

Abstract
The organizing power of energy flow is hypothesized to be the origin of biological complexity and its decline the basis of “complex” diseases and aging. Energy flow through organic systems creates nucleic acids, which store information, and the annual accumulation of information generates today's complexity. Energy flow through our bodies is mediated by the mitochondria, symbiotic bacteria whose genomes encompass the mitochondrial DNA (mtDNA) and more than 1000 nuclear genes. Inherited and/or epigenomic variation of the mitochondrial genome determines our initial energetic capacity, but the age-related accumulation of somatic cell mtDNA mutations further erodes energy flow, leading to disease. This bioenergetic perspective on disease provides a unifying pathophysiological and genetic mechanism for neuropsychiatric diseases such as Alzheimer and Parkinson Disease, metabolic diseases such as diabetes and obesity, autoimmune diseases, aging, and cancer.
It was assumed that if we sequenced the human genome, defined all of the common single-nucleotide polymorphisms (SNPs) and their haplotype associations, and performed genome-wide association studies (GWAS) between the SNPs and the various “complex” diseases, we would identify the responsible genes, understand the causes, and find the cures. However, after enormous effort, we have yet to explain more than 20% of common disease risk.
Thomas Kuhn, in his book The Structure of Scientific Revolutions (Kuhn 1996), argued that when a scientific endeavor invests increasing effort in a problem with diminishing returns, the difficulty may lie with the core assumptions (paradigms) on which the work was based. Assuming that this argument is valid in the current circumstance, what are the basic assumptions upon which Western biomedical science and these efforts were based? There are three: that evolution occurs through natural selection of variant forms, that organ-specific symptoms are the result of organ-specific defects, and that genes are located on the chromosomal DNA and inherited according to the laws of Mendel. All three of these paradigms are rooted in observations on the structure of organisms, thus generating an anatomical perspective on biology and medicine.
However, recent discoveries on the role of bioenergetics in human evolution and disease suggest that these classical paradigms are incomplete. Life requires not only anatomy but also the vital force that animates anatomy. Therefore, life is the interplay among structure (anatomy), energy (the vital force), and information to encode the structural and energetic systems.
Anatomical observations (i.e., the beaks of Darwin's finches) were the basis on which the theory of evolution by natural selection was derived. Whereas this can explain how different forms can exploit adjacent niches, it cannot explain the evolution of complex forms. The Second Law of Thermodynamics requires that complex forms decay because entropy increases. Hence, simple forms—viruses and bacteria—should destroy complex forms: humans. Yet humans exist and persist.
We persist because we counter entropy by maintaining a constant flow of energy through our bodies. That is, life is in a state of nonequilibrium thermodynamics, not equilibrium thermodynamics as required by the Second Law (Morowitz 1968).
Energy flow has the inherent property of generating structure in matter, for example the formation of convection currents in heated liquids. For terrestrial life, high-energy photons emanating from the sun are absorbed by the biosphere, the energy is used to build and maintain structures, and the spent energy is radiated out into space as near-infrared photons. For humans, we eat food that contains reducing equivalents, breathe oxygen, and react to the reducing equivalents and the oxygen within our mitochondria to generate the energy necessary to build, maintain, and animate our structures.
Whereas energy flow explains the maintenance of complexity, it does not explain the origin of complexity. For this, we need a new biological principle. This can be derived from the inherent feature of carbon-based molecules to form nucleic acids when they are exposed to energy flow (Ricardo and Szostak 2009). Nucleic acids, in turn, are digital systems that can encode information. Hence, each year energy flow through the biosphere creates more nucleic acids that add more information to the system. The more information, the greater the complexity that can be encoded. Therefore, after 3.5 billion years of energy flow, sufficient information has accumulated to encode the complex structures of higher organisms and the defense systems they use to combat entropy. Natural selection's function, then, is to compare similar systems and, through competition, choose the most energy efficient of the alternatives. Therefore, in biology the sum of energy flow is proportional to the sum of organismal information, which adds up to complexity (Wallace 2010).
