

Abstract
The molecular mechanisms underlying how sleep fragmentation (SF) influences cancer growth and progression remain largely elusive. Here, we present evidence that SF reduced ROS production by downregulating gp91phox expression and activity in TC1 cell tumor associated macrophages (TAMs), while genetic ablation of phagocytic Nox2 activity increased tumor cell proliferation, motility, invasion, and extravasation in vitro. Importantly, the in vivo studies using immunocompetent syngeneic murine tumor models suggested that Nox2 deficiency mimics SF-induced TAMs infiltration and subsequent tumor growth and invasion. Taken together, these studies reveal that perturbed sleep could adversely affect innate immunity within the tumor by altering Nox2 expression and activity, and indicate that selective potentiation of Nox2 activity may present a novel therapeutic strategy in the treatment of cancer.
Keywords: cancer, NADPH oxidase, reactive oxygen species, sleep apnea, tumor associated macrophage
Abbreviations: ANOVA, Analysis of variance; FBS, fetal bovine serum; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; MFI, median fluorescence intensities; Nox2, NADPH Oxidase Type 2; PMA, phorbol 12-myristate 13-acetate; ROS, reactive oxygen species; rpm, revolutions per minute; SE, standard error; SF, sleep fragmentation; TAMs, tumor associated macrophages; TLR-4, toll like receptor 4; WT, wild type
Introduction
For now several decades, the epidemiological associations between curtailed sleep duration and cancer have been quite extensively investigated.1-2 However, the adverse effects of perturbed sleep, a much more prevalent condition, as illustrated by unrefreshing sleep, SF, or sleep discontinuity have only recently gained attention as potentially underlying important modulatory effects on biological pathways involved in tumor growth and invasion.3 In this context, evidence of altered innate immunity by SF that involved TLR4-mediated pathways was apparent, and promoted accelerated proliferation and invasiveness of syngeneic murine tumor models.3
Recent studies have implicated reactive oxygen species (ROS) in general, and NADPH oxidases (Nox) in particular, as playing dual and divergent roles in oncological processes. The generation of ROS following activation of Nox is not only tightly regulated, but is further coupled with a spatially-restricted compartmentalization of ROS, thereby warranting the specificity of the Nox enzymes in cellular and sub-cellular signaling pathways.4 On the one hand, Nox may be involved in preserving homeostatic cellular redox levels, and alternatively Nox-derived ROS may enhance the risk for genomic instability and cancer.5-7 High levels of ROS are frequently present in tumors, and in some cases, the source of these ROS has been traced to Nox isoform deregulation, such that targeting of selective Nox isoforms either with inhibitors or using transgenic approaches has been shown to impair tumor growth in vivo.8-9 The driving assumption posited that production of ROS by both cancer cells and cancer-associated stromal elements may provide mimicry of the innate immune response that ultimately promotes tumor growth and invasiveness.10 Notwithstanding, recent studies would suggest that contrary to these assumptions, anti-oxidants may in fact adversely affect outcomes in oncological processes.11-13 One of the putative explanations for such discrepant findings could reside in the aforementioned compartmentalization of ROS, such that disruption of ROS in specific cellular or sub-cellular regions could lead to divergent effects on tumor growth and invasion.14
In a series of studies exploring some of the deleterious consequences of SF, we identified the presence of increased Nox2-derived ROS and oxidative stress as major pathophysiological determinants of end-organ morbidity.15-17 Based on such findings and the promoting effects of SF on tumor growth and invasion,3 we hypothesized that genetic ablation of Nox2 would potentially elucidate the role of this important source of ROS in the oncogenic processes modulated by SF, and potentially abolish the pro-tumorigenic effects of SF.
Results
Nox2-deficiency mediates SF accelerated tumor growth
Tumors grow faster and are more likely to be invasive to surrounding tissues in mice exposed to SF, as previously reported.3 These observations led us to further explore the nature of the molecular mechanisms underlying the pro-cancerigenic activities of SF. Given the critical role of ROS in the pathogenesis of most cancers,18-19 we hypothesized that Nox2-mediated oxidative stress contributes to SF-induced tumor growth and progression. To test this hypothesis, we used a gp91phox-/Y mouse model, which lacks the catalytic core subunit (gp91) of the Nox2 enzyme complex and is therefore deficient in Nox2 activity. Increased tumor growth rate (Fig. 1A), tumor weight (Fig. 1B) and invasiveness (Fig. 1C) were consistently observed across WT mice exposed to SF and gp91phox-/Y mice exposed to either SC or SF, as compared to WT mice exposed to SC. Taken together, these findings suggested that contrary to our initial hypothesis Nox2-deficiency underlies components of SF-induced accelerated tumor growth and invasiveness.
