA confer viability to 3-deoxy-d-manno-oct-2-ulosonic acid-depleted Escherichia coli. Mol Microbiol. 2008;67:633–48. doi: 10.1111/j.1365-2958.2007.06074.x. [DOI] [PubMed] [Google Scholar]
- 13.Meredith TC, Woodard RW. Escherichia coli YrbH is a d-arabinose 5-phosphate isomerase. J Biol Chem. 2003;278:32771–7. doi: 10.1074/jbc.M303661200. [DOI] [PubMed] [Google Scholar]
- 14.Meredith TC, Woodard RW. Identification of GutQ from Escherichia coli as a d-arabinose 5-phosphate isomerase. J Bacteriol. 2005;187:6936–42. doi: 10.1128/JB.187.20.6936-6942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid a modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirokawa G, Nijman RM, Raj VS, Kaji H, Igarashi K, Kaji A. The role of ribosome recycling factor in dissociation of 70S ribosomes into subunits. RNA. 2005;11:1317–28. doi: 10.1261/rna.2520405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janosi L, Ricker R, Kaji A. Dual functions of ribosome recycling factor in protein biosynthesis: disassembling the termination complex and preventing translational errors. Biochimie. 1996;78:959–69. doi: 10.1016/S0300-9084(97)86718-1. [DOI] [PubMed] [Google Scholar]
- 18.Inokuchi Y, Hirashima A, Sekine Y, Janosi L, Kaji A. Role of ribosome recycling factor (RRF) in translational coupling. EMBO J. 2000;19:3788–98. doi: 10.1093/emboj/19.14.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamoto A, Terui Y, Yamamoto T, Kasahara T, Nakamura M, Tomitori H, et al. Enhanced biofilm formation and/or cell viability by polyamines through stimulation of response regulators UvrY and CpxR in the two-component signal transducing systems, and ribosome recycling factor. Int J Biochem Cell Biol. 2012;44:1877–86. doi: 10.1016/j.biocel.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Touzé T, Tran AX, Hankins JV, Mengin-Lecreulx D, Trent MS. Periplasmic phosphorylation of lipid A is linked to the synthesis of undecaprenyl phosphate. Mol Microbiol. 2008;67:264–77. doi: 10.1111/j.1365-2958.2007.06044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freddolino PL, Amini S, Tavazoie S. Newly identified genetic variations in common Escherichia coli MG1655 stock cultures. J Bacteriol. 2012;194:303–6. doi: 10.1128/JB.06087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen KF. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol. 1993;175:3401–7. doi: 10.1128/jb.175.11.3401-3407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishop RE. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol Microbiol. 2005;57:900–12. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- 24.Brozek KA, Raetz CR. Biosynthesis of lipid a in Escherichia coli. Acyl carrier protein-dependent incorporation of laurate and myristate. J Biol Chem. 1990;265:15410–7. [PubMed] [Google Scholar]
- 25.Carty SM, Sreekumar KR, Raetz CR. Effect of cold shock on lipid a biosynthesis in Escherichia coli. Induction at 12 degrees C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J Biol Chem. 1999;274:9677–85. doi: 10.1074/jbc.274.14.9677. [DOI] [PubMed] [Google Scholar]
- 26.Clementz T, Zhou Z, Raetz CR. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid a. Acylation by MsbB follows laurate incorporation by HtrB. J Biol Chem. 1997;272:10353–60. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 27.Vorachek-Warren MK, Carty SM, Lin S, Cotter RJ, Raetz CR. An Escherichia coli mutant lacking the cold shock-induced palmitoleoyltransferase of lipid a biosynthesis: absence of unsaturated acyl chains and antibiotic hypersensitivity at 12 degrees C. J Biol Chem. 2002;277:14186–93. doi: 10.1074/jbc.M200408200. [DOI] [PubMed] [Google Scholar]
- 28.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CR. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid a. J Biol Chem. 2002;277:14194–205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Z, Lin S, Cotter RJ, Raetz CR. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-l-arabinose, phosphoethanolamine and palmitate. J Biol Chem. 1999;274:18503–14. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 30.Gibbons HS, Kalb SR, Cotter RJ, Raetz CR. Role of Mg2+ and pH in the modification of Salmonella lipid a after endocytosis by macrophage tumour cells. Mol Microbiol. 2005;55:425–40. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 31.Doerrler WT, Gibbons HS, Raetz CR. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J Biol Chem. 2004;279:45102–9. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 32.Herrera CM, Hankins JV, Trent MS. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid a promoting resistance to antimicrobial peptides. Mol Microbiol. 2010;76:1444–60. doi: 10.1111/j.1365-2958.2010.07150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21 (DE3) and comparison of the E. coli B and K-12 genomes. J Mol Biol. 2009;394:653–80. doi: 10.1016/j.jmb.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 34.Yoon SH, Han MJ, Jeong H, Lee CH, Xia XX, Lee DH, et al. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012;13:R37. doi: 10.1186/gb-2012-13-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, et al. Biological activities of lipopolysaccharides are determined by the shape of their lipid a portion. Eur J Biochem. 2000;267:2008–13. doi: 10.1046/j.1432-1327.2000.01204.x. [DOI] [PubMed] [Google Scholar]
- 36.Walsh C, Gangloff M, Monie T, Smyth T, Wei B, McKinley TJ, et al. Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J Immunol. 2008;181:1245–54. doi: 10.4049/jimmunol.181.2.1245. [DOI] [PubMed] [Google Scholar]