The anatomical perspective also dominates Western medical thought and hasdone sosince the seminal description of human anatomy by Vesalius 450 years ago. Following Vesalius's example, subsequent physicians and medical investigators have specialized in subregions of the human body, leading to today's organ-based clinical subspecialists: neurologists, ophthalmologists, cardiologists, nephrologists, and so on. This organ specialization has led to an unstated corollary that organ-specific symptoms are the result of organ-specific problems. However, in the past 20 years, we have observed many patients with organ-based symptoms who have been found to have systemic mitochondrial defects. Therefore, systemic energetic defects can also cause organ-specific symptoms (Wallace 2005, 2007).
The anatomical perspective also solidified our confidence in the predominance of Mendel's laws of inheritance. Mendel observed that certain traits showed a deterministic pattern of transmission correctly interpreted as individuals having two copies of each hereditary factor, one contributed from each parent in their sex cells. Early 20th-century Drosophila geneticists studying the inheritance of fly morphological traits realized that Mendel's laws coincided with the inheritance patterns they observed. Subsequently, the chromosomes were also shown to follow the same hereditary pattern. These observations led to a powerful synthesis. Morphological traits are encoded by genes that are located on chromosomes that are inherited according to the laws of Mendel. Because all of the anatomical genes are encoded by the chromosomal genes, this paradigm was completely internally consistent.
However, deterministic genetics became increasingly problematic when it was applied to humans. Many familial traits do not exactly conform to Mendel's laws. Deviations were then given terms such as “variable penetrance and expressivity” and “delayed-onset-and-progressive-course,” which had no mechanistic explanation. The problems became unresolvable when the prevailing paradigms were applied to the causes of the age-related maladies such as neuropsychiatric disorders (Alzheimer disease [AD] and Parkinson disease [PD]), metabolic disorders (Type II diabetes and obesity), and autoimmune diseases (multiple sclerosis and Type I diabetes). To cope with the inadequacy of Mendel's laws for these diseases, they were declared to be “complex,” the result of the interaction of multiple genes and a variable environment. However, the number of genes was not defined and the nature of the “environment” was not specified, making this hypothesis impossible to disprove.
Our perception of complex diseases began to change when it was discovered that there are genes that are not inherited according to Mendel's laws. These non-Mendelian genes are encoded by the mitochondrial DNA (mtDNA), and the mtDNA encodes the core genes for bioenergetics. Moreover, non-DNA sequence changes were found to be maintained during cell division and even transmitted across generations, leading to the concept that the epigenome contributed to complex diseases. This concept came together with bioenergetics when it was realized that all reactions that changed the epigenome were mediated by energy-rich mitochondrial intermediates (Wallace 2005, 2007; Wallace and Fan 2010; Wallace et al. 2010).
Consequently, it is now clear that the anatomical perspective of biomedical science has a complement, the bio-energetic perspective of biology and medicine (Fig. 1). In the realm of evolutionary theory, the origin of species by natural selection is complemented by the origin of complexity through the information-generating capacity of energy flow. In medicine, the concept that organ-specific defects cause organ-specific symptoms is complemented by the concept that systemic energy defects can also result in organ-specific symptoms. Finally, in the domain of genetics, the deterministic laws of Mendelian anatomical genetics are complemented by the stochastic rules of mtDNA bioenergetic genetics and epigenomics modulated by energy flow.
Figure 1.

The anatomical and bioenergetic complementarities in evolution, medicine, and genetics. Comparison of the three pairs of complementary paradigms derived from the anatomical perspective and the bioenergetic perspective.