Figure 1.
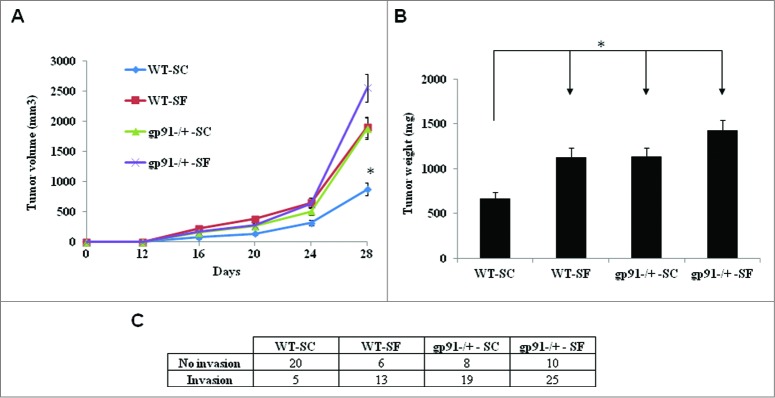
Loss of NADPH oxidase 2 (Nox2) and sleep fragmentation (SF) accelerate tumor growth. (A) Analysis of tumor growth curves showed accelerated growth of TC1 tumors in Nox2-deficient mice (gp91phox-/Y-SC, n = 27 and gp91phox-/Y-SF, n = 35) and C57BL/6J mice (WT-SF, n = 39) when compared with control group WT-SC (n = 37). Data are shown as mean ± standard error (SE). *p < 0.05 (two-way ANOVA). (B) Tumor weight was increased at day 28 after innoculation in gp91phox-/Y-SC (n = 27), gp91phox-/Y-SF (n = 35) and WT-SF (n = 39) groups as compared with WT–SC group (n = 37). *p < 0.05 is considered significant (one-way ANOVA). (C) The number of cases manifesting tumor invasion beyong the capsular boundaries toward adjacent tissues was significantly increased in groups gp91phox-/Y-SC, gp91phox-/Y-SF and WT-SF vs. WT-SC (p < 0.05).
gp91phox downregulation contributes to SF-reduced ROS production in TAMs
To determine whether the tumor-promoting effects of gp91phox deletion were related to a reduction in ROS production, intracellular ROS levels in TAMs were measured by using a fluorogenic ROS indicator (H2DCFDA). As expected, ROS was significantly reduced in TAMs isolated from tumor-bearing gp91phox-/Y mice exposed to either SC or SF (Fig. 2A). Most importantly, SF also reduced ROS production in TAMs derived from WT mice (Fig. 2A), and Nox activities in WT TAMs were markedly attenuated in SF-exposed WT mice (Fig. 2B). Further analysis of gp91 protein revealed significantly reduced gp91phox expression in the tumors of WT mice exposed to SF (Fig. 2C), suggesting that the reduced ROS production in tumors of SF-exposed mice were mediated, at least in part, by downregulation of gp91phox expression.
Figure 2.

SF reduces reactive oxygen species (ROS) production in tumor-associated macrophages (TAMs) as well as gp91phox downregulation in expression and activity. (A) Flow cytometric analysis showed reduced levels of ROS in TAMs isolated from TC1 tumor-bearing gp91phox-/Y-SC (n = 12), gp91phox-/Y-SF (n = 18) and WT-SF (n = 18) mice compared with WT-SC (n = 13) mice. Left panel, a representative flow cytometric histogram of DCF-positive cells (gated on cells expressing CD11b and F4/80). Right panel, ROS levels were quantified as the median fluorescence intensity (MFI) of DCF-positive cells among CD11b+ and F4/80+ cells. Data are plotted as fold changes18 compared with WT-SC. *p< 0.05. (B) Summary of Nox enzymatic activity experiments in TAMs isolated from wild type mice exposed to SC (n = 6) or SF (n = 6) illustrating significant reductions in Nox activity in tumors from mice exposed to SF conditions (*p < 0.05 vs. SC). (C) Immunoblots of tumor lysates revealed marked reduction in gp91phox protein expression in WT-SF tumors (n = 3) when compared with WT-SC (n = 3) (left panel). Band intensities were quantified and data are expressed as fold changes over β-actin (right panel). *p < 0.05.
