

Abstract
Angiogenesis (the formation of blood vessels from existing blood vessels) plays a critical role in many diseases such as cancer, benign tumors, and macular degeneration. There is a need for cell culture methods capable of dissecting the intricate regulation of angiogenesis within the microenvironment of the vasculature. We have developed a microscale cell-based assay that responds to complex pro- and anti-angiogenic soluble factors with an in vitro readout for vessel formation. The power of this system over traditional techniques is that we can incorporate the whole milieu of soluble factors produced by cells in situ into one biological readout (vessel formation), even if the identity of the factors is unknown. We have currently incorporated macrophages, endothelial cells, and fibroblasts into the assay, with the potential to include additional cell types in the future. Importantly, the microfluidic platform is simple to operate and multiplex to test drugs targeting angiogenesis in a more physiologically relevant context. As a proof of concept, we tested the effect of an enzyme inhibitor (targeting matrix metalloproteinase 12) on vessel formation; the triculture microfluidic assay enabled us to capture a dose-dependent effect entirely missed in a simplified coculture assay (p<0.0001). This result underscores the importance of cell-based assays that capture chemical cross-talk occurring between cell types. The microscale dimensions significantly reduce cell consumption compared to conventional well plate platforms, enabling the use of limited primary cells from patients in future investigations and offering the potential to screen therapeutic approaches for individual patients in vitro.
Standard techniques for drug and toxicant testing are predicated on the concept that assays quantify the effect of a chemical agent on a biological target using a measurable readout. This concept breaks down in complex processes involving multiple cell types where the effect of the chemical agent on the target cell type of interest can be markedly different depending on the milieu of signals secreted by other cell types. In such cases, assays must be revamped to model the biological complexity required to capture context-dependent effects of the chemical agent. Considering this need, we have developed a microfluidic multiculture assay to study angiogenesis, the formation of blood vessels from existing blood vessels. Angiogenesis is a critical process in development, wound healing, and normal homeostasis.1 However, aberrant angiogenesis contributes to many human disease processes such as cancer,2 benign tumors,3 and macular degeneration.4 The dysregulation of angiogenesis in disease results from the interplay of competing pro- and anti-angiogenic factors secreted by many different cell types in the microenvironment of the vasculature.5 Studying these competing effects and signaling between multiple cell types may prove critical for understanding disease etiology and progression and the development of new therapies targeting angiogenesis and microenvironment. Our microfluidic assay models signaling among key cellular players in angiogenesis, and can be used both as a tool to test the effect of chemical agents (such as potential drugs) in the context of relevant cellular signals and for fundamental biology experiments aimed at disentangling how angiogenesis is regulated by interactions between cell types in the vascular microenvironment.
Microfluidic systems have been used to elegantly address questions in angiogenesis that are inaccessible with conventional culture platforms.6,7 Previous microfluidic models of angiogenesis have enabled researchers to study the effect of fluid flow and shear8 and interactions with three dimensional architectures.9,10,11,12,13,14 In some previous models, exogenous extracellular matrix (ECM) components are added in side channels, allowing vessels to sprout into the matrix.8,14 Other approaches utilize exogenous ECM components to template lumen structures on which endothelial cells are seeded.9–11 With the development of more complex ‘organ on a chip’ platforms that aim to recapitulate tissue- or organ-level functionality,15 many groups are pursuing methods to create functional artificial blood vessels to sustain the in vitro tissues and organs.12 The overall device design and decision to include exogenous ECM components into microfluidic angiogenesis models is driven by the ultimate biological questions that the platform will be used to address. In the present manuscript, our goal was to develop a simple, arrayable platform to study the effects of soluble factor signaling between cell types on angiogenesis. We incorporated an established tubule formation assay that includes a feeder layer of fibroblasts in mixed culture with endothelial cells and hence does not require the addition of exogenous ECM components as established by Bishop et al.16 Microculture platforms show great promise for studying soluble factor signaling between cell types in a controlled manner.17,18,19,20,21 The microscale dimensions of these systems offer increased sensitivity in capturing paracrine signaling due to reduced culture volumes, diffusion distances, and the convection-free culture environment produced.17,18 In addition to increased sensitivity, the reduced volumes inherent to microculture systems enable the use of limited or rare cells, such as primary cells from patient samples.22,23,24
Here, we developed a microfluidic method to study the effects of soluble factor signaling on endothelial tubule formation, an important step in and established in vitro indicator of angiogenesis.16,25,26 Historically, this tubule formation assay has been conducted with mixed cultures of endothelial cells and fibroblasts.16,25,26 To better mimic the in vivo microenvironment, we incorporated macrophages into the assay, utilizing advances in microfluidic cell culture to precisely position the cells and enable soluble factor communication between macrophages and the endothelial/fibroblast mixed culture. Macrophages are important mediators of angiogenesis, and they are known to secrete both pro- and anti-angiogenic factors.27,28,29 We studied the net effect of these factors on tubule formation. As a proof of concept, we then focused on the macrophage-secreted factor matrix metalloproteinase 12 (MMP12), an anti-angiogenic factor of emerging importance in several diseases.30,31 Specifically, we tested the hypothesis that MMP12 secreted by macrophages suppresses tubule formation and that this can be mimicked by exogenous MMP12 and rescued by MMP12 inhibitor. This hypothesis is consistent with several clinical and in vivo studies that correlate MMP12 with a reduction in angiogene-sis,30,32,33 but our microfluidic study is the first to address this hypothesis directly in vitro by modeling interactions between cell types. These results underscore our ability to use the microfluidic multiculture model to dissect complicated interactions among multiple cell types and test the effects of these interactions on biological function (tubule formation) within microchannels. Importantly, our microscale system uses only 600 primary endothelial cells and several thousand fibroblasts and macrophages, enabling studies with limited patient samples in the future.