- 37.Cognet I, de Coignac AB, Magistrelli G, Jeannin P, Aubry JP, Maisnier-Patin K, et al. Expression of recombinant proteins in a lipid a mutant of Escherichia coli BL21 with a strongly reduced capacity to induce dendritic cell activation and maturation. J Immunol Methods. 2003;272:199–210. doi: 10.1016/S0022-1759(02)00506-9. [DOI] [PubMed] [Google Scholar]
- 38.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263:256–73. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 39.Beck WH, Adams CP, Biglang-Awa IM, Patel AB, Vincent H, Haas-Stapleton EJ, et al. Apolipoprotein A-I binding to anionic vesicles and lipopolysaccharides: role for lysine residues in antimicrobial properties. Biochim Biophys Acta. 1828;2013:1503–10. doi: 10.1016/j.bbamem.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emancipator K, Csako G, Elin RJ. In vitro inactivation of bacterial endotoxin by human lipoproteins and apolipoproteins. Infect Immun. 1992;60:596–601. doi: 10.1128/iai.60.2.596-601.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001;2:338–45. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- 42.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87:989–99. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 43.Tsan MF, Gao B. Pathogen-associated molecular pattern contamination as putative endogenous ligands of Toll-like receptors. J Endotoxin Res. 2007;13:6–14. doi: 10.1177/0968051907078604. [DOI] [PubMed] [Google Scholar]
- 44.Caparon MH, Rust KJ, Hunter AK, McLaughlin JK, Thomas KE, Herberg JT, et al. Integrated solution to purification challenges in the manufacture of a soluble recombinant protein in E. coli. Biotechnol Bioeng. 2010;105:239–49. doi: 10.1002/bit.22542. [DOI] [PubMed] [Google Scholar]
- 45.Ding JL, Ho B. Endotoxin detection - from Limulus amebocyte lysate to recombinant factor C. Subcell Biochem. 2010;53:187–208. doi: 10.1007/978-90-481-9078-2_9. [DOI] [PubMed] [Google Scholar]
- 46.Brandenburg K, Howe J, Gutsman T, Garidel P. The expression of endotoxic activity in the Limulus test as compared to cytokine production in immune cells. Curr Med Chem. 2009;16:2653–60. doi: 10.2174/092986709788682001. [DOI] [PubMed] [Google Scholar]
- 47.Gutsmann T, Howe J, Zähringer U, Garidel P, Schromm AB, Koch MH, et al. Structural prerequisites for endotoxic activity in the Limulus test as compared to cytokine production in mononuclear cells. Innate Immun. 2010;16:39–47. doi: 10.1177/1753425909106447. [DOI] [PubMed] [Google Scholar]
- 48.Rueda F, Cano-Garrido O, Mamat U, Wilke K, Seras-Franzoso J, Garcia-Fruitos E, et al. Production of functional inclusion bodies in endotoxin-free Escherichia coli. Appl Microbiol Biotechnol. 2014;98:9229–38. doi: 10.1007/s00253-014-6008-9. [DOI] [PubMed] [Google Scholar]
- 49.Akashi S, Nagai Y, Ogata H, Oikawa M, Fukase K, Kusumoto S, et al. Human MD-2 confers on mouse toll-like receptor 4 species-specific lipopolysaccharide recognition. Int Immunol. 2001;13:1595–9. doi: 10.1093/intimm/13.12.1595. [DOI] [PubMed] [Google Scholar]
- 50.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohto U, Fukase K, Miyake K, Shimizu T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc Natl Acad Sci U S A. 2012;109:7421–6. doi: 10.1073/pnas.1201193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scior T, Lozano-Aponte J, Figueroa-Vazquez V, Yunes-Rojas JA, Zähringer U, Alexander C. Three-dimensional mapping of differential amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic activation by lipid a, antagonism by eritoran and species-dependent activities of lipid IVA in the mammalian LPS sensor system. Comput Struct Biotechnol J. 2013;7:e201305003. doi: 10.5936/csbj.201305003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meredith TC, Mamat U, Kaczynski Z, Lindner B, Holst O, Woodard RW. Modification of lipopolysaccharide with colanic acid (M-antigen) repeats in Escherichia coli. J Biol Chem. 2007;282:7790–8. doi: 10.1074/jbc.M611034200. [DOI] [PubMed] [Google Scholar]
- 55.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller JH. A short course in bacterial genetics. Cold Spring Harbor, N.Y., Cold Spring harbor laboratory press. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 57.Lee DJ, Bingle LE, Heurlier K, Pallen MJ, Penn CW, Busby SJ, et al. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol. 2009;9:252. doi: 10.1186/1471-2180-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 59.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989;171:4617–22. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blom J, Jakobi T, Doppmeier D, Jaenicke S, Kalinowski J, Stoye J, et al. Exact and complete short-read alignment to microbial genomes using graphics processing unit programming. Bioinformatics. 2011;27:1351–8. doi: 10.1093/bioinformatics/btr151. [DOI] [PubMed] [Google Scholar]
- 61.Galanos C, Lüderitz O, Westphal O. A new method for the extraction of R lipopolysaccharides. Eur J Biochem. 1969;9:245–9. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 62.Kondakova A, Lindner B. Structural characterization of complex bacterial glycolipids by fourier transform mass spectrometry. Eur J Mass Spectrom (Chichester, Eng) 2005;11:535–46. doi: 10.1255/ejms.721. [DOI] [PubMed] [Google Scholar]
- 63.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 64.Pupo E, Lindner B, Brade H, Schromm AB. Intact rough- and smooth-form lipopolysaccharides from Escherichia coli separated by preparative gel electrophoresis exhibit differential biologic activity in human macrophages. FEBS J. 2013;280:1095–111. doi: 10.1111/febs.12104. [DOI] [PubMed] [Google Scholar]