Ablation of phagocytic Nox2 activity mediates SF-driven tumor cell proliferation, motility, invasion and extravasation in vitro
Based on aforementioned findings, we reasoned that the in vitro effects of Nox2 loss on the biology of tumor cells may mimic their in vivo counterparts. To explore this possibility, analysis of the effects of Nox2 deficiency in TAMs on the malignant phenotype of TC1 tumor cells was performed. As shown in Fig. 3, when cultured with TC1 tumor cells, TAMs from tumors harvested from gp91phox-/Y-SC, gp91phox-/Y-SF and WT-SF mice resulted in significant increases of TC1 cell proliferation, motility, invasion and extravasation when compared with TAMs derived from WT-SC mice.
Figure 3.

Genetic ablation of phagocytic Nox2 activity underlies components of SF-induced increases in tumor cell proliferation and invasion, but not migration, in vitro. (A) When co-cultured with TC1 tumor cells in monolayer, TAMs from tumors harvested from gp91phox-/Y-SC (n =10), gp91phox-/Y-SF (n = 15) and WT-SF (n = 13) mice increased TC1 cell proliferation compared with WT-SC (n = 7). Results are expressed as fold changes over TC1 cell single culture group (n = 12). *p < 0.05. (B) Compared with WT–SC (n = 5), TAMs from gp91phox-/Y-SF (n = 8) and WT-SF (n = 9), but not gp91phox-/Y-SC (n = 5) mice were associated with increased numbers of TC1 cells migrating through polycarbonate membrane pores. The values shown represent fold changes compared with TC1 cell alone (n = 8). *p < 0.05. (C) TC1 cells displayed increased invasion when co-cultured with TAMs isolated from gp91phox-/Y-SC (n = 7), gp91phox-/Y-SF (n = 11) and WT-SF (n = 11) tumors than with TAMs from WT-SC (n = 8) tumors. Data are expressed as fold changes compared to the number of cells measured in the TC1 cell alone group (n = 12). *p < 0.05.
Nox2 deficiency mediates SF-triggered TAMs induction
Given our previous studies demonstrated that SF accelerates tumor growth and progression through recruitment and polarity shift of TAMs,3 we hypothesized that SF-induced changes of TAMs phenotypes may be associated with the reduced expression of Nox2. To test this hypothesis, flow cytometric analysis of TAMs and their polarity revealed that although the overall number of TAMs was increased in all experimental groups compared to WT-SC mice, (Fig. 5A), SF-induced TAM polarity shift toward to M2 was only observed in WT mice, and was not apparent among gp91phox -/Y mice.
Figure 5.
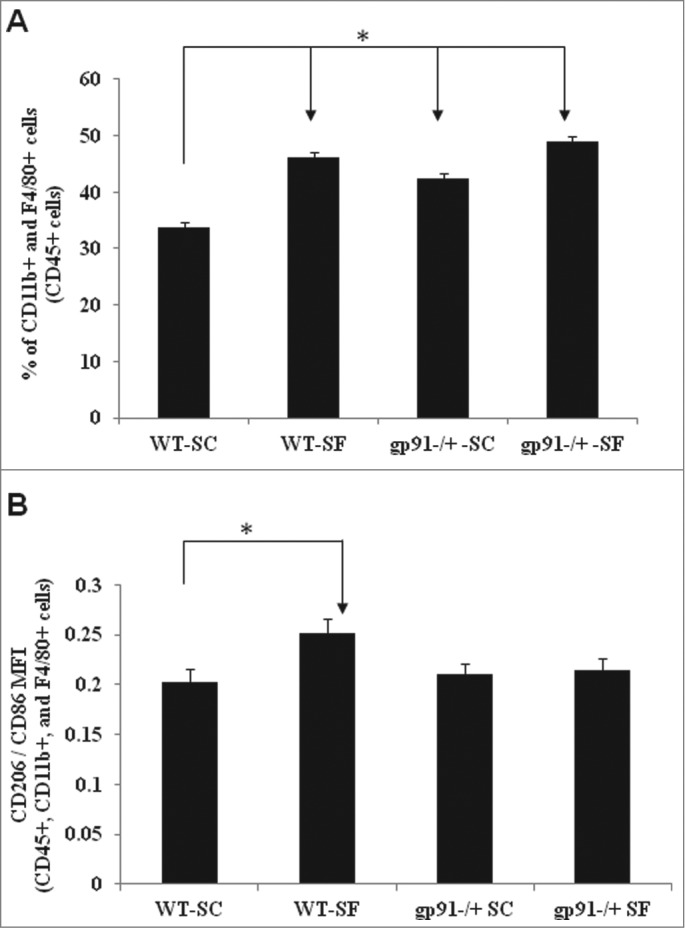
Nox2 deficiency increases TAMs but does not underlie SF-induced changes in TAM polarity. Significant increases in the number of infiltrated TAMs (A) along with no changes in TAM polarity (B) emerged tumors from gp91phox-/Y-SC (n = 25), gp91phox-/Y-SF (n = 32) and WT-SF (n = 17) mice vs. WT-SC (n = 18) mice. Data are expressed as either percentages of the immediate parent population (TAM) or the ratio of CD206 MFI to CD86 MFI (polarity). *p < 0.05.
Discussion
In this study and contrary to our initial hypothesis, we found that ROS levels and Nox2 activity are reduced in syngeneic solid TC1 cell tumors when mice are subjected to SF. Furthermore, transgenic ablation of Nox2 resulted in enhanced oncogenic properties of TC1 cell tumors, as evidenced by increased proliferation, migration, extravasation, and invasion. Taken together, these findings suggest that Nox2 plays an important role in anti-tumoral host responses, and that some of the cellular pathways altered by SF in its effect on tumoral properties most likely involved downregulation of Nox2 expression and activity.
Our current study recapitulates our previous findings indicating that chronically fragmented sleep, a highly prevalent condition associated with a multiplicity of human disorders, promotes enhanced tumor growth and invasiveness in mice, and that SF induces changes in TAM polarity with a shift toward increased M2-like macrophages.3 It is important to note however that the importance of the temporal sequence of events, i.e., tumor preceding SF or SF preceding cancer onset is unknown at this stage, and that current work assumes the latter option based on epidemiological trends whereby sleep disorders are usually chronically established before patients seek medical attention.20 Furthermore, the current study focused on altered tumor properties in the context of SF, but did not investigate whether SF (as seen in sleep apnea or periodic leg movement disorder of sleep) or any other chronic sleep disturbances associated with curtailed sleep (insomnia, behaviorally or lifestyle restricted sleep) alter tumorigenesis. However, the paramount observation of the present study is the increased tumor proliferation and marked changes in invasiveness induced by Nox2 ablation, in the absence of TAM polarity changes. As such, these observations provide important observations in a murine model on the potential anti-tumoral role played by Nox2, and how perturbed sleep could be adversely affecting innate immunity within the tumor by altering Nox2 expression and activity.
The mechanisms mediating the increased tumor proliferative rates induced by SF exposures are unclear and likely diverse. The potential attenuation of innate immune responses against tumor cells was somewhat anticipated, considering the currently available literature on the immunomodulatory effects associated with disruption of sleep homeostasis.21-22 In this context however, the absence of SF-induced changes in TAM polarity among Nox2 null mice were unexpected and quite surprising, particularly when considering the previously reported absence of SF-induced shifts in macrophage markers and in total numbers of macrophages within visceral adipose tissues during SF.17 Notwithstanding, recent evidence from our laboratory has implicated hypothalamic changes, particularly altered leptin receptor sensitivity and increased protein tyrosine phosphatase 1B (PTP1B), as underlying orexigenic behaviors that clearly reflect more extensive metabolic derangements in the context of chronic SF.23 Thus, the absence of obesity-promoting effects of SF and of metabolic changes within adipose tissues in gp91phox-/Y mice should have protected, rather than accelerated tumoral growth, based on the putative contributions of peri-tumoral adipose tissues to oncogenic processes, such as enhanced proliferation and invasion.24 Furthermore, altered sleep in the host may trigger a complex set of biologically-relevant pathways that promote among many other changes the reduced expression and activity of Nox2, the latter emerging as an anti-tumoral agent. In this context, both SF-exposed mice and gp91phox-/Y mice manifested increased frequency of tumor invasion to surrounding tissues (Fig. 1).
As with any physiologically vital, evolutionarily higher function such as sleep, transfer of the experimental framework from an in vivo murine model to an in vitro model is obviously unfeasible, thereby hampering our ability to study the mechanisms underlying the changes in Nox2 expression and activity associated with SF. To partially overcome these limitations, we conducted in vitro experiments with naïve TC1 cells exposed to TAMs derived from the four experimental groups to determine whether the in vivo differences in tumor proliferation and invasion associated with SF and with Nox2 deficiency could be recapitulated. In this setting, we found that reduced Nox2 activity in TAMs (i.e., SF and/or gp91phox-/Y) accelerated TC1 proliferation and invasion, but did not account for the effects of SF on TC1 cell migration (Fig. 3) or extravasation (Fig. 4). Based on these findings, we surmise that the compartmentalized reduction in ROS generation by Nox2 activity as induced by SF or by genetic ablation of Nox2 alters specific functional components of TAM-TC1 cell interactions, such that current studies provide initial directions of specific areas for which increased Nox2 activity may be desirable in tumor therapeutics.
Figure 4.

Endothelial disruption induced by TAMs alone (A) and in co-culture with TC1 tumor cells (B). Time-course of normalized endothelial resistance (left) and their values 4h after adding cells to the endothelial monolayer (right). Upper panel represent the extravasation changes (reduced resistance) induced by addition of TAMs and lower panels shows changes after addition of both TC1 cells and TAMs. Data are presented as the mean ± SE (n = 6). *p < 0.05.
In summary, ROS derived from host-dependent Nox2 activity appear to play anti-tumoral functions primarily involving the restraint of tumor cell proliferation and invasion. Furthermore, the accelerated growth of tumors and their enhanced invasiveness occurring in the context of sleep perturbations mimicking those occurring in many human disorders appear to be mediated at least in part by reduced Nox2 expression and activity.
Materials and Methods
Cell culture, animals, and reagents
The murine lung epithelial cell carcinoma line,TC1, and primary murine brain endothelial cells, bEnd.3, were maintained as recommended.3,25 Male C57BL/6J (WT) and hemizygous gp91phox-/Y (gp91−/+) mice were purchased from Jackson Laboratory and housed at the same environmental conditions in the animal facility at the University of Chicago. gp91phox-/Y mice were further bred in house, and routinely genotyped by the standard genotyping protocol provided by Jackson Laboratory. All animal experimental procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Antibodies used in this study were obtained from Santa Cruz (β-actin; sc-47778), Biolegend (CD45-APC - 103112, F4/80-PE - 123109, F4/80-PB - 123132, CD45-FITC - 103108, CD11b-PB - 101224, CD11b-APC - 101212), and BD Biosciences (gp91; 611414). The EasySep mouse CD11b positive selection kit was purchased from STEMCELL (19761A). Live/dead fixable Aqua dead cell stain kit (L34957) and CM-H2DCFDA kit (C6827) were purchased from Life Technologies, while Zombie NIR Fixable viability kit was purchased from Biolegend (423105). Collagenase type I was obtained from Worthington Biochemical Corporation (SM2P13900), and Matrigel was acquired from Trevigen (3445-005-01).
Sleep fragmentation (SF) paradigm and TC1 tumor model
The device used to induce SF in mice was originally designed and developed in our laboratory and employs intermittent tactile stimulation of freely behaving mice in a standard laboratory mouse cage, using a near-silent motorized mechanical sweeper (Lafayette Instruments, Model 80391).15,26,27 3-mo-old WT and gp91phox-/Y mice were exposed to either SF or normal sleep conditions (SC; the SF cage was used but the sweeper remained immobile). To induce moderate to severe SF, we chose a 2-min interval between each sweep, implemented during the light period (7:00 am–7:00 pm). Depending on experimental needs, four or five mice were housed in each SF cage to prevent isolation stress, with matched number of mice housed in paired control cages. After 7 d of exposure during which mice had ad libitum access to food and water, all mice corresponding to four experimental groups (WT-SC, WT-SF, gp91phox-/Y -SC, and gp91phox-/Y -SF) were inoculated with 200 μL of 1 × 105 TC1 tumor cells by subcutaneous injection into the right middle flank through a 25-gauge needle. After the injection, the mice were returned to their cages followed by weekly monitoring for the presence of subcutaneous tumors. Palpable tumors routinely developed within two weeks, at which time tumor size (width and length) were measured three times per week using precision calipers. Four weeks after tumor implantation, the mice were euthanized by CO2, followed by cervical dislocation. The entire tumors were surgically removed, measured, and weighted, while the presence of invasion toward the skeletal muscle was visually assessed. The majority of tumors was immediately processed either for analysis of tumor-infiltrating cells or TAMs isolation. The remaining tissues were frozen in liquid nitrogen and kept at −80°C prior to analysis.