A BIOENERGETIC PARADIGM FOR COMPLEX DISEASES, AGING, AND CANCER
Once we realize that the central principle of biology is energy flow and its capacity to create information, we realize that the “complex” diseases could be reinterpreted within a single energy-based pathophysiological and genetic system (Fig. 2). For higher animals, the mitochondrion lies at the nexus of the environment and the body. The mitochondria process the available calories from the environment and convert them into appropriate forms of energy for use by our cells and tissues. This is accomplished by the central mitochondrial energy transformation system oxidative phosphorylation (OXPHOS). Mitochondrial OXPHOS powers a wide range of energy-based cellular functions, including maintaining the oxidation–reduction balance of the cell, generating a capacitance across the mitochondrial inner membrane, converting the potential energy of the capacitance into chemical energy in the form of ATP, using the mitochondrial capacity to import calcium to modulate cellular and mitochondrial reactions, producing reactive oxygen species (ROS) as signal transduction molecules, and initiating cell death by activation of the mitochondrial permeability transition pore (mtPTP) to destroy those cells with defective mitochondrial energy production.
Figure 2.
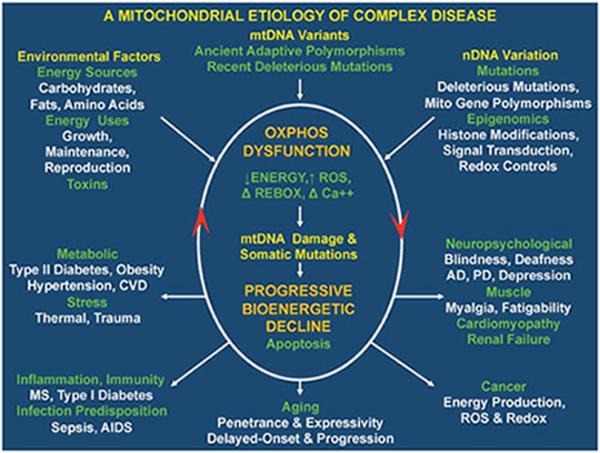
Integrated mitochondrial paradigm to explain the genetic and phenotypic complexities of the “complex” human diseases. An integrated model for the genetics and pathophysiology of complex diseases, aging, and cancer. The top of the figure indicates the three types of variation that impact individual mitochondrial oxidative phosphorylation (OXPHOS) robustness and hence risk for developing disease symptoms. These include nuclear DNA (nDNA) variation encompassing DNA sequence changes and epigenomic modification of gene regulation and signal transduction pathways, mitochondrial DNA (mtDNA) variation including recent deleterious mutations and ancient adaptive polymorphisms, and environmental influences encompassing the availability and demand for calories and inhibition of mitochondrial function by environmental insults. The central oval encompasses the pathophysiological basis of disease processes and the basis of disease progression. The primary defect is reduction in the energy transformation capacity of OXPHOS. This can result in reduced energy output, increased reactive oxygen species (ROS) production, altered redox status, and altered calcium homeostasis. The decline in OXPHOS efficiency can in turn perturb mitochondrial biogenesis, increase ROS production, impair mitophagy, and so on, resulting in the progressive increase in mtDNA damage and somatic mutations and further decline in mitochondrial function. Once mitochondrial function falls below the bioenergetic threshold of a tissue, symptoms ensue. Continued energetic failure can initiate cell destruction by apoptosis or necrosis. The lower five derived disease categories summarize the phenotypic outcomes of perturbed mitochondrial energy transformation. The bottom arrow shows the effect of the stochastic accumulation of somatic mtDNA mutations, resulting in delay-onset and progressive course of diseases and aging. The right arrow indicates clinical problems that can result from reduced energy production in the most energetic tissues, the brain, heart, muscle, and kidney. The number and severity of symptoms in these organs reflect the degree and specific nature of the mitochondrial defect. The arrow to the left indicates the metabolic effects of mitochondrial dysfunction that result in the perturbation of the body's energy balance. This results in the symptoms of the metabolic syndrome. The arrow toward the lower right corner indicates mitochondrial alterations that are critical for cancer initiation, promotion, and metastasis. The arrow to the lower left corner outlines the hypothesized inflammatory and autoimmune responses that may result from the chronic introduced into the bloodstream of the mitochondria's bacteria-like DNA and N-formyl methionine proteins.
The mitochondrial genome, which is the sum of all of the genes necessary for producing the structure and function of the mitochondrion, is encoