

Abstract
Background/Objectives
We previously demonstrated that reducing cardiac aldehydic load by aldehyde dehydrogenase 2 (ALDH2), a mitochondrial enzyme responsible for metabolizing the major lipid peroxidation product, protects against acute ischemia/reperfusion injury and chronic heart failure. However, time-dependent changes in ALDH2 profile, aldehydic load and mitochondrial bioenergetics during progression of post-myocardial infarction (post-MI) cardiomyopathy is unknown and should be established to determine the optimal time window for drug treatment.
Methods
Here we characterized cardiac ALDH2 activity and expression, lipid peroxidation, 4-hydroxy-2-nonenal (4-HNE) adduct formation, glutathione pool and mitochondrial energy metabolism and H2O2 release during the 4 weeks after permanent left anterior descending (LAD) coronary artery occlusion in rats.
Results
We observed a sustained disruption of cardiac mitochondrial function during the progression of post-MI cardiomyopathy, characterized by >50% reduced mitochondrial respiratory control ratios and up to 2 fold increase in H2O2 release. Mitochondrial dysfunction was accompanied by accumulation of cardiac and circulating lipid peroxides and 4-HNE protein adducts and down-regulation of electron transport chain complexes I and V. Moreover, increased aldehydic load was associated with a 90% reduction in cardiac ALDH2 activity and increased glutathione pool. Further supporting an ALDH2 mechanism, sustained Alda-1 treatment (starting 24hrs after permanent LAD occlusion surgery) prevented aldehydic overload, mitochondrial dysfunction and improved ventricular function in post-MI cardiomyopathy rats.
Conclusion
Taken together, our findings demonstrate a disrupted mitochondrial metabolism along with an insufficient cardiac ALDH2-mediated aldehyde clearance during the progression of ventricular dysfunction, suggesting a potential therapeutic value of ALDH2 activators during the progression of post-myocardial infarction cardiomyopathy.
Keywords: myocardial infarction, 4-hydroxinonenal, oxidative stress, bioenergetics, aldehyde dehydrogenase 2
Introduction
Despite the advances in clinical interventions and pharmacological therapies, cardiovascular diseases remain the main cause of mortality worldwide [1, 2]. Therefore, the discovery of novel therapies that will improve cardiovascular diseases outcome remains a major priority. Cardiac mitochondria, the main source of ATP as well as reactive oxygen species, are critical organelles for normal functioning of the heart [3, 4]. Disruption of mitochondrial metabolism seems to be a critical contributor to the pathophysiology of acute [5, 6] and chronic cardiovascular diseases [7, 8]. Therefore, mitochondria have become a very attractive target for novel cardioprotective therapies [9].
We previously found that increased myocardium aldehydic load, characterized by mitochondrial dysfunction-mediated accumulation of 4-hydroxy-2-nonenal (4-HNE), plays a detrimental role during acute ischemia-reperfusion injury and chronic heart failure [10–12]. The highly reactive carbonyl compounds form adducts with proteins and DNA, and negatively affects their biological function. Activation of aldehyde dehydrogenase 2 (ALDH2), the main enzyme that catalyzes 4-HNE metabolism, is sufficient to protect the heart against acute ischemia-reperfusion injury [10].
To date, it is not known whether disrupted mitochondrial bioenergetics and consequent accumulation of lipid peroxidation-derived reactive aldehydes are involved in the progression of post-myocardial infarction cardiomyopathy induced in the days and weeks after permanent left anterior descending coronary artery occlusion. Here we characterize the time-dependent changes in mitochondrial energy metabolism, H2O2 release, lipid peroxidation, aldehydic load profile and ALDH2 activity during the progression of post-myocardial infarction cardiomyopathy in rats.
Methods
Ethical approval
The animal care and protocols in this study were reviewed and approved by the Ethical Committee of Biomedical Sciences Institute of University of São Paulo (20012/36). This study was conducted in accordance with the ethical principles in animal research adopted by the Brazilian Society of Laboratory Animal Science (www.cobea.org.br). At the end of the protocol, all rats were anesthetized with sodium pentobarbital (100mg/kg IP) and euthanized by decapitation.
Animals and study design
The present investigation was carried out in male Wistar rats (250–300g) maintained in a 12:12h light-dark cycle and temperature-controlled environment (22°C) with free access to standard laboratory chow (Nuvital Nutrientes, Curitiba, PR, Brazil) and tap water. Animals used for the time-window study were assigned into four experimental groups: post-MI cardiomyopathy rats evaluated at weeks 1 (n=8), 2 (n=13) and 4 (n=11) and sham (control, n=9), according to the experimental model depicted in Figure 1A. Animals used for the pharmacological ALDH2 activation study (using Alda-1) were assigned into three experimental groups: sham (control, n=8), placebo-treated post-MI cardiomyopathy (n=8) and Alda-1-treated post-MI cardiomyopathy (n=8). 45% of rats died immediately after LAD occlusion and therefore were excluded from the study. Data related to all the other rats in each group are included in the analyses.
Figure 1. Cardiac function and morphology during the progression of post-MI cardiomyopathy.