EXPERIMENTAL SECTION
Microfluidic device fabrication
Microfluidic devices were fabricated following standard soft lithography techniques.34 High resolution mask files (.pdf) and a schematic detailing all dimensions are included in the Supporting Information. Key dimensions are also summarized here for convenience: culture channel width = 1 mm, culture channel height = 330 μm, communication channel width = 500 μm, communication channel height = 30 μm, input port diameter = 1 mm, output port diameter = 4 mm, input/output port heights = 830 μm, and total device length = 12.7 mm.
SU-8 master fabrication
As shown in Figure 1A, the device contains features of three different heights (communication channels, culture channels, and input/output ports), requiring three SU-8 fabrication steps. The procedures followed for each SU-8 layer are as follows: Communication channel layer (30 μm): a thin layer of SU8-50 (Microchem) was loaded onto a 6 inch silicon wafer (E&M, Ashiya, Japan), spin coated at 500 rpm for 10 s and increased to 3000 rpm for 45 s, followed by a 10 min pre-bake at 95 °C. The layer was then UV cross-linked under the corresponding mask (300 mW/cm2) and post-baked for 10 min at 95 °C. Culture channel layer (300 μm): on top of the communication channel layer, SU-8 100 was loaded and spin coated at 500 rpm for 10 s, then 1100 rpm for 45 s, followed by a 30 min pre-bake at 65 °C and a 3 h pre-bake at 95 °C. The layer was UV cross-linked (700 mW/cm2) and post-baked for 30 min at 65 °C and 90 min at 95 °C. Input/output port layer (500 μm): an excess amount of SU-8 100 was loaded on top of the first two layers, spin coated at 500 rpm for 120 s, followed by a 30 min pre-bake at 65 °C and a 4 h pre-bake at 95 °C. The layer was UV cross-linked (1000 mW/cm2) and post-baked for 20 min at 65 °C and 3 h at 95 °C. The fully patterned wafers were submerged in SU-8 developer (propylene glycol monomethyl ether acetate, MicroChem) for development. Finally, the wafer was washed and dried, providing a template for PDMS device fabrication.
Figure 1.

Microfluidic device design and workflow. A) Schematic showing two channels for seeding macrophages (left channel) and HUVEC + NHDF mixture (right channel) connected by a series of communication channels to allow soluble factor signaling. The output is larger than the input to facilitate passive pumping.39 B) Photograph showing an array of 14 microculture devices filled with red dye for visualization. C) Device workflow showing cell seeding (via pipette), polarization, and ICC. Inset shows an example of CD31 (green) and DAPI (blue) stained microchannel.
Polydimethylsiloxane (PDMS) device fabrication
PDMS (Sylgard 184, Dow and Corning) was mixed in a 10:1 ratio (base:curing agent), degassed, poured onto the SU-8 master, and baked for 4 h at 80 °C. The cured PDMS device was then removed from the master and treated with a Soxhlet extractor containing 100% ethanol for 4 hours (6 to 8 cycles) to purge the devices of uncrosslinked PDMS and linkers.35,36 Devices were then dried, sterilized via autoclave, and then passively bonded on a cell culture treated polystyrene OmniTray (Nunc).
Cell culture
Maintenance of cells in standard flask/petri dish culture
The human monocytic cell line THP-1 (ATCC, TIB-202) was cultured in RPMI medium with 10% Fetal Bovine Serum (FBS, Gibco), penicillin (100 units/mL), and streptomycin (100 μg/mL). Human Umbilical Vein Endothelial Cells (HUVEC, Lonza) were cultured in Endothelial Cell Growth Media Kits (EGM - 2 Bulletkit, Lonza). Primary neonatal Normal Human Dermal Fibroblasts (NHDF, ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, containing 1000 mg/L glucose, L-glutamine, and sodium bicarbonate), 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL). All cells were maintained in 37 °C incubator with 5% carbon dioxide. Cells were used at the following passage numbers: THP-1 P3-9, HUVEC P3-P6, and NHDF P7-P10.
Cell culture in microchannels
Before seeding cells into the microchannels, channels were filled with 15 μL of above mentioned THP-1 culture media (the volume of each culture compartment was 1.5 μL, and the remaining media filled the input and output ports as shown in Figure 1B). The timeline for cell culture in microchannels is shown in Figure 1C. THP-1 cells were seeded at 6000 cells per channel, differentiated, and polarized based on methods established by Tjiu et al.; this protocol has been employed previously to yield M2 Tumor Associated Macrophages (TAMs).37 Specifically, the cells were seeded in the microchannel in the presence of 0.1 μg/ml phorbol-12-myristate-13-acetate (PMA, Sigma Aldrich) and cultured overnight. This process facilitates cell adhesion and differentiation to M0 macrophages. Once adhesion was confirmed, the cells were cultured with 0.1 μg/mL interleu-kin-4, interleukin-13, and PMA for another 24 hours.37 After differentiation, the macrophages were allowed to rest two days in original THP-1 media detailed above. After maturation, the other chamber of this device was coated with 10% fibronectin (Sigma Aldrich) in PBS for one hour in a 37 °C. The chamber was then washed with PBS two times and a mixture of 2400 NHDFs and 600 HUVECs was seeded (without the addition of exogenous extracellular matrix components) based on previous studies.16,38 All cell types were maintained in EGM - 2 Bulletkit (Lonza) with an additional 10% FBS. Where indicated, cells were treated with a selective MMP12 inhibitor (CAS 1258003-93-8, Millipore) and/or MMP12 protein (Sigma Aldrich, catalytic domain human, recombinant, expressed in E. coli, 50 ng/mL), which was added in the cell culture media and applied to both culture channels at the time of the first media change 20 h after cell seeding (and freshly added to media on subsequent days during daily media changes). The triculture continued for 3 days, with daily media changes, before immunocytochemistry.
Note concerning experimental design used for controls: In the macrophage-only culture, we included media in the other channel that normally contains HUVECs + NHDFs. Similarly, in the coculture condition, we seeded HUVECs + NHDFs into one channel and added only media in the other channel. We treated the ‘empty’ channel containing only media in both of these cases exactly as if cells were present (for example, in the case of the ‘empty’ channel normally containing macrophages, the channel was treated with the same differentiation/polarization reagents and media was exchanged prior to seeding HUVECs + NHDFs).
Immunocytochemistry (ICC)
Cells were fixed and then stained through standard ICC staining procedures. Specifically, each channel was first washed with PBS at 37 °C three times (each time of operation, the entire volume of the channel was aspirated by vacuum and then refilled with PBS). Then the PBS was removed and replaced by 37 °C 4% paraformaldehyde (diluted from 16% PFA, EM Grade, VMR). The devices with PFA were then incubated for 20 minutes. After incubation, the devices were washed with PBS three times. After aspirating PBS from the channels, 0.2% Triton-X-100 (diluted in PBS) was added to the channels. The channels were then left in room temperature for 10 minutes. Triton-X-100 was replaced by 10% FBS in PBS blocking buffer. With the buffer in the channels, the microfluidic devices were left overnight at 4 °C.
CD-31 primary antibody (mouse anti human CD-31, MCA1738, AbD Serotec) was diluted in the above mentioned FBS blocking buffer (with 1:50 dilution) and applied to the channels to replace the blocking buffer within 16 h of time. With the primary antibody in the channel, the devices were put into 37 °C incubator for 1 h. The channels were then washed with 2% Triton-X-100 three times. Goat antimouse secondary antibody (green, 488 nm) and DAPI were diluted 1:400 and applied to the channels. The devices were then incubated at room temperature for 1 h, protected from light. Finally, the secondary was washed two times with PBS and replaced by mounting medium (Vectashield).
Quantification of soluble factors
Conditioned media was removed from microchannels at the end of the culture period and stored at −80 °C prior to analysis. Soluble factor levels (presented in Figure 2) were analyzed with Milliplex Map Human Angiogenesis/Growth Factor Magnetic Bead Panel 1 and Milliplex Map Human MMP Panel 1 Magnetic Bead Kits (Millipore). Specifically, media from four replicate channels was collected and mixed together as one sample. Samples were analyzed from three independent experiments (Figure 2B–G) or four independent experiments (Figure 2A). Data were analyzed using Milliplex Analyst v5.1 using a four parameter logistic curve. Factors reported in Figure 2 showed quality controls within the expected range (as indicated by the manufacturer) and levels of soluble factors measured were within the quantifiable region of the standard curve (as determined using Milliplex Analyst v5.1 software); other factors from the panel that did not meet these criteria were not reported.
Figure 2.
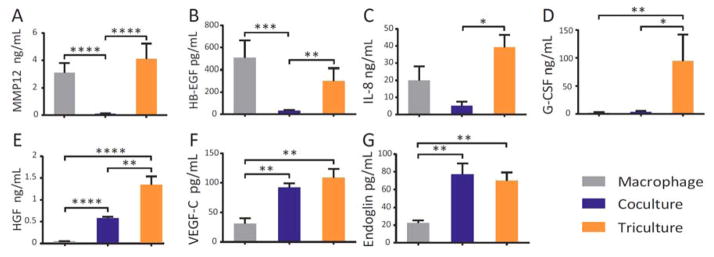
Quantification of soluble factors in different microenvironmental conditions: macrophage monoculture, coculture (mixture of fibroblasts and endothelial cells), and triculture (mixture of fibroblasts and endothelial cells in soluble factor communication with macrophages). Error bars represent the standard error of the mean (SEM) of three independents experiments. Repeated measures Analysis of Variance (ANOVA) indicated significant differences within culture conditions for each factor (p<0.05); p-values are indicated for Tukey’s posthoc tests as follows: * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001.
Image processing
Images were acquired using a Nikon Eclipse fluorescence microscope and processed with ImageJ. First, pictures were converted to 8 bit. A POV was then selected for each picture (398×926 pix), which covers >90% of entire view of each channel. The POV was then cropped using ImageJ. The images of CD31 and DAPI staining were then overlaid through image-J (image - color merge channels). CD31 images were set to be green and DAPI images were set to be blue. After merging the green and blue images, the green color balance of all images was post polished modified through ImageJ (image - adjust - color balance); maximum intensity of all images was adjusted down to 152 rather than the full 255.
Calculation of tubule index
To calculate the tubule index (ratio of perimeter to area of CD31 positive cell clusters), each image was analyzed using default functions from ImageJ (see Supporting Information, Figure S1). Specifically, each image was first converted to 8 bit prior to calculation. A default threshold of “Li Dark” was chosen to convert the images into black and white. The perimeters and areas of the fluorescent signals of the endothelial islands or tubule network in each picture were then calculated. Specifically, we performed the “fill holes” function. The original image was subtracted from the images with filled holes to produce the image of the holes and images were added or subtracted following the scheme shown in Figure S1 to obtain the perimeter, area, and finally the perimeter/area ratio.
Statistics
Statistical analysis was performed using GraphPad Prism 6 software. Analysis of Variance (ANOVA) was used to compare groups (details of the ANOVA test are indicated in the captions of Figures 2–4). When necessary, data were log transformed in order to better meet the assumptions of ANOVA. Tukey’s posthoc tests were used to evaluate the significance of pairwise comparisons.
Figure 4.

Exogenous MMP12 protein decreased tube formation and MMP12 inhibitor partially rescues tube formation in coculture. A–E) Fluorescence micrographs of the mixed endothelial/fibroblast culture are representative of four channels from three independent experiments (green=CD31, blue=DAPI). F–G) Quantification of the degree of tubule character and number of branch points. Error bars represent the SEM of three independent experiments. Datasets were analyzed using 2-way ANOVA tests (showing a significant interaction effect, p<0.01 for F and p<0.0001 for G); p-values are indicated for pairwise comparisons between groups (blue and green bars) at each inhibitor concentration as follows: * p≤0.05, *** p≤0.001, **** p≤0.0001 (Tukey’s posthoc tests).
RESULTS AND DISCUSSION
Microfluidic triculture platform design
Each microculture device consists of two parallel culture channels (330 μm tall, 1.5 μL culture volume per channel) connected by communication channels (30 μm tall) that allow soluble factor communication between the two culture regions (Fig. 1A and B). The differential height of the culture channels and the communication channels allows each culture channel to be addressed separately to selectively seed different cell types in each channel without cross contamination of the other culture channel. The short distance between the two culture channels (500 μm) enables efficient diffusion and communication between cells in the connected culture channels, a consideration that is particularly important for studying bidirectional signaling and signaling involving short-lived molecules. Using this platform, we investigated interactions between endothelial cells and two key players that regulate angiogenesis in the body, fibroblasts and macrophages. Importantly, this culture platform enabled us to study soluble factor profiles arising from precisely controlled multi-cell cultures and determine the effects on tubule formation, a morphological readout. Hence we can establish links between soluble factor communication and biological function.
We incorporated an established tubule formation assay into one channel of our microculture device. The assay involves mixed culture of primary human umbilical vein endothelial cells (HUVEC) and primary normal human dermal fibroblasts (NHDF).16,26 Mixed culture with primary fibroblasts stimulates the endothelial cells to form tubules without exogenous extracellular matrix;16 for this reason, endothelial cells were seeded as a mixture with fibroblasts whereas macrophages were seeded in a separate compartment (since the goal was to observe the effects of soluble factor signals from macrophages, not processes mediated by physical contract). In the presence of angiogenesis inhibitors, the endothelial cells assemble into islands, rather than tubules, providing a morphological readout for anti-angiogenic processes.16,26 As shown in the schematic (Fig. 1C), we first loaded the left channel with THP-1 monocytes (subsequently differentiated into macrophages and polarized following a protocol adapted from Tjiu et al.37). We then seeded HUVECs and NHDFs as a mixture in the right channel. Changes in tubule formation in response to soluble factors produced by macrophages were visualized using immunocytochemical (ICC) staining for the endothelial marker CD31 in the right channel (Fig. 1C). Importantly, device operation does not require specialized equipment such as syringe pumps. The device can be loaded using a conventional pipette based on passive surface-tension driven flows39 and stored within a humidified cell culture tray in an incubator during the multiday culture period, making this technology accessible to all biology laboratories. This platform is also compatible with high throughput liquid handling systems.40
Soluble factor quantification in micro triculture
We quantified levels of known pro-angiogenic soluble factors using a bead-based ELISA system (Luminex) (Fig. 2B–G).41,42,43,44,45,46 In triculture (mixture of fibroblasts and endothelial cells in one compartment in soluble factor communication with macrophages in adjacent compartment) HB-EGF, IL-8, G-CSF, HGF increased significantly compared to levels detected in coculture (mixture of fibroblasts and endothelial cells in the absence of macrophages). Furthermore, for a subset of these factors (G-CSF and HGF), concentrations measured in triculture were higher than the sum of the concentrations measured in coculture + macrophage monoculture, suggesting a synergistic relationship observed only when all three cell types were present at the same time. This underscores the power of the micro triculture assay to capture bidirectional and dynamic interactions that would be missed in simple conditioned media studies, a conventional method for testing soluble factor interactions.
In addition to pro-angiogenic factors, we also quantified levels of the anti-angiogenic factor MMP12.31,30,33 We measured significantly higher MMP12 concentrations in triculture than coculture, consistent with previous reports that MMP12 is produced largely by macrophages.47 MMP12 has emerged as an important anti-angiogenic factor in a number of diseases.31,30,33 An in vitro multiculture assay to study the effects of MMP12 produced in situ would enable future investigations of angiogenesis targeting specific disease mechanisms. Hence, we designed experiments to decipher the effect of MMP12 on tubule formation in our micro triculture model. These experiments serve as a proof of concept to demonstrate the utility of the microfluidic triculture platform for studying the role of a specific soluble factor and evaluating the effect of potential treatments in a platform that captures additional biological complexity in comparison to traditional methods.
Studying vessel formation in different soluble factor environments
Soluble factor profiling indicated that macrophages contribute both pro- and anti-angiogenic soluble factors (Fig. 2), and we studied the net effect of these two competing sets of factors on tubule formation. Further, our microculture assay is a powerful tool for deciphering relationships among this complex milieu of factors as we can easily manipulate the culture condition (coculture vs. triculture) and treat with selective inhibitors to systematically eliminate the effects of individual soluble factors. As illustrated in Fig. 3A–B, triculture with macrophages indeed had a profound effect on endothelial cell morphology. When in soluble factor communication with macrophages (triculture, Fig. 3B), endothelial cells formed islands instead of tubules observed in standard fibroblast-endothelial mixed coculture (Fig. 3A). To test the hypothesis that MMP12 (an anti-angiogenic factor secreted largely by macrophages) prevents tubule formation in triculture, we used a pharmacological loss-of-function approach (i.e., a selective MMP12 inhibitor). In support of our hypothesis, we observed a striking increase in tubule formation when MMP12 inhibitor was added in triculture (Fig 3C–E). Notably, in the presence of 20 nM MMP12 inhibitor we observed a greater extent of tubule formation in triculture than observed in coculture, suggesting that when MMP12 is inhibited, the effects of pro-angiogenic factors secreted by macrophages dominate. The communication channels are large enough to enable cells to migrate through, and we have observed tubules growing into the communication channels, but the tubules do not extend into the macrophage-containing channel. Depending on the biological question under investigation the migration of cells into the communication channels could be quantified and used as an additional functional readout in future studies.
Figure 3.

Microfluidic triculture assay probes the role of MMP12 in angiogenesis under different culture conditions. A) As shown previously by Bishop et al.16 endothelial cells form tubules in coculture with fibroblasts. B) Tubule formation is prevented in triculture and C–E) rescued with MMP12 inhibitor. Fluorescence micrographs (in A–E) show the right channel containing mixed endothelial/fibroblast culture; ICC shows the endothelial cell marker CD31 (green), enabling visualization of tubules and islands. Fibroblast and endothelial nuclei (DAPI) are shown in blue. Images are representative of four channels from three independent experiments. F–G) Quantification of the degree of tubule character and the number of branch points. Error bars represent the SEM of three independent experiments. Datasets were analyzed using 2-way ANOVA tests (showing a significant interaction effect, p<0.0001 for F and G); p-values are indicated for pairwise comparisons between co- and triculture at each inhibitor concentration as follows: ** p≤0.01, *** p≤0.001, **** p≤0.0001 (Tukey’s posthoc tests). All pairwise comparisons among the coculture groups are not significant; all pairwise comparisons among the triculture groups are significant (p≤0.0001 in F and p≤0.001 in G).
