

Abstract
Harnessing the immune adjuvant properties of natural killer T (NKT) cells is an effective strategy to generate anticancer immunity. The objective of this study was to increase the potency and durability of vaccine-induced immunity against B cell lymphoma by combining α-galactosylceramide (α-GalCer)-loaded tumor cell vaccination with an agonistic antibody targeting the immune checkpoint molecule 4–1BB (CD137). We observed potent synergy when combining vaccination and anti-4–1BB antibody treatment resulting in significantly enhanced survival of mice harboring Eμ-myc tumors, including complete eradication of lymphoma in over 50% of mice. Tumor-free survival required interferon γ (IFNγ)-dependent expansion of CD8+ T cells and was associated with 4–1BB-mediated differentiation of KLRG1+ effector CD8+ T cells. 'Cured' mice were also resistant to lymphoma re-challenge 80 days later indicating successful generation of immunological memory. Overall, our results demonstrate that therapeutic anticancer vaccination against B cell lymphoma using an NKT cell ligand can be boosted by subsequent co-stimulation through 4–1BB leading to a sustainable immune response that may enhance outcomes to conventional treatment.
Keywords: α-galactosylceramide, B cell lymphoma, cancer vaccine, hematological malignancies, immunotherapy, immune checkpoint molecule, monoclonal antibody, NKT cells, 4–1BB
Abbreviations: α-GalCer, α-galactosylceramide; cIg: isotype control immunoglobulin; CRs, complete responses; DPEC, double positive effector cells; EEC, early effector cells; KO, knock-out; mAb, monoclonal antibody; MPEC, memory precursor effector cells; SLEC, short-lived effector cells; WT, wild-type.
Introduction
Immunotherapy that can evoke a durable antitumor immune response against hematological cancers may complement conventional treatments by providing ongoing protection against tumor development or post-treatment relapse. Autologous tumor cell vaccination approaches utilizing the synthetic glycolipid immune adjuvant, α-galactosylceramide (α-GalCer), targeting the immunoregulatory properties of Natural Killer T (NKT) cells have proven to be effective at controlling tumor growth by promoting the generation and activities of Natural Killer (NK) cells and T cells.1-7 With the aim to boost antitumor immunity against non-Hodgkin's B cell lymphomas (NHL), we recently showed in the Eμ-myc mouse model of NHL that a vaccine composed of irradiated, α-GalCer-loaded lymphoma cells was sufficient to significantly inhibit growth of established tumors and prolong survival.5 CD8+ T cells were important for the observed therapeutic efficacy, however there was no evidence for prolonged CD8+ T-cell activation or memory formation. This deficiency in developing a persistent vaccine-induced immune response may explain the rapid outgrowth of Eμ-myc lymphoma after an initial period of growth suppression.
Immune checkpoint molecules have been identified as critical regulators of antitumor immunity.8 Tumor necrosis factor receptor superfamily 9 (TNFRSF9), better known as 4-1BB (or CD137) is an inducible, co-stimulatory molecule expressed on a range of leukocytes including activated T cells and NK cells.9-12 Stimulation of 4–1BB by ligand binding increases the proliferation, activation, function and survival of these immune cells.10,11, 13,14 The therapeutic application of agonistic anti-4–1BB antibodies in combination with other anti-cancer agents are reported to be effective against many types of malignancies,15-23 including B cell lymphomas.24,25
Modulating CD8+ T-cell immunity via immunotherapy aims to harness both their potent anticancer effector mechanisms and capacity to form a long-lived memory cell pool, thereby providing persistent immunological protection. Four subsets of CD8+ effector T cells with distinct effector mechanisms and capacities to become memory cells have been previously identified on the basis of the expression of 2 surface markers: killer cell lectin-like receptor G1 (KLRG1) and interleukin 7 receptor α (IL-7Rα), also called CD127.26-28 In response to infection or vaccination, naïve CD8+ T cells become activated and form a pool of early effector cells (EEC) that are KLRG1lowCD127low in phenotype. These EECs then develop into memory precursor effector cells (MPEC) which are KLRG1lowCD127high or short-lived effector cells (SLEC) identified as KLRG1highCD127low, depending on inflammatory mediators and T cell receptor (TCR) engagement.26,29,30 SLEC and MPEC CD8+ T cells are similar in effector functions represented by their ability to produce effector molecules, such as IFNγ, tumor necrosis factor α (TNFα), and granzyme B.29,31 However, MPEC have a higher potential to become memory cells and are capable of initiating a recall response through rapid proliferation and differentiation into other subsets.26,27 Double positive effector cells (DPEC) co-express KLRG1 and IL-7Rα (KLRG1highCD127high). DPECs are long-lived,32 have characteristics of poly-functional effector cells and retain proliferative capacity in response to viral challenge.26-28,33 DPECs have also been observed within tumors of 4–1BB agonist antibody-treated mice,34 however their significance to antitumor immunity is currently unknown.
In this study, we show that combination immunotherapy incorporating an NKT cell-targeting vaccine together with an agonistic anti-4–1BB antibody led to complete eradication of established Eμ-myc lymphomas in over 50% of mice. This enhanced antitumor immune response was a result of effective generation, differentiation and expansion of effector CD8+ T cells, defined by KLRG1 and IL-7Rα co-expression.
Results
Therapeutic vaccination with irradiated, α-GalCer-loaded tumor cells in combination with agonistic anti-4–1BB antibody treatment leads to clearance of Eμ-myc lymphoma
Previously we reported transient therapeutic efficacy of an irradiated, α-GalCer-loaded tumor cell vaccine against murine Eμ-myc B cell lymphoma.5 An agonistic anti-4–1BB mAb (clone 3H3) was administered to determine whether vaccine-induced anti-tumor immunity could be enhanced by co-stimulation through 4–1BB (Fig. 1A). Anti-4–1BB mAb treatment alone was sufficient to suppress the growth of established 4242 tumors, to a similar extent as single vaccination, leading to enhanced survival (Fig. 1B). Therapeutic efficacy was significantly enhanced by the combination of vaccine with anti-4–1BB mAb treatment, leading to long-term inhibition of Eμ-myc tumor growth as evinced by tumor-free survival in 50% of mice beyond 100 d (Figs. 1B–C). When surviving mice were re-challenged with 4242 Eμ-myc tumors at day 80, protection against tumor outgrowth was evident, despite receiving no additional treatments. Fifty percent of re-challenged mice were completely protected against Eμ-myc tumor development and remained tumor-free for at least a further 100 d (Fig. 1D). Additionally, the combination therapy was also very effective against Eμ-myc tumor clone 299, in which 100% of mice achieved complete remissions (Fig. S1). When treatment was delayed until day 15, a time of significantly disseminated tumor burden, combination therapy continued to suppress lymphoma outgrowth, whereas vaccine treatment alone was no longer effective (Fig. S1C). Collectively this data shows that treatment utilizing an NKT cell-targeting vaccine in conjunction with an agonistic anti-4–1BB mAb leads to more effective and persistent antitumor immunity.
Figure 1.