(A) Schematic panel. Twelve-week old rats were subjected to permanent left anterior descending coronary artery occlusion surgery to induce post-MI cardiomyopathy. Animals were assigned into four experimental groups: control (sham, n=9) and post-MI cardiomyopathy rats at weeks 1 (n=8), 2 (n=13) and 4 (n=11) after LAD occlusion surgery. (B) Left ventricle fractional shortening; (C) Ejection fraction; (D) Left ventricle end-diastolic diameter (LVEDD); (E) Left ventricle end-systolic diameter (LVESD); (F) Left ventricle end-diastolic posterior wall thickness (PWth) and (G) Left ventricle end-diastolic inter-ventricular septum thickness (IVSth) in control (sham, white bar) and post-MI cardiomyopathy rats at different weeks after LAD occlusion (gray bars). Data are means ± SEM. *, p<0.05 vs. control (sham) rats. The observer was blinded to the experimental conditions.
Post-myocardial infarction cardiomyopathy animal model
Post-MI cardiomyopathy was induced by ligation of the left anterior descending coronary artery (LAD), as previously described [13]. We have chosen this model since post-myocardial infarction cardiomyopathy is the underlying etiology of heart failure in nearly 70% of patients [14]. Male Wistar rats were anesthetized with 3% isoflurane, endotracheally intubated, and mechanically ventilated with room air (respiratory rate of 60–70 breaths/min and tidal volume of 2.5 mL). Left thoracotomy between the fourth and fifth ribs was performed and the LAD was ligated. After the surgery, animals were monitored daily. Left thoracotomy with equal procedure duration to that of heart failure group, but without LAD ligation, was undertaken in the sham group (control).
Treatment with Alda-1
Post-MI cardiomyopathy rats were treated with Alda-1 (selective ALDH2 activator) for 4 weeks. Continuous infusion of Alda-1 (10 mg/kg per day) was achieved using Alzet osmotic pumps (2ML4 and 2ML2) and began 24 hours after LAD occlusion surgery and ended 4 weeks later. A group of rats implanted with pumps containing the vehicle alone (50% polyethylene glycol and 50% dimethyl sulfoxide by volume) served as the control. Subcutaneous pump implantation was performed in 3% vaporized isoflurane-anesthetized rats. This concentration provided deep anaesthesia, allowing mini-pump implantation without any sigh of pain, such as withdrawal reflex. The pumps were inserted in the back of animals after making a sub-scapular incision.
Two-dimensional guided M-mode echocardiography
Non-invasive cardiac function was assessed by M-mode echocardiography in anesthetized (isoflurane 3%) sham and post-MI cardiomyopathy rats. Briefly, rats were positioned in the supine position with front paws wide open and ultrasound transmission gel was applied to the precordium. Transthoracic echocardiography was performed using an Acuson Sequoia model 512 echocardiographer equipped with a 14-MHz linear transducer as described elsewhere [15]. Left ventricle systolic function was estimated by fractional shortening (FS) as follows: FS (%) = [(LVEDD – LVESD)/LVEDD] x 100, where LVEDD is the left ventricular end-diastolic diameter, and LVESD is the left ventricular end-systolic diameter. The observer was blinded to the experimental conditions.
Infarct size measurement
Forty-eight hours after the end of the protocol, all rats were killed and their tissues were harvested. Cardiac chambers were then fixed by immersion in 4% buffered formalin and embedded in paraffin for routine histological processing. Sections (4 μm) were stained with Masson’s trichrome for the quantification of myocardial infarct area as described elsewhere [16]. The myocardial infarcted area was expressed as a percentage of total surface area of the left ventricle. The observer was blinded to the experimental conditions.
Mitochondrial isolation
Cardiac mitochondria were isolated as described elsewhere [17]. Briefly, heart samples were minced and homogenized in isolation buffer (300 mM sucrose, 10 mM Hepes, 2 mM EGTA, pH 7.2, 4°C) containing 0.1 mg·mL−1 of type I protease (bovine pancreas) to release mitochondria from within muscle fibers and later washed in the same buffer in the presence of 1 mg·mL−1 bovine serum albumin. The suspension was homogenized in a 40 mL tissue grinder and centrifuged at 950 g for 5 min. The resulting supernatant was centrifuged at 9500 g for 10 min. The mitochondrial pellet was washed, resuspended in isolation buffer and submitted to a new centrifugation (9500 g for 10 min). The mitochondrial pellet was washed and the final pellet was resuspended in a minimal volume of isolation buffer.
Mitochondrial H2O2 release and O2 consumption
Mitochondrial H2O2 release was determined by measuring the oxidation of Amplex Red in the presence of horseradish peroxidase using a spectrophotometer with 563 nm excitation and 587 nm of emission [8]. Mitochondrial O2 consumption was monitored using a computer-interfaced Clark-type electrode (Hansatech Instruments) operating with continuous stirring at 37 °C [8]. Succinate, malate and glutamate (2 mmol/L of each) were used as substrates and ADP (1 mmol/L) was added to induce State 3 respiratory rate. A subsequent addition of oligomycin (1 μg/mL) was used to determine State 4 rate. The respiratory control ratio was calculated by dividing State 3 by State 4 oxygen consumption rates.
Enzymatic activity of ALDH2
ALDH2 enzymatic activity was determined by measuring the conversion of NAD+ to NADH, as described elsewhere [10]. The assays were carried out at 25°C in 50 mM sodium pyrophosphate buffer (pH 9.5) in the presence of 10 mM acetaldehyde. Measurement of mitochondrial ALDH2 activity in the rat myocardium was determined by directly adding 400 mg of the mitochondrial fraction of the myocardium to the reaction mix and reading absorbance at 340 nm for 10 min.