Inhibitor dose-response testing
We tested the effect of MMP12 inhibitor across a range of concentrations. To quantify the effect on tubule formation, we analyzed fluorescence micrographs of cultures stained for the endothelial marker CD31 (see SI for image analysis workflow). We defined a measure of tubule-character, the ‘tubule index’, which is the ratio of the perimeter to the area of CD31 positive cell assemblies (tubules and/or islands). A high tubule index corresponds to fluorescence micrographs containing a greater degree of tubule-character, whereas a low tubule index corresponds to island-character. We observed significant differences between co- and triculture at 0, 6, and 20 nM MMP12 inhibitor (Fig. 3F). Further, we observed a significant dose-dependent effect within the triculture group, with MMP12 inhibitor treatment significantly increasing the degree of tubule formation, whereas MMP12 inhibitor did not affect tubule formation in coculture (Fig. 3F). These results demonstrate how the added complexity of the triculture system can capture a dose-response entirely missed in coculture.
For doses of MMP12 inhibitor where tubule-character was predominate (tubule index >0.14 observed at 2, 6, and 20 nM MMP12 inhibitor, but not at 0 nM), we further quantified the number of branch points present in the tubule network (Fig. 3G). We observed a significant increase in the number of branch points in triculture vs. coculture at doses of 6 and 20 nM MMP12 inhibitor, consistent with the trend observed in the tubule index value. Taken together, the dose-dependent responses observed with both tubule index and number of branch points as morphological readouts for angiogenesis indicate the importance of incorporating relevant aspects of the microenvironment, such as macrophages, when determining the angiogenic effects of compounds (such as potential drugs) since no significant response was observed across the dose range tested in the absence of macrophages (coculture).
Investigating effect of exogenous factors on vessel formation
Having observed that the anti-angiogenic factor MMP12 is secreted by macrophages (Fig. 2) and prevents tubule formation in micro triculture with endothelial cells and fibroblasts (Fig. 3), we tested the hypothesis that this can be both mimicked by exogenous MMP12 and rescued by simultaneous addition of MMP12 inhibitor in coculture in the absence of macrophages. Indeed, when exogenous MMP12 was added to the microfluidic coculture system, islands were observed and tubule formation was significantly reduced (Fig. 4A–B). The islands were smaller than islands previously observed in the presence of macrophages (Fig. 3B); we attribute this difference to additional stimulatory factors secreted by macrophages. When MMP12 inhibitor was added in addition to exogenous MMP12 protein (Fig. 4C–E), we observed restoration of tubule formation with increasing MMP12 inhibitor concentration. At 200 nM MMP12 inhibitor, the extent of tubule formation in cultures with exogenous MMP12 was not significantly different from baseline levels in cultures without exogenous MMP12 (based on tubule index and number of branch points, Fig. 4F–G), although some islands were observed among the tubules in the fluorescence micrograph (Fig 4F), suggesting that the exogenous MMP12 was not fully inhibited at this concentration of MMP12 inhibitor.
An important difference between the triculture experiment (Fig. 3) and the experiment in which coculture is supplemented with MMP12 protein (Fig. 4) is that in triculture the net effects of both anti-angiogenic factors (such as MMP12) and pro-angiogenic factors secreted by macrophages can be observed. Hence, as MMP12 inhibitor concentration is increased in triculture (Fig. 3), the effects of pro-angiogenic factors dominate, and tubule formation increases in triculture compared to coculture. In Fig. 4, without the added complexity of macrophages in culture, we simply observe a return to baseline conditions when increasing MMP12 inhibitor concentration.
CONCLUSIONS
In summary, we have developed an assay that can be used for both fundamental biology investigations to understand mechanisms regulating angiogenesis and also to test potential drugs to target angiogenesis in disease. The in vitro model enables us to decipher which cell types (or combinations of cell types) within the microenvironment produce specific pro- and anti-angiogenic signals and to directly test mechanistic hypotheses with loss- or gain-of-function approaches, such as the selective MMP12 inhibitor employed in these studies. Our assay enabled the first in vitro multiculture investigation of the emerging anti-angiogenic factor MMP12 secreted by macrophages in situ. Underscoring the importance of biological context, we observed a significant increase in tubule formation when MMP12 inhibitor was added in triculture, but the MMP12 inhibitor did not affect tubule formation in coculture.
Our platform holds great potential for drug testing as it captures complex biological signaling processes but is simple to operate and multiplex with liquid handling systems.40 The microscale dimensions significantly reduce cell consumption compared to conventional well plate platforms, enabling the use of limited primary cells from patients and offering the potential to screen therapeutic approaches for individual patients in vitro. Importantly, the system models dynamic and bidirectional signaling between cell types, enabling potential drug molecules to be tested in the context of endogenous signals that may modulate the drug effect. In the future, additional culture compartments could be added to accommodate other cell types (such as epithelial cells from carcinomas), providing a modular method to incorporate different microenvironmental aspects into the assay.
Supplementary Material
Acknowledgments
We thank Dr. Steve Hayes, Tristan Nicholson, Dr. Erwin Berthier, Dr. Lauren Bischel, and Dr. Scott Berry helpful discussions; Dr. Kyung Sung for assistance performing the bead-based ELISA; and Dr. Glen Leverson for biostatistics advice. This work was funded by NIH R01EB010039, NIH R01 01 CA155192, R01 DK093690, U54 DK104310, the University of Wisconsin-Madison Carbone Cancer Center Support Grant NIH P30 CA014520, Wisconsin Multidisciplinary K12 Urologic Research Career Development Program K12DK100022, University of Wisconsin-Madison Molecular and Environmental Toxicology Center NIH Grant T32ES007015, a Hilldale Undergraduate/Faculty Research Fellowship, and NSERC Discovery Grant 436117-2013.
Footnotes
Supporting Information Available
High resolution mask files (.pdf) used to make the SU8 molds, a schematic detailing all device dimensions, and a description and illustration of the image analysis workflow used to quantify tubule index. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carmeliet P. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Nature medicine. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 3.Shih SJ, Dall’Era MA, Westphal JR, Yang J, Sweep CG, Gandour-Edwards R, Evans CP. Prostate cancer and prostatic diseases. 2003;6:131–137. doi: 10.1038/sj.pcan.4500637. [DOI] [PubMed] [Google Scholar]
- 4.Ng EW, Adamis AP. Canadian journal of ophthalmology Journal canadien d’ophtalmologie. 2005;40:352–368. doi: 10.1016/S0008-4182(05)80078-X. [DOI] [PubMed] [Google Scholar]
- 5.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Physiological reviews. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stroock AD, Fischbach C. Tissue engineering Part A. 2010;16:2143–2146. doi: 10.1089/ten.tea.2009.0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young EW. Journal of laboratory automation. 2013;18:427–436. doi: 10.1177/2211068213495206. [DOI] [PubMed] [Google Scholar]
- 8.Song JW, Munn LL. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen DH, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6712–6717. doi: 10.1073/pnas.1221526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bischel LL, Young EW, Mader BR, Beebe DJ. Biomaterials. 2013;34:1471–1477. doi: 10.1016/j.biomaterials.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DH, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Nature materials. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Markov DA, Wikswo JP, McCawley LJ. Biomedical microdevices. 2011;13:837–846. doi: 10.1007/s10544-011-9554-2. [DOI] [PubMed] [Google Scholar]
- 14.Sudo R, Chung S, Zervantonakis IK, Vickerman V, Toshimitsu Y, Griffith LG, Kamm RD. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:2155–2164. doi: 10.1096/fj.08-122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young EW. Integrative biology: quantitative biosciences from nano to macro. 2013;5:1096–1109. doi: 10.1039/c3ib40076j. [DOI] [PubMed] [Google Scholar]
- 16.Bishop ET, Bell GT, Bloor S, Broom IJ, Hendry NF, Wheatley DN. Angiogenesis. 1999;3:335–344. doi: 10.1023/a:1026546219962. [DOI] [PubMed] [Google Scholar]
- 17.Domenech M, Yu H, Warrick J, Badders NM, Meyvantsson I, Alexander CM, Beebe DJ. Integrative biology: quantitative biosciences from nano to macro. 2009;1:267–274. doi: 10.1039/b823059e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domenech M, Bjerregaard R, Bushman W, Beebe DJ. Integrative biology: quantitative biosciences from nano to macro. 2012;4:142–152. doi: 10.1039/c1ib00104c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spencer KH, Kim MY, Hughes CC, Hui EE. Integrative biology: quantitative biosciences from nano to macro. 2014;6:382–387. doi: 10.1039/c3ib40211h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang JD, Berry SM, Powers GL, Beebe DJ, Alarid ET. Integrative biology: quantitative biosciences from nano to macro. 2013;5:807–816. doi: 10.1039/c3ib20265h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carney CM, Muszynski JL, Strotman LN, Lewis SR, O’Connell RL, Beebe DJ, Theberge AB, Jorgensen JS. Biology of reproduction. 2014;91:85. doi: 10.1095/biolreprod.114.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, Hutchison JB, Rothberg JM, Link DR, Perrimon N, Samuels ML. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14195–14200. doi: 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes D, Pettigrew D, Reccius CH, Gwyer JD, van Berkel C, Holloway J, Davies DE, Morgan H. Lab on a chip. 2009;9:2881–2889. doi: 10.1039/b910053a. [DOI] [PubMed] [Google Scholar]