Flow cytometric analysis of tumor-associated macrophages (TAMs)
Tumors were minced in very small pieces with a scalpel, and incubated in RPMI1840 medium containing 1 mg/mL collagenase type I for 1-h at 37°C with a gentle agitation (40 rpm). Following digestion, the tissues were passed through a 100-μm cell strainer to obtain a single cell suspension. For TAMs isolation, CD11b+ cells were enriched by using EasySep mouse CD11b positive selection kit according to the manufacturer instructions, and isolated cells (purity: 95%) were immediately used. For analysis of TAMs, viable cells were initially identified using Aqua-fluorescent reactive dye, and then CD11b+ and F4/80+ cells within the CD45+ leukocyte population were defined as TAMs. To determine whether Nox2 deficiency changed TAM polarity, further flow cytometric analysis of TAMs was done by using a M1 marker CD86 (105028) and a M2 marker CD206 (141708). All dyes and antibodies were incubated for 30 min in the dark on ice or at 4°C. For analysis of ROS levels in TAMs, live cells were selected by using Zombie NIR Fixable dye staining, and then ROS levels of CD11b+ and F4/80+ cells were measured with a fluorescent DCF dye. All dyes and antibodies were incubated for 30 min in the dark at 37°C. Flow cytometry analyses were performed using a Canto II instrument (BD Biosciences) and results were analyzed using Flowjo flow cytometry analysis software (Treestar Inc.). Data were expressed as either percentages of the immediate parent population or fold changes compared to the corresponding control.
NADPH oxidase activity
1 × 106 TAMs were incubated in RPMI-1640 medium containing 2% FBS, 20 mM HEPES, and 5 μM lucigenin. Superoxide-dependent reduction of lucigenin was initialized by the addition of phorbol 12-myristate 13-acetate (PMA, 100 nM). Light emission was recorded using a VICTOR3V plate reader (Perkin Elmer) at 30-sec intervals for 30 min and specific NOX activities were determined by subtracting the background level of luminescence of paired samples containing 10μM diphenyliodonium.
Western blot analysis
Total protein was extracted from frozen tumor tissues using standard techniques as previously described.28 In brief, tissues were homogenized in lysis buffer, incubated for 30 min at 4°C, and then centrifuged at 32,000 × g at 4°C for 10 min. The protein concentration of the supernatant was determined by Bio-Rad protein assay as directed. For western blot analysis, 10 μg protein were fractionated on SDS-12% polyacrylamide gels and transferred onto nylon membranes. The membranes were blocked with 5% skim milk in phosphate-buffered saline with 0.05% Tween 20 (PBS-T) at room temperature for 2 h with gentle agitation, followed by 2-h incubation at room temperature with a 1:1000 (vol/vol) dilution of primary antibody in PBS-T. The blots were washed in PBS-T three times for 5 min each time with gentle agitation and then incubated with a 1:20,000 dilution (vol/vol) of secondary antibody for 2-h at room temperature with gentle agitation. Target proteins were visualized using a Clarity western ECL substrate (Biorad; 170-5060), and images were captured using a Gel Doc imaging system (Biorad). Band intensities were quantified by densitometry using Image Lab 4.1 software (Biorad) and normalized to β-actin.
Cell growth, motility, and invasion assay
For cell growth assay, 5 × 104 TC1 cells were cultured in complete medium either with or without the addition of 2 × 105 CD11b+ cells (ratio 1:4) in 12-well plates. After 2 d, cells were stained with CD45 antibody, and the absolute TC1 cell number was calculated by multiplying the percentage of CD45-cells to the total absolute cell counts. Data were expressed as fold changes compared with TC1 control.
Cell motility was assessed using transwell migration assay as previously described.25 In brief, 5 × 104 TC1 cells (with and without 2 × 105 CD11b+ cells) were placed in a transwell chamber with a 8 μm pore membrane, and allowed to migrate toward 3% fetal bovine serum (FBS) for 24 h. The number of TC1 cells migrating to the lower surface of the membrane was counted by a flow cytometry using the CD45-cell gating approach described above.