Immunoblotting
Protein levels were evaluated by immunoblotting in total extracts from the ventricular remote area [18]. Briefly, samples were subjected to SDS-PAGE in polyacrylamide gels (6–15%) depending upon protein molecular weight. After electrophoresis, proteins were electrotransferred to nitrocellulose membranes (BioRad Biosciences; Piscataway, NJ). Equal gel loading and transfer efficiency were monitored using 0.5% Ponceau S staining of blot membrane. Anti-cytochrome c, NDUFA9 (complex I) and IF1 (complex V) mouse antibodies (Abcam; Cambridge, UK), anti-GAPDH mouse antibody (Advanced Immunochemical Inc; Long Beach, CA), anti-4-HNE-protein adducts rabbit antibody (Millipore, MA) and anti-ALDH2 goat antibody (Santa Cruz Biotechnology, Santa Cruz, CA) were used for immunoblotting followed secondary probing with HRP-conjugated goat anti-rabbit, mouse and rabbit anti-goat IgG antibodies. All data were normalized by internal controls of GAPDH.
Circulating 4-HNE levels
4-HNE levels were determined using a fluorometric reverse-phase high-pressure liquid chromatography (Shimadzu LC-20, equipped with RF20A detector), as previously described [19]. Fresh blood proteins were initially precipitated with trichloroacetic acid (10% v/v) and supernatant 4-HNE was derivatized with 1,3-cyclohexanedione (CHD) in solution containing CHD 1.25%, NH4Ac 20% and Thiourea 6% at 60°C for 1 hour. The solution was filtered and assayed for 4-HNE.
Lipid peroxidation
Lipid hydroperoxides were evaluated by the modified ferrous oxidation-xylenol orange technique (FOX2) [20]. Heart samples were homogenized (1:20w/v) in cold potassium phosphate buffer (50mM, pH7.4) and immediately centrifuged at 12,000g for 20min at 4°C. Cardiac and serum proteins were precipitated with trichloroacetic acid (10% w/v) and supernatant was mixed with FOX reagent (250 μM ammonium ferrous sulfate, 100 μM xylenol orange and 25 mM H2SO4) and incubated for 30min at room temperature. The absorbance of the sample was read at 560nm.
Glutathione pool
Heart glutathione levels were measured using the glutathione reductase enzyme recycling method, as previously described [21]. The assay is based on the reaction of reduced glutathione (GSH) with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) that produces the oxidized glutathione–TNB adduct (GS–TNB) and the TNB chromophore, which has a maximal absorbance at 412 nm. The disulfide product (GS–TNB) is then reduced by glutathione reductase in the presence of NADPH, recycling GSH back into the reaction. The rate of formation of TNB, measured at 412 nm, is proportional to the concentration of glutathione in the sample.
Statistical analysis
Data are presented as means ± standard error of the mean (SEM). Data normality was assessed through Shapiro-Wilk’s test. One-way analysis of variance (ANOVA) was used to analyze data presented in Tables 1–3 and Figures 1, 2, 3, 4, 5A–B and D, 6 and 7. Whenever significant F-values were obtained, Bonferroni adjustment was used for multiple comparison purposes. Linear regression was used to assess the association between variables in Figures 5C and 5E. GraphPad Prism Statistics was used for the analysis. Statistical significance was considered achieved when the value of P was <0.05.
Table 1.
Glutathione levels during progression of post-MI cardiomyopathy
| Parameter | Sham | Post-MI cardiomyopathy
|
||
|---|---|---|---|---|
| week 1 | week 2 | week 4 | ||
|
|
|
|||
| GSH (nmol/mg) | 3.41±0.20 | 4.52±0.11* | 4.43±0.35* | 4.63±0.37* |
| GSSG (nmol/mg) | 0.39±0.02 | 0.53±0.02* | 0.50±0.04* | 0.64±0.05* |
| GSH/GSSG | 8.90±0.71 | 8.64±0.27 | 9.15±0.97 | 7.30±0.31 |
|
|
|
|||
Measurements of reduced glutathione (GSH), oxidized glutathione (GSSG) and reduced:oxidized glutathione ratio in cardiac sample from Sham (n=5), 1 week post-MI (n=6), 2 weeks post-MI (n=7) and 4 weeks post-MI (n=5) animals. Data were analyzed by one-way ANOVA followed by post-hoc Duncan. Error bars indicate SEM.
p<0.05 vs. control (sham rats).
Table 3.