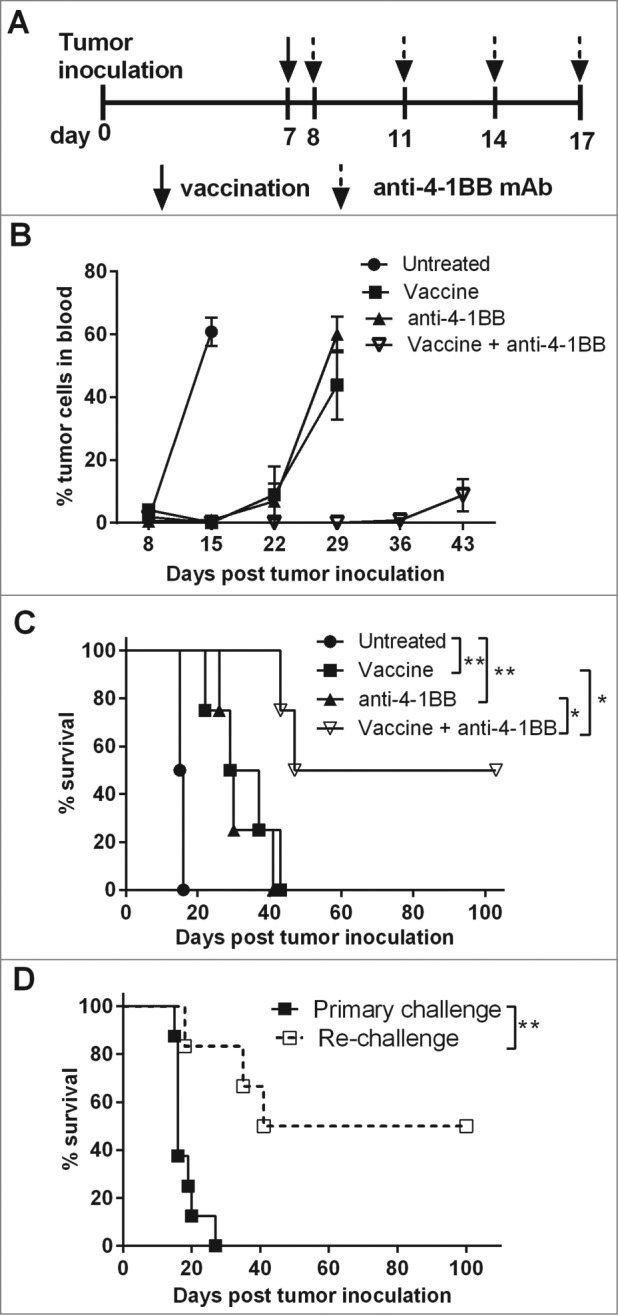
Enhanced suppression of established Eμ-myc 4242 tumor growth in mice treated with a combination of α-GalCer-loaded tumor vaccine and anti-4–1BB antibody. (A) C57BL/6 wild-type (WT) mice were challenged with 1 × 105 GFP-expressing Eμ-myc 4242 tumor cells and then treated with single vaccination and/or anti-4–1BB mAb on the days indicated. (B) Mean ± SEM (n = 4 per group) Eμ-myc 4242 tumor burden in blood after designated treatments. (C) Overall survival of mice after receiving the indicated treatments. **P < 0.01; *P < 0.05, log-rank test. (D) Mice that had previously been treated with vaccine + anti-4–1BB mAb and showed tumor-free survival of at least 75 d were re-challenged with 1 × 105 Eμ-myc 4242 tumor cells and overall survival is shown compared to naive mice that received an equivalent number of tumor cells as a primary challenge (n = 6–8). **P < 0.01, log-rank test. Representative data from 3 independent experiments is shown for A–C. Pooled data from 2 independent experiments are shown for D.
α-GalCer-loaded tumor cell vaccination leads to rapid induction of 4–1BB surface expression on a range of activated lymphocytes
Lymphocyte activation and 4–1BB surface expression was assessed following vaccination in lymphoma-bearing mice to predict which cells were likely to be targeted by anti-4–1BB mAb treatment. Excluding tumor cells from analysis, the overall percentage of peripheral blood cells expressing surface 4–1BB increased from 0.53 ± 0.08% to 2.84 ± 0.58% (mean ± SEM) within 24 h of vaccination in tumor-bearing mice (Fig. 2A). NK cells and non-CD8+ T cells contributed mostly to the total 4–1BB positive cell population at day 1 post vaccination in tumor-bearing mice (Fig. 2B, upper graph). These cells along with B cells also contributed to the 4–1BB positive population at day 13 when overall expression levels had returned back to baseline (Fig. 2A and 2B, lower graph). The percentage of activated CD69+ 4–1BB+ cells were significantly increased in all lymphocyte populations examined (Fig. 2C). The lymphocyte subsets exhibiting the greatest fold increase in the mean percentages of CD69+ 4–1BB+ cells after vaccination were NK cells (24.8 fold) followed by CD8+ T cells (6.8 fold).
Figure 2.

Vaccination increases the percentages of activated, 4–1BB-expressing lymphocytes. C57BL/6 wild-type (WT) mice were either challenged with 1 × 105 Eμ-myc 4242 tumor cells or left tumor free, a group of each were then vaccinated on day 7 with α-GalCer-loaded tumor cells only. (A) The percentage of total peripheral blood cells (excluding tumor cells) that expressed surface 4–1BB on the indicated day post-vaccination. (B) The proportions of the indicated lymphocyte populations that constitute the total 4–1BB-expressing cell population in (A). Top pie chart refers to populations from the day 1 tumor-vaccine group. Bottom pie chart shows the equivalent group on day 13. 'Other' T cells refer to the CD8+ negative T cell population, which also excludes natural killer T (NKT) cells. (C) The percentages of activated 4–1BB+CD69+ lymphocytes 1 day after vaccination (+Vacc) compared to untreated tumor-bearing mice (Unt) and non-tumor-bearing controls (No tumor). Data in A-C show means ± SEM, n = 4–6 per group; *P < 0.05; **P < 0.01; ***P < 0.001, unpaired t-test.
Combination immunotherapy drives IFNγ-dependent expansion of CD8+ T cells in tumor-bearing organs
Both NK cells and CD8+ T cells are important antitumor effector cells in vaccine-induced immunity against Eμ-myc lymphoma, and the effectiveness of vaccination is dependent on IFNγ production.5 The numbers of these cells were monitored at sites of lymphoma burden following anti-4–1BB mAb treatment with, or without, prior vaccination. Two weeks after treatment initiation, anti-4–1BB mAb significantly reduced the numbers of NK cells, particularly in the spleen and blood (Fig. 3A). Conversely, anti-4–1BB mAb treatment alone was sufficient to increase CD8+ T-cell numbers in each organ and, the combination of vaccine and antibody treatment significantly increased the expansion of CD8+ T cells in the lymph nodes and spleen (Fig. 3B). This expansion was significantly inhibited in IFNγ knockout (KO) mice, indicating that optimal CD8+ T-cell expansion following anti-4–1BB mAb treatment is dependent on IFNγ production (Fig. 3C). In addition, tumor antigen-specific CD8+ T cells also expanded in response to combination treatment (Fig. S2).
Figure 3.

Combination therapy increases the expansion of CD8+ T cells in tumor-bearing organs. C57BL/6 wild-type (WT) mice were challenged with 1 × 105 Eμ-myc 4242 tumor cells and given the indicated treatments commencing on day 7 (n = 6 per group). The absolute numbers of NK cells (A) and CD8+ T cells (B) at day 19 post-tumor inoculation are shown for blood (left column), inguinal lymph node (middle column) and spleen (right column). Representative data from 3 independent experiments is shown. (C) WT or IFN-γ knockout (KO) mice inoculated with 1 × 105 Eμ-myc 4242 tumor cells were treated commencing on day 7 with vaccine plus anti-4–1BB mAb or left untreated and CD8+ T cells enumerated on day 15 (n = 4 , per group). All data show means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant, unpaired t-test.
Direct 4–1BB signaling promotes CD8+ T-cell proliferation and effector function in vitro
CD8+ T cells were purified from spleens of Eμ-myc lymphoma-bearing WT and IFNγKO mice to determine whether agonistic anti-4–1BB mAb treatment in vitro could directly promote CD8+ T-cell activity. Some mice received vaccination 24 h prior to organ harvest to determine the requirement for prior activation of CD8+ T cells for the effects of anti-4–1BB mAb signaling. At this time-point, both WT and IFNγKO mice had similar tumor burdens. IFN-γ secretion was used as a functional CD8+ T-cell response to antibody treatment. As expected, splenocytes isolated from vaccinated mice secreted significant amounts of IFNγ ex vivo, which was potentiated by the addition of anti-4–1BB mAb to the culture (Fig. 4A). Anti-4–1BB mAb given directly to CD8+ T cells promoted a significant increase in IFNγ production (Fig. 4B). IFNγ levels were further elevated if CD8+ T cells were previously exposed to vaccination suggesting a combined effect of vaccination and anti-4–1BB mAb treatment (Fig. 4B). Both in vivo vaccine treatment and in vitro anti-4–1BB mAb treatment enhanced the proliferation of CD8+ T cells to a similar extent over 3 d of culture (Fig. 4C). Combining these therapies provided no additional proliferative effect, possibly because CD8+ T cells were already reaching maximal detectable proliferation levels with individual treatments. Vaccine-induced CD8+ T cell proliferation was dependent on IFNγ, as shown by the lack of enhanced proliferation among CD8+ T cells isolated from vaccine-treated IFNγKO mice (Fig. 4C). Furthermore, anti-4–1BB mAb treatment also induced sub-optimal proliferation of IFNγKO cells (Fig. 4C). Taken together, these data suggest that 4–1BB signaling directly on CD8+ T cells from lymphoma-bearing mice is capable of upregulating proliferative capacity and effector function. Vaccination enhances this effect, likely via induction of IFNγ, as previously shown in vivo.5
Figure 4.

Direct 4–1BB signaling promotes CD8+ T cell effector function and proliferation in vitro. C57BL/6 wild-type (WT) mice or interferon γ (Ifn) knockout (IFNγKO) mice were challenged with 1 × 105 Eμ-myc 4242 tumor cells, some of which were then vaccinated on day 7 (n = 3 per group). Whole splenocytes (A) or MACS-purified CD8 T cells (B, C) were isolated one day post-vaccination and cultured in vitro for 3 d, supplemented with IL-2, with or without addition of 5 μg/mL anti-4–1BB monoclonal antibody (mAb). (A–B) IFNγ secretion into the culture supernatant was measured by ELISA. (C) Cell proliferation was calculated from levels of carboxyfluorescein succinimidyl ester (CFSE) dilution indicated as the percentage of CFSE low cells relative to the undivided CFSE-labeled lymphocyte peak. All data show mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant, unpaired t-test).
CD8+ T cells are required for enhanced antitumor activity mediated by the addition of anti-4–1BB mAb treatment
To determine whether NK cells and/or CD8+ T cells contribute to the enhanced therapeutic efficacy conferred by combination treatment, these cells were depleted from Eμ-myc-tumor-bearing mice 1 d prior to treatment. This early depletion of either NK cells or CD8+ T cells was sufficient to completely abrogate the therapeutic effect (Fig. 5A–B). Early depletion of these lymphocytes may simply be inhibiting the vaccine-induced component of therapy, as previously shown.5 To distinguish the degree to which each population contributes to the prolonged therapeutic effect mediated by anti-4–1BB mAb treatment, depletion of NK cells and CD8+ T cells was delayed in subsequent experiments until 6 d after vaccination. In this setting, depletion of CD8+ T cells significantly abrogated the capacity for combination therapy to suppress tumor outgrowth, whereas NK cell depletion at this later point had no effect on tumor growth suppression (Fig. 5C). Furthermore, transfer of CD8+ T cells isolated from combination treated mice was sufficient to enhance the survival of naïve mice challenged with Eμ-myc tumors (median survival - 63 days), as compared with mice receiving CD8+ T cells transferred from untreated donors (median survival - 20 days) (Fig. 5D). Therefore, engaging CD8+ T cells appears to be critical for the enhanced therapeutic effect of combination therapy.
Figure 5.
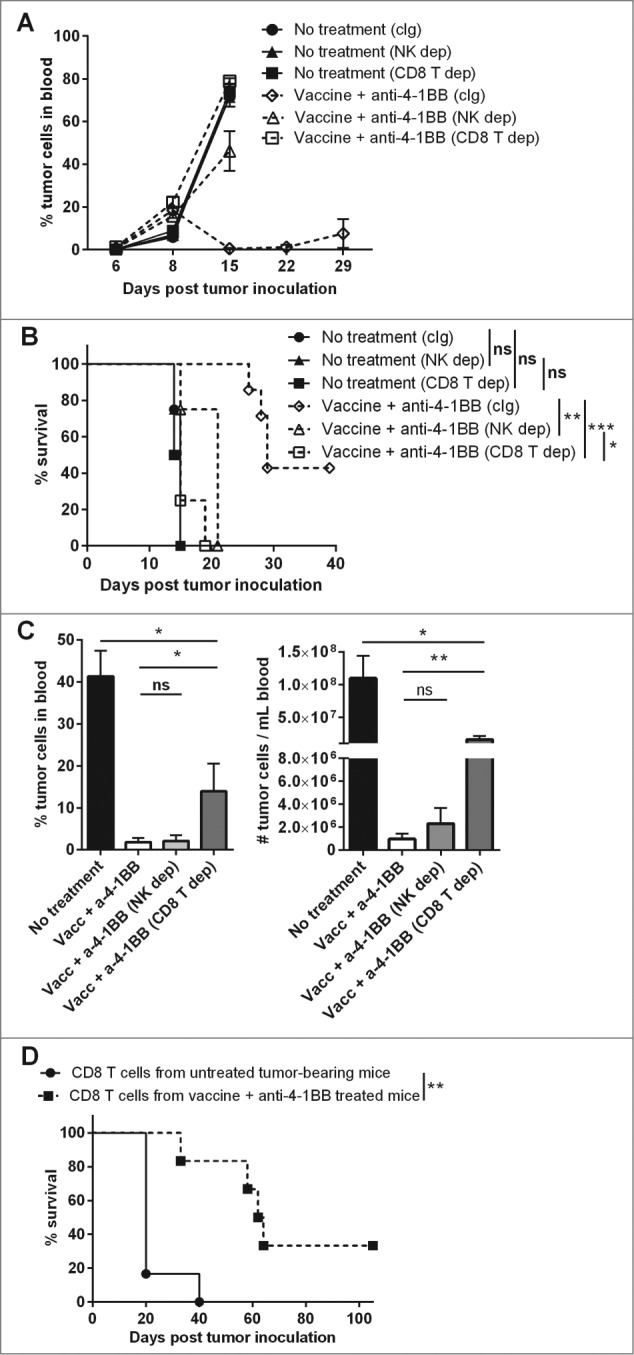
CD8+ T cells are required for enhanced treatment efficacy mediated by anti-4–1BB mAb. C57BL/6 wild-type (WT) mice were challenged with 1 × 105 Eμ-myc 4242 tumor cells and treated on d 6 onwards with a combination of vaccine and anti-4–1BB monoclonal antibody (mAb), or left untreated (n = 4–7 per group). As indicated, untreated and treated groups received mAb-based depletion of natural killer (NK) cells with anti-asialo-GM1 (NK dep), CD8+ T cells with anti-CD8β (CD8 T dep) or an isotype control mAb, 2A3 (cIg). For (A-B) cell depletion commenced on day 5. For (C) cell depletion was delayed and commenced on day 11. (A) Eμ-myc tumor burden in blood over time (mean ± SEM). (B) Overall survival of mice after receiving the indicated treatments (***P < 0.001; **P < 0.01; *P < 0.05; ns = not significant, log-rank test). (C) The percentage (left graph) and numbers (right graph) of tumor cells in blood at day 19 of mice receiving the indicated treatments (means ± SEM). (**P < 0.01; *P < 0.05; unpaired t-test). (D) Overall survival of mice that received 1 × 105 CD8+ T cells isolated from untreated or combination treated donor mice 1 d prior to Eμ-myc 4242 tumor cell inoculation; **P < 0.01, log-rank test. Data in A, B and C are representative of 2 independent experiments.
Anti-4–1BB mAb treatment induces the differentiation of IFNγ-producing KLRG1+ CD127+ effector CD8+ T cells
To address the capacity for anti-4–1BB mAb treatment to differentiate CD8+ T-cell populations, the generation of CD8+ T-cell subsets defined by KLRG1 and CD127 expression was assessed in lymphoma-bearing mice after treatment (Fig. 6A). Treatment with anti-4–1BB mAb alone significantly increased the proportion of KLRG1+ CD127+ (DPEC), and to a lesser extent KLRG1+ CD127- (SLEC) CD8+ T cells, which occurred in both CD62L+ (Fig. 6B) and CD62L- subsets (Fig. 6C). An equivalent decrease in the proportion of KLRG1- CD127+ (MPEC) CD8+ T cells was observed over this period (Figs. 6B–C). Vaccination did not further increase differentiation conferred by anti-4–1BB mAb treatment alone. Functionally, the majority of SLEC and DPEC CD8+ T cells produced IFN-γ following combination therapy and at significantly greater levels than KLRG1 negative subsets (MPEC and EEC) (Figs. 7A-B). In addition, the KLRG1 positive subsets (SLEC and DPEC) contained a significant proportion of IFNγ-producing cells that were also actively proliferating in response to combination therapy, as measured by in vivo BrdU incorporation (Fig. 7C). Of note, the expansion of individual DPEC and SLEC CD8+ T cell subsets was inhibited in IFNγKO mice (Fig. S3A). However, the differentiation of CD8+ T cells into DPEC and SLEC populations was not overtly affected by the absence of IFN-γ (Fig. S3B). Finally, CD8+ T-cell subsets generated in combination therapy treated mice and individually transferred into naïve mice did not suppress Eμ-myc tumor growth or prolong survival, indicating the absence of any protective anti-tumor effect when these CD8+ T-cell subsets were given in isolation (Fig. S4).
Figure 6.
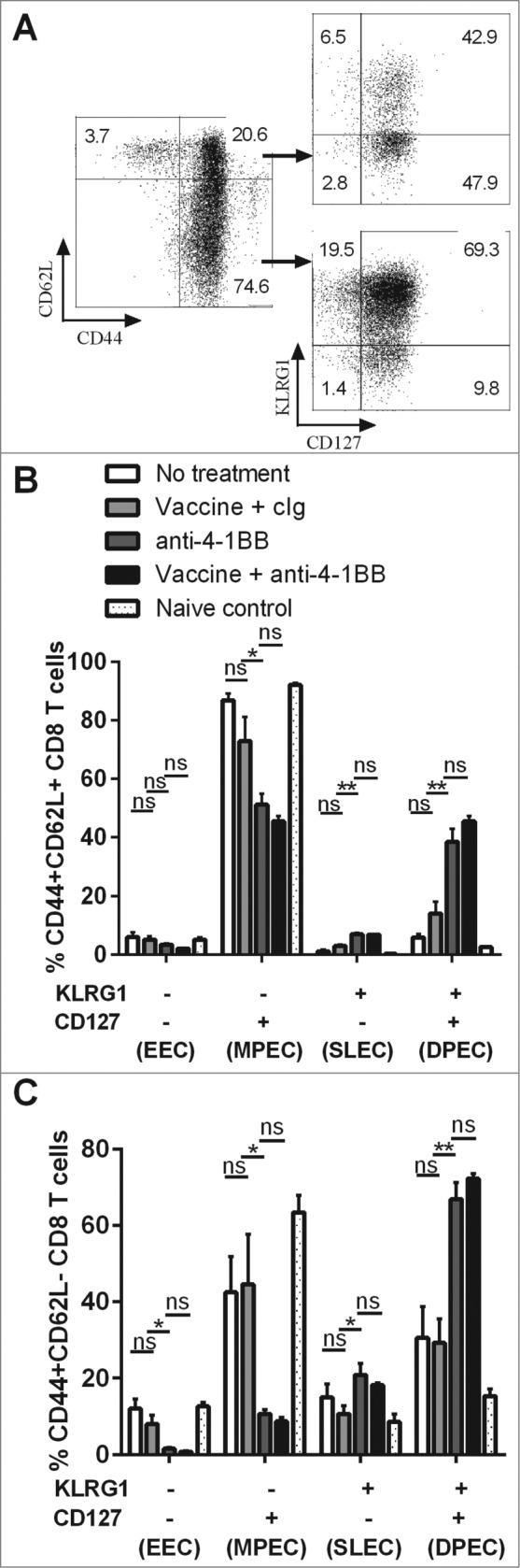
Differentiation of CD8+ Teffector cell subsets by anti-4–1BB mAb treatment. C57BL/6 wild-type (WT) mice were challenged with 1 × 105 Eμ-myc 4242 tumor cells and given the indicated treatments commencing on day 6 (n = 4 per group). Peripheral blood lymphocytes from treated and untreated mice were subject to immunofluorescence staining and cytofluorimetric analysis. (A) Representative flow cytometry plots analyzing lymphocytes from the blood of a tumor-bearing mouse treated with combination therapy showing the gating strategy used to identify CD8+ T cells subsets. (B) The proportions of CD44+ CD62L+ CD8+ T cells and (C) the proportions of CD44+ CD62L- CD8+ T cells expressing surface KLRG1 and/or CD127 from blood cells derived from tumor-bearing mice at day 21 after receiving the indicated treatment. Data in B and C show means ± SEM; *P < 0.05; **P < 0.01; ns = not significant, unpaired t-test. Representative data from 3 independent experiments are shown.
Figure 7.
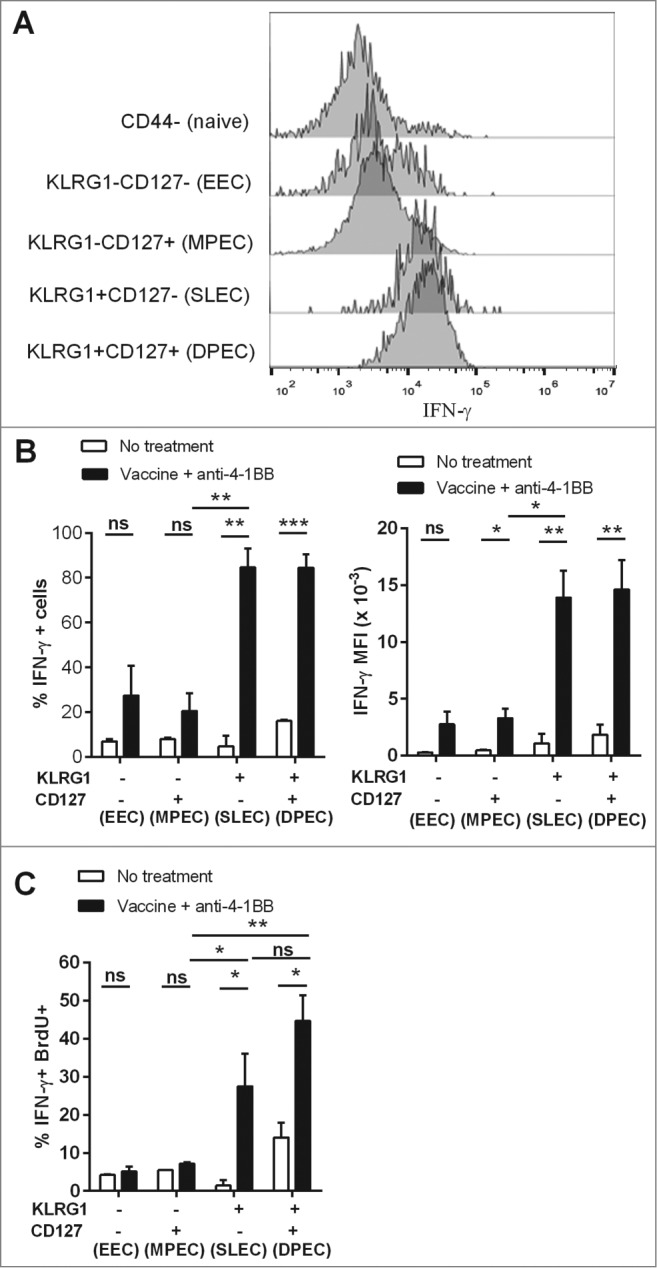
KLRG1+ CD8+ T cell subsets from combination treated mice have enhanced proliferation and IFNγ production. C57BL/6 wild-type (WT) mice were challenged with 1 × 105 Eμ-myc 4242 tumor cells and given combination treatment commencing on day 6 (n=4 per group), or left untreated. Splenic preparations from treated and untreated mice were subject to immunofluorescence staining and cytofluorimetric analysis. (A) Representative flow cytometry histograms of intracellular interferon γ (IFNγ) levels in the different activated CD8+ T-cell subsets (gated from total CD44+ cells) and naive CD8+ T cells (CD44-) isolated from the spleen of a combination treated tumor-bearing mouse at day 19. (B) The percentage of each CD8+ T cell subset producing IFNγ (left graph) and mean fluorescent intensity (MFI) of IFNγ expression on IFNγ+ cells (right graph) from the spleens of untreated or combination treated mice at day 19 post-tumor inoculation. (C) The percentage of CD8+ T-cell subsets that produced IFNγ and incorporated BrdU in vivo. Data in B and C show means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant, unpaired t-test.
Discussion
An ongoing challenge is the development of combination immunotherapeutic strategies that reduce the incidence of tumor resistance and combat counter-regulatory mechanisms within the context of a suppressed immune system, an environment that naturally jeopardizes the effectiveness of antitumor immunity. We have recently developed a whole tumor cell-based therapeutic vaccine against NHL that targets the immune adjuvant properties of NKT cells.5 Initial suppression of Eμ-myc B cell lymphoma upon single vaccination with α-GalCer-loaded, irradiated tumor cells was found to be due to elicitation of a potent innate immune response, evinced by rapid NKT cells and NK cell activation and IFNγ production.5 We also discovered that CD8+ T cells were important for the observed therapeutic efficacy, however we could not find strong evidence for prolonged CD8+ T-cell activation or memory formation. This lack of effective generation or persistence of CD8+ T-cell immunity led us to investigate the combination of vaccine with 4–1BB co-stimulation using an agonistic anti-4–1BB mAb. Previous reports have indicated that targeting 4–1BB can promote the proliferation, activity and survival of lymphocytes, including CD8+ T cells.10,11,13,14 In addition, Teng et al., 2007 showed that injecting agonistic anti-4–1BB mAb together with soluble α-GalCer suppressed growth of mammary tumors and renal cell carcinomas in BALB/c mice in an NKT cell and in a CD8+ T cell-dependent manner, although in this experimental model anti-DR5 mAb was also required for effective therapy.35 In the current study, vaccination stimulated an increase in 4–1BB surface expression on a range of activated lymphocytes, with the greatest induction observed in NK cells and CD8+ T cells, provided strong rationale for this combinatorial immunotherapy approach. Of note, ‘other’ T cells (non-CD8+, non-NKT) were also shown to express 4–1BB in this setting and we have not ruled out the possibility that CD4+ T cells may therefore contribute to antitumor immunity elicited by this combination immunotherapeutic approach, as previously shown for other α-GalCer-based tumor cell vaccinations.1,3,7 However, our previous work in the Eμ-myc lymphoma model did not reveal a significant role for CD4+ T cells in vaccine-induced immunity 5 such that CD4+ T cells were not investigated further in the current study.
We observed potent treatment synergy with the vaccine / anti-4–1BB mAb combination resulting in significantly enhanced survival of mice harboring Eμ-myc tumors, including durable complete responses (CRs) in 50% to 100% of mice, depending on the clone of Eμ-myc tumor targeted. Ongoing studies investigating the efficacy of this combination treatment strategy against a range of spontaneous Eμ-myc lymphoma clones arising in transgenic mice may help decipher why some tumors are more sensitive to combination immunotherapy than others.
Elimination of lymphoma burden in combination treated mice correlated with increased numbers and persistence of CD8+ T cells located in tumor-bearing organs. Consistent with our previous observations,4,5 α-GalCer-driven vaccination alone was insufficient to effectively drive subsequent CD8+ T-cell expansion in vivo. Anti-4–1BB mAb treatment, however, led to increases in CD8+ T-cell numbers in blood, spleen and lymph nodes of tumor-bearing mice, which was significantly enhanced with vaccination. This suggests that 4–1BB signaling promotes the proliferation and survival of activated CD8+ T cells that are generated by α-GalCer vaccination. CD8+ T-cell expansion conferred by combination treatment was largely dependent upon the presence of IFNγ, providing a critical link between vaccine-induced IFNγ production and expansion of CD8+ T cells in response to anti-4–1BB mAb treatment. CD8+ T cells from lymphoma-bearing mice proliferated and produced IFNγ in response to anti-4–1BB mAb treatment in vitro. This data does not rule out the possibility that anti-4–1BB mAb may also indirectly promote CD8+ T-cell activity in vivo via signaling through other 4–1BB positive cells; however it shows that CD8+ T cells taken from the tumor environment have the capacity to respond directly to anti-4–1BB mAb.
Consistent with a recent report showing 4–1BB-mediated expansion of human tumor antigen-specific CD8+ T cells,36 OT-I T cell antigen-specific responses to Eμ-myc lymphoma cells expressing ovalbumin expanded to a similar degree as the total CD8+ T cell pool in response to anti-4–1BB mAb treatment. The extent to which this combination therapy drives antigen-specific versus non-antigen specific bystander CD8+ T-cell expansion could not be resolved in this study as the repertoire of tumor-reactive T cells against Eμ-myc lymphomas is unknown. Notably, although vaccination induced rapid NK cell activation, NK cells were substantially depleted in all major secondary lymphoid organs following anti-4–1BB mAb treatments, consistent with that observed in another study utilizing in vivo treatment of anti-4–1BB mAb in mice.37 This result also indicated that generation and persistence of CD8+ T cells, rather than NK cells, were important for the sustained therapeutic response elicited by the combination therapy approach. We observed that combination therapy failed to suppress Eμ-myc tumor growth when NK cells and CD8+ T cells were depleted prior to commencement of therapy, while at the later stages of therapy only CD8+ T-cell depletion led to loss of tumor control. We suggest from these observations that a bi-phasic immune response is elicited by combination treatment. Initially, administration of α-GalCer-loaded tumor cells induces rapid activation of NKT cells and NK cells, which are responsible for high levels of IFNγ production, killing of lymphoma cells and priming of CD8+ T cells via NKT cell-mediated maturation of antigen presenting cells (APCs).2,5,7 Then, co-stimulation provided by agonistic anti-4–1BB mAb treatment drives the expansion of CD8+ T cells and promotes their activity and survival, processes critical for durable immune protection against the lymphoma.
KLRG1 expression on T cells has been identified as a useful marker for monitoring the antitumor response elicited by anti-4–1BB mAb therapy.38,39 KLRG1+ CD8+ T cells induced by 4–1BB agonistic antibody express elevated levels of cytotoxicity-associated molecules, such as granzymes, perforin and Fas-Ligand, factors previously shown to correlate with enhanced killing of B16 melanoma cells.34 In the Eμ-myc lymphoma model, we found that anti-4–1BB mAb treatment induced the expression of KLRG1 in CD8+ T cells, indicating differentiation into effector cells, and that a large proportion of these cells retained co-expression of CD127 (Fig. 6). This occurred for both classical effector memory (CD44+ CD62L−) and central memory (CD44+ CD62L+) phenotypes. These KLRG1+ CD127+ CD8+ T cells, referred to as DPEC,26 resembled the phenotype of SLEC cells in this model with comparable levels of IFNγ production and proliferative capacity. Interestingly, IFNγ was not required for differentiation of CD8+ T cells into these subsets, but was critical for their expansion. Although the potential of DPEC cells to contribute to the memory pool of antitumor CD8+ T cells remains to be elucidated, we propose that they may represent a long-lived effector cell population with sustained proliferative capacity and memory potential.28
Eu-myc tumor-targeted immunological memory was successfully generated by our combinatorial immunotherapeutic approach as shown by resistance to tumor re-challenge more than 80 d after mice had cleared their primary tumor. Memory formation specifically within the CD8+ T-cell pool was evinced by the protective effect afforded by transfer of bimodal therapy-induced CD8+ T cells into naive mice challenged with Eμ-myc lymphoma. Transfer of CD8+ T cells alone was sufficient to control tumor growth and significantly enhance survival in this setting. We were unable to reproduce this protective effect using individual CD8+ T effector cell subsets (based on KLRG1 and CD127 expression) were transferred at equivalent numbers suggesting either that lymphoma control is mediated by multiple CD8+ T-cell subsets, or alternatively, that in vitro manipulation required for lymphocyte isolatation disrupted their functional capacity or viability in vivo.
Targeting 4–1BB co-stimulation has proven to be an effective form of immunomodulation to promote antitumor immunity and potentiate the therapeutic effects of tumor- and immune-targeting antibodies against lymphoma24,25,40 and other cancers.21,22 In this study we show that anti-4–1BB mAb treatment significantly improves the efficacy of a cellular vaccine strategy that targets the immune adjuvant properties of NKT cells. We believe the efficacy of this combination strategy is a result of engaging cells of both the innate and adaptive arms of the antitumor immune response allowing for a rapid, multi-pronged attack of the tumor and generation of a durable immune response by formation of immunological memory. The outcome is complete clearance of established and aggressive myc-oncogene driven B cell lymphomas in mice and we predict these outcomes will extend to other cancers that are susceptible to immune-mediated killing.
The frequency and functional capacity of human NKT cells in hematological malignancies remains inconclusive and therefore the translatability of this vaccine approach requires further investigation. Most previous studies addressing this have focused on ‘NKT-like’ cells rather than specifically identifying classical CD1d-restricted Vα24+/Vβ11+ NKT cells,41 making it difficult to draw conclusions. At least in the settings of multiple myeloma42 and B cell chronic lymphocytic leukemia,43 NKT cell frequency appears comparable to those of healthy individuals and their function is either intact or, if defective, reparable by intervention.
Methods
Mice
Inbred C57BL/6 wildtype (WT) mice were purchased from the Animal Resources Center - ARC (Perth, Australia). CD45.1 congenic OT-I mice (B6.SJLxOT-I) and Ifnγ gene-targeted knockout (IFN-γKO) mice, both on the C57BL/6 background, were bred and maintained onsite at the Translational Research Institute Biological Research Facility. Mice were used at the ages of 6–10 weeks, sex-matched and housed under specific pathogen-free conditions. All experiments were conducted following the animal ethics guidelines provided by the National Health and Medical Research Council of Australia and approved by the University of Queensland – Health Sciences Animal Ethics Committee.
B cell lymphoma tumor model
Eμ-myc transgenic mice develop B cell lymphomas due to constitutive expression of c-Myc and is a model used to represent human NHL.44 Eμ-myc clones 299 and 4242 used for transplantation were freshly isolated lymphomas from lymph nodes of Eμ-myc transgenic mice, and stably transduced GFP or ovalbumin (OVA)-expressing lines were engineered by retroviral transduction of these cells as previously described.45 Eμ-myc cells were maintained in high-glucose modified DMEM media supplemented with 10% fetal calf serum (FCS) and 20 μM 2-mercaptoethanol. WT or IFNγKO mice were inoculated intravenously (i.v.) with 1 × 105 tumor cells, unless otherwise indicated. Tumor growth was monitored by measuring GFP+ cell events in the peripheral blood by flow cytometry.
Reagents and flow cytometry
α-GalCer was purchased from Avanti Polar Lipids (Alabaster, Alabama). α-GalCer-loaded CD1d tetramer was generously provided by D. Godfrey (University of Melbourne, Australia). Fluorochrome-conjugated anti-mouse monoclonal antibodies (mAbs) to CD3e (145–2C11), CD8α (53–6.7), CD8β (YTS156.7.7), CD19 (1D3 or 6D5), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD62L (MEL-14), CD69 (H1.2F3), CD127 (A7R34), CD137/4-1BB (17B5), IFNγ (XMG1.2), KLRG1 (2F1/KLRG1), NK1.1 (PK136) and TCRβ (H57–597) and associated isotype control antibodies were purchased from Biolegend (San Diego, CA), BD Biosciences (San Diego, CA), or eBioscience (San Diego, CA). Cells were antibody-labeled at predetermined optimal concentrations of mAb for 25 min at 4°C in PBS containing 2% FCS and 2 mM EDTA. Flow-count fluorospheres (Beckman Coulter) were added to the samples to calculate cell numbers upon acquisition. Intracellular cytokine staining was preceded by addition of Golgiplug (BD Biosciences) to the cells for 4 h to prevent cytokine release from the Golgi/endoplasmic reticulum complex. Cells were permeabilised and fixed using BD Cytofix/Cytoperm kit, following the manufacturer's instructions (BD Biosciences). Labeled cells were acquired on Gallios (Beckman Coulter), LSR-II or FACS Canto flow cytometers (BD Biosciences) and analyzed using Kaluza version 1.2 (Beckman Coulter) or FlowJo version 7.6.3 (Tree Star, Ashland, OR) software.
Combination immunotherapy
For vaccine preparation, autologous Eμ-myc tumor cells were loaded with 500 ng/mL of α-GalCer overnight in culture. The α-GalCer-loaded tumor cells were then irradiated with 5000 cGy to arrest proliferation of the tumor cells prior to administration. The vaccine treatment regimen comprised of single i.v. administration of 5 × 105 cells per recipient animal, given upon detection of disseminated tumor burden (∼1–5% GFP+ events in peripheral blood). For therapeutic antibody treatments, mice received 3 or 4 intraperitoneal (i.p.) injections of 100 μg anti-4–1BB (3H3) mAb or 100 μg control Ig (cIg; Rat IgG2a, 2A3) (both from Bio-X-cell, West Lebanon, NH) at day +1, day +4, day +7, and day +10 relative to vaccination.
Lymphocyte depletion in vivo
For in vivo depletion of NK cells and CD8+ T cells, 100 μg anti-asialo-GM1 (Wako Chemicals, Richmond, VA) or 200 μg anti-CD8β (53.5.8) (Bio-X-cell, West Lebanon, NH) mAbs, respectively, were administered by i.p. injection, to combination therapy recipient mice either at day -1, day +1, and day +8, or at day +6, day +9, day +12 relative to vaccination. For controls, equivalent doses of 2A3 cIg were administered.
Detection of IFNγ
IFNγ levels in mouse sera and culture supernatants were detected using an ELISA kit from R&D Systems (Minneapolis, MN) according to the manufacturer's instructions. Acquisition and analysis was performed on a Multiskan FC Photometer (Thermo Scientific, Waltham, MA).
CD8+ T-cell proliferation, isolation and adoptive transfer
To assess CD8+ T-cell proliferation in vivo, 200 μL of 5-bromodeoxyuridine (BrdU) labeling reagent (Life Technologies, Carlsbad, CA) was administered by i.p. injection. After 2 h, splenocytes were harvested, fixed and permeabilised using Nuclear Factor Fixation and Permeabilisation buffers (Biolegend). The samples were then labeled with anti-BrdU mAb (BU20A; eBioscience) and analyzed by cytofluorimetric analysis. For in vitro assays, splenocytes or purified CD8+ T cells were labeled with 2.5 μM of carboxyfluorescein succinimidyl ester (CFSE) and 3 d later CFSE dilution was measured by fluorescence cytometry. Proliferation was calculated as the percentage of CFSE low cells relative to the undivided CFSE-labeled immune cell peak fluorescence. For purification and transfer of total CD8+ T cells, a magnetic-activated cell sorting (MACS) CD8+ T-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany), was used to isolate an 'untouched' CD8+ T-cell population with > 90% purity. Purified cells were injected i.v. at equal numbers to groups of recipient mice 1 d prior to tumor challenge.
Statistical analysis
Results are expressed as the mean ± SEM. Kaplan–Meier plots were used to analyze mouse survival and a log-rank test was performed to assess the statistical significance of differences between survival curves. For all other data for which statistics were performed, a 2-tailed, unpaired t test was used for assessment of differences between groups (GraphPad Prism 5 Software, San Diego, CA). P values <0.05 were considered significant.
Supplementary Material
Acknowledgments
The authors thank Rebecca West for maintenance of the mice, Michelle Kappler and Ben Harvie for mouse technical assistance and Dalia Khalil for FACS sorting assistance. Prof Ricky Johnstone (Peter MacCallum Cancer Center) for providing the Eμmyc tumor cells and Prof Dale Godfrey (Melbourne University) for providing CD1d-tetramer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Project Grant (APP1044355) from the National Health and Medical Research Council (NHMRC) of Australia and a Grant-in-Aid awarded by the Leukemia Foundation of Australia. S.R.M. was supported by an NHMRC Career Development Fellowship (APP1061429).
Authorship Contributions
TK designed and performed experiments, data analysis and assisted with writing of the manuscript. BLD and RCR assisted with experimentation, data analysis and provided experimental feedback. GRL provided critical input throughout the study and critical review of the manuscript. SRM conceived the study, designed and supervised the experiments, assisted with data analysis and wrote the manuscript.
References
- 1. Chung Y, Qin H, Kang CY, Kim S, Kwak LW, Dong C. An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood 2007; 110:2013-9; PMID:; http://dx.doi.org/ 10.1182/blood-2006-12-061309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gibbins JD, Ancelet LR, Weinkove R, Compton BJ, Painter GF, Petersen TR, Hermans IF. An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood; 2014.124(19):2953-63; PMID:; http://dx.doi.org/ 10.1182/blood-2014-04-568956 [DOI] [PubMed] [Google Scholar]
- 3. Hunn MK, Farrand KJ, Broadley KW, Weinkove R, Ferguson P, Miller RJ, Field CS, Petersen T, McConnell MJ, Hermans IF. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin Cancer Res 2012; 18:6446-59; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0704 [DOI] [PubMed] [Google Scholar]
- 4. Mattarollo SR, Steegh K, Li M, Duret H, Foong Ngiow S, Smyth MJ. Transient Foxp3(+) regulatory T-cell depletion enhances therapeutic anticancer vaccination targeting the immune-stimulatory properties of NKT cells. Immunol Cell Biol 2013; 91:105-14; PMID:; http://dx.doi.org/ 10.1038/icb.2012.58 [DOI] [PubMed] [Google Scholar]
- 5. Mattarollo SR, West AC, Steegh K, Duret H, Paget C, Martin B, Matthews GM, Shortt J, Chesi M, Bergsagel PL, et al. . NKT cell adjuvant-based tumor vaccine for treatment of myc oncogene-driven mouse B-cell lymphoma. Blood 2012; 120:3019-29; PMID:; http://dx.doi.org/ 10.1182/blood-2012-04-426643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimizu K, Goto A, Fukui M, Taniguchi M, Fujii S. Tumor cells loaded with α-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol 2007; 178:2853-61; http://dx.doi.org/ 10.4049/jimmunol.178.5.2853 [DOI] [PubMed] [Google Scholar]
- 7. Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-presentation of glycolipid from tumor cells loaded with α-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med 2007; 204:2641-53; PMID:; http://dx.doi.org/ 10.1084/jem.20070458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim DH, Chang WS, Lee YS, Lee KA, Kim YK, Kwon BS, Kang CY. 4-1BB engagement costimulates NKT cell activation and exacerbates NKT cell ligand-induced airway hyperresponsiveness and inflammation. J Immunol 2008; 180:2062-8; PMID:; http://dx.doi.org/ 10.4049/jimmunol.180.4.2062 [DOI] [PubMed] [Google Scholar]
- 10. Melero I, Hirschhorn-Cymerman D, Morales-Kastresana A, Sanmamed MF, Wolchok JD. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin Cancer Res 2013; 19:1044-53; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vinay DS, Kwon BS. Immunotherapy of cancer with 4-1BB. Mol Cancer Ther 2012; 11:1062-70; PMID:; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0677 [DOI] [PubMed] [Google Scholar]
- 12. Melero I, Murillo O, Dubrot J, Hervas-Stubbs S, Perez-Gracia JL. Multi-layered action mechanisms of CD137 (4-1BB)-targeted immunotherapies. Trends Pharmacol Sci 2008; 29:383-90; PMID:; http://dx.doi.org/ 10.1016/j.tips.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 13. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13:227-42; PMID:; http://dx.doi.org/ 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev 2009; 229:192-215; PMID:; http://dx.doi.org/ 10.1111/j.1600-065X.2009.00765.x [DOI] [PubMed] [Google Scholar]
- 15. Kim JA, Averbook BJ, Chambers K, Rothchild K, Kjaergaard J, Papay R, Shu S. Divergent effects of 4-1BB antibodies on antitumor immunity and on tumor-reactive T-cell generation. Cancer Res 2001; 61:2031-7; PMID: [PubMed] [Google Scholar]
- 16. Miller RE, Jones J, Le T, Whitmore J, Boiani N, Gliniak B, Lynch DH. Four-1BB-specific monoclonal antibody promotes the generation of tumor-specific immune responses by direct activation of CD8 T cells in a CD40-dependent manner. J Immunol 2002; 169:1792-800; http://dx.doi.org/ 10.4049/jimmunol.169.4.1792 [DOI] [PubMed] [Google Scholar]
- 17. Murillo O, Arina A, Hervas-Stubbs S, Gupta A, McCluskey B, Dubrot J, Palazón A, Azpilikueta A, Ochoa MC, Alfaro C, et al. . Therapeutic antitumor efficacy of anti-CD137 agonistic monoclonal antibody in mouse models of myeloma. Clin Cancer Res 2008; 14:6895-906; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu D, Gu P, Pan PY, Li Q, Sato AI, Chen SH. NK and CD8+ T cell-mediated eradication of poorly immunogenic B16-F10 melanoma by the combined action of IL-12 gene therapy and 4-1BB costimulation. Int J Cancer 2004; 109:499-506; PMID:; http://dx.doi.org/ 10.1002/ijc.11696 [DOI] [PubMed] [Google Scholar]
- 19. Ju SA, Lee SC, Kwon TH, Heo SK, Park SM, Paek HN, Suh JH, Cho HR, Kwon B, Kwon BS, et al. . Immunity to melanoma mediated by 4-1BB is associated with enhanced activity of tumour-infiltrating lymphocytes. Immunol Cell Biol 2005; 83:344-51; PMID:; http://dx.doi.org/ 10.1111/j.1440-1711.2005.01330.x [DOI] [PubMed] [Google Scholar]
- 20. John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, Stewart TJ, Westwood JA, Guo ZS, Bartlett DL, et al. . Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res 2012; 72:1651-60; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2788 [DOI] [PubMed] [Google Scholar]
- 21. Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, Mueller A, Sagiv-Barfi I, Marabelle A, Lira R, et al. . Targeting CD137 enhances the efficacy of cetuximab. J Clin invest 2014; 124:2668-82; PMID:; http://dx.doi.org/ 10.1172/JCI73014 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, Colevas AD, Weng WK, Clarke MF, Carlson RW , et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest 2012; 122:1066-75; PMID:; http://dx.doi.org/ 10.1172/JCI61226 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, Duret H, Yagita H, Johnstone RW, Smyth MJ, et al. . Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res 2012; 72:3163-74; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0210 [DOI] [PubMed] [Google Scholar]
- 24. Houot R, Goldstein MJ, Kohrt HE, Myklebust JH, Alizadeh AA, Lin JT, Irish JM, Torchia JA, Kolstad A, Chen L, et al. . Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 2009; 114:3431-8; PMID:; http://dx.doi.org/ 10.1182/blood-2009-05-223958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, Müller A, Pachynski R, Czerwinski D, Coutre S, et al. . CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood 2011; 117:2423-32; PMID:; http://dx.doi.org/ 10.1182/blood-2010-08-301945 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, Lefrançois L. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J Immunol 2011; 187:4967-78; http://dx.doi.org/ 10.4049/jimmunol.1102335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rubinstein MP, Lind NA, Purton JF, Filippou P, Best JA, McGhee PA, Surh CD, Goldrath AW. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood 2008; 112:3704-12; PMID:; http://dx.doi.org/ 10.1182/blood-2008-06-160945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steffensen MA, Holst PJ, Steengaard SS, Jensen BA, Bartholdy C, Stryhn A, Christensen JP, Thomsen AR. Qualitative and quantitative analysis of adenovirus type 5 vector-induced memory CD8 T cells: not as bad as their reputation. J Virol 2013; 87:6283-95; PMID:; http://dx.doi.org/ 10.1128/JVI.00465-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007; 27:281-95; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol 2010; 184:6719-30; http://dx.doi.org/ 10.4049/jimmunol.0904089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 2008; 205:625-40; PMID:; http://dx.doi.org/ 10.1084/jem.20071641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jabbari A, Harty JT. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J Exp Med 2006; 203:919-32; PMID:; http://dx.doi.org/ 10.1084/jem.20052237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cush SS, Flano E. KLRG1+NKG2A+ CD8 T cells mediate protection and participate in memory responses during gamma-herpesvirus infection. J Immunol 2011; 186:4051-8; http://dx.doi.org/ 10.4049/jimmunol.1003122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, Al-Shamkhani A, Sun JC, Allison JP. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med 2013; 210:743-55; PMID:; http://dx.doi.org/ 10.1084/jem.20121190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Teng MW, Westwood JA, Darcy PK, Sharkey J, Tsuji M, Franck RW, Porcelli SA, Besra GS, Takeda K, Yagita H, et al. . Combined natural killer T-cell based immunotherapy eradicates established tumors in mice. Cancer Res 2007; 67:7495-504; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0941 [DOI] [PubMed] [Google Scholar]
- 36. Choi BK, Lee SC, Lee MJ, Kim YH, Kim YW, Ryu KW, Lee JH, Shin SM, Lee SH, Suzuki S, et al. Four-1BB-based isolation and expansion of CD8+ T cells specific for self-tumor and non-self-tumor antigens for adoptive T-cell therapy. J Immunother 2014; 37:225-36; PMID:; http://dx.doi.org/ 10.1097/CJI.0000000000000027 [DOI] [PubMed] [Google Scholar]
- 37. Choi BK, Kim YH, Kim CH, Kim MS, Kim KH, Oh HS, Lee MJ, Lee DK, Vinay DS, Kwon BS. Peripheral 4-1BB signaling negatively regulates NK cell development through IFN-gamma. J Immunol 2010; 185:1404-11; http://dx.doi.org/ 10.4049/jimmunol.1000850 [DOI] [PubMed] [Google Scholar]
- 38. Choi BK, Kim YH, Kang WJ, Lee SK, Kim KH, Shin SM, Yokoyama WM, Kim TY, Kwon BS. Mechanisms involved in synergistic anticancer immunity of anti-4-1BB and anti-CD4 therapy. Cancer Research 2007; 67:8891-9; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1056 [DOI] [PubMed] [Google Scholar]
- 39. Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PloS one 2011; 6:e19499; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0019499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westwood JA, Matthews GM, Shortt J, Faulkner D, Pegram HJ, Duong CP, Chesi M, Bergsagel PL, Sharp LL, Huhn RD, et al. . Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers. Leuk Res 2014; 38:948-54; PMID:; http://dx.doi.org/ 10.1016/j.leukres.2014.05.010 [DOI] [PubMed] [Google Scholar]
- 41. Ghalamfarsa G, Hadinia A, Yousefi M, Jadidi-Niaragh F. The role of natural killer T cells in B cell malignancies. Tumour Biol 2013; 34:1349-60; PMID:; http://dx.doi.org/ 10.1007/s13277-013-0743-x [DOI] [PubMed] [Google Scholar]
- 42. Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, Krasovsky J. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med 2003; 197:1667-76; PMID:; http://dx.doi.org/ 10.1084/jem.20021650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weinkove R, Brooks CR, Carter JM, Hermans IF, Ronchese F. Functional invariant natural killer T-cell and CD1d axis in chronic lymphocytic leukemia: implications for immunotherapy. Haematologica 2013; 98:376-84; PMID:; http://dx.doi.org/ 10.3324/haematol.2012.072835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985; 318:533-8; PMID:; http://dx.doi.org/ 10.1038/318533a0 [DOI] [PubMed] [Google Scholar]
- 45. Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, Williams S, Wiegmans AP, Dear AE, Scott CL, et al. . Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A 2007; 104:8071-6; PMID:; http://dx.doi.org/ 10.1073/pnas.0702294104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.