

Abstract
Hereditary angioedema (HAE) is characterized as an episodic swelling disorder with autosomal dominant inheritance. Clinical features include nonpitting edema of external or mucosal body surfaces, and patients often present with swelling of the extremities, abdominal pain, and swelling of the mouth and throat, which can lead to asphyxiation. Patients with HAE classically have no associated urticaria, which is often referred to as nonhistaminergic angioedema. Treatment for HAE involves long-term prophylaxis, short-term prophylaxis, and management of acute attacks. Up until the past few years, acute HAE episodes were predominately treated with supportive measures. Three classes of medications have recently been approved by the US Food and Drug Administration (FDA) for the management of acute HAE attacks. Ecallantide, a recombinant protein that acts as a reversible inhibitor of kallikrein, is currently indicated for acute attacks of HAE in those aged ≥12 years. In two randomized, double-blind, placebo-controlled, multicenter trials, EDEMA3 and EDEMA4, patients treated with 30 mg of ecallantide demonstrated statistically significant improvement in symptoms compared to those on placebo. In addition to its use as treatment for HAE, ecallantide has been used off label in the management of nonhistaminergic angioedema, not due to HAE. Ecallantide has shown promise in the treatment of these other forms; however, data are limited to mainly case reports at this time. Ecallantide is generally a safe and well-tolerated medication; however, based on reports of anaphylaxis, ecallantide does contain a black box warning. Due to the risk of anaphylaxis, ecallantide cannot be self-administered and must be given by a health care professional. Overall, ecallantide is a safe and effective medication for the treatment of acute attacks of HAE.
Keywords: idiopathic angioedema, ACE-Inhibitor induced angioedema, nonhistaminergic angioedema, bradykinin, acquired angioedema, Kallikrein, 7- C1-Inhibitor
Introduction
Angioedema is defined by swelling of the skin or mucosal tissues, resulting from loss of vascular integrity and extravasation of fluid into the interstitium. Different forms of angioedema are classified based on the mechanisms causing angioedema. The two basic categories are histaminergic angioedema and nonhistaminergic angioedema. Histaminergic angioedema is typically associated with urticaria and histamine release. These patients generally respond to antihistamine therapy. Nonhistaminergic angioedema is refractory to antihistamines because these forms are mediated primarily by bradykinin, not histamine. Types of nonhistaminergic angioedema include hereditary angioedema (HAE), some forms of idiopathic angioedema, angiotensin-converting enzyme (ACE) inhibitor-associated angioedema, and acquired angioedema.1
HAE is a genetic condition characterized by episodes of subcutaneous and submucosal edema involving the face, tongue, larynx, arms, hands, trunk, legs, bowel, and/or genitalia.2 HAE has an estimated worldwide prevalence of 1 in 10,000 to 1 in 150,000.3 In a nationwide survey in Denmark, the minimal prevalence of HAE was 1.41:100,000.4 Prevalence studies in Spain showed similar results, with minimal prevalence of 1.09:100,000.5 A recent study revealed a 1:90,000 prevalence rate in Greece.6 This disorder results in debilitating – and even life-threatening – symptoms, primarily asphyxiation from laryngeal edema, but patients can also develop significant hypotension from abdominal attacks due to fluid shifts into the peritoneal cavity or interstitium.2
Because HAE is a rare disease, this disorder does not cause significant burden from a population perspective, but individuals with HAE can suffer a substantial amount in terms of socioeconomic burden. HAE can cause significant impairment in terms of both quality of life and increased medical costs. In a cross-sectional survey of 164 patients with HAE in Spain, Germany, and Denmark, investigators examined the impact that HAE had on individuals with the diagnosis and their caregivers’ lives.7 They surveyed those who had experienced at least one attack over a 6-month period of time. They found that 51% believed HAE hindered their career/educational advancement.7 Those who were in school or work estimated missing, on average, 20 days of work or school per year.7
Onset of symptoms typically occurs in childhood, including the 1st year of life, but there is often a significant delay in diagnosis such that most patients are not diagnosed until adulthood.8 In a study of 221 patients with HAE, mean age of disease onset was 11.2 years of age, with a range from 1 year to 40 years.9 Patients often experience prodromal symptoms, which can include erythema marginatum, fatigue, headaches, myalgias, numbness, tingling, and gastrointestinal complaints. Symptoms involving the skin and the gastrointestinal tract are the most common.9 Abdominal attacks tend to be painful. Consensus data from 323 patients diagnosed with HAE in the UK demonstrated that peripheral swelling accounted for 58% of attacks.10 Laryngeal attacks were less common, but 19% of the 323 patients had experienced at least one episode of laryngeal swelling over the previous year.10 Nearly half of patients with HAE will experience at least one episode of laryngeal edema in their lifetime.9,11 In a study involving 123 patients with HAE, 49.6% experienced laryngeal edema.11
There are three forms of HAE that are currently described in the literature. Type 1 HAE is characterized by a mutation in one of the two alleles of the C1-inhibitor (C1-INH) gene, leading to low levels of functional C1-INH protein. In type 1, serum levels of C1-INH are often <30% of normal. On the other hand, type 2 HAE is characterized by a mutation in one of the C1-INH alleles, which affects the active site of the protein. These patients often have normal or elevated amounts of C1-INH protein circulating, but levels of function are similar to those in patients with type 1 HAE because half the protein synthesized is nonfunctional. Autosomal dominant inheritance of mutations in the C1-INH gene (SERPING1) is responsible for most cases of HAE. Spontaneous mutations can occur and are estimated to be responsible for 25% of cases.12 Type 3 HAE is now typically called HAE with normal C1-INH.13 This form of HAE is rare and defined by family history of nonhistaminergic angioedema but with normal C1-INH levels and function.14 Findings suggest that this form of HAE is related to other enzymes involved in the proteolytic cascade, with evidence of increased bradykinin activity, similar to HAE types 1 and 2.14 For example, mutations in factor XII (fXII) or the Hageman factor gene were identified in several individuals with HAE with normal C1-INH, which were not observed in 145 healthy controls.15 Some forms of idiopathic angioedema are similar to HAE with normal C1-INH, except that patients falling under this category lack a family history of angioedema. It should be noted that there are some forms of idiopathic angioedema that are mediated by histamine, referred to as histaminergic angioedema. Other forms are unresponsive to high-dose antihistamines and are commonly referred to as nonhistaminergic idiopathic angioedema. ACE inhibitor-associated angioedema results from ACE-inhibitor use causing decreased degradation of bradykinin. Acquired angioedema tends to occur in older patients with monoclonal antibodies to C1-INH or from malignancy. In addition, some patients with autoimmune disorders such as systemic lupus erythematosus appear to consume sufficient C1-INH to induce an HAE-like syndrome. These patients have low levels of C4 and low levels of C1q, which is the third protein that forms the C1 complex along with C1r and C1s and which is at normal levels in patients with true HAE. Patients with acquired disease may have normal levels of C1-INH, although levels are usually low, but their functional level is always low. Unlike patients with true HAE, their C3 levels may be low.16
Because most cases of HAE are due to low functional levels of C1-INH, C1 protein in the complement pathway is able to continuously cleave and consume the next two proteins, C4 and C2. Therefore, patients with HAE types 1 and 2 almost always have low levels of C4 and C2, even between attacks.17 C1-INH levels, C1-INH function, and C4 measurements are the usual diagnostic tests. In the case where C4, C1-INH antigen, and C1-INH function are low, the patient most likely has type 1 HAE. If C4 is low, C1-INH is normal, and C1-INH function is low, then the patient most likely has type 2 HAE (Table 1).
Table 1.
Diagnosis of types 1, 2, 3, and idiopathic HAE
| Laboratory test | Type 1 HAE |
Type 2 HAE |
Type 3 and idiopathic HAE |
|---|---|---|---|
| C4 concentration | Low | Low | Normal |
| C1-INH concentration | Low | Normal/high | Normal |
| C1-INH function | Low | Low | Normal |
Abbreviations: C1-INH, C1-inhibitor; HAE, hereditary angioedema.
C1-INH is also involved in regulating the proteolytic cascades of the contact system and coagulation cascades. Uninhibited activation of the contact system plays an important role in the pathophysiology of HAE. The contact pathway activates fXIIa, which subsequently leads to activation of plasma kallikrein (Figure 1). Factor XII is converted to fXIIa, which then converts prekallikrein to kallikrein. Kallikrein cleaves high-molecular-weight kininogen (HMWK) to bradykinin. Bradykinin cleaves factor XII producing fXIIa, forming a strong positive feedback loop, amplifying its effects.18 Patients with HAE do not have enough functional C1-INH to keep up with this activation, resulting in uncontrolled production of bradykinin. Bradykinin then binds to its transmembrane B1 or B2 receptors. Both receptor subtypes are G-protein-coupled receptors and can be found on vascular endothelium and smooth muscle. The B2 receptors are constitutively expressed and are responsible for mediating most of the kinins’ vascular and metabolic actions. The B1 receptor is inducible and can be detected in certain states, including tissue damage and inflammation.19 When bradykinin binds with its receptors, it results in the release of endothelium-derived mediators, including nitric oxide, prostacyclin, and endothelium-derived hyperpolarizing factor.16 Thus, bradykinin is a potent vasodilator leading to increased vascular permeability.20 Bradykinin is degraded by ACE (kinase II) and by multiple other kinases.16 Uncontrolled bradykinin activity leads to the symptoms of nonhistaminergic angioedema, resulting in edema and pain. Various treatments for nonhistaminergic angioedema attempt to target portions of the contact system to prevent unrestrained activation of bradykinin.
Figure 1.
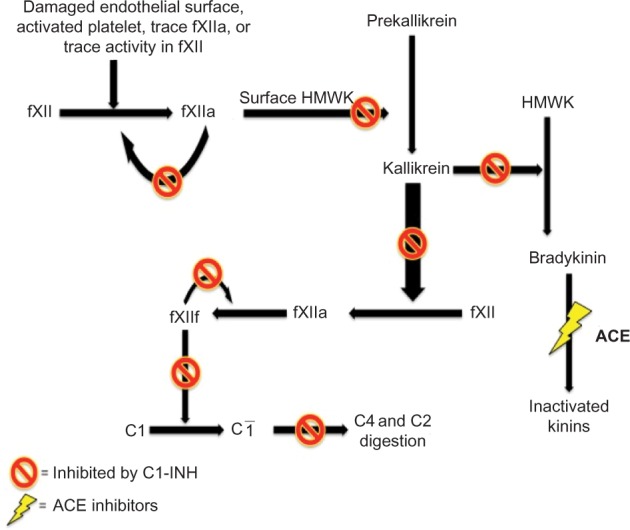
The pathophysiology of bradykinin formation via the contact pathway.