Mitochondrial respiratory rates and State-dependent H2O2 release
|
|
Mitochondrial respiratory rates
|
State-dependent H2O2 release
|
||||
|---|---|---|---|---|---|---|
| Parameter | Sham | Post-MI Placebo | Post-MI Alda-1 | Sham | Post-MI Placebo | Post-MI Alda-1 |
|
|
|
|
||||
| State 2 | 97±10 | 50±11* | 74±12 | 1.6±0.3 | 3.7±0.4* | 2,1±0.4‡ |
| State 3 | 277±31 | 135±27* | 190±33 | 0.2±0.1 | 0.6±0.2* | 0.3±0.1 |
| State 4 | 82±7 | 60±9* | 67±11 | 1.7±0.4 | 0.7±0.2* | 1.1±0.3 |
|
|
|
|
||||
Measurements of oxygen consumption rates and State-dependent H2O2 release were performed in cardiac mitochondria isolated from Sham (n=5), post-MI placebo (4 wks after MI, n=5) and post-MI Alda-1 treated (4 wks after MI, n=6) animals incubated in experimental buffer at 37oC. Animals were treated for 4wks, starting 24hrs after permanent LAD occlusion surgery. Succinate (2mM), malate (2mM) and glutamate (2mM) were used as respiratory substrate. ADP (1mM) was added to induce State 3 respiratory rates (shown in nmol O2 mg−1 min−1). A subsequent addition of oligomycin (1ug/mL) was used to determine State 4 rates. Data were analyzed by one- or two-way ANOVA followed by post-hoc Duncan. Error bars indicate SEM.
p<0.05 vs. control (sham);
p<0.05 vs. Post-MI Placebo rats.
Figure 2. Mitochondrial oxygen consumption rates during the progression of post-MI cardiomyopathy.

(A) Respiratory control ratio (State3/State4) and (B–D) State-dependent oxygen consumption rates in cardiac mitochondria isolated from control (sham, white bars, n=9) and post-MI cardiomyopathy rats at weeks 1 (n=8), 2 (n=13) and 4 (n=11) after LAD occlusion surgery. Succinate, malate and glutamate (2 mM of each) were used as substrates (State 2) and ADP (1 mM) was added to induce State 3 respiratory rate. Addition of oligomycin (1 μg·mL−1) was used to determine State 4 respiratory rates. Data are means ± SEM. *, p<0.05 vs. control (sham) rats.
Figure 3. Mitochondrial ROS release and protein levels of respiratory complexes during the progression of post-MI cardiomyopathy.

(A–C) State-dependent H2O2 release from isolated cardiac mitochondria; (D) NDUFA9 (mitochondrial complex I), (E) Cytochrome c and (F) IF1 (mitochondrial complex V) protein levels; (G) Representative western blots showing the level of cardiac mitochondrial complex I, cytochrome c, complex V in control (sham, white bars, n=6–9) and post-MI cardiomyopathy rats at weeks 1 (n=6–8), 2 (n=6–13) and 4 (n=6–11) after LAD occlusion surgery. Succinate, malate and glutamate (2 mM of each) were used as substrates (State 2) and ADP (1 mM) was added to induce State 3 respiratory rate. Addition of oligomycin (1 μg·mL−1) was used to determine State 4 respiratory rates. Data are means ± SEM. *, p<0.05 vs. control (sham) rats. Protein expression was normalized by GAPDH and expressed as % sham. &, p<0.05 vs. post-MI cardiomyopathy at week 4 after LAD occlusion surgery.
Figure 4. Cardiac and circulating aldehydic load during the progression of post-MI cardiomyopathy.

(A) Cardiac lipid peroxides; (B) Serum lipid peroxides; (C) Cardiac 4-HNE-protein adducts; (D) Serum 4-HNE levels, (E) Representative western blots showing the level of cardiac 4-HNE-protein adducts in control (sham, white bars, n=6–9) and post-MI cardiomyopathy rats at weeks 1 (n=6–8), 2 (n=6–13) and 4 (n=6–11) after LAD occlusion surgery. Protein expression was normalized by GAPDH and expressed as % sham. Data are means ± SEM. *, p<0.05 vs. control (sham) rats.
Figure 5. Cardiac ALDH2 profile and glutathione levels during the progression of post-MI cardiomyopathy.

(A) Cardiac ALDH2 activity (input: delta ALDH2 activity); (B) Cardiac ALDH2 protein level and representative western blots showing the level of cardiac ALDH2; (C) Correlation between ventricular ejection fraction and cardiac ALDH2 activity; (D) Total glutathione levels; (E) Correlation between ALDH2 activity and total glutathione levels in control (sham, white bars, n=9) and post-MI cardiomyopathy rats at weeks 1 (n=8), 2 (n=13) and 4 (n=11) after LAD occlusion surgery. Cardiac ALDH2 protein level was normalized by GAPDH and expressed as % sham. Data are means ± SEM. *, p<0.05 vs. control (sham) rats.
Figure 6. Sustained ALDH2 activation reduces aldehydic load and improves mitochondrial metabolism in post-MI cardiomyopathy in rats.

Twelve-week old rats were subjected to permanent left anterior descending coronary artery occlusion surgery to induce post-MI cardiomyopathy. Animals were assigned into three experimental groups: control (sham), vehicle-treated and Alda-1-treated post-MI cardiomyopathy rats (n=8 for each experimental condition). Post-MI cardiomyopathy rats were treated for 4 weeks, starting 24hrs after permanent LAD occlusion surgery. Continuous infusion of Alda-1 (10 mg/kg per day) or vehicle (50%DMSO was achieved using Alzet osmotic pumps (2ML4 and 2ML2) and began 24hrs after LAD occlusion surgery and ended 4 weeks later. (A) ALDH2 activity; (B) ALDH2 activity (% sham group); (C) ALDH2 protein levels; (D) Cardiac lipid peroxides; (E) Serum lipid peroxides; (F) Cardiac 4-HNE protein adducts; (G) NDUFA9 (mitochondrial complex I), (H) Cytochrome c and (I) IF1 (mitochondrial complex V) protein levels; (J) Mitochondrial respiratory control ratio; (K) Cardiac glutathione pool and (L–M) Representative western blots showing the level of cardiac 4-HNE-protein adducts, ALDH2, mitochondrial complex I, cytochrome c and complex V in control (sham, white bars), vehicle-treated (gray bars) and Alda-1-treated post-MI cardiomyopathy rats (red bars). Protein expression was normalized by GAPDH and expressed as % sham. Data are means ± SEM. *, p<0.05 vs. control (sham) rats; #, p<0.05 vs. vehicle-treated post-MI cardiomyopathy rats.
Figure 7. Sustained ALDH2 activation improves ventricular function in post-MI cardiomyopathy in rats.

(A) Left ventricle fractional shortening; (B) Ejection fraction; (C) Left ventricle end-diastolic diameter (LVEDD); (D) Left ventricle end-systolic diameter (LVESD); (E) Left ventricle end-diastolic posterior wall thickness (PWth) and (F) Left ventricle end-diastolic inter-ventricular septum thickness (IVSth) in control (sham, white bars), vehicle-treated (gray bars) and Alda-1-treated post-MI cardiomyopathy rats (red bars). Data are means ± SEM. *, p<0.05 vs. control (sham) rats; #, p<0.05 vs. vehicle-treated post-MI cardiomyopathy rats. The observer was blinded to the experimental conditions.
Results
In this study, we determined the time-window of changes in cardiac mitochondrial energy metabolism, H2O2 release, local and circulating aldehydic load and ALDH2 during the progression of post-MI cardiomyopathy in rats. Rats underwent permanent coronary artery ligation or sham surgery and were euthanized at 1, 2 and 4 weeks. The echocardiographic data are provided in Figure 1. Compared to sham-treated rats, the post-MI cardiomyopathy rats (referred to as post-myocardial infarction cardiomyopathy) studied at each time point had lower ventricular fractional shortening and ejection fraction (Figures 1B–C). These changes were accompanied by left ventricle dilation and posterior wall hypertrophy from weeks 1 and 4, respectively (Figure 1D–F). Infarct size, measured by masson trichrome staining, was similar at all time points (35±4% of the left ventricle).
Considering that disrupted mitochondrial bioenergetics profile represents a key underlying process of cardiovascular diseases [7], we set out to characterize mitochondrial functions during the progression of the post-MI cardiomyopathy. To assess mitochondrial function, we measured oxygen consumption and H2O2 release in isolated mitochondria from the sham and post-MI cardiomyopathy groups at weeks 1, 2 and 4 after surgery.
Our results indicate that post-MI cardiomyopathy rats displayed a significant decrease in the efficiency of mitochondrial oxidative phosphorylation at all time points as compared with control (sham) rats; as measured by respiratory control ratio (State 3/State 4; Figure 2A). The decrease in respiratory control ratio was mainly due to a reduction of State 3 respiratory rate (the oxygen consumption rate maximized by the addition of ADP; Figure 2C), while respiration in the absence of oxidative phosphorylation (State 4) was only significantly affected at weeks 1 and 4 after LAD surgery (Figure 2D). Isolated mitochondria from dysfunctional hearts also presented reduction in basal respiratory rate as compared with sham animals (Figure 2B).
Since mitochondria are the main source of ROS, we further evaluated cardiac mitochondrial H2O2 release during the progression of ventricular dysfunction. As shown in Figure 3A, failing hearts presented increased mitochondrial State 2 H2O2 release at weeks 1 and 4 relative to the sham group. Cardiac mitochondrial State 3 H2O2 release was also elevated in the post-MI cardiomyopathy group at all time points (Figure 3B), a result in line with the finding of reduced respiratory rates under these conditions (Figure 2C), since respiratory inhibition is often accompanied by enhanced mitochondrial oxidant generation [22]. Finally, mitochondria isolated from post-MI cardiomyopathy rats presented reduced State 4 H2O2 release at week 4. Disruption of mitochondrial metabolism and increased H2O2 release were accompanied by down-regulation of cardiac mitochondrial electron transport chain complexes I and V, without affecting cytochrome c levels, during the progression of post-MI cardiomyopathy (Figures 3D–F).
We next measured lipid peroxide levels during the progression of the post-MI cardiomyopathy, since increased mitochondrial pro-oxidant levels result in exacerbated lipid peroxidation through its reaction with the polyunsaturated fatty acids of biological membranes. Both cardiac and circulating lipid peroxide levels increased in failing hearts at all time points (Figure 4A–B). Moreover, excessive lipid peroxidation contributed to the establishment of aldehydic overload profile by accumulating both cardiac 4-HNE protein adducts and circulating 4-HNE levels (Figure 4C–D).
Considering that aldehydic overload profile is characterized by a disrupted equilibrium of aldehyde generation and removal, we next determined the activity and level of the main enzyme related to cardiac 4-HNE metabolism, ALDH2. Rats with post-MI cardiomyopathy presented a significant reduction of cardiac ALDH2 activity at all time points compared with sham-operated rats. As shown in Figure 5A, ALDH2 activity decreased by 81±3% 1 week after the LAD occlusion surgery. This reduction was significantly different from sham animals until week 4 (Figure 5A). Post-MI cardiomyopathy had no effect on cardiac ALDH2 protein expression levels during the progression of cardiac dysfunction (Figure 5B). Of interest, cardiac ALDH2 activity and ventricular ejection fraction displayed a positive correlation during the progression of post-MI cardiomyopathy (Figure 5C).
