

Abstract
Background
Fungal skin infections associated with Ophidiomyces ophiodiicola, a member of the Chrysosporium anamorph of Nannizziopsis vriesii (CANV) complex, have been linked to an increasing number of cases of snake fungal disease (SFD) in captive snakes around the world and in wild snake populations in eastern North America. The emergence of SFD in both captive and wild situations has led to an increased need for tools to better diagnose and study the disease.
Results
We developed two TaqMan real-time polymerase chain reaction (PCR) assays to rapidly detect O. ophiodiicola in clinical samples. One assay targets the internal transcribed spacer region (ITS) of the fungal genome while the other targets the more variable intergenic spacer region (IGS). The PCR assays were qualified using skin samples collected from 50 snakes for which O. ophiodiicola had been previously detected by culture, 20 snakes with gross skin lesions suggestive of SFD but which were culture-negative for O. ophiodiicola, and 16 snakes with no clinical signs of infection. Both assays performed equivalently and proved to be more sensitive than traditional culture methods, detecting O. ophiodiicola in 98% of the culture-positive samples and in 40% of the culture-negative snakes that had clinical signs of SFD. In addition, the assays did not cross-react with a panel of 28 fungal species that are closely related to O. ophiodiicola or that commonly occur on the skin of snakes. The assays did, however, indicate that some asymptomatic snakes (~6%) may harbor low levels of the fungus, and that PCR should be paired with histology when a definitive diagnosis is required.
Conclusions
These assays represent the first published methods to detect O. ophiodiicola by real-time PCR. The ITS assay has great utility for assisting with SFD diagnoses whereas the IGS assay offers a valuable tool for research-based applications.
Keywords: Chrysosporium anamorph of Nannizziopsis vriesii (CANV), Emerging disease, Ophidiomyces ophiodiicola, Real-time PCR, Snake fungal disease
Background
Skin infections are among the most common diseases observed in captive snakes [1,2]. While fungi are often implicated in many of these infections through histopathologic analyses, identifying the exact causative agent can be difficult. Traditionally, culture analyses were the standard for determining the likely causes of dermatomycoses, but such analyses are complicated by the abundance and diversity of fungi naturally found on the skin of reptiles [2,3]. In addition, relatively little work has been conducted to identify which of these fungi are medically important [2]. The veterinary literature contains numerous case reports implicating particular fungi as the causes of dermatitis in snakes, but few offer compelling evidence that the fungi grown in culture truly represent the agent observed in supporting histopathology [2]. In many cases, the etiology of skin infections in snakes has likely been incorrectly attributed to common environmental microorganisms and such misdiagnoses may lead to unsuccessful treatment regimens and serve to confound our understanding of the causes of such infections in captive snakes.
Fungi formerly grouped within the Chrysosporium anamorph of Nannizziopsis vriesii (CANV) complex are among the few species that have been repeatedly associated with dermatological disease in reptiles (summarized by [2,4]), and at least one species has been shown through infection trials to act as a primary pathogen in healthy chameleons [5]. Recent phylogenetic analyses of the CANV complex have revealed several new taxa [6,7]. One of these species, Ophidiomyces (formerly Chrysosporium) ophiodiicola, was described for the first time in 2009 after its isolation from a captive snake with a facial granuloma [8]. However, upon re-examining isolates formerly classified as CANV, Sigler et al. [6] found that O. ophiodiicola has been associated with emerging skin infections in captive snakes for the last several decades. Many additional cases of dermatitis associated with O. ophiodiicola are thought to have been incorrectly attributed to bacteria or other fungi [2], and O. ophiodiicola may be one of the most common, albeit overlooked, causes of skin infections in captive snakes.
Since 2006, O. ophiodiicola has also been isolated from wild snakes with severe, and often fatal, infections in the eastern U.S. [9,10]. These emerging infections, referred to as snake fungal disease (SFD), are currently considered a serious threat to some snake populations [9,11]. However, recent attempts to study the prevalence, distribution, and impacts of SFD on wild snakes have been hampered by the lack of rapid, cost-effective, and reliable laboratory tests to detect O. ophiodiicola.
Confirming presence of O. ophiodiicola in association with skin lesions can be problematic due to the difficulty of isolating the fungus in culture. Isolation is particularly challenging when small samples such as biopsies or scale clippings are collected, and euthanasia of the animal is frequently required to obtain larger samples sufficient for confirming the presence of O. ophiodiicola. Even when the fungus is isolated in culture, identification of O. ophiodiicola based on morphological characteristics is often unreliable. Furthermore, the fungus is relatively slow-growing such that the process of successful isolation and identification can take weeks and may be dependent upon laboratory expertise. Given the recent emergence of SFD in wild snakes and the clinical importance of fungal dermatitis in captive snakes, a rapid and more sensitive tool for the detection of O. ophiodiicola is needed.
Here we describe the development and qualification of two TaqMan real-time polymerase chain reaction (PCR) assays that reliably detect O. ophiodiicola from small pieces of skin tissue collected from live snakes. The PCR tests target either the multi-copy internal transcribed spacer region (ITS) or intergenic spacer region (IGS) of O. ophiodiicola and are extremely sensitive, highly specific, and yield results in less than 24 hours. These assays offer utility in assisting with diagnosis of SFD in both wild and captive snakes in addition to providing an important research tool for better understanding the biology of the fungus and ecology of this disease.
Methods
DNA extraction, amplification, and sequencing
All fungal cultures were grown at 24°C on Sabouraud dextrose agar containing chloramphenicol and gentamicin or dermatophyte test medium. After 7 to 21 days (depending on the growth rate of a particular isolate), approximately 5 to 10 mg of mycelia were scraped off of the medium and placed into 600 μl lyticase solution [1 M sorbitol, 100 mM EDTA, 200 U lyticase (Sigma-Aldrich, St. Louis, MO), and 14 mM beta-mercaptoethanol], ground with a pestle, and incubated at 30°C and 500 rpm for 1 hour. Fungal protoplasts were pelleted by centrifugation at 500 x g for 10 min, the supernatant removed, and genomic DNA (gDNA) extracted using the Gentra®Puregene® Tissue Kit (Qiagen Inc., Valencia, CA) according to the “solid tissues protocol” and omitting proteinase K and RNase treatments.
The gDNA extracted from isolates of O. ophiodiicola used in this study (see Table 1) was quantified at the University of Wisconsin Biotechnology Center using an Invitrogen™ Qubit™ Fluorometer (Life Technologies, Carlsbad, CA). To ensure that the gDNA extracted from all other fungal isolates (i.e., non-O. ophiodiicola isolates listed in Tables 1 and 2) was of sufficient quantity and quality for downstream PCR applications, the internal transcribed spacer region (ITS) was amplified using the fungal-specific primers ITS1-F and ITS4 [12] under the following cycling conditions: 94°C for 3 min; 40 cycles of 94°C for 1 min, 53°C for 1 min, and 72°C for 3 min; and a final extension at 72°C for 10 min. GoTaq® Flexi DNA polymerase (Promega Corporation, Madison, WI) was used for the PCR reactions following the manufacturer’s instructions, and 5 μl template (diluted 1:10) was added per 25 μl reaction. All such reactions yielded strong bands of the correct size, indicating that fungal gDNA had been successfully extracted.
Table 1.
Fungal isolates for which ribosomal RNA regions were analyzed to develop Ophidiomyces ophiodiicola -specific PCR assays
| GenBank Accession No.* | ||||
|---|---|---|---|---|
| Isolate | Species | ITS Reference | ITS | IGS |
| CBS 122913 | Ophidiomyces ophiodiicola | [8] | EU715819 | KP691505 |
| UAMH 6218 | Ophidiomyces ophiodiicola | [6] | KF477227 | KP691506 |
| UAMH 6642 | Ophidiomyces ophiodiicola | [6] | KC884267 | KP691507 |
| UAMH 6688 | Ophidiomyces ophiodiicola | [6] | KF477228 | KP691508 |
| UAMH 9985 | Ophidiomyces ophiodiicola | [6] | KF477230 | KP691509 |
| UAMH 10296 | Ophidiomyces ophiodiicola | [6] | KF477232 | KP691510 |
| UAMH 10768 | Ophidiomyces ophiodiicola | [6] | KF477234 | KP691511 |
| UAMH 10769 | Ophidiomyces ophiodiicola | [6] | KF477235 | KP691512 |
| UAMH 10949 | Ophidiomyces ophiodiicola | [6] | KF477236 | KP691513 |
| UAMH 11295 | Ophidiomyces ophiodiicola | [6] | KF477237 | KP691514 |
| UAMH 10212 | Chrysosporium indicum | this study | KP691483 | KP691515 |
| CBS 629.79 | Chrysosporium sp. | this study | KP691484 | KP691516 |
| UAMH 10352 | Nannizziopsis guarroi | [6] | KF477208 | KP691517 |
| UAMH 10417 | Nannizziopsis infrequens | [6,22] | AY744467 | KP691518 |
| UAMH 3527 | Nannizziopsis vriesii | [6] | KF477198 | KP691519 |
| UAMH 10439 | Paranannizziopsis australasiensis | [6] | KF477218 | KP691520 |
| UAMH 10693 | Paranannizziopsis californiensis | [6] | KF477224 | KP691521 |
| UAMH 8392 | Pseudoamauroascus australiensis | [23] | AJ131787 | KP691522 |
CBS = isolates obtained from the Centraalbureau voor Schimmelcultures.
UAMH = isolates obtained from the University of Alberta Microfungus Collection and Herbarium.
ITS = internal transcribed spacer region of the ribosomal RNA gene complex.
IGS = intergenic spacer region (3′ and 5′ ends) of the ribosomal RNA gene complex.
*All IGS DNA sequences were newly generated for this study.
Table 2.
Fungal isolates from snakes that were used to assess specificity of Ophidiomyces ophiodiicola PCR assays
| Isolate | Species* | ITS GenBank Accession No. |
|---|---|---|
| 44736-30-02-02 | Alternaria sp. | KP691485 |
| 24411-01-01-02 | Arthroderma/Trichophyton sp. | KP691486 |
| 44736-44-01-01 | Arthroderma/Trichophyton sp. | KP691487 |
| 24833-01-01-01A | Ascomycete sp. | KP691488 |
| 24821-01-02-01 | Aspergillus sp. | KP691489 |
| 44736-02-01-01 | Beauveria sp. | KP691490 |
| 24392-01-03-04 | Bionectria sp. | KP691491 |
| 24266-04-05-01 | Cladosporium sp. | KP691492 |
| 24392-01-02-02 | Hypocreales sp. | KP691493 |
| 24415-01-02-02 | Isaria sp. | KP691494 |
| 24266-01-03-02 | Metarhizium sp. | KP691495 |
| 24266-01-01-01A | Mucor sp. | KP691496 |
| 44736-32-02-01 | Myriodontium sp. | KP691497 |
| 44736-01-01-01 | Paecilomyces sp. | KP691498 |
| 44736-43-05-04A | Penicillium sp. | KP691499 |
| 24281-01-01-01 | Penicillium sp. | KP691500 |
| 44736-32-01-01 | Pseudogymnoascus sp. | KP691501 |
| 44736-03-02-01 | Purpureocillium lilacinum | KP691502 |
| 24281-01-02-01 | Trichocomaceae sp. | KP691503 |
| 24826-01-01-01A | Trichoderma sp. | KP691504 |
*Identifications were based on DNA sequencing of the internal transcribed spacer region (ITS).
To generate sequence data for the IGS of O. ophiodiicola isolates, primers CNL12 [13] and CNS1 [14] were used to amplify the approximately 2,800 bp region under the following cycling conditions: initial denaturation at 98°C for 2 min; 47 cycles of 98°C for 10 sec, 50.5°C for 30 sec, and 72°C for 7 min; and a final extension of 72°C for 7 min. Products were gel-purified using the QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA) and sequenced from both ends with primers CNL12 and CNS1. A primer walking strategy was employed in an attempt to sequence the entire IGS. However, repetitive runs of guanine and cytosine nucleotides made it difficult to sequence through certain regions, resulting in successful sequencing of approximately 330 nucleotides at the 5′-end of IGS and approximately 875 nucleotides at the 3′-end. The 5′- and 3′-ends of the IGS for several fungi that are closely related to O. ophiodiicola (Table 1) were also amplified and sequenced as described above. All newly generated sequences for this study (which included sequences from 10 isolates of O. ophiodiicola and eight species of closely related fungi) were deposited in GenBank (Table 1).
Primer and probe design
The ITS and IGS of multiple strains of O. ophiodiicola, along with selected near relatives (Table 1), were aligned using MegAlign™ (Lasergene® 10, DNASTAR, Madison, WI). For the ITS alignment, ITS-1 demonstrated the greatest interspecific variability, but this region also contained numerous single nucleotide polymorphisms (SNPs) between strains of O. ophiodiicola and appeared to be an unsuitable target for the development of an O. ophiodiicola-specific PCR capable of detecting all strains of the fungus. ITS-2 was more conserved among different strains of O. ophiodiicola but still demonstrated interspecific divergence. Thus, ITS-2 was targeted for assay development. For the IGS alignment, both the 5′- and 3′-ends of the IGS sequence of O. ophiodiicola were highly divergent from those of its nearest neighbors’ (excluding portions of the sequences that represented and flanked the 3′-end of the 28S ribosomal RNA (rRNA) gene and the 5′-end of the 18S rRNA gene; these areas were avoided for assay development). Different strains of O. ophiodiicola shared high identity within the IGS, but intraspecific variation was observed at numerous SNPs throughout the regions sequenced. Areas of the IGS that were most conserved among all evaluated strains of O. ophiodiicola were targeted for development of an IGS PCR.
Primers and probes were designed using the online PrimerQuest® program (Integrated DNA Technologies, Coralville, IA) [15]. The qPCR option was selected and design parameters were modified such that the program chose primers with a minimum calculated melting temperature (Tm) of 57°C, an optimum Tm of 60°C, and a maximum Tm of 65°C; probe Tm was set between 64°C and 75°C, with an optimum Tm of 70°C. Several primer/probe sets meeting these criteria were identified for both ITS and IGS; however, in each case the binding sites for the set either spanned one to several SNPs among strains of O. ophiodicola or resided within an area where the oligos might bind to DNA from non-target species. Thus, binding positions of some oligos were modified slightly from the original PrimerQuest® output such that they would bind more specifically to DNA from O. ophiodiicola while also residing within regions conserved among all analyzed strains of the fungus. Potential formation of secondary structure, self-binding, and Tm were also considered when designing the oligos. The newly designed primer and probe sequences for the ITS assay were: Oo-rt-ITS-F (forward primer) 5′-GAGTGTATGGGAATCTGTTTC-3′; Oo-rt-ITS-R (reverse primer) 5′-GGTCAAACCGGAAAGAATG-3′; Oo-rt-ITS-P (probe) 5′-(FAM)TCTCGCTCGAAGACCCGATCG(BHQ-1)-3′. The newly designed primer and probe sequences for the IGS assay were: Oo-rt-IGS-F (forward primer) 5′-CGGGTGAATTACCCAGTT-3′; Oo-rt-IGS-R (reverse primer) 5′-AGCCATCCTTCCCTACAT-3′; Oo-rt-IGS-P (probe) 5′-(FAM)ATACTCTCCGGGCGCTTGTCTTCC(BHQ-1)-3′ (Figure 1). All primers and probes were searched against the GenBank database using the Basic Local Alignment Search Tool (BLAST) [16]. No sequences in GenBank (excluding those of O. ophiodiicola) were a perfect match to the primers or probes, and those sequences that were most similar to a given oligo did not demonstrate significant similarity with the other two oligos in the set.
Figure 1.
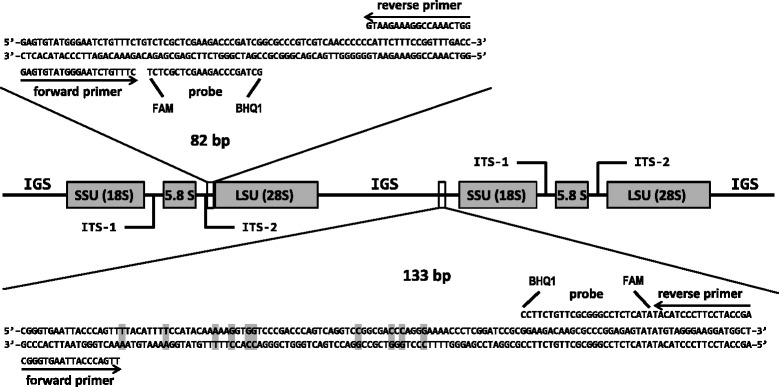
Diagram of fungal ribosomal RNA regions and areas targeted for Ophidiomyces ophiodiicola-specific real-time PCR assays. One assay targets the 3′-end of the internal transcribed spacer region 2 (ITS-2) while the second assay targets the 3′-end of the intergenic spacer region (IGS). Expanded sequences (representing the type isolate of O. ophiodiicola) show the amplicons produced by each PCR and the primer and probe binding sites. Shaded boxes within the sequences represent nucleotide positions that vary between strains of O. ophiodiicola.
Assay development
Real-time PCR was performed on the Applied Biosystems® 7500 Fast Real-Time PCR System. Master mix (QuantiFast Probe PCR + ROX Vial Kit, Qiagen, Inc, Valencia, CA) was prepared according to the manufacturer’s specifications. Included in the 25 μl reaction volume was 12.5 μl 1X QuantiFast Probe Master Mix, 0.5 μl 1X ROX dye solution, 5 μl template DNA, 0.5 μl of each 20 μM primer stock solution (final reaction concentration 0.4 μM), 0.25 μl 20 μM probe stock solution (final reaction concentration 0.2 μM), and 0.5 μl of 20 μg/μl Bovine Serum Albumin (BSA) solution (Sigma-Aldrich, St. Louis, MO; final reaction concentration of 0.4 μg/μl) to mitigate possible PCR inhibitors. Oligos were synthesized by a commercial company (BioSearch Technologies, Inc., Novata CA). Standard cycling conditions were used for both assays: 95°C for 3 minutes, 95°C for 3 seconds, 60°C for 30 seconds for a total of 40 cycles. To standardize cycle threshold (Ct) values across PCR runs, the threshold was set at four percent of the maximum background-subtracted fluorescence (ΔRn). At this threshold, sample amplification curves approached maximum efficiency, negative controls were below the threshold, and weak positives exceeded the threshold. Efficiency along individual amplification curves was determined by calculating the slope of five consecutive ΔRn fluorescence readings [17]. Samples that crossed the threshold at ≤ 36 cycles for the ITS assay and ≤ 38 cycles for the IGS assay were considered positive for diagnostic purposes. These threshold cutoffs were selected because standard curve results indicated that the assays performed inconsistently when template DNA concentrations yielded Ct values above 36 and 38 for each assay, respectively. For each plate run (including those for assay qualification), a negative control (water added in place of template) and standard curve of gDNA (5 X 105, 5 X 104, 5 X 103, 5 X 102, and 50 fg) isolated from the type isolate of O. ophiodiicola (CBS 122913) were included.
The efficiency, precision, and detection limit of the PCR assays were evaluated based on a ten-fold standard curve dilution series of gDNA (5.0 ag to 5.0 ng) isolated from the type isolate of O. ophiodiicola. Efficiency was calculated from the slope of the best fit line of Ct value plotted against the log-transformed quantity (fg) of DNA template, (efficiency = −1 + 10 (−1/slope)). The standard curve was run in triplicate, and precision was evaluated based on the expectation that the standard deviations not exceed one Ct value. The detection limit was the gDNA concentration at which all three replicates reliably amplified.
Assay qualification
Consistent detection assays should amplify and capture all O. ophiodiicola genetic variants with minimal differences in Ct values among those variants. To assess consistency of the real-time PCR assays, 5 pg of gDNA from seven isolates (CBS 122913, UAMH 6218, UAMH 6642, UAMH 6688, UAMH 9985, UAMH 10296, UAMH 10768; Table 1) representing all known ITS and IGS sequence variants of O. ophiodiicola were tested in duplicate.
To ensure that the assays were specific to O. ophiodiicola, the primer and probe sets were tested against a panel of eight closely related fungal species (Table 1). These included several reptile-infecting taxa for which phylogenetic relationships were recently resolved [6], a Chrysosporium sp. isolated from a snake (CBS 629.79), and other near neighbors within the Onygenales group. The assays were also screened against representative isolates of 20 fungal species that were commonly isolated from the skin of snakes submitted to the U.S. Geological Survey – National Wildlife Health Center (NWHC) for diagnostic evaluation (Table 2).
The sensitivity of the real-time PCR assays were determined using tissue samples collected from 70 wild snakes with skin lesions from the eastern half of the U.S. Samples originated from submissions to the NWHC that included snakes carcasses found in the wild, snakes that were humanely euthanized, and non-lethally collected skin biopsies. These samples ranged from deep dermal granulomas and full cross-sections of infected skin (collected from dead snakes or as biopsies) to superficial epidermal crusts (collected as scale clippings or from lesions on shed skin). Corresponding sections of tissue from 50 of the 70 snakes had tested positive for O. ophiodiicola by culture analyses conducted at the NWHC. Sections of snout, dorsal, and ventral skin from 16 snakes without signs of skin infection and that had tested negative for the presence of O. ophiodiicola by culture analyses were analyzed as disease-negative samples. DNA was extracted from tissues approximately 2 mm3 in size using the Gentra®Puregene® Tissue Kit, “solid tissues protocol”. For the lysis procedure, 0.5 mg/ml proteinase K was added to the lysis buffer containing tissue, and the mixture was digested for 1.5 to 3 hours. Tissues were then macerated with a pestle and further digested overnight. The RNase treatment step was omitted, but the procedure was otherwise carried out according to the manufacturer’s instructions. A negative control (no tissue added) extraction was performed to ensure that reagents were not contaminated. Extracted DNA was diluted ten-fold and the diluted template used for real-time PCR as described above. To test for inhibition, the snout, dorsal, and ventral skin DNA extracts of four randomly selected PCR- and culture-negative snakes were spiked with 5 pg O. ophiodiicola gDNA. The Ct values of the spiked samples were then compared to Ct values of the 5 pg standard curve replicates on the same plate to ensure that amplification was equivalent. The sensitivity of each PCR was compared to that of the culture-based analyses with McNemar’s Test in SigmaPlot 11.2 (Systat Software, Inc., San Jose, CA).
Amplified DNA from 5 randomly selected PCR- and culture-positive samples and all culture-negative samples that produced positive PCR results were sequenced to confirm that the amplicons matched the targeted region. Briefly, amplicons were cloned using the Invitrogen™ TOPO® TA Cloning® Kit with the pCR™ 2.1-TOPO® vector and TOP10 competent cells (Life Technologies, Carlsbad, CA), the clones screened by PCR with vector-specific primers, and the resulting PCR products sequenced according to the manufacturer’s instructions.
Ethics statement
All carcass collection, euthanasia, and biopsy sampling procedures were performed under the authority of other agencies in accordance with applicable state laws; specimens were submitted for routine diagnostic analyses that did not require further review by NWHC institutional committees. Collection of scale clippings from live snakes by NWHC personnel was conducted under NWHC Institutional Animal Care and Use Committee protocol EP130117 and was done in compliance with all necessary permissions and permitting requirements. Animals sampled for this study were wild and not client-owned; thus no owner consent was required.
Results
Assay efficiency and precision
The standard curve efficiency of both the ITS and IGS PCR assays were within the accepted range of 90%-110% [18]. Specifically, the ITS standard curve was 101.41% efficient with an R2-value of 0.998. The IGS standard curve had an efficiency of 95.02% and an R2-value of 0.997 (Figure 2). All standard curve replicates were precise, varying from one another by less than one Ct value (standard deviations for replicates of the ITS assay ranged from 0.08 to 0.49; those of the IGS assay were between 0.09 and 0.44). The detection limit for both assays was 5 fg of purified gDNA. The ITS assay, on average, yielded Ct values that were 2.44 cycles lower than that of the IGS assay.
Figure 2.
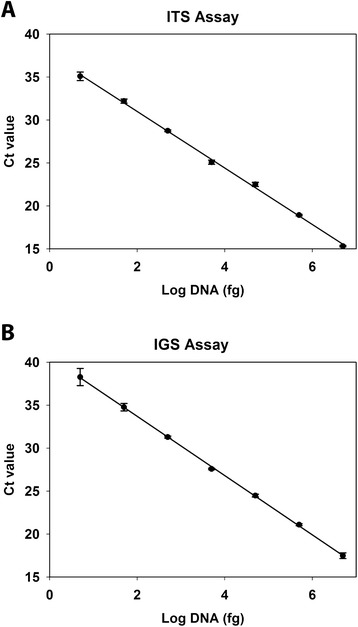
Standard curves for Ophidiomyces ophiodiicola real-time PCR assays. Genomic DNA (gDNA) isolated from a pure culture of the type isolate of O. ophiodiicola was quantified, serially diluted, and used as template. All samples were run in triplicate with all points depicting mean Ct values and error bars representing standard deviation. The assays targeting (A) the internal transcribed spacer region (ITS assay) and (B) intergenic spacer region (IGS assay) of O. ophiodiicola were both linear over seven logs, ranging from 5 fg to 5 ng template gDNA (ITS assay: R2 = 0.998; IGS assay: R2 = 0.997).
Assay specificity and sensitivity
The specificities of the ITS and IGS assays were tested using a panel of eight fungi closely related to O. ophiodiicola and 20 additional fungi that are commonly associated with snake skin. No discernible amplification was detected when DNA extracted from these fungi were used as template. The assays did, however, amplify target DNA from all known genetic variants of O. ophiodiicola. The Ct values among these variants were very similar (standard deviations of 0.63 and 0.64 for the ITS and IGS assays, respectively), indicating that performance of the PCR did not vary by strain.
Both assays also performed identically when they were used to screen snake tissue samples for the presence of O. ophiodiicola. Forty-nine of the 50 (98%) tissue samples that were culture-positive for O. ophiodiicola were also positive by PCR. The remaining sample showed no amplification after 40 cycles with the ITS assay, but had a Ct of 39.12 with the IGS assay (above the 38 cycle cut-off). Eight of the 20 tissue samples from snakes that had gross lesions suggestive of SFD but that were culture-negative for O. ophiodiicola were PCR-positive. One additional sample had a Ct of 38.76 with the IGS assay, but all other samples yielded no amplification. Of the 48 total samples from animals without clinical signs of SFD (3 pieces of skin from each of 16 snakes), a single sample (snout skin from one snake) was PCR-positive by both assays. An additional sample (dorsal body skin) from the same animal, as well as ventral body skin from a second animal, demonstrated amplification with the ITS assay, but had Ct values over 36. No amplification was observed in the negative extraction control sample. The inability of the two assays to perform equivalently at high Ct values and the questionable clinical significance of extremely low levels of O. ophiodiicola DNA further demonstrated the practicality of setting the Ct threshold at 36 (ITS assay) and 38 (IGS assay) for diagnostic purposes. When the two detection methods were compared, the PCR assays proved to be more sensitive than culture-based detection methods (χ2 = 4.900, p = 0.027).
For all runs, negative controls performed as expected and standard curves were linear. The Ct values of the spiked negative extracts were within one Ct of the standard curve replicates containing the same amount of gDNA; thus, there was no evidence of inhibition. All PCR products that were sequenced were 100% identical to the desired ITS or IGS amplicons, indicating the positive results were not due to cross-amplification of non-target DNA.
Discussion
Advances in diagnostics and research related to O. ophiodiicola and its associated infections have been hampered by lack of reliable and sensitive techniques to detect the fungus. We designed two real-time PCR methods to rapidly identify the fungus in clinical samples. Both PCR assays proved to be more sensitive than culture-based methods for detecting O. ophiodiicola. Specifically, PCR yielded negative results for only 2% of samples that were culture-positive for O. ophiodiicola; and detected the fungus in 8 of 20 (40%) culture-negative samples that had been collected from snakes with clinical signs suggestive of SFD. In four of the remaining 12 snakes with grossly visible skin infections that were culture- and PCR-negative, fungi possessing morphological characteristics inconsistent with O. ophiodiicola were observed by histopathology; in three additional samples, fungi consistent with O. ophiodiicola were seen but could not be definitively identified microscopically; and the five remaining samples consisted of scale clippings and thus were unsuitable for histopathologic examination. Since no “gold standard” method for detection of O. ophiodiicola currently exists, it is difficult to ascertain whether other microorganisms caused most of the infections that were PCR-negative. Bacteria, other fungi, and noninfectious processes are known to cause clinical signs that are grossly indistinguishable from those associated with O. ophiodiicola [2], and it is likely that at least some PCR-negative samples represented infections caused by other etiological agents. In histopathology, the cutaneous fungal invasion that characterizes SFD is often localized. Thus, it is also possible that dividing tissues for different types of analyses may have resulted in allocation of tissue for PCR that did not capture the active lesion or contained insufficient numbers of fungal elements. This likely explains the 2% false-negative rate for culture-positive samples and highlights the importance of targeting an active area of the lesion for PCR testing. Nonetheless, these data indicate that the PCR assays represent the most sensitive tool currently available to detect O. ophiodiicola.
In our PCR screen, one of 16 (~6%) snakes without clinical signs of SFD tested positive for O. ophiodiicola (Ct values of 29.16 and 30.94 for the ITS and IGS PCR assays, respectively). To rule out contamination as a source of these PCR-positive results, we sampled additional tissues from the same animal (i.e., chin skin, spectacle, and two pieces of snout skin). PCR indicated that O. ophiodiicola was also present on the chin skin and spectacle, suggesting that contamination was an unlikely explanation for the positive results. A sample from one additional snake without lesions that was classified as PCR-negative also showed evidence of amplification above the diagnostic threshold cut-off. Sequenced amplicons of the PCR-positive snake without clinical signs of infection and of the additional culture-negative snake with a Ct value above the diagnostic cut-off (neither of which were suspected to have had SFD) were a 100% match to O. ophiodiicola. Whether such detections represent early stages of colonization/infection by the fungus or indicate the potential of some snakes to harbor low levels of O. ophiodiicola in the absence of disease manifestation is unclear, but the latter seems more plausible. Paré et al. [3] reported culturing O. ophiodiicola from shed skin of less than 1% of captive snakes without clinical signs of skin infection, and the authors speculated that the fungus occurs only rarely on asymptomatic animals. However, the low detection rate in that study may have been due to the insensitivity of culture techniques, particularly when the fungus is at low abundance. With its much greater sensitivity, PCR has great utility in research aimed at better parsing out the association between O. ophiodiicola and disease, prevalence of the fungus on asymptomatic animals, and the importance of pathogen load in disease development.
The PCR assays also provide an important tool to assist with identification of O. ophiodiicola-associated infections in the clinical setting. The rapid turnaround time of PCR (less than 24 hours) can provide important preliminary results that allow for immediate treatment of animals suspected to have SFD, can be used to help verify whether fungal hyphae observed in histopathology are O. ophiodiicola, and will provide an alternative to morphological- or DNA sequencing-based identification of pure cultures of O. ophiodiicola. Although the fungus may occasionally be detected on the skin of snakes that do not have SFD, animals with gross lesions tend to have heavier burdens of O. ophiodiicola. For example, based on average Ct values, snakes with infections known to be associated with O. ophiodiicola (i.e., culture-positive) had approximately 1,000 times more O. ophiodiicola DNA detected in their tissues than PCR-positive snakes without clinical signs of SFD. When the active lesion is sampled, Ct values can generally be expected to be low if O. ophiodiicola is associated with the infection. Furthermore, snakes with severe skin infections typically have multiple lesions on the body that can be non-lethally sampled. By sampling more than one lesion (or several areas of a single large lesion), clinicians can be confident that PCR results represent true-positive or true-negative detections.
While the PCR assays we describe are useful diagnostic tools, it is important to note that histopathology in conjunction with PCR is necessary to make definitive diagnoses that accurately distinguish disease from mere presence of the pathogen. However, there are applications in which simply confirming the presence of O. ophiodiicola (irrespective of actual disease) may be useful. For example, PCR would provide a much more sensitive method to screen for asymptomatic carriers of the fungus during the importation and quarantine process at zoological parks and reptile collections as well as to screen environmental samples for potential contamination with O. ophiodiicola. For such applications, laboratories may wish to set the PCR threshold at 40 or more cycles to detect very low levels of the fungus. In addition, multiple samples should be tested to reduce the likelihood of false-negative results.
Both PCR assays described in this report target portions of the rRNA gene complex. This gene region occurs as multiple copies within the fungal genome, affording high sensitivity to assays that target the rRNA gene complex. For example, Muller et al. [19] estimated that the lower limit of detection of a real-time PCR assay specific to the IGS of Pseudogymnoascus destructans (the causative agent of bat white-nose syndrome) was approximately 0.1 genome equivalents. At this time, neither the number of copies of rRNA genes on, nor the size of, the O. ophiodiicola genome is known, making it difficult to predict the lower limit of detection in genome equivalents. However, detection limits based on purified gDNA were similar between our assays and that of Muller et al. [19]. Different strains of O. ophiodiicola apparently have a similar number of rRNA gene region copies as Ct values were consistent between them when known amounts of gDNA were used as template. This indicates that the PCR assays have great potential for research that requires quantification of the fungus in samples. However, more work is necessary to more definitively determine how Ct values translate to cell-equivalents.
The overall performance of the ITS and IGS assays were similar, but each assay is optimally suited for different applications. The ITS assay is the more robust option for diagnostic work, particularly for laboratories that may be receiving samples from captive snakes. Although both assays performed well against all currently known strains of O. ophiodiicola, there are undoubtedly more strains yet to be characterized. The more highly conserved nature of ITS between strains, however, makes the ITS assay more likely to detect novel and potentially divergent genetic variants of O. ophiodiicola. Such variants are more likely to be encountered in captive snakes or wild snakes from outside eastern North America (since most characterized strains upon which the assays were designed originated in the eastern U.S.). The IGS exhibits greater DNA sequence variation between strains, increasing the possibility that novel genetic variants may have SNPs in oligo binding sites within the IGS. Thus while there is no evidence to indicate that the IGS assay is unsuitable for diagnostic work, the use of the ITS assay for this purpose represents the more conservative approach.
For screening environmental samples for O. ophiodiicola, the IGS assay is recommended. While both assays discriminated between O. ophiodiicola and its closest known relatives, we cannot rule out the possibility that there are other uncharacterized environmental fungi that are genetically similar to O. ophiodiicola and that could cross-react with the ITS assay primers and probe. For example, CANV complex species within polytypic genera (e.g., Nannizziopsis and Paranannizziopsis) may share over 97% DNA sequence identity in the ITS [6]; if such small differences in ITS sequences existed between O. ophiodiicola and other hypothetical species of Ophidiomyces, the ITS assay would be unlikely to differentiate them. While sequencing of amplicons in this study indicated no evidence that such closely-related fungi exist on the skin of snakes, environmental samples may harbor much more diverse fungal communities. For example, the original ITS-based PCR used to detect P. destructans performed well when testing bat tissues, but was unsuitable for discriminating the pathogen from a wide variety of closely-related, nonpathogenic, and previously undiscovered Pseudogymnoascus spp. in soil samples [20,21]; this subsequently prompted the development of a more specific PCR assay targeting the IGS of P. destructans which exhibits much higher interspecific variation than the ITS [19]. Such high variation in the region makes it highly unlikely that the IGS assay described herein would cross-amplify DNA from fungi other than O. ophiodiicola, including very closely related hypothetical taxa that are yet to be discovered. The IGS assay also has utility for research that requires differentiation among strains of O. ophiodiicola – because the amplicon produced by the IGS PCR encompasses numerous SNPs (Figure 1), sequencing the amplicon can distinguish between some genetic variants of the fungus.
Conclusions
The increase of CANV-associated infections in captive reptiles and the emergence of SFD in wild snakes have elicited concern over the future health of both captive and wild snake populations. As clinicians attempt to diagnose an increasingly common disease and researchers attempt to better understand the disease system, enhanced methods to detect the fungus linked to these infections will be essential. The PCR assays described herein provide important tools to advance SFD diagnostics, support surveillance efforts, and aid researchers trying to better understand the biology of O. ophiodiicola, the intricacies of host-pathogen dynamics, and the drivers of SFD emergence.
Acknowledgements
This work was funded by the U.S. Geological Survey and the U.S. Fish and Wildlife Service. We thank D. Earl Green, Susan Knowles, and Julia Lankton for collecting tissue samples during routine necropsies and for providing histopathology results, and Katie Schmidt, Stephanie Steinfeldt, Brenda Berlowski-Zier, Dottie Johnson, and Nathan Ramsay for technical support. Finally, we are grateful to the numerous state and federal agencies and private individuals that submitted samples to the NWHC and made this work possible. The use of trade, product or firm names is for descriptive purposes only and does not imply endorsement by the U.S. government.
Abbreviations
- BHQ-1
Black Hole Quencher®-1
- BLAST
basic local alignment search tool
- BSA
bovine serum albumin
- CANV
Chrysosporium anamorph of Nannizziopsis vriesii
- CBS
Centraalbureau voor Schimmelcultures
- Ct
cycle threshold
- DNA
deoxyribonucleic acid
- ΔRn
background-subtracted fluorescence
- FAM
6-carboxyfluorescein
- gDNA
genomic deoxyribonucleic acid
- IGS
intergenic spacer region
- ITS
internal transcribed spacer region
- NWHC
U.S. Geological Survey - National Wildlife Health Center
- PCR
polymerase chain reaction
- RNA
ribonucleic acid
- rRNA
ribosomal ribonucleic acid
- ROX
6-carboxyl-x-rhodamine
- SFD
snake fungal disease
- SNPs
single nucleotide polymorphisms
- Tm
melting temperature
- UAMH
University of Alberta Microfungus Collection and Herbarium
Footnotes
Elizabeth Bohuski and Jeffrey M Lorch contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EB assisted with experimental design, performed most of the experimental work, analyzed data, and contributed to the writing process. JML designed the assays, assisted with experimental work and data analysis, and drafted the manuscript. KMG and DSB provided essential intellectual contributions to this work, assisted with experimental design, and provided critical revisions to the manuscript content. All authors read and approved the final manuscript.
Contributor Information
Elizabeth Bohuski, Email: ebohuski@usgs.gov.
Jeffrey M Lorch, Email: jmlorch@wisc.edu.
Kathryn M Griffin, Email: kgriffin@usgs.gov.
David S Blehert, Email: dblehert@usgs.gov.
References
- 1.Hoppmann E, Barron HW. Dermatology in reptiles. J Exot Pet Med. 2007;16:210–24. doi: 10.1053/j.jepm.2007.10.001. [DOI] [Google Scholar]
- 2.Paré JA, Jacobson ER. Mycotic diseases of reptiles. In: Jacobson ER, editor. Infectious Diseases and Pathology of Reptiles: Color Atlas and Text. Boca Raton, FL: CRC Press, Taylor and Francis; 2007. pp. 527–70. [Google Scholar]
- 3.Paré JA, Sigler L, Rypien KL, Gibas CC. Cutaneous mycobiota of captive squamate reptiles with notes on the scarcity of Chrysosporium anamorph of Nannizziopsis vriesii. J Herpetol Med Surg. 2003;13:10–5. [Google Scholar]
- 4.Mitchell MA, Walden MR. Chrysosporium anamorph Nannizziopsis vriesii, an emerging fungal pathogen of captive and wild reptiles. Vet Clin North Am Exot Anim Pract. 2013;16:659–68. doi: 10.1016/j.cvex.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Paré JA, Coyle KA, Sigler L, Maas AK, Mitchell RL. Pathogenicity of the Chrysosporium anamorph of Nannizziopsis vriesii for veiled chameleons (Chamaeleo calyptratus) Med Mycol. 2006;44:25–31. doi: 10.1080/13693780500165461. [DOI] [PubMed] [Google Scholar]
- 6.Sigler L, Hambleton S, Paré JA. Molecular characterization of reptile pathogens currently known as members of the Chrysosporium anamorph of Nannizziopsis vriesii complex and relationship with some human-associated isolates. J Clin Microbiol. 2013;51:3338–57. doi: 10.1128/JCM.01465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stchigel AM, Sutton DA, Cano-Lira JF, Cabañes FJ, Abarca L, Tintelnot K, et al. Phylogeny of chrysosporia infecting reptiles: proposal of the new family Nannizziopsiaceae and five new species. Persoonia. 2013;31:86–100. doi: 10.3767/003158513X669698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajeev S, Sutton DA, Wickes BL, Miller DL, Giri D, Van Meter M, et al. Isolation and characterization of a new fungal species, Chrysosporium ophiodiicola, from a mycotic granuloma of a black rat snake (Elaphe obsoleta obsoleta) J Clin Microbiol. 2009;47:1264–8. doi: 10.1128/JCM.01751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allender MC, Dreslik M, Wylie S, Phillips C, Wylie DB, Maddox C, et al. Chrysosporium sp. infection in eastern massasauga rattlesnakes. Emerg Infect Dis. 2012;17:2383–4. doi: 10.3201/eid1712.110240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.U.S. Geological Survey - National Wildlife Health Center . Snake Fungal Disease in the United States. U.S. Geological Survey - National Wildlife Health Center Wildlife Health Bulletin 2013–2. 2013. [Google Scholar]
- 11.Clark RW, Marchand MN, Clifford BJ, Stechert R, Stephens S. Decline of an isolated timber rattlesnake (Crotalus horridus) population: interactions between climate change, disease, and loss of genetic diversity. Biol Conserv. 2011;144:886–91. doi: 10.1016/j.biocon.2010.12.001. [DOI] [Google Scholar]
- 12.Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol. 1993;2:113–8. doi: 10.1111/j.1365-294X.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 13.Anderson JB, Stasovski E. Molecular phylogeny of northern hemisphere species of Armillaria. Mycologia. 1992;84:505–16. doi: 10.2307/3760315. [DOI] [Google Scholar]
- 14.White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M, Gelfand D, Sninsky J, White T, editors. PCR Protocols: a Guide to Methods and Applications. San Diego, CA: Academic Press; 1990. pp. 315–22. [Google Scholar]
- 15.Integrated DNA Technologies SciTools® Web Tools. [http://www.idtdna.com/pages/scitools]
- 16.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 17.Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Life Technologies. Real-time PCR: Understanding Ct. Carlsbad, CA: Life Technologies Corporation; 2011 [http://tools.lifetechnologies.com/content/sfs/brochures/cms_053906.pdf]
- 19.Muller LK, Lorch JM, Lindner DL, O’Connor M, Gargas A, Blehert DS. Bat white-nose syndrome: a real-time TaqMan polymerase chain reaction test targeting the intergenic spacer region of Geomyces destructans. Mycologia. 2013;105:253–9. doi: 10.3852/12-242. [DOI] [PubMed] [Google Scholar]
- 20.Lorch JM, Gargas A, Meteyer CU, Berlowski-Zier BM, Green DE, Shearn-Bochsler V, et al. Rapid polymerase chain reaction diagnosis of white-nose syndrome in bats. J Vet Diagn Invest. 2010;22:224–30. doi: 10.1177/104063871002200208. [DOI] [PubMed] [Google Scholar]
- 21.Lindner DL, Gargas A, Lorch JM, Banik MT, Glaeser J, Kunz TH, et al. DNA-based detection of the fungal pathogen Geomyces destructans in soils from bat hibernacula. Mycologia. 2011;103:241–6. doi: 10.3852/10-262. [DOI] [PubMed] [Google Scholar]
- 22.Brandt ME, Gaunt D, Igbal N, McClinton S, Hambleton S, Sigler L. False-positive Histoplasma capsulatum Gen-Probe chemiluminescent test result caused by a Chrysosporium species. J Clin Microbiol. 2005;43:1456–8. doi: 10.1128/JCM.43.3.1456-1458.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cano J, Solé M, Pitarch LB, Guarro J. Pseudoamauroascus, a new genus of the Onygenales (Ascomycota) Stud Mycol. 2002;47:175–9. [Google Scholar]