Notes: Functioning C1-INH inhibits the formation of bradykinin. ACE inhibitors impair the degradation of bradykinin.
Abbreviations: ACE, angiotensin-converting enzyme; C1-INH, C1-inhibitor; fXII, factor XII; HMWK, high-molecular-weight kininogen.
Current therapies for HAE are divided into management of acute attacks and short- and long-term prophylaxes. Long-term prophylaxis may be initiated in patients who have severe, frequent episodes of angioedema. Medications in this category include androgens, plasma-derived C1-INH (pdC1-INH), and tranexamic acid. Short-term prophylaxis can be considered for surgeries, especially those requiring endotracheal intubation and/or manipulation of the upper airway,14 stressful periods, pregnancy, and childbirth. Prophylactic treatment options at the time of pregnancy would likely involve pdC1-INH because of the better safety profile. In the pediatric population, tranexamic acid would likely be the treatment of choice for long- and short-term prophylaxes, especially when pdC1-INH is unavailable. One needs to use androgens with caution during childhood because of potential adverse side effects. Of note, long-term prophylaxis has been shown to decrease the number of attacks that patients have; however, they have not been proven to place patients in complete remission.21
Over the past several years, significant advances in the management of acute attacks have been made. Management used to consist of mainly supportive measures (fluids, pain control, endotracheal intubation, or tracheostomy if needed) and possible fresh-frozen plasma transfusions. Recently, the development of medications that target specific points along the proteolytic cascade leading to the activation of bradykinin is changing how acute attacks are being managed (Figure 2). Medications currently approved for the treatment of acute HAE attacks include various forms of C1-INH (both plasma-derived and recombinant), icatibant (a reversible, competitive antagonist of the bradykinin type 2 receptor), and ecallantide (a reversible kallikrein inhibitor).
Figure 2.
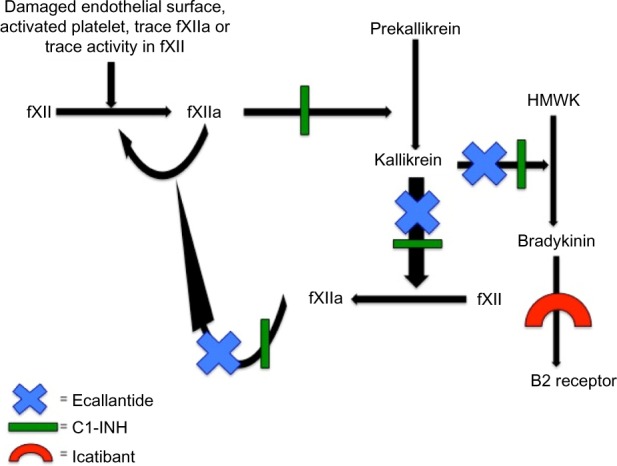
Mechanisms of action of the drugs used for treatment of acute HAE attacks.
Abbreviations: C1-INH, C1-inhibitor; fXII, factor XII; HAE, hereditary angioedema; HMWK, high-molecular-weight kininogen.
Ecallantide
Ecallantide is a small, 60-amino acid, recombinant protein inhibitor of plasma kallikrein, which was first approved by the FDA in December 2009 for treatment of acute attacks of HAE in patients aged ≥16 years and was approved more recently for patients aged ≥12 years. Ecallantide was discovered by phage display technology and is synthesized by the yeast strain Pichia pastoris.22 It inhibits kallikrein by blocking the plasma-binding site, thereby stopping production of bradykinin and, consequently, its effects. It also reduces further activation of fXIIa, halting the positive feedback mechanism leading to more kallikrein production (Figure 2).
Pharmacokinetics and metabolism
Recommended dosing of ecallantide is 30 mg delivered subcutaneously. A single dose of ecallantide has a mean maximum plasma concentration (Cmax) of 586 ng/mL in healthy volunteers. After 30 mg subcutaneous (SQ) dosing, mean (Cmax) values were observed at 2–3 hours postadministration.23 Mean elimination half-life was 2±0.5 hours after the dose was given. Ecallantide has rapid distribution in the vascular compartment after intravenous or SQ dosing, followed by rapid clearance. Because of its short half-life, ecallantide is not used in prophylactic therapy. An additional dose of 30 mg may be administered within a 24-hour period should the reaction persist.24 No data are available regarding those with hepatic or renal impairment or on the drug–drug interactions of ecallantide.24 However, as ecallantide is not metabolized through the cytochrome P-450 system, interactions with medications metabolized by the cytochrome P-450 pathway are not anticipated.25
Pharmacodynamics
Ecallantide reversibly binds to plasma kallikrein, preventing conversion of HMWK to bradykinin. While SQ administration of ecallantide alters the contact pathway, it does not appear to affect the complement or coagulation pathways. Intravenous administration demonstrated transient prolongation of activated partial thromboplastin time, but this was not shown after SQ administration.24,25
Clinical outcomes
Clinical efficacy of ecallantide for treatment of acute HAE was first established in the EDEMA studies, conducted by Dyax, with three Phase II trials (EDEMA0, 1, and 2), followed by two Phase III trials (EDEMA3 and 4). EDEMA2 was influential in establishing the optimal dose and administration of ecallantide. It was a dose-ranging study, examining patients with HAE presenting within 4 hours of an acute attack. The primary efficacy variables were the proportion of patients with a successful outcome (defined as resolution of attack within 4 hours of receiving an ecallantide dose and maintained for 24 hours) and partial response (defined as initial response but relapse between 4 hours and 24 hours of dosing).26 Safety was also assessed in this trial by monitoring for production of antibodies to ecallantide or P. pastoris, electrocardiograms, physical examinations, and adverse outcomes.26
Results from EDEMA3 and EDEMA4 led to the approval of ecallantide use in acute attacks of HAE. EDEMA3 was a randomized, double-blind, placebo-controlled study performed on HAE patients presenting within 8 hours of initiation of a moderate-to-severe attack. Patients received either placebo or 30 mg SQ ecallantide. Patients were observed for 4 hours after receiving the dose and had as many as three follow-up visits. The primary efficacy measurement in this study was the treatment outcome score (TOS) and the secondary efficacy measurement was the mean symptom complex severity (MSCS) score. Similar safety assessments were followed in EDEMA3 as were conducted in EDEMA2. An open-label extension phase was added, giving patients in the double-blind phase the choice to continue in the study. During this open-label portion, patients would present to the study site within 8 hours of an acute attack and receive 30 mg SQ ecallantide. If patients had a partial response, then a randomized blinded dose of placebo or ecallantide would be administered. Efficacy end points were the same as in the double-blind phase. Outcomes of EDEMA3 were influenced by errors in two patients. One patient was randomized to placebo and received ecallantide and the other patient was randomized to ecallantide but received placebo. This error led to significant changes in data interpretation; however, if the intention to treat is defined as treated as opposed to randomized, then this study demonstrated statistical significance between placebo and ecallantide.
EDEMA4 had the same design as EDEMA3, except that the primary efficacy measurement was the MSCS and the secondary efficacy measurement was the TOS. A total of 96 patients aged ≥10 years with known HAE type 1 or type 2 were enrolled in the study and 95 completed the study. Both the MSCS score and TOS indicated statistically significant greater improvement in symptoms in the treatment group when compared with the placebo group 4 hours after dosing. EDEMA4 also had an open-label extension similar to EDEMA3. The results from EDEMA4 demonstrate that ecallantide is an effective treatment for acute attacks of HAE and acts by both preventing progression and greatly reducing HAE symptoms.27 In addition to the individual results from EDEMA3 and EDEMA4, integrated analysis of the two trials further illustrated the effectiveness of ecallantide in relieving acute HAE attacks.
Since the EDEMA trials, multiple investigators have conducted further analysis of these studies examining the efficacy of ecallantide, proportion of relapse or rebound after ecallantide, and factors associated with the need for a second dose of ecallantide. Prospective analysis of EDEMA3 double blind (EDEMA3-DB) and EDEMA4 trials concluded that ecallantide is a safe and effective treatment for acute attacks of HAE regardless of anatomic location of the angioedema.28 Sheffer et al29 analyzed four clinical studies (EDEMA2, EDEMA3, EDEMA4, and DX-88/19) to examine the efficacy of ecallantide for treatment specifically of laryngeal HAE attacks. The analysis included 98 patients treated with 30 mg SQ ecallantide for 220 laryngeal attacks. Their analysis concluded that 80% of patients experienced sustained improvement after receiving ecallantide and 63% achieved complete or near-complete resolution of symptoms within 4 hours of dosing.29
Bernstein et al30 performed a post hoc analysis of EDEMA3-DB and EDEMA4 to investigate the likelihood of relapse or rebound symptoms after ecallantide treatment. Rebound was defined as initial improvement at the 4-hour mark after receiving ecallantide, followed by worsening of symptoms at the 24-hour mark beyond the subject’s initial severity rating. Relapse was defined as initial improvement at the 4-hour mark after receiving ecallantide, followed by worsening of symptoms at the 24-hour mark but not beyond the initial severity rating. Of the 42 patients treated with ecallantide included in analysis, one patient demonstrated likely relapse (2.4%), with an additional three patients showing possible relapse or rebound (7.2%). They concluded there was little evidence to suggest that rebound occurred, and a small proportion experienced relapse after treatment with ecallantide.30 Pooled clinical data from EDEMA2, DX-88/19, and EDEMA4 revealed that 12% of ecallantide-treated patients received a second ecallantide dose. Peripheral attacks were the only factor found to be associated with the need for a second dose of ecallantide.31 These data suggest that peripheral HAE attacks may be slower to respond to ecallantide treatment. Additional evaluation of these data also found no increase in treatment-related adverse events with a second dose of ecallantide.31
Ecallantide has been used in other forms of nonhistaminergic angioedema. Some forms of idiopathic angioedema are characterized by angioedema that is not responsive to antihistamines and corticosteroids, with normal levels and function of C1-INH and no family history of angioedema. As previously mentioned, some forms of idiopathic angioedema are mediated by histamine and those can be effectively treated with antihistamines in most cases. Successful treatment of nonhistaminergic idiopathic angioedema with ecallantide has been described in case reports. One case describes a 16-year-old female diagnosed with idiopathic angioedema presenting with an acute attack of angioedema. She was treated with 30 mg SQ ecallantide and had improvement of her symptoms within 30 minutes and resolution within 4 hours of receiving treatment.1 A second case reports an 8-year-old with idiopathic angioedema, who was treated with 30 mg SQ ecallantide, resulting in symptom improvement at 2 hours and complete resolution of symptoms within 11 hours.32 These results suggest that some cases of idiopathic angioedema are mediated by bradykinin, similar to HAE, indicating that ecallantide may be effective in these forms of angioedema; however, not all cases are mediated by bradykinin and, in those cases, ecallantide would likely not be effective.
ACE inhibitor-associated angioedema is thought to be secondary to impaired degradation of bradykinin (Figure 1).16 Inhibition of ACE results in impaired breakdown of bradykinin and substance P (another vasoactive molecule); however, not all patients taking ACE inhibitors develop angioedema, suggesting that certain patients have some unknown factor that predisposes them to develop angioedema.33 Recent research has identified that that genetic variations in the gene encoding neprilysin (MME), an enzyme that degrades bradykinin and substance P, were highly correlated with ACE inhibitor-induced angioedema.34 Ecallantide’s role in treating ACE inhibitor-associated angioedema is limited to case studies and one small trial. A case report involving a patient admitted to the hospital with ACE inhibitor-associated angioedema describes gradual worsening of symptoms over a period of 2 days, requiring intubation. On day 2, the patient received a dose of ecallantide and improved over 8 hours, allowing for extubation.35 There was a double-blinded, placebo-controlled, multicenter study, enrolling 76 patients and examining the use of ecallantide as treatment for ACE inhibitor-associated angioedema.36 The primary end point of this study was meeting a set of predetermined discharge criteria within 6 hours of receiving ecallantide or placebo.36 The results demonstrated no statistical significance in favor of using ecallantide over placebo; however, investigators noted that the majority of patients experiencing mild-to-moderate angioedema will meet discharge criteria within 6 hours of presentation, regardless of the intervention or lack of intervention.36
Acquired angioedema can be seen in cases of severe illness, such as autoimmune disease, or malignancy, and it results from decreased C1-INH or impaired function of C1-INH. Cicardi and Zanichelli37 describe two patients with acquired angioedema who were both successfully treated with ecallantide. In the Phase II clinical trial of ecallantide, inclusion criteria included those with acquired angioedema.38 Two patients, both with acquired angioedema related to underlying monoclonal gammopathy of undetermined significance, were treated with ecallantide and experienced significant symptom reduction within a few hours of treatment.39 Evidence for ecallantide use in acquired angioedema is limited to a small number of patients and its use is not currently approved by the FDA for acquired angioedema.
Safety, tolerability, and home infusion programs
The following safety and tolerability data are composed from the EDEMA3 and EDEMA4 studies,27,40 the US manufacturer’s prescribing information,41 the FDA review,26 and briefing documents.41 In 255 patients with HAE who were treated with ecallantide, the following reactions were observed as treatment-related adverse events (ordered from most prevalent to least prevalent): headache, nausea, fatigue, diarrhea, upper respiratory tract infection, injection site reactions, nasopharyngitis, vomiting, pruritus, abdominal pain, and pyrexia (Table 2).41 Treatment-related adverse events occurred in 11.1% of those treated with ecallantide and 13.9% of those receiving placebo in the EDEMA3 study.40 In the EDEMA4 study, 8% of those treated with ecallantide experienced treatment-related adverse events compared to 15% of patients in the placebo arm.27 Anaphylaxis occurred in 4% of patients with HAE.41 For this reason, ecallantide is only approved for administration by a health care professional in a setting with appropriate medical support.41 Patients exposed to ecallantide are at risk for developing anti-ecallantide antibodies. Anti-P. pastoris immunoglobulin (Ig) E antibodies were identified in 20.2% of patients tested and anti-ecallantide IgE antibodies were found in 4.7% of patients tested.41 Long-term effects of these antibodies are unknown, although elevated IgE levels to any agent could lead to increased risk of an allergic reaction. Craig et al42 conducted a study that specifically examined anaphylaxis associated with ecallantide. The study included 230 individuals treated with ecallantide and eight (3.5%) had anaphylactic reactions within an hour of receiving ecallantide. No one had a reaction on first exposure. Additionally, they did not find a correlation between having anti-P. pastoris IgE or anti-ecallantide antibodies and developing a type 1 hypersensitivity reaction to ecallantide.42
Table 2.
Adverse reactions occurring at ≥3% than placebo in two placebo-controlled clinical trials in patients with HAE treated with ecallantide
| Adverse reactions | Ecallantide (N=100), n (%)* |
Placebo (N=100), n (%) |
|---|---|---|
| Headache | 8 (8%) | 6 (7%) |
| Nausea | 5 (5%) | 1 (1%) |
| Diarrhea | 4 (4%) | 3 (4%) |
| Pyrexia | 4 (4%) | 0 |
| Injection site reaction | 3 (3%) | 1 (1%) |
| Nasopharyngitis | 3 (3%) | 0 |
Notes:
Patients experiencing the same adverse reaction more than one time were only counted once in that category. Adverse reactions of ecallantide compared to placebo from prescribing information package insert. Data from US FDA.26
Abbreviation: HAE, hereditary angioedema.
Home infusion programs using ecallantide were instituted in an attempt to provide more convenient treatment but still maintain the same level of safety and effectiveness an established health care center would provide. A retrospective analysis was performed by a home infusion company on the treatment of nonlaryngeal HAE attacks, demonstrating this as an effective delivery method for treatment.43 Patients enrolled in this program had to have a diagnosis of HAE and a referral from their provider. Additionally, they were screened for comorbidities and had to live within a specified distance from the nursing services. Pediatric patients were ineligible. Patients call the service over the telephone and would receive treatment within 4 hours. The study examined data from September 2011 to December 2012, which included 158 patients for a total of 1,177 nursing visits. The mean time for nursing response was 64 minutes, and patients reported symptom relief at a mean time of 42 minutes after ecallantide administration. There were nine patients who experienced allergic reactions, seven of which were classified as anaphylaxis. All of the reactions resolved without serious sequelae, but eight of the nine were sent to the emergency department. The article concluded that home nursing infusion of ecallantide is an effective and safe treatment for HAE acute attacks.43 In addition to improving patient satisfaction, home infusion programs likely save a substantial amount of health care dollars. The mean cost of treatment for HAE acute attack in an emergency department is $2,603, while treatment using home care infusion is $150.44
Use in special populations
The FDA extended approval of ecallantide use from 16 years and older down to 12 years and older in 2014. Specific examination of pediatric patients from EDEMA2, EDEMA3, EDEMA4, and DX-88/19 revealed significant decrease in symptoms in those treated with ecallantide compared with those on placebo.8 Patients included in the analysis were aged 9–17 years old. Only one serious treatment-emergent adverse event occurred, which was a staphylococcal cellulitis infection distant to the site of ecallantide administration. This was judged to be unrelated to treatment.8 Ecallantide is a pregnancy category C as assigned by the FDA. Animal studies demonstrated developmental toxicity.41 Use of ecallantide in breastfeeding women is cautioned against as it is not known whether ecallantide is excreted in breast milk and it is not known what effects it may have on a developing, growing infant.
Three different targets for treatment of acute HAE attack
Within the past several years, medications targeting three different pharmacologic sites along the contact proteolytic cascade have been approved for use in treatment of acute HAE attacks. As in many other disease processes, it is advantageous to have drugs that work via different mechanisms. Some patients may respond more favorably to a particular medication or may require treatment with an alternate class of medication if the first therapy fails to alleviate symptoms. C1-INH is the only therapy used in treatment of acute attacks and short- and long-term prophylaxes. As ecallantide and icatibant work via a different mechanism than C1-INH medications, those who are on long- or short-term prophylaxes with C1 inhibitors may benefit from ecallantide or icatibant for relief of an acute attack. Burke et al45 describe a case involving a 49-year-old woman with known HAE who was receiving Cinryze™ for short-term prophylaxis for a procedure and who developed an acute HAE attack 1.5 hours postoperatively. She was successfully treated with ecallantide.45 Further research is needed to determine whether ecallantide can be used in short-term prophylaxis for perioperative procedures. Currently, ecallantide and two pdC1-INH medications (Cinryze™ and Berinert®) are approved for use in children ≥12 years, while icatibant is only approved for those who are ≥18. Ruconest®, a recombinant C1-INH, can also be used in adolescents and adults. Both icatibant and ecallantide are administered subcutaneously, giving them a theoretical advantage of easier administration over intravenous agents.46 While icatibant and pdC1-INH medications can be self-administered, ecallantide and Ruconest cannot because they both have the risk of anaphylaxis.
Conclusion
HAE can be a debilitating and even life-threatening disease characterized by deficiency or decreased function of C1-INH. Management for HAE can involve medications for long-term prophylaxis, short-term prophylaxis, and acute attacks. While long- and short-term prophylaxes have been shown to decrease symptom severity and frequency, these treatments have not eliminated acute attacks of HAE and have their own side effects and associated problems with long-term therapies (cost, compliance, etc). Over the past 5 years, the management of acute attacks of HAE has greatly changed with the advent and FDA approval of novel medications. New therapies target specific points of the contact cascade, inhibiting its continuous activation. Currently, the World Allergy Organization consensus guidelines recommend the use of C1-INH, ecallantide, or icatibant for acute treatment of HAE attacks.14
The efficacy and safety of ecallantide have been demonstrated in multiple trials, particularly in the EDEMA trials, which ultimately led to FDA approval. Ecallantide is approved for use in those with HAE who are ≥12 years. While ecallantide is not approved for other forms of nonhistaminergic angioedema, case studies have demonstrated promise for ecallantide treatment in these populations. Ecallantide is administered in doses of 30 mg delivered subcutaneously. Overall, it is a safe medication; however, due to the risk of anaphylaxis, a health care professional must administer it. Some patients treated with ecallantide have developed antibodies to ecallantide and P. pastoris. The significance of these antibodies is not known at this time.
Head-to-head trials comparing C1-INH, ecallantide, and icatibant have not been conducted. In general, having three medications with different mechanisms of action will likely prove beneficial as further research may reveal varied responses to therapies among patient populations. Each medication has its own advantages and disadvantages. The advantages of ecallantide include SQ administration and approved use in younger patients, with its main disadvantage being risk of anaphylaxis, thereby inhibiting self-administration.
In summary, ecallantide is a novel treatment for acute attacks of HAE and has proven highly effective and safe. Further postmarketing research of long-term effects of ecallantide’s use and comparison with other agents approved for acute treatment of HAE are needed and will help further shape management of acute attacks.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Berry A, Firszt R. Successful treatment of idiopathic angioedema with ecallantide. J Allergy Clin Immunol. 2013;1(3):297–298. doi: 10.1016/j.jaip.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Zuraw BL. Hereditary angioedema. N Engl J Med. 2008;359(10):1027–1036. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 3.Cicardi M, Agostoni A. Hereditary angioedema. N Engl J Med. 1996;334(25):1666–1667. doi: 10.1056/NEJM199606203342510. [DOI] [PubMed] [Google Scholar]
- 4.Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol. 2009;161(5):1153–1158. doi: 10.1111/j.1365-2133.2009.09366.x. [DOI] [PubMed] [Google Scholar]
- 5.Roche O, Blanch A, Caballero T, Sastre N, Callejo D, Lopez-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94(4):498–503. doi: 10.1016/S1081-1206(10)61121-0. [DOI] [PubMed] [Google Scholar]
- 6.Psarros F, Koutsostathis N, Farmaki E, Speletas MG, Germenia AE. Hereditary angioedema in Greece: the first results of the Greek hereditary angioedema registry. Int Arch Allergy Immunol. 2014;164(4):326–332. doi: 10.1159/000366276. [DOI] [PubMed] [Google Scholar]
- 7.Aygören-Pürsün E, Bygum A, Beusterien K, et al. Socioeconomic burden of hereditary angioedema: results from the hereditary angioedema burden of illness study in Europe. Orphanet J Rare Dis. 2014;9:99. doi: 10.1186/1750-1172-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macginnitie AJ, Davis-Lorton M, Stolz LE, Tachdjian R. Use of ecallantide in pediatric hereditary angioedema. Pediatrics. 2013;132(2):e490–e497. doi: 10.1542/peds.2013-0646. [DOI] [PubMed] [Google Scholar]
- 9.Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs and course. Am J Med. 2006;119(3):267–274. doi: 10.1016/j.amjmed.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 10.Jolles S, Williams P, Carne E, et al. A UK national audit of hereditary and acquired angioedema. Clin Exp Immunol. 2013;175(1):59–67. doi: 10.1111/cei.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bork K, Hardt J, Schicketanz K, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163(10):1229–1235. doi: 10.1001/archinte.163.10.1229. [DOI] [PubMed] [Google Scholar]
- 12.Sardana N, Craig T. Advances in management and treatment of hereditary angioedema. Pediatrics. 2011;128(6):1173–1180. doi: 10.1542/peds.2011-0546. [DOI] [PubMed] [Google Scholar]
- 13.Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33(Suppl 1):S145. doi: 10.2500/aap.2012.33.3627. [DOI] [PubMed] [Google Scholar]
- 14.Craig T, Aygören-Pürsün E, Bork K, et al. WAO guideline for the management of hereditary angioedema. World Allergy Organ J. 2012;5:182–199. doi: 10.1097/WOX.0b013e318279affa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343(4):1286–1289. doi: 10.1016/j.bbrc.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 16.Bas M, Adams V, Suvorava T, Niehues T, Hoffmann TK, Kojda G. Nonallergic angioedema: role of bradykinin. Eur J Allergy Clin Immunol. 2007;62(8):842–856. doi: 10.1111/j.1398-9995.2007.01427.x. [DOI] [PubMed] [Google Scholar]
- 17.Gompels MM, Lock RJ, Morgan JE, Osborne J, Brown A, Virgo PF. A multicentre evaluation of the diagnostic efficiency of serological investigations for C1 inhibitor deficiency. J Clin Pathol. 2002;55(2):145. doi: 10.1136/jcp.55.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cicardi M, Johston DT. Hereditary and acquired complement component 1 esterase inhibitor deficiency: a review for the Hematologist. Acta Haematol. 2012;127(4):208–220. doi: 10.1159/000336590. [DOI] [PubMed] [Google Scholar]
- 19.Kastin AJ, editor. Handbook of Biologically Active Peptides. 2nd ed. Massachusetts: Academic Press; 2013. [Google Scholar]
- 20.Bouillet L, Mannic T, Arboleas M, et al. Hereditary angioedema: key role for kallikrein and bradykinin in vascular endothelial-cadherin cleavage and edema formation. J Allergy Clin Immunol. 2011;128(1):232–234. doi: 10.1016/j.jaci.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 21.Frank MM. Update on preventative therapy (prophylaxis) for hereditary angioedema. Immunol Allergy Clin North Am. 2013;33(4):495–503. doi: 10.1016/j.iac.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Zuraw B, Yasothan U, Kirkpatrick P. Ecallantide. Nat Rev Drug Discov. 2010;9(3):189–190. doi: 10.1038/nrd3125. [DOI] [PubMed] [Google Scholar]
- 23.Farkas H, Varga L. Ecallantide is a novel treatment for attacks of hereditary angioedema due to C1 inhibitor deficiency. Clin Cosmet Investig Dermatol. 2011;4:61–68. doi: 10.2147/CCID.S10322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garnock-Jones KP. Ecallantide in acute hereditary angioedema. Drugs. 2010;70(11):1423–1431. doi: 10.2165/11205850-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Martello JL, Woytowish MR, Chambers H. Ecallantide for treatment of acute attacks of hereditary angioedema. Am J Health Syst Pharm. 2012;69(8):651–657. doi: 10.2146/ajhp110227. [DOI] [PubMed] [Google Scholar]
- 26.US FDA. Center for Drug Evaluation and Research Application 125277: Ecallantide Summary Review of Regulatory Action [online] 2009. [Accessed October 16, 2014]. Available from: http://www.accessdata.fda.gov.ezproxy.lib.utah.edu/drugsatfda_docs/nda/2009/125277s000SumR.pdf.
- 27.Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(6):523–529. doi: 10.1016/j.anai.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Stolz LE, Sheffer AL. Prospective, double-blind, placebo-controlled trials of ecallantide for acute attacks of hereditary angioedema. Expert Rev Clin Immunol. 2012;8(1):25–32. doi: 10.1586/eci.11.81. [DOI] [PubMed] [Google Scholar]
- 29.Sheffer AL, MacGinnitie AJ, Campion MC, Stolz LE, Pullman WE. Outcomes after ecallantide treatment of laryngeal hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2013;110(3):184–188. doi: 10.1016/j.anai.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Bernstein JA, Shea EP, Koester J, Iarrobino R, Pullman WE. Assessment of rebound and relapse following ecallantide treatment for acute attacks of hereditary angioedema. Eur J Allergy Clin Immunol. 2012;67(9):1173–1180. doi: 10.1111/j.1398-9995.2012.02864.x. [DOI] [PubMed] [Google Scholar]
- 31.Li HH, Campion M, Craig TJ, et al. Analysis of hereditary angioedema attacks requiring a second dose of ecallantide. Ann Allergy Asthma Immunol. 2013;110(3):168–172. doi: 10.1016/j.anai.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Dy TB, Rasheed M, Parikh P, Bernstein B. Resolution of an acute attack of idiopathic angioedema with ecallantide. Am Allergy Asthma Immunol. 2013;111(3):224–226. doi: 10.1016/j.anai.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 33.Baram M, Kommuri A, Sellers SA, Cohn JR. ACE inhibitor-induced angioedema. J Allergy Clin Immunol. 2013;1(5):442–445. doi: 10.1016/j.jaip.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Pare G, Kubo M, Byrd JB, et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genomics. 2013;23(9):470–478. doi: 10.1097/FPC.0b013e328363c137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohn JR, McDaniel C, Richards N, Au C, Baram M. Prompt reversal of airway obstruction secondary to angiotensin converting enzyme inhibitor (ACEI) induced angioedema by ecallantide: a case report. J Allergy Clin Immunol. 2012;129(Suppl 2):AB220. [Google Scholar]
- 36.Lewis L, Graffeo C, Crosley P, et al. Ecallantide for the acute treatment of angiotensin converting enzyme inhibitor-induced angioedema: a multicenter randomized controlled trial. Ann Emerg Med. 2013;62(Suppl 2):S132. doi: 10.1016/j.annemergmed.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Cicardi M, Zanichelli A. Acquired angioedema. Allergy Asthma Clin Immunol. 2010;6(1):14. doi: 10.1186/1710-1492-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalbitor® (Ecallantide) The Safety and Efficacy of Ecallantide in Patients with Acquired Angioedema. Cambridge, MA: Dyax Corp; 2014. [Google Scholar]
- 39.Patel NS, Fung SM, Zanichelli A, Cicardi M, Cohn JR. Ecallantide for treatment of acute attacks of acquired C1 esterase inhibitor deficiency. Allergy Asthma Proc. 2013;34(1):72–77. doi: 10.2500/aap.2013.34.3620. [DOI] [PubMed] [Google Scholar]
- 40.Levy R, McNeil D, Li H, et al. Results of a phase 3 double-blind, placebo-controlled trial, EDEMA3: a stubby of subcutaneous DX-88 (ecallantide) in patients with hereditary angioedema [abstract no 15] J Allergy Clin Immunol. 2008;121(Suppl 2A):A6. [Google Scholar]
- 41.KALBITOR® (Ecallantide) Injection, for Subcutaneous Use [Prescribing Information] [online] Cambridge, MA: Dyax Corp; 2014. [Accessed October 18, 2014]. Available from: http://www.kalbitor.com/hcp/rems/pdf/KalbitorFullPrescribing-Information.pdf. [Google Scholar]
- 42.Craig TJ, Li HH, Riedl M, et al. Characterization of anaphylaxis after ecallantide treatment of hereditary angioedema attacks. J Allergy Clin Immunol Pract. 2015;3(2):206–212. doi: 10.1016/j.jaip.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Kuhlen J, Guyer A, Morphew T, Tachdjian R, Banerji A. Assessment of home infusion program for treating nonlaryngeal hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2014;112(5):471–472. doi: 10.1016/j.anai.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Wilson DA, Bork K, Shea EP, Rentz AM, Blaustein MB, Pullman WE. Economic costs associated with acute attacks and long-term management of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(4):314e–320e. doi: 10.1016/j.anai.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 45.Burke S, Petrov A, Johnson J, MacGinnitie A. Successful use of ecallantide to treat a hereditary angioedema attack after failure of C1 inhibitor prophylaxis. Ann Allergy Asthma Immunol. 2011;107(2):181–182. doi: 10.1016/j.anai.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 46.Thomas M, Shah S. New treatment options for acute edema attacks caused by hereditary angioedema. Am J Health Syst Pharm. 2011;68(22):2129–2138. doi: 10.2146/ajhp100718. [DOI] [PubMed] [Google Scholar]