Since increased glutathione levels may counteract damaging effects induced by accumulation of reactive aldehydes in ALDH2 transgenic mice [23], we measured cardiac glutathione pools during the progression of ventricular dysfunction. The progression of post-MI cardiomyopathy was characterized by a significant increase of cardiac glutathione pools at all time points after LAD surgery, which inversely correlated with ALDH2 activity (Figures 5D–E and Table 1). However, increase of glutathione pools did not confer resistance against post-MI cardiomyopathy in rats.
Finally, we determined whether sustained ALDH2 activation positively affects aldehyde load profile and mitochondrial metabolism with further impact on ventricular function during the progression of post-MI cardiomyopathy. Sustained Alda-1 treatment (a selective ALDH2 activator) over 4 weeks (starting 24hrs after permanent LAD occlusion) significantly increased ALDH2 activity without affecting its protein levels (Figure 6A–C). Moreover, ALDH2 activation reduced cardiac and systemic aldehydic load (Figures D–F and Table 2), preserved mitochondrial bioenergetics status and prevented both down-regulation of cardiac mitochondrial electron transport chain complexes (I and V) and excessive H2O2 release in post-MI cardiomyopathy rats (Figures 6G–M and Table 3). Of interest, Alda-1-treated post-MI rats presented better left ventricle fraction shortening and ejection fraction relative to vehicle-treated post-MI rats (Figure 7A–B). ALDH2 activation also resulted in a cardiac anti-remodeling effect, characterized by reduced left ventricular diastolic diameter and posterior wall thickness (Figure 7C–F).
Table 2.
Glutathione levels in placebo and Alda-1-treated post-MI cardiomyopathy rats
| Parameter | Sham | Post-MI cardiomyopathy
|
|
|---|---|---|---|
| Placebo | Alda-1 | ||
|
|
|
||
| GSH (nmol/mg) | 3.06±0.44 | 4.29±0.21* | 3.31±0.14‡ |
| GSSG (nmol/mg) | 0.36±0.03 | 0.61±0.05* | 0.47±0.04 |
| GSH/GSSG | 8.34±0.82 | 7.14±0.48 | 7.56±1.15 |
|
|
|
||
Measurements of reduced glutathione (GSH), oxidized glutathione (GSSG) and reduced:oxidized glutathione ratio in cardiac sample from Sham (n=5), post-MI placebo (4 wks after MI, n=5) and post-MI Alda-1 treated (4 wks after MI, n=7) animals. Animals were treated for 4wks, starting 24hrs after permanent LAD occlusion surgery. Data were analyzed by one-way ANOVA followed by post-hoc Duncan. Error bars indicate SEM.
p<0.05 vs. control (sham);
p<0.05 vs. Post-MI Placebo rats.
Discussion
Most of cardiac damage occurring following acute ischemia-reperfusion injury is believed to be due to mitochondrial dysfunction-mediated ROS production [9, 24]. More recently, we have shown that lipid peroxidation-generated reactive aldehydes are key players in the propagation of ROS-mediated cardiac damage upon acute ischemia-reperfusion [10, 11, 25]. In the present study, using an in vivo rat model of post-MI cardiomyopathy, we have extended the aforementioned findings and demonstrated that disruption of mitochondrial bioenergetics profile, oxidative stress and exacerbated lipid peroxidation continue over 4 weeks after acute myocardial infarction. Of interest, this pro-oxidant scenario contributed to the accumulation of cardiac 4-HNE-protein adducts and increased circulating 4-HNE, the major aldehyde derived from lipid hydroperoxides [7]. Reduction of cardiac ALDH2 activity, the main enzyme in the detoxification of 4-HNE, is likely contributing to the aldehydic overload profile seen during the progression of ventricular dysfunction. In fact, cardiac ALDH2 activity was positively associated with ventricular ejection fraction during the progression of post-MI cardiomyopathy; and sustained ALDH2 activation prevented aldehydic overload profile, mitochondrial dysfunction and significantly improved ventricular function in these animals.
Aldehydes are highly diffusible and reactive agents generated during numerous pathological processes [26, 27]. For example, 4-hydroxy-2-nonenal (4-HNE) accumulates rapidly upon mitochondrial dysfunction-induced oxidative stress [12] and forms protein adducts via Michaels addition, inhibiting key metabolic proteins [including components of mitochondrial electron transport chain], and ultimately affecting the function of the cell [28, 29]. Over time, sustained lipid peroxidation and further accumulation of 4-HNE lead to impaired mitochondrial metabolism, at least in part, through down-regulation of mitochondrial electron transport chain components [30, 31]. However, the molecular mechanism behind 4-HNE-mediated down-regulation of mitochondrial proteins is still unknown. Here, we demonstrated that the progression of post-MI cardiomyopathy is characterized by accumulation of cardiac 4-HNE-protein adducts (Figures 4C and E) and down-regulation of mitochondrial electron transport chain complexes (I and V). 4-HNE modified proteins are significantly elevated in myocardial biopsies from patients with hypertrophic and dilated cardiomyopathy compared to health subjects [19, 32]. Indeed, the long-term use of carvedilol, which improves ventricular function, is associated with a significant reduction of cardiac 4-HNE adducts in patients with dilated cardiomyopathy [32]. To date, there is no data regarding on 4-HNE adduct levels in patients with post-MI cardiomyopathy.
Beside its ability to form adduct with proteins, circulating 4-HNE levels have been considered a potential biomarker for congestive heart failure and alcoholic cardiomyopathy [33, 34]. Our findings demonstrated for the first time that circulating 4-HNE levels increase both early- and at a late-stage post-MI cardiomyopathy in rats (Figures 4D). 4-HNE detoxification is a likely cardioprotective mechanism mediated by oxidative conversion of 4-HNE to 4-hydroxynoneic acid via ALDH2 and glutathione-S-transferase (GST)-mediated glutathione (GSH). Here, we demonstrated that cardiac ALDH2 activity is impaired during the progression of post-MI cardiomyopathy (Figure 5A). 4-HNE inactivates ALDH2 at high concentrations, resulting in drastic inhibition of the enzyme, in vitro [11]. It is therefore likely that impaired 4-HNE detoxification and accumulation of this very reactive aldehyde induce inactivation of ALDH2 in post-MI cardiomyopathy. However, further studies are needed to determine a causal relationship between accumulation of 4-HNE and ALDH2 inactivation, in vivo.
Transgenic mice overexpressing the inactive ALDH2 mutation (ALDH2*2) exhibit reduced mitochondrial oxygen consumption and accumulation of cardiac 4-HNE [23]. Of interest, these mice present a compensatory increase in cardiac glutathione levels, which confers cardioprotection against acute ischemia-reperfusion injury. We therefore evaluated whether post-MI-induced ALDH2 inhibition results in increased cardiac glutathione pools. Our findings demonstrated that reduced ALDH2 activity is accompanied by a significant increased of cardiac GSH levels during the progression of post-MI cardiomyopathy (Figures 5D–E and Table 1). However, unlike transgenic mouse overexpressing ALDH2*2, this higher GSH level seen in post-MI neither reduced cardiac aldehydic load nor avoided the progression of ventricular dysfunction (Figures 4C–E). Therefore, ALDH2 seems to be a key enzyme in the 4-HNE metabolism during progression of post-MI cardiomyopathy.
In fact, sustained ALDH2 activation (starting 24 hrs after permanent LAD occlusion) prevented aldehydic overload, mitochondrial dysfunction, down-regulation of electron transport chain complexes (I and V) and improved ventricular function in post-MI cardiomyopathy in rats (Figures 6 and 7). We have recently shown that reducing cardiac aldehydic overload through ALDH2 improves heart failure outcome [12]. Moreover, either pharmacological activation or genetic over-expression of ALDH2 induces cardioprotection against acute ischaemia-reperfusion injury [11] and aldehydic overload cardiomyopathy [35]. It seems that both reduced generation and efficient removal of 4-HNE are likely to be of crucial importance for cardiomyocyte survival.
In summary, our data suggest that mitochondrial energy metabolism dysfunction and further uncontrolled oxidative stress can yield excessive lipid peroxidation, 4-HNE generation and ALDH2 inactivation during the progression of post-MI cardiomyopathy (Figure 7). In fact, pharmacological ALDH2 activation prevents aldehydic overload and improves post-MI cardiomyopathy outcome. It is therefore likely that the impaired 4-HNE metabolism and accumulation of 4-HNE adducts observed during post-MI cardiomyopathy occurs as a dual consequence of increased generation of aldehydes from lipid peroxidation and impaired metabolism of these aldehydes due to inactivation of ALDH2.
Figure 8. Proposed model for aldehydic overload during the progression of post-MI cardiomyopathy.
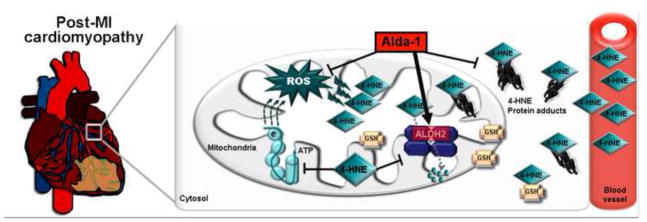
Mitochondrial energy metabolism dysfunction and further uncontrolled oxidative stress can yield excessive lipid peroxidation, 4-HNE generation and ALDH2 inactivation during the progression of post-MI cardiomyopathy. Impaired 4-HNE metabolism and accumulation of 4-HNE adducts observed during post-MI cardiomyopathy occurs as a dual consequence of increased generation of aldehydes from lipid peroxidation and impaired metabolism of these aldehydes due to inactivation of ALDH2. Pharmacological ALDH2 activation improves post-MI cardiomyopathy outcome through increased removal of 4-HNE.
Acknowledgments
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo, São Paulo (FAPESP #2012/05765-2, 2010/51906-1; 2013/07937-8), Conselho Nacional de Pesquisa e Desenvolvimento – Brasil (CNPq #470880/2012-0, 301105/2012-0, 407306/2013-7, 302898/2013-1; 573530/2008-4) to JCBF and AJK and National Institutes of Health NIAAA 11147 to DMR. KMSG holds master’s fellowship from FAPESP.
Footnotes
All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Disclosure: DM-R is founder of ALDEA Pharmaceuticals. However, none of the research is supported by or in collaboration with the company. Other authors have no disclosure.
References
- 1.Ferreira JC, Koyanagi T, Palaniyandi SS, et al. Pharmacological inhibition of betaIIPKC is cardioprotective in late-stage hypertrophy. J Mol Cell Cardiol. 2011;51:980–987. doi: 10.1016/j.yjmcc.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figueira TR, Barros MH, Camargo AA, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 4.Palaniyandi SS, Qi X, Yogalingam G, Ferreira JC, Mochly-Rosen D. Regulation of mitochondrial processes: a target for heart failure. Drug Discov Today Dis Mech. 2010;7:e95–e102. doi: 10.1016/j.ddmec.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Disatnik MH, Ferreira JC, Campos JC, et al. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem. 2013;288:18947–18960. doi: 10.1074/jbc.M113.466870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campos JC, Gomes KM, Ferreira JC. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem Toxicol. 2013;62:107–119. doi: 10.1016/j.fct.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 8.Campos JC, Queliconi BB, Dourado PM, et al. Exercise training restores cardiac protein quality control in heart failure. PLoS One. 2012;7:e52764. doi: 10.1371/journal.pone.0052764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2012;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med. 2011;3:107ra111. doi: 10.1126/scitranslmed.3002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomes KM, Campos JC, Bechara LR, et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodeling. Cardiovasc Res. 2014 doi: 10.1093/cvr/cvu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johns TN, Olson BJ. Experimental myocardial infarction. I. A method of coronary occlusion in small animals. Ann Surg. 1954;140:675–682. doi: 10.1097/00000658-195411000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gheorghiade M, Bonow RO. Chronic heart failure in the United States: a manifestation of coronary artery disease. Circulation. 1998;97:282–289. doi: 10.1161/01.cir.97.3.282. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira JC, Boer BN, Grinberg M, Brum PC, Mochly-Rosen D. Protein quality control disruption by PKCbetaII in heart failure; rescue by the selective PKCbetaII inhibitor, betaIIV5-3. PLoS One. 2012;7:e33175. doi: 10.1371/journal.pone.0033175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palaniyandi SS, Ferreira JC, Brum PC, Mochly-Rosen D. PKCbetaII inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses. J Cell Mol Med. 2011;15:1769–1777. doi: 10.1111/j.1582-4934.2010.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancherini DV, Queliconi BB, Kowaltowski AJ. Pharmacological and physiological stimuli do not promote Ca(2+)-sensitive K+ channel activity in isolated heart mitochondria. Cardiovasc Res. 2007;73:720–728. doi: 10.1016/j.cardiores.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 18.Bueno CR, Jr, Ferreira JC, Pereira MG, Bacurau AV, Brum PC. Aerobic exercise training improves skeletal muscle function and Ca2+ handling-related protein expression in sympathetic hyperactivity-induced heart failure. J Appl Physiol. 2010;109:702–709. doi: 10.1152/japplphysiol.00281.2010. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura K, Kusano KF, Matsubara H, et al. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J Card Fail. 2005;11:117–123. doi: 10.1016/j.cardfail.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Hermes-Lima M, Willmore WG, Storey KB. Quantification of lipid peroxidation in tissue extracts based on Fe(III)xylenol orange complex formation. Free Radic Biol Med. 1995;19:271–280. doi: 10.1016/0891-5849(95)00020-x. [DOI] [PubMed] [Google Scholar]
- 21.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 22.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Endo J, Sano M, Katayama T, et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res. 2009;105:1118–1127. doi: 10.1161/CIRCRESAHA.109.206607. [DOI] [PubMed] [Google Scholar]
- 24.Bolli R, Jeroudi MO, Patel BS, et al. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci U S A. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice. J Mol Cell Cardiol. 2010;48:757–764. doi: 10.1016/j.yjmcc.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev. 2014;94:1–34. doi: 10.1152/physrev.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira JC, Mochly-Rosen D. Nitroglycerin use in myocardial infarction patients. Circ J. 2012;76:15–21. doi: 10.1253/circj.cj-11-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulteau AL, Lundberg KC, Humphries KM, et al. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 29.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Faerber G, Barreto-Perreia F, Schoepe M, et al. Induction of heart failure by minimally invasive aortic constriction in mice: reduced peroxisome proliferator-activated receptor gamma coactivator levels and mitochondrial dysfunction. J Thorac Cardiovasc Surg. 2011;141:492–500. 500 e491. doi: 10.1016/j.jtcvs.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 31.Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura K, Kusano K, Nakamura Y, et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation. 2002;105:2867–2871. doi: 10.1161/01.cir.0000018605.14470.dd. [DOI] [PubMed] [Google Scholar]
- 33.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol. 2009;54:2187–2196. doi: 10.1016/j.jacc.2009.04.100. [DOI] [PubMed] [Google Scholar]
- 34.Mak S, Lehotay DC, Yazdanpanah M, Azevedo ER, Liu PP, Newton GE. Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J Card Fail. 2000;6:108–114. [PubMed] [Google Scholar]
- 35.Ma H, Yu L, Byra EA, et al. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. J Mol Cell Cardiol. 2010;49:322–329. doi: 10.1016/j.yjmcc.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]