

Abstract
American foulbrood is the most destructive brood disease of honeybees (Apis mellifera) globally. The absence of a repeatable, universal typing scheme for the causative bacterium Paenibacillus larvae has restricted our understanding of disease epidemiology. We have created the first multilocus sequence typing scheme (MLST) for P. larvae, which largely confirms the previous enterobacterial repetitive intergenic consensus (ERIC)–polymerase chain reaction (PCR)-based typing scheme's divisions while providing added resolution and improved repeatability. We have used the new scheme to determine the distribution and biogeography of 294 samples of P. larvae from across six continents. We found that of the two most epidemiologically important ERIC types, ERIC I was more diverse than ERIC II. Analysis of the fixation index (FST) by distance suggested a significant relationship between genetic and geographic distance, suggesting that population structure exists in populations of P. larvae. Interestingly, this effect was only observed within the native range of the host and was absent in areas where international trade has moved honeybees and their disease. Correspondence analysis demonstrated similar sequence type (ST) distributions between native and non-native countries and that ERIC I and II STs mainly have differing distributions. The new typing scheme facilitates epidemiological study of this costly disease of a key pollinator.
Introduction
Paenibacillus larvae, a Gram-positive spore-forming bacterium, causes American foulbrood (AFB), which is the most destructive brood disease of the honeybee (Apis mellifera). AFB is contagious because of the large numbers of highly resistant spores that are produced and efficiently transmitted by contaminated adult bees within and between colonies (Lindström, 2008; Lindström et al., 2008a,b,). Only the spores are infective and are fed to bee brood by nurse bees in contaminated larval food (glandular secretions and processed honey) (Yue et al., 2008). Once infected, larvae usually die within 3–12 days (Genersch et al., 2005; Rauch et al., 2009). Paenibacillus larvae spores are able to remain infective for more than 35 years in old hives and are resistant to extremes of temperature (Hasemann, 1961). This makes the control of the disease difficult because human activity can spread the disease over long distances and previously dormant strains may cause an outbreak several years after the original outbreak.
Antibiotics only affect the vegetative stage of the bacterium, masking the symptoms of AFB; they have no effect on the infective spores (Genersch and Otten, 2003). In many countries, burning infected colonies and hive materials is thought to be the most effective way of preventing the spread of AFB. Therefore, whether AFB is ignored or treated, the colony will be killed, which leads to considerable economic loss to global apiculture.
AFB is found on every continent where honeybees are kept (Matheson, 1993). The disease is spread by both humans and bees, and it is spread predominantly via horizontal routes although it has been shown to spread vertically (Fries et al., 2006; Lindström et al., 2008a). Horizontal transmission of AFB can occur by several means because of humans through the movement of contaminated honey or the movement of contaminated hives or equipment (Genersch, 2010). AFB can also be spread horizontally by bees, either through the movement of adult bees between colonies (drifting) or the behaviour of foragers (robbing) (Lindström et al., 2008a). Using genetic markers to identify outbreaks caused by closely related strains can give important evidence to confirm the source and routes of disease transmission. The shortcomings of phenotypically based typing methods for P. larvae (Genersch et al., 2006) have led to the development of molecular typing methods based on the microbial DNA sequence (Alippi and Aguilar, 1998; Genersch and Otten, 2003; Genersch et al., 2006; Antúnez et al., 2007; Pentikäinen et al., 2008). Four genotypes of P. larvae have been identified based on repetitive-element polymerase chain reaction (PCR) (rep-PCR) using enterobacterial repetitive intergenic consensus (ERIC) primers (Genersch et al., 2006). These four genotypes were shown to form two clusters using pulsed-field gel electrophoresis (PFGE) (Genersch et al., 2006). ERIC types differ in phenotype including morphology (Genersch et al., 2006), metabolic capacity (Neuendorf et al., 2004) virulence (Genersch et al., 2005; Rauch et al., 2009) and virulence factors (Poppinga et al., 2012; Fünfhaus et al., 2013). The above typing schemes have been used to categorize crude patterns of P. larvae distribution in Europe (Genersch and Otten, 2003; Pentikäinen et al., 2008; Loncaric et al., 2009; Di Pinto et al., 2011), the Americas (Alippi et al., 2004; Antúnez et al., 2007) and Australasia (Alippi et al., 2004). However, rep-PCR methods have the disadvantage that they are difficult to repeat or to standardize between laboratories and therefore comparisons between different studies is difficult (Rusenova et al., 2013). Recently, it was shown that the four ERIC genotypes can also be discriminated via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Schäfer et al., 2014), providing a cost-effective, reliable and highly reproducible alternative tool for P. larvae ERIC typing. The advantage of the established ERIC scheme for P. larvae (Genersch et al., 2006) is that it allows grouping into biologically relevant genotypes differing in practically important phenotypic features. However, it does not give enough resolution to be used effectively as an epidemiological tool to study disease spread. Therefore, a state-of-the-art method providing sufficient resolution to be informative to epidemiological studies is urgently needed in order to enhance differentiation beyond the four ERIC genotypes.
The utilization of sequence-based typing would allow a single, universal and unambiguous scheme that would help us to better understand the global spread of this damaging disease. Multilocus sequence typing (MLST) can provide a standardized set of strain types that can be used to study bacterial population structure and evolution at both a global and a more local scale. MLST is based on unambiguous DNA sequences and allelic profiles can be shared between laboratories using online databases such as PubMLST (Jolley and Maiden, 2010). MLST schemes have become the gold standard for epidemiological studies, providing insight into the epidemiology of human pathogens such as the Bacillus cereus group (Helgason et al., 2004) to which P. larvae is related. Generally, MLST schemes consist of short regions of six or seven housekeeping genes that evolve at a slow even pace across all strains (Maiden, 2006).
Here, we report the development of a novel seven genes MLST scheme to enhance differentiation within the species P. larvae and we use this scheme to identify global patterns in the population structure of P. larvae.
Results
Loci discovery and primer design
In total, 31 primer pairs, including non-coding loci, were tested against P. larvae isolates representing all four ERIC types. The majority of loci were rejected because of low diversity between test isolates (Table S1). Of the remaining loci, seven offered the largest diversity within the 294 isolates of P. larvae tested: clpC (catabolite control protein A), ftsA (cell division protein), glpF (glycerol uptake facilitator protein), glpT (glycerol-3-phosphate permease), Natrans (forward sodium dependant transporter), sigF (sporulation sigma factor F) and rpoB (RNA polymerase beta subunit) (Table 1).
Table 1.
MLST primer sequences
| Gene | Forward primer | Reverse primer | Annealing temperature (°C) |
|---|---|---|---|
| clpC | 5′TTTGGAAGATTTACTGAACGA3′ | 5′ATCAGAACCGGGTTATTTTT3′ | 52 |
| ftsA | 5′AAATCGGTGAGGAAGACATT3′ | 5′TGCCAATACGGTTTACTTTA3′ | 52 |
| glpF | 5′GTCAGCGGGGCTCATTTA3′ | 5′TGCTTACGATGAGAAATCCT3′ | 52 |
| glpT | 5′GGATTGAAAAACTTGAAACG3′ | 5′CATGCTGAGAGAAATCTTCC3′ | 52 |
| Natrans | 5′GCTTCGGTAATGGTAACCTA3′ | 5′TTGAACCCATTGTAAATTCC3′ | 52 |
| rpoB | 5′ATAACGCGAGACATTCCTAA3′ | 5′GAACGGCATATCTTCTTCAG3′ | 52 |
| sigF | 5′GTCACTGAAGGAATTGGCTA3′ | 5′TATCTGGTTACGGATGGACT3′ | 52 |
Fragment length and G + C content for the seven selected loci ranged from 271 bp (glpF) to 541 bp (clpC) and 43.8% to 48.7% respectively (Table 2). The percentage of variable sites ranged from 0.65 (ftsA) to 2.0 (Natrans) resulting in 4–6 alleles per locus (Table 2). The dN/dS values were all lower than 1 for all genes, except glpF, which contained a deletion. The ratio of non-synonymous and synonymous substitutions (dN/ds) measures the level of selection in a protein-coding gene. To ensure consistency in an MLST, it is preferable that each locus is under stabilizing selection. However, genes that are under positive selection may give more resolution to the scheme (Maiden, 2006). The ratio of dN/dS indicates purifying selection (negative selection) if values are < 1, positive selection if values are > 1 and neutral evolution if values are close to 1. A value approaching 1 may also indicate a combination of positive and purifying selection.
Table 2.
Feature summary of seven loci selected for P. larvae typing scheme
| Locus | Sequence length (bp)a | Number of alleles | Mean G + Cb content | Number of polymorphic sitesa | Number of non-synonymous substitutions | dN/dS ratio |
|---|---|---|---|---|---|---|
| clpC | 541 | 5 | 48.7 | 4(0.74) | 3 | 0.4183 |
| ftsA | 464 | 4 | 46.0 | 3(0.65) | 2 | 0.7756 |
| glpF | 271 | 6 | 45.2 | 5(1.85) | 4 | 1.4835 |
| glpT | 502 | 4 | 47.4 | 9(1.80) | 6 | 0.5569 |
| Natrans | 490 | 6 | 46.8 | 10(2.00) | 6 | 0.2180 |
| rpoB | 339 | 5 | 48.7 | 4(1.50) | 2 | 0.4276 |
| sigF | 345 | 4 | 43.8 | 4(1.20) | 1 | 0.1607 |
Percentage of polymorphic sites in parentheses.
Guanine-cytosine content.
The index of association (IA) was significantly different from 0 when only one representative of each sequence type (ST) was included in the computation (1.16; P = 0.000), indicating limited recombination events and a clonal populations structure in P. larvae. The IA measures the extent of linkage equilibrium within a population by quantifying the amount of recombination among a set of sequences and detecting association between alleles at different loci (Smith et al., 2014).
Typed isolates included 173 ERIC type I, 92 ERIC type II, 3 ERIC type III and 7 ERIC type IV and 19 isolates where ERIC type was not determined because of a shortage of DNA (Table S2). At least one isolate of each ST was ERIC typed. The 7-gene MLST scheme resolved the 294 P. larvae isolates into 21 different STs (Fig. 1 and Fig. S2). The allele sequences have been submitted to the EMBL database under Accession numbers HG530076 to HG530109. The entire scheme is available at http://pubMLST.org/plarvae/ (Jolley and Maiden, 2010). ST designations represented a single ERIC grouping except for a single ERIC III isolate that grouped with ST8 (Figs 1 and 2). Isolates from ERIC I were separated into 16 STs, whereas ERIC II isolates were only separated into three separate STs. The Chao1 estimates suggest this difference in observed diversity was unlikely to be due to a biased sampling effort. After an initial increase, the mean Chao1 estimate for all geographical regions became relativley level as sample size increased (Fig. 3), therefore we compared the ST diversity estimates at the highest sample size for each ERIC type (Hughes et al., 2001). Total diversity of ERIC types is significantly different as estimated by Chao1 (Fig. 3). Chao1 estimates that ERIC I has 18.49 STs [95% confidence intervals (CIs) 16.37 and 32.91], and ERIC II has 3 STs (CIs 3 and 4.49). The STs were roughly split following the pattern of the ERIC typing scheme with different ERIC I and II types forming distinct groups with both the eburst and phylogenetic analysis (Figs 1 and 2; Table S2). ERIC III and IV isolates were less distinct with one isolate (P6266) LMG 16252) ERIC typed as ERIC III but sequence typing as ST8 and grouping with ERIC IV isolates (Fig. 1, Table S2). All isolates within ERIC I and II are linked by single loci variants, whereas the ERIC III and ERIC IV STs differ from each other at two loci. ERIC I and ERIC II isolates differ from one another by at least four loci (link not shown in Fig. 1) and ERIC III isolates differ from ERIC I isolates by at least six loci (Fig. 1). The ERIC III ST 9 is made up of only two isolates found in Chile (Table S2).
Fig 1.
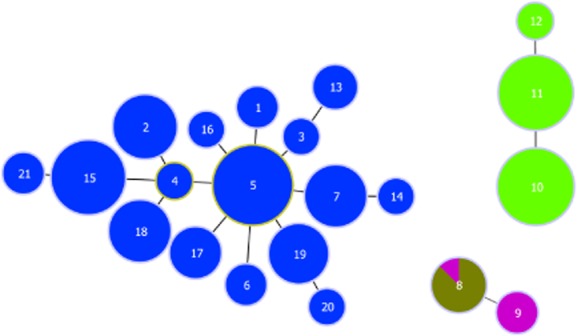
eburst diagram of P. larvae MLST scheme. The numbers in the circles represent the ST of the isolates (Table 2). The size of the circle represents the number of isolates with that type (Table 2). STs with variation at more than three loci are not connected. Clockwise from left, groups are: isolates typed as ERIC I, isolates typed as ERIC II and isolates typed as ERIC III and IV.
Fig 2.
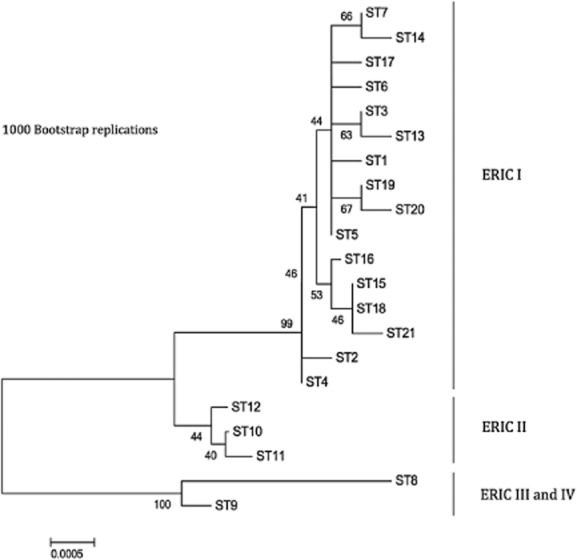
Neighbour-joining tree of all sequence types. The evolutionary history was inferred using the neighbour-joining method. The optimal tree with the sum of branch length = 0.01386545 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site.
Fig 3.
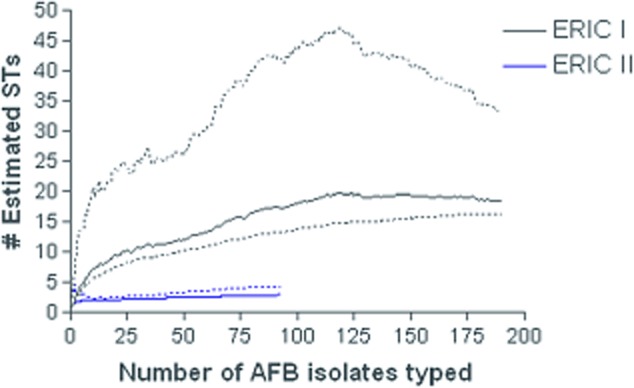
Chaol estimates of ERIC I and ERIC II ST richness as a function of sample size. Dotted lines are 95% CIs and were calculated with the variance formula derived by Chao (1987). The lower solid line represents ERIC II and the upper solid line represents ERIC I.
Population structure and biogeography
The two most common and widespread types of ERIC I were ST 15 and 5, which were each found causing disease in multiple countries and across five continents (Table S2). Contrastingly of the two most common STs of ERIC II, ST10 was well distributed and ST 11 was found only in Germany and countries to the east of Germany. In this study, no ERIC II isolates were collected from countries to the west of Germany within the native honeybee range.
The relationship between fixation index (FST) and geographic distance of the global populations of P. larvae were not significant (P = 0.996, r2 = 5.506 × 10−7), suggesting no relationship between genetic and geographic distance when considering the sampled global population of P. larvae. However, when the analysis was restricted to isolates collected from within the native range of honeybees [Europe, Africa and Eastern Asia; 226 of 294 isolates (see Table S2)] a significant relationship between genetic and geographic distance was detected (P = 0.01, r2 = 0.122; Fig. 4).
Fig 4.
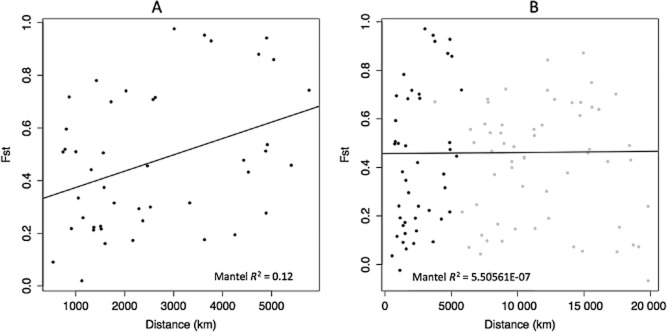
FST by distance.A. Describes the FST by distance of P. larvae populations within the native range of the host (Apis mellifera).B. Describes the FST by distance of P. larvae populations both within (dark grey dots) and outside of the native range of the host (light grey dots).
Correspondence analysis (CA)
The ordination graph (Fig. 5) describing the results of the CA shows a clear split in the distribution of the two ERIC types. In addition, Fig. 5 shows no split in the distribution of countries, whether they were in the native range of honeybees or not. This suggests that most STs are found in both the native range and the countries outside of the native range.
Fig 5.
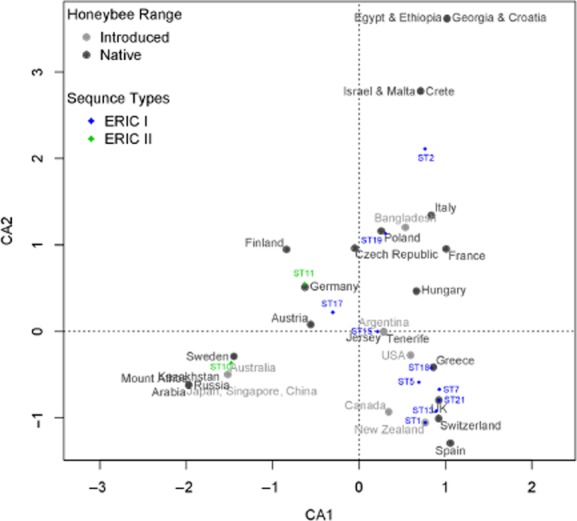
Correspondence analysis ordination graph. The CA ordination graph illustrates the associations among countries of isolate origins and MLST STs. Filled circles represent countries (dark grey represents native host range, light grey represents introduced host range) Diamonds represent STs.
The proportion of variance explained by the first two eigenvectors was 0.4026. In the ordination graph describing the results of the CA (Fig. 5), STs that have similar distribution are represented by points closer together in space and the proximity of STs to countries indicate that those STs are associated with that country. The first axis highlights the difference in distribution between ERIC I STs and ERIC II STs. The ERIC II STs (ST 10 and ST 11) cluster on the left (negative values) of CA1, and the ERIC I STs on the right, with the exception of ST17. ST10 groups with Arabia, Mount Athos, Kazakhstan, Russia, Sweden and Australia as well as mixed origin isolates from China, Singapore and Japan. ST11 is associated with Germany and Finland. Native range designation did not appear to influence the distribution of the points on the ordination plot, i.e. countries where honeybees have been introduced grouped with countries where honeybees are native.
Rarefaction
Rarefaction curves comparing the sampling effort for each continent show steep curves for all except Europe. However, when only data from Europe were analysed at country level, it was clear that only Germany and the UK were sampled to adequately describe the resident ST populations. This suggests that further sampling in other countries would yield previously unidentified STs.
Discussion
AFB is a serious disease of honeybee brood with near global distribution; however, disease epidemiology is poorly understood, in part due to an absence of a repeatable method to discriminate between strain types. We have developed the first MLST scheme, which ratifies and extends the established four-group ERIC typing scheme. The new scheme describes 21 different P. larvae STs, therefore providing improved resolution when compared with ERIC typing. Although other typing schemes, such as PFGE (Pentikäinen et al., 2008) offer greater resolution, i.e. more types, than MLST, these methods are difficult to repeat, preventing comparisons between published studies. MLST is now the gold standard for epidemiological studies (Francisco et al., 2012) and results can be applied to derive epidemiological meaning both locally and internationally.
Our MLST scheme was able to identify a significant relationship between geographic distance and genetic distance among P. larvae populations within the native range of the honeybee (Fig. 4A). This relationship is surprising given the history of human movement of bees within Europe in particular (De la Rúa et al., 2009). It might be expected that because of past honeybee movements, mixing of P. larvae STs might be equally distributed throughout the native range; however, this is not the case. A detectable link between physical and genetic distance remains despite these large-scale movements and endemic populations of P. larvae still appear to exist within the native range. Such a result could suggest that certain P. larvae strains are adapted to local honeybee populations. Honeybees are known to differ in their ability to resist pathogens (Jensen et al., 2009; Büchler et al., 2010), and AFB-tolerant honeybees have been bred in Argentina (Spivak and Reuter, 2001). However, little is known more generally about the susceptibility of honeybee races to different strains of P. larvae. A more comprehensive understanding of inter-race susceptibility of A. mellifera is required to understand whether host factors are in part responsible for the significant pathogen population structure within the native range of A. mellifera.
The commercial interest in honeybee hive products has lead to the spread of A. mellifera far beyond its native geographic range. This global industry has facilitated the spread of honeybee pests and pathogens such as the ectoparastic mite Varroa destructor (Solignac et al., 2005), Nosema ceranae. (Klee et al., 2007) and Israel acute paralysis virus (Palacios et al., 2008) and it is known that P. larvae spores can remain infective in honey (Morse, 1992; Govan et al., 1999; Hansen et al., 2003). Our data indicate that when AFB has been moved outside the native honeybee range, any evidence of significant pathogen population structure breaks down (Fig. 4B). This finding suggests that the international trade in honeybees and their hive products may have moved P. larvae multiple times in a non-systematic manner to infect honeybees beyond the native range. Despite haphazard movements of host and pathogen, there remain some interesting links between historic honeybee movements and ordination graph observations linking STs to locations (Fig. 5). Some historic honeybee movements are recorded in the literature, and it is possible to trace, for example, honeybee imports into USA and New Zealand back over 400 years to their origins in Europe (Donovan, 1980; Goulson, 2003). It is perhaps unsurprising that the CA (Fig. 5) groups countries together from within and out with the native host range group, given that there are traceable links and potential disease transmission routes between these countries (know import/exports of bees and hive materials).
Novel types were sometimes identified only outside the native range of A. mellifera with ST3 only found in New Zealand and ST4, ST9, ST16 only identified in the Americas (Table S2). There are two plausible explanations for this observation. First, the presence of unique types outside the native range could suggest that these types have evolved since becoming isolated from the founder pathogen population. Second and perhaps more likely, our sampling scheme was more intensive in some countries (such as the UK and Germany) and superficial in other countries within the native range (Table S2). Therefore, it is likely that our sampling scheme was not sufficiently exhaustive to detect the full extent of ST diversity within the native range of A. mellifera.
Paenibacillus larvae isolates classed as ERIC I were more diverse than those classed as ERIC II, containing 16 unique STs compared with only 3 for ERIC II (Fig. 1). The results of the Choa1 estimate (Fig. 3) show that although the sampling was uneven with more ERIC I isolates typed than ERIC II, for these data, the Chao1 estimator levels off (Fig. 3), suggesting that the Chao1 estimate is relatively independent of sample size. ERIC I and II isolates differ phenotypically in many ways for example endospore resistance to temperature (Forsgren et al., 2008), rates of sporulation (Saville, 2011), time to host death (Genersch et al., 2005; 2006; Rauch et al., 2009) and it was suggested that they employ different strategies for killing larvae (Poppinga et al., 2012; Fünfhaus et al., 2013; Djukic et al., 2014). Perhaps these differences account for the disparity in diversity, as some STs may be better able to spread to new areas.
It had previously been assumed that ERIC II is confined to Europe and that it is not rare in Germany or Austria (Genersch, 2010). However, our data suggest that ERIC II STs are much more widely distributed than previously thought, being present in Asia, North America and Australasia as well as Europe, although in this study, no ERIC II isolates were found in Europe in countries west of Germany. However, there is evidence that ERIC II is present in Germany in areas that border France and Belgium (Saarland, see Table S2), which might suggest its presence in these countries. This study has split ERIC II into three new STs (Fig. 1) with ST10 and ST11 being common and ST12 being identified once. Considering the prevalence of ST10, it seems unusual that ST11 remains localized. These differences in prevalence and distribution between ERIC II STs could reflect a difference in phenotype, rather than an artefact of sampling bias, as there was no previous method to discern among ERIC II strains. Future work could include genomic and phenotypic comparisons between ERIC II STs, to identify reasons for the observed differences in distribution.
Interestingly, ERIC I and II have mainly different distributions as indicated in the ordination graph, ST17 is the only ERIC I isolate on the left side of the graph (Fig. 5). No ERIC II STs (ST10, ST11) were found in Africa or in the west of Europe in the native range of A. mellifera (Table S2). The fact that they were not found in Africa and much of the west of Europe may be explained by poor sampling effort. However, the UK was more thoroughly sampled and still no instances of ERIC II STs were found. One suggestion for their differing distribution might be that ERIC II STs are host specific. There are around a dozen subspecies of Apis mellifera, and they are split into four groups M, W, C and O (African, Western Europe, Eastern Europe and Western Asia respectively). It is thought that Apis mellifera originated in Africa, and from there, it is likely that there was one expansion into western Europe and either one or two expansions into the east (eastern Europe and Asia) (Whitfield et al., 2006). This means that there is a closer relationship between the western European group of subspecies and African subspecies than between eastern and western European subspecies. This may explain the split in the distribution of ERIC types with STs of ERIC II only being found in Germany and countries to the east within Europe and Asia. The C group subspecies Apis mellifera carnica has almost completely replaced the W group Apis mellifera mellifera in Central European countries such as Germany, whereas in Poland, where no ERIC II was found, the majority of bees are still A. m. mellifera (Meixner et al., 2007). The native range of A. m. mellifera is from the UK to Scandinavia and from France to Poland. However, Italian (C group) and Carnelian (carnica) bees have been transported around Europe in the A. m. mellifera range and hybridization has occurred; in fact, it is thought that in Scandinavia and the UK, the bees are a mixture of all three subspecies (De La Rúa et al., 2001; Jensen et al., 2005) and although both Scandinavia and the UK are thought to have a similar mix of subspecies, ERIC II STs were found in Scandinavia. As we have no information on the host subspecies in our data set, it is impossible to determine whether different P. larvae STs affect A. mellifera subspecies differently. Future work could involve testing this theory by infecting larvae of the honeybee subspecies with a range of STs of P. larvae.
Although local epidemiological observations were not the primary purpose of this study, our data offer some evidence for the hitherto unknown origin of the 2010 AFB outbreak in Jersey, an island in the English Channel. Our scheme matched one Jersey isolate (Table S2) with a ST that was only found in France (ST6), Jersey's closest neighbour, providing evidence of a potential transmission route and demonstrating the potential power of MLST to inform disease aetiology when coupled to more extensive local sampling efforts.
In summary, we have developed an important new tool for describing the genetic structure of P. larvae, which raises unanswered questions about differential host susceptibilities to P. larvae STs. Future proofing in an age of rapid advancement in sequencing technologies is an important consideration, and MLST is compatible with methods that could potentially supersede, such as whole genome sequencing (Larsen et al., 2012). National laboratories responsible for the control of AFB can now use this scheme to gather comprehensive data on ST locations and expand the online database http://pubMLST.org/plarvae/ (Jolley and Maiden, 2010) to build a comprehensive multinational data set to better understand the distribution and transmission networks of AFB on a global scale. Our scheme therefore provides the first universal method for the description of strains of P. larvae and will increase our understanding of the epidemiology of this damaging and costly disease at many spatial scales.
Experimental procedures
MLST development
Whole genome comparisons were made between two genetically diverse strains of P. larvae (Genersch et al., 2006) (p6678 and p6993; LMG 16241 and LMG 16247, ERIC I and ERIC IV respectively). Housekeeping genes used in a previous typing scheme for the related B. cereus group (Helgason et al., 2004) were found to have no variation when compared between these two isolates, so novel regions with 80–90% similarity were identified. Pairwise comparisons were made using the online program doubleact and the result visualized using Artemis Comparison Tool (Carver et al., 2005) and mega version 5 (Tamura et al., 2011).
Two further genomes were later compared [DSM25719 (ERIC I), NCBI acc. No. ADFW01000002; DSM25430 (ERIC II), NCBI acc. No. NC_023134]. This led to the discovery of additional suitable loci, which were identified using the B. cereus Group Typing Database (University of Oslo, 2012) and by targeting genes likely involved in observed phenotypic differences in sporulation frequency between ERIC types (Saville, 2011).
Primer design
Primers were designed to candidate loci using primer 3 (v0.4.0) (Rozen and Skaletsky, 2000), and oligocalc (Kibbe, 2007) with an optimum melting temperature of 55°C (± 5°C). In total, 31 primer pairs were tested including those for non-coding loci to give a scheme composed of seven coding loci. The primer sets were used to amplify a panel of P. larvae isolates of all four ERIC types. Any primer set that did not add extra resolution to the scheme was rejected (Table S1).
The PCR reaction conditions were as follows: After the initial activation step (3 min, 95°C), 35 cycles at 95°C for 30 s, 52 °C for 30 s and 72°C for 1 min were run followed by a final elongation step at 72°C for 10 min.
Global isolates
In total, 294 P. larvae isolates from existing national and international culture collections were recovered on either blood agar or brain heart infusion agar and PLA (de Graaf et al., 2013). Briefly, spore containing honey was either heated at 90°C for 6 min prior to plating (Genersch and Otten, 2003) or plated directly without heat treatment (Forsgren et al., 2008) because temperatures above 90°C or an incubation time of 10 min could have negatively affected germination of ERIC II strains (Forsgren et al., 2008). Isolates were collected from 38 countries from across six continents: Africa (n = 5), Australasia (n = 26), Asia (n = 27), Europe (n = 199), North America (n = 16) and South America (n = 4) as well as some of mixed origin and some from culture collections (n = 17) (Table S2).
DNA was extracted from P. larvae cultures using a simple Chelex method. Bacteria were transferred to 300 μl 6% Chelex®100 and heated to 56°C for 20 min followed by boiling for 8 min. DNA extracts were stored at −20°C until required.
ERIC typing was completed using the method described in Genersch and colleagues (2006).
Sequencing
PCR products were purified using Qiagen® PCR purification and sequenced on the ABI 3730xl 96-capillary DNA Analysers. Sequences were aligned using clustalw in mega version 5 (Tamura et al., 2011) and allele types were counted and numbered in order of discovery as described by Aanensen and Spratt (2005). An allele was identified as a sequence or sequences with one or more genuine nucleotide difference from previously assigned sequences. The combination of allele numbers for each of the target genes gives the allelic profile or ST of an isolate. All putative loci were amplified from a panel of P. larvae isolates representing all four ERIC types. Loci that failed to add resolution to the scheme were rejected, and all isolates were typed using the final MLST scheme.
MLST analysis
The ratio of non-synonymous and synonymous substitutions (dN/ds) of MLST gene fragments was determined using the modified Nei–Gojobori method (Nei and Gojobori, 1986) in the program start2 (Jolley et al., 2001).
Recombination was tested using the IA with the program start2 (Jolley et al., 2001).
Population structure and biogeography
phyloviz (Francisco et al., 2012) was used to analyse allelic profiles using the goeburst algorithm (Feil et al., 2004; Francisco et al., 2009). The program was used to discover clonal complexes and infer founder clones (Francisco et al., 2009). The most parsimonious patterns of descent of all isolates in each clonal complex from the predicted founder(s) were calculated as previously described (Francisco et al., 2009; 2012,). A phylogeny was also constructed from an alignment of 2948 sites representing the concatenated sequences of each ST. The sequences were aligned using clustalw, as implemented in mega 5.2 (Tamura et al., 2011). The phylogenetic analysis was then carried out using the neighbour-joining method and the Maximum Composite Likelihood model as implemented in mega 5.2 (1000 replications).
To test the population structure of P. larvae among different countries, pairwise FST was calculated using the haploDiv command in the r package diversity (Keenan et al., 2013) and bootstrapped 95% CIs (500 repeats) were calculated. The 40 single nucleotide polymorphisms (SNPs) identified in the concatenated MLST gene sequences were used to derive pairwise FST between populations. Populations were taken as all samples from a single country, see Table S2. Countries where there were samples from fewer than five isolates were discounted (see Table S2) or grouped: Samples from Bangladesh, Japan, China, Singapore and Mongolia became Asia. Geographic Distances were taken as the great circle distance between the centre point location for each country or group of countries. A Mantel test (Mantel, 1967) with 1000 replications was used to determine whether the correlation between physical distance and FST was significantly different from a random sample of the data. All results were visualized using r (version 2.15.2) (R Core Team, 2012).
CA was also applied to the data. The CA takes into account the STs present in each country to investigate associations between STs (which types were commonly found together) and patterns in their distribution (which countries are associated with the STs).
Finally, rarefaction curves were constructed to compare the sampling efforts between different continents and between countries within Europe. r library vegan (Oksanen et al., 2013) was used to carry out these final two analyses.
Acknowledgments
B. J. M. was funded by a BBSRC CASE studentship in partnership with Bee Disease Insurance and the National Bee Unit. G.E.B. was funded jointly by a grant from BBSRC, Defra, NERC, the Scottish Government and the Wellcome Trust, under the Insect Pollinator Initiative (Grant BB/I000801/1). E.G was funded by grants from the Ministries for Agriculture from the Federal States of Brandenburg (MIL) and Sachsen-Anhalt (MLU), Germany. L.P. and A.F. were funded by a grant from the German Research Foundation (DFG, grant GE 1365/1-1) Thanks to Dr. Aileen Mill, Dr. Theo Allnutt and Dr. Ian Cleasby for help and advice on statistical analyses. The authors gratefully acknowledge the cooperation of all collaborators who provided isolates to culture collections for use in this study.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1. Genes for which primers were designed and then rejected.
Table S2. Origins of isolates typed with MLST scheme.
References
- Aanensen DM, Spratt BG. The multilocus sequence typing network: mlst. net. Nucleic Acids Res. 2005;33:W728–W733. doi: 10.1093/nar/gki415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alippi AM, Aguilar OM. Characterization of isolates of Paenibacillus larvae subsp. larvae from diverse geographical origin by the polymerase chain reaction and BOX primers. J Invertebr Pathol. 1998;72:21–27. doi: 10.1006/jipa.1998.4748. [DOI] [PubMed] [Google Scholar]
- Alippi AM, Reynaldi FJ, Lopez AC, De Giusti MR, Aguilar OM. Molecular epidemiology of Paenibacillus larvae larvae and incidence of American foulbrood in Argentinean honeys from Buenos Aires province. J Apic Res. 2004;43:135–143. [Google Scholar]
- Antúnez K, Piccini C, Castro-Sowinski S, Rosado AS, Seldin L, Zunino P. Phenotypic and genotypic characterization of Paenibacillus larvae isolates. Vet Microbiol. 2007;124:178–183. doi: 10.1016/j.vetmic.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Büchler R, Berg S, Le Conte Y. Breeding for resistance to Varroa destructor in Europe. Apidologie. 2010;41:393–408. [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis comparison tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Chao A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics. 1987;43:783–791. [PubMed] [Google Scholar]
- De la Rúa P, Jaffé R, Dall'Olio R, Muñoz I, Serrano J. Biodiversity, conservation and current threats to European honeybees. Apidologie. 2009;40:263–284. [Google Scholar]
- De La Rúa P, Galián J, Serrano J, Moritz RFA. Genetic structure and distinctness of Apis mellifera L. populations from the Canary Islands. Mol Ecol. 2001;10:1733–1742. doi: 10.1046/j.1365-294x.2001.01303.x. [DOI] [PubMed] [Google Scholar]
- Di Pinto A, Novello L, Terio V, Tantillo G. ERIC-PCR genotyping of Paenibacillus larvae in Southern Italian honey and brood combs. Curr Microbiol. 2011;63:416–419. doi: 10.1007/s00284-011-9996-z. [DOI] [PubMed] [Google Scholar]
- Djukic M, Brzuszkiewicz E, Fünfhaus A, Voss J, Gollnow K, Poppinga L, et al. How to kill the honey bee larva: genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS ONE. 2014;9:e90914. doi: 10.1371/journal.pone.0090914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan BJ. Interactions between native and introduced bees in New Zealand. NZ J Ecol. 1980;3:104–116. [Google Scholar]
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsgren E, Stevanovic J, Fries I. Variability in germination and in temperature and storage resistance among Paenibacillus larvae genotypes. Vet Microbiol. 2008;129:342–349. doi: 10.1016/j.vetmic.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Francisco AP, Bugalho M, Ramirez M, Carriço JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics. 2009;10:152. doi: 10.1186/1471-2105-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço JA. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics. 2012;13:87. doi: 10.1186/1471-2105-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries I, Lindstrom A, Korpela S. Vertical transmission of American foulbrood (Paenibacillus larvae) in honey bees (Apis mellifera. Vet Microbiol. 2006;114:269–274. doi: 10.1016/j.vetmic.2005.11.068. [DOI] [PubMed] [Google Scholar]
- Fünfhaus A, Poppinga L, Genersch E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood: two novel Paenibacillus larvae toxins. Environ Microbiol. 2013;15:2951–2965. doi: 10.1111/1462-2920.12229. [DOI] [PubMed] [Google Scholar]
- Genersch E. American foulbrood in honeybees and its causative agent, Paenibacillus larvae. J Invertebr Pathol. 2010;103:S10–S19. doi: 10.1016/j.jip.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Genersch E, Otten C. The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie. 2003;34:195–206. [Google Scholar]
- Genersch E, Ashiralieva A, Fries I. Strain- and genotype-specific differences in virulence of Paenibacillus larvae subsp. larvae, a bacterial Pathogen causing American foulbrood disease in honeybees. Appl Environ Microbiol. 2005;71:7551–7555. doi: 10.1128/AEM.71.11.7551-7555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genersch E, Forsgren E, Pentikäinen J, Ashiralieva A, Rauch S, Kilwinski J, Fries I. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int J Syst Evol Microbiol. 2006;56:501–511. doi: 10.1099/ijs.0.63928-0. [DOI] [PubMed] [Google Scholar]
- Goulson D. Effects of introduced bees on native ecosystems. Ann Rev Ecol Evol Syst. 2003;34:1–26. [Google Scholar]
- Govan VA, Allsopp MH, Davison S. A PCR detection method for rapid identification of Paenibacillus larvae. Appl Environ Microbiol. 1999;65:2243–2245. doi: 10.1128/aem.65.5.2243-2245.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf DC, Alippi AM, Antúnez K, Aronstein KA, Budge G, De Koker D, et al. Standard methods for American foulbrood research. J. Apic. Res. 2013;52:1–28. doi: 10.3896/IBRA.1.52.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen H, Brødsgaard CJ, Kryger P, Nicolaisen M. A scientific note on the presence of Paenibacillus larvae larvae spores in sub-Saharan African honey. Apidologie. 2003;34:471–472. [Google Scholar]
- Hasemann L. How long can spores of American foulbrood live? Am Bee J. 1961;101:298–299. [Google Scholar]
- Helgason E, Tourasse NJ, Meisal R, Caugant DA, Kolsto A-B. Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol. 2004;70:191–201. doi: 10.1128/AEM.70.1.191-201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2001;67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AB, Palmer KA, Boomsma JJ, Pedersen BV. Varying degrees of Apis mellifera ligustica introgression in protected populations of the black honeybee, Apis mellifera mellifera, in northwest Europe. Mol Ecol. 2005;14:93–106. doi: 10.1111/j.1365-294X.2004.02399.x. [DOI] [PubMed] [Google Scholar]
- Jensen AB, Pedersen BV, Eilenberg J. Differential susceptibility across honey bee colonies in larval chalkbrood resistance. Apidologie. 2009;40:524–534. [Google Scholar]
- Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Feil EJ, Chan M-S, Maiden MCJ. Sequence type analysis and recombinational tests (START) Bioinformatics. 2001;17:1230–1231. doi: 10.1093/bioinformatics/17.12.1230. [DOI] [PubMed] [Google Scholar]
- Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol. 2013;4:782–788. [Google Scholar]
- Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee J, Besana AM, Genersch E, Gisder S, Nanetti A, Tam DQ, et al. Widespread dispersal of the microsporidian Nosema ceranae, an emergent pathogen of the western honey bee, Apis mellifera. J Invertebr Pathol. 2007;96:1–10. doi: 10.1016/j.jip.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström A. Distribution of Paenibacillus larvae spores among adult honey bees (Apis mellifera) and the relationship with clinical symptoms of American Foulbrood. Microb Ecol. 2008;56:253–259. doi: 10.1007/s00248-007-9342-y. [DOI] [PubMed] [Google Scholar]
- Lindström A, Korpela S, Fries I. Horizontal transmission of Paenibacillus larvae spores between honey bee (Apis mellifera ) colonies through robbing. Apidologie. 2008a;39:515–522. [Google Scholar]
- Lindström A, Korpela S, Fries I. The distribution of Paenibacillus larvae spores in adult bees and honey and larval mortality, following the addition of American foulbrood diseased brood or spore-contaminated honey in honey bee (Apis mellifera) colonies. J Invertebr Pathol. 2008b;99:82–86. doi: 10.1016/j.jip.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Loncaric I, Derakhshifar I, Oberlerchner JT, Köglberger H, Moosbeckhofer R. Genetic diversity among isolates of Paenibacillus larvae from Austria. J Invertebr Pathol. 2009;100:44–46. doi: 10.1016/j.jip.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Maiden MCJ. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- Matheson A. World bee health report. Bee World. 1993;74:176–212. [Google Scholar]
- Meixner MD, Worobik M, Wilde J, Fuchs S, Koeniger N. Apis mellifera mellifera in eastern Europe – morphometric variation and determination of its range limits. Apidologie. 2007;38:191–197. [Google Scholar]
- Morse KSR. American foulbrood incidence in some US and Canadian honeys. Apidologie. 1992;23:2951–2965. [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Neuendorf S, Hedtke K, Tangen G, Genersch E. Biochemical characterization of different genotypes of Paenibacillus larvae subsp. larvae, a honey bee bacterial pathogen. Microbiology. 2004;150:2381–2390. doi: 10.1099/mic.0.27125-0. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, et al. 2013. Vegan: community ecology package. R package version 2.0-6 URL http://vegan.r-forge.r-project.org.
- Palacios G, Hui J, Quan PL, Kalkstein A, Honkavuori KS, Bussetti AV, et al. Genetic analysis of Israel acute paralysis virus: distinct clusters are circulating in the United States. J Virol. 2008;82:6209–6217. doi: 10.1128/JVI.00251-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentikäinen J, Kalliainen E, Pelkonen S. Molecular epidemiology of Paenibacillus larvae infection in Finland. Apidologie. 2008;40:73–81. [Google Scholar]
- Poppinga L, Janesch B, Fünfhaus A, Sekot G, Garcia-Gonzalez E, Hertlein G, et al. Identification and functional analysis of the S-layer protein SplA of Paenibacillus larvae, the causative agent of American foulbrood of honey bees. PLoS Pathog. 2012;8:e1002716. doi: 10.1371/journal.ppat.1002716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2012. R: a language and environment for statistical computing Vienna, Austria. URL http://www.R-project.org.
- Rauch S, Ashiralieva A, Hedtke K, Genersch E. Negative correlation between individual-insect-level virulence and colony-level virulence of Paenibacillus larvae, the etiological agent of American foulbrood of honeybees. Appl Environ Microbiol. 2009;75:3344–3347. doi: 10.1128/AEM.02839-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA, editors. Bionformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Rusenova N, Parvanov P, Stanilova S. Molecular typing of Paenibacillus larvae strains isolated from Bulgarian apiaries based on Repetitive element Polymerase Chain Reaction (Rep-PCR) Curr Microbiol. 2013;66:574–577. doi: 10.1007/s00284-013-0318-5. [DOI] [PubMed] [Google Scholar]
- Saville B. York, UK: University of York; 2011. Differentiation of virulent and biological control Paenibacillus larvae strains associated with American foulbrood in bee hives. PhD Thesis. [Google Scholar]
- Schäfer MO, Genersch E, Fünfhaus A, Poppinga L, Formella N, Bettin B, Karger A, et al. Rapid identification of differentially virulent genotypes of Paenibacillus larvae, the causative organism of American foulbrood of honey bees, by whole cell MALDI-TOF mass spectrometry. Vet Microbiol. 2014;170:291–297. doi: 10.1016/j.vetmic.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Smith JM, Smith NH, O'Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci USA. 1993;90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solignac M, Cornuet JM, Vautrin D, Le Conte Y, Anderson D, Evans J, et al. The invasive Korea and Japan types of Varroa destructor, ectoparasitic mites of the Western honeybee (Apis mellifera), are two partly isolated clones. Proc Biol Sci. 2005;272:411–419. doi: 10.1098/rspb.2004.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak M, Reuter GS. Resistance to American foulbrood disease by honey bee colonies Apis mellifera bred for hygienic behavior. Apidologie. 2001;32:555–565. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Oslo. 2012. Bacillus cereus group multilocus and multidata typing [WWW document]. URL http://mlstoslo.uio.no.
- Whitfield CW, Behura SK, Berlocher SH, Clark AG, Johnston JS, Sheppard WS, et al. Thrice out of africa: ancient and recent expansions of the honey bee, Apis mellifera. Science. 2006;314:642–645. doi: 10.1126/science.1132772. [DOI] [PubMed] [Google Scholar]
- Yue D, Nordhoff M, Wieler LH, Genersch E. Fluorescence in situ hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American foulbrood of honeybees (Apis mellifera. Environ Microbiol. 2008;10:1612–1620. doi: 10.1111/j.1462-2920.2008.01579.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes for which primers were designed and then rejected.
Table S2. Origins of isolates typed with MLST scheme.