A cell invasion assay was carried out as described above for cell motility, except that the chambers were coated with 1 mg/mL Matrigel, the number of TC1 cells was increased to 2 × 105, and cells were allowed to migrate toward 10% FBS for 24 h. Data were expressed as fold changes compared with TC1 control.
Transendothelial extravasation assay
The endothelial disruption induced by isolated TAMs either alone or in co-culture with tumor cells (TC1) was evaluated employing an electrical substrate-impedance sensing system (ECIS, Applied Biophysics), as previously described.25 Briefly, 15 × 104 mouse brain primary endothelial bEnd.3 cells were seeded in each well (8W10E arrays, Applied Biophysics) for 24 h. Then, 1.5 × 104 TC1 cells and 5 × 104 TAMs isolated from tumors of animals (WT and gp91phox-/Y) exposed to either SC or SF were added in isolation or together. A control group was also performed by adding only medium. This assay measures the changes in electrical resistance in real-time associated with endothelial disruption of tight junctions elicited by tumor cells and/or TAMs. Resistance values (Ohms) were normalized relative to a previous undisturbed confluent endothelial monolayer just before the addition of TAMs and/or TC1 cells.
Data Analysis
Data are reported as mean ± SE unless stated otherwise. Numerical data were analyzed using the appropriated statistical methods as indicated, with a two-tailed p value < 0.05 being considered as achieving statistical significance.
Acknowledgments
JZ, YW, and DG participated in the conceptual framework of the project, performed experiments, analyzed data, and drafted components of the manuscript. IA, SXZ, AC, and ZQ, performed experiments and analyzed data. DG conceptualized the project, provided critical input in all phases of the experiments, analyzed data, drafted the ulterior versions of the manuscript, and is responsible the manuscript content, and financial aspects of the project. All authors have reviewed and approved the final version of the manuscript. No competing financial interests exist.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
DG is supported by NIH grants HL-065270 and HL-086662.
References
- 1. Lu Y, Tian N, Yin J, Shi YH, Huang ZP. Association between sleep duration and cancer risk: a meta-analysis of prospective cohort studies. Plos One 2013; 8(9):e74723; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0074723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haus EL, Smolensky MH. Shift work and cancer risk: Potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med Rev 2013; 17(4):273-84; PMID:; http://dx.doi.org/ 10.1016/j.smrv.2012.08.003 [DOI] [PubMed] [Google Scholar]
- 3. Hakim F, Wang Y, Zhang SX, Zheng J, Yolcu ES, Carreras A, Khalyfa A, Shirwan H, Almendros I, Gozal D. Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling. Cancer Res 2014; 74(5):1329-37; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Sign 2009; 11(6):1289-99; PMID:; http://dx.doi.org/ 10.1089/ars.2008.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weaver AM. Regulation of cancer invasion by reactive oxygen species and Tks family scaffold proteins. Sci Signal 2009; 2(88):pe56; PMID:; http://dx.doi.org/ 10.1126/scisignal.288pe56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hayes P, Knaus UG. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid Redox Signal 2013; 18(15):1937-45; PMID:; http://dx.doi.org/ 10.1089/ars.2012.4895 [DOI] [PubMed] [Google Scholar]
- 7. Weyemi U, Redon CE, Parekh PR, Dupuy C, Bonner WM. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anti-Cancer Agent Med Chem 2013; 13(3):502-14; PMID: [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang CX, Lan T, Hou JC, Li J, Fang RD, Yang ZC, Zhang M, Liu J, Liu B. NOX4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget 2014; 5(12):4392-405; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki S, Shiraga K, Sato S, Punfa W, Naiki-Ito A, Yamashita Y, Shirai T, Takahashi S. Apocynin, an NADPH oxidase inhibitor, suppresses rat prostate carcinogenesis. Cancer Sci 2013; 104(12):1711-17; PMID:; http://dx.doi.org/ 10.1111/cas.12292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs "fertilizer." Cell Cycle 2011; 10(15):2440-9; PMID:; http://dx.doi.org/ 10.4161/cc.10.15.16870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet 2004; 364(9441):1219-28; PMID:; http://dx.doi.org/ 10.1016/S0140-6736(04)17138-9 [DOI] [PubMed] [Google Scholar]
- 12. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 2007; 297(8):842-57; PMID:; http://dx.doi.org/ 10.1001/jama.297.8.842 [DOI] [PubMed] [Google Scholar]
- 13. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 2014; 6(221):221ra15; PMID:; http://dx.doi.org/ 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- 14. Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med 2014; 371(2):177-8; PMID:; http://dx.doi.org/ 10.1056/NEJMcibr1405701 [DOI] [PubMed] [Google Scholar]
- 15. Nair D, Zhang SX, Ramesh V, Hakim F, Kaushal N, Wang Y, Gozal D. Sleep fragmentation induces cognitive deficits via nicotinamide adenine dinucleotide phosphate oxidase-dependent pathways in mouse. Am J Respir Crit Care Med 2011; 184(11):1305-12; PMID:; http://dx.doi.org/ 10.1164/rccm.201107-1173OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khalyfa A, Wang Y, Zhang SX, Qiao Z, Abdelkarim A, Gozal D. Sleep fragmentation in mice induces nicotinamide adenine dinucleotide phosphate oxidase 2-dependent mobilization, proliferation, and differentiation of adipocyte progenitors in visceral white adipose tissue. Sleep 2014; 37(5):999-1009; PMID:; http://dx.doi.org/ 10.5665/sleep.3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang SX, Khalyfa A, Wang Y, Carreras A, Hakim F, Neel BA, Brady MJ, Qiao Z, Hirotsu C, Gozal D. Sleep fragmentation promotes NADPH oxidase 2-mediated adipose tissue inflammation leading to insulin resistance in mice. Int J Obes (Lond) 2014; 38(4):619-24; PMID:; http://dx.doi.org/ 10.1038/ijo.2013.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Afanas'ev I. Reactive oxygen species signaling in cancer: comparison with aging. Aging Dis 2011; 2(3):219-30; PMID: [PMC free article] [PubMed] [Google Scholar]
- 19. Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol 2009; 15(14):1702-7; PMID:; http://dx.doi.org/ 10.3748/wjg.15.1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rahaghi F, Basner RC. Delayed Diagnosis of Obstructive Sleep Apnea: don't Ask, Don't Tell. Sleep Breath. 1999; 3(4):119-124; PMID:; http://dx.doi.org/ 10.1007/s11325-999-0119-z [DOI] [PubMed] [Google Scholar]
- 21. Prather AA, Hall M, Fury JM, Ross DC, Muldoon MF, Cohen S, Marsland AL. Sleep and antibody response to hepatitis B vaccination. Sleep 2012; 35(8):1063-9; PMID:; http://dx.doi.org/ 10.5665/sleep.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuo TH, Williams JA. Increased sleep promotes survival during a bacterial infection in Drosophila. Sleep 2014; 37(6):1077-86, 86A–86D; PMID:NOT_FOUND [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hakim F, Wang Y, Carreras A, Hirotsu C, Zhang J, Peris E, Gozal D. Chronic sleep disruption during the sleep period induces hypothalamic endoplasmic reticulum stress and PTP1b-mediated leptin resistance in mice. Sleep 2015; 38(1):31-40; PMID:; http://dx.doi.org/10.5665/sleep.4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res 2013; 19(22):6074-83; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Almendros I, Wang Y, Becker L, Lennon FE, Zheng J, Coats BR, Schoenfelt KS, Carreras A, Hakim F, Zhang SX, et al. Intermittent hypoxia-induced changes in tumor-associated macrophages and tumor malignancy in a mouse model of sleep apnea. Am J Respir Crit Care Med 2014; 189(5):593-601; PMID:; http://dx.doi.org/ 10.1164/rccm.201310-1830OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Carreras A, Lee S, Hakim F, Zhang SX, Nair D, Ye H, Gozal D. Chronic sleep fragmentation promotes obesity in young adult mice. Obesity (Silver Spring) 2014; 22(3):758-62; PMID:; http://dx.doi.org/ 10.1002/oby.20616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramesh V, Nair D, Zhang SXL, Hakim F, Kaushal N, Kayali F, Wang Y, Li RC, Carreras A, Gozal D. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-α pathway. J Neuroinflamm 2012; 9:91; PMID:; http://dx.doi.org/ 10.1186/1742-2094-9-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zheng J, Ather JL, Sonstegard TS, Kerr DE. Characterization of the infection-responsive bovine lactoferrin promoter. Gene 2005; 353(1):107-17; PMID:; http://dx.doi.org/ 10.1016/j.gene.2005.04.016 [DOI] [PubMed] [Google Scholar]