- 24.Young EW, Pak C, Kahl BS, Yang DT, Callander NS, Miyamoto S, Beebe DJ. Blood. 2012;119:e76–85. doi: 10.1182/blood-2011-10-384347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooley LS, Handsley MM, Zhou Z, Lafleur MA, Pennington CJ, Thompson EW, Poschl E, Edwards DR. Journal of cell science. 2010;123:3808–3816. doi: 10.1242/jcs.064279. [DOI] [PubMed] [Google Scholar]
- 26.Sarkanen JR, Mannerstrom M, Vuorenpaa H, Uotila J, Ylikomi T, Heinonen T. Frontiers in pharmacology. 2010;1:147. doi: 10.3389/fphar.2010.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh H, Takagi H, Takagi C, Suzuma K, Otani A, Ishida K, Matsumura M, Ogura Y, Honda Y. Investigative ophthalmology & visual science. 1999;40:1891–1898. [PubMed] [Google Scholar]
- 28.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Cancer research. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 29.Lamagna C, Aurrand-Lions M, Imhof BA. Journal of leukocyte biology. 2006;80:705–713. doi: 10.1189/jlb.1105656. [DOI] [PubMed] [Google Scholar]
- 30.Margheri F, Serrati S, Lapucci A, Chilla A, Bazzichi L, Bombardieri S, Kahaleh B, Calorini L, Bianchini F, Fibbi G, Del Rosso M. Arthritis and rheumatism. 2010;62:2488–2498. doi: 10.1002/art.27522. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Wang JJ, Peng Q, Chen C, Humphrey MB, Heinecke J, Zhang SX. PloS one. 2012;7:e52699. doi: 10.1371/journal.pone.0052699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerkela E, Ala-Aho R, Jeskanen L, Rechardt O, Grenman R, Shapiro SD, Kahari VM, Saarialho-Kere U. The Journal of investigative dermatology. 2000;114:1113–1119. doi: 10.1046/j.1523-1747.2000.00993.x. [DOI] [PubMed] [Google Scholar]
- 33.Serrati S, Cinelli M, Margheri F, Guiducci S, Del Rosso A, Pucci M, Fibbi G, Bazzichi L, Bombardieri S, Matucci-Cerinic M, Del Rosso M. The Journal of pathology. 2006;210:240–248. doi: 10.1002/path.2048. [DOI] [PubMed] [Google Scholar]
- 34.Duffy DC, McDonald JC, Schueller OJ, Whitesides GM. Analytical chemistry. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 35.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET, Beebe DJ. Lab on a chip. 2009;9:2132–2139. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Chaudhury MK, Owen MJ. Journal of Colloid and Interface Science. 2000;226:231–236. doi: 10.1016/j.jcis.2005.06.068. [DOI] [PubMed] [Google Scholar]
- 37.Tjiu JW, Chen JS, Shun CT, Lin SJ, Liao YH, Chu CY, Tsai TF, Chiu HC, Dai YS, Inoue H, Yang PC, Kuo ML, Jee SH. The Journal of investigative dermatology. 2009;129:1016–1025. doi: 10.1038/jid.2008.310. [DOI] [PubMed] [Google Scholar]
- 38.Echeverria V, Worzella T, Skoien A, Lamers C, Meyvantsson I, Goulter A, Bowen W, Hayes S. 2008 [Google Scholar]
- 39.Walker G, Beebe DJ. Lab on a chip. 2002;2:131–134. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- 40.Meyvantsson I, Warrick JW, Hayes S, Skoien A, Beebe DJ. Lab on a chip. 2008;8:717–724. doi: 10.1039/b715375a. [DOI] [PubMed] [Google Scholar]
- 41.Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. The Journal of cell biology. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ongusaha PP, Kwak JC, Zwible AJ, Macip S, Higashiyama S, Taniguchi N, Fang L, Lee SW. Cancer research. 2004;64:5283–5290. doi: 10.1158/0008-5472.CAN-04-0925. [DOI] [PubMed] [Google Scholar]
- 43.Witzenbichler B, Asahara T, Murohara T, Silver M, Spyridopoulos I, Magner M, Principe N, Kearney M, Hu JS, Isner JM. The American journal of pathology. 1998;153:381–394. doi: 10.1016/S0002-9440(10)65582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.ten Dijke P, Goumans MJ, Pardali E. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 45.Natori T, Sata M, Washida M, Hirata Y, Nagai R, Makuuchi M. Biochemical and biophysical research communications. 2002;297:1058–1061. doi: 10.1016/s0006-291x(02)02335-5. [DOI] [PubMed] [Google Scholar]
- 46.Simonini A, Moscucci M, Muller DW, Bates ER, Pagani FD, Burdick MD, Strieter RM. Circulation. 2000;101:1519–1526. doi: 10.1161/01.cir.101.13.1519. [DOI] [PubMed] [Google Scholar]
- 47.Newby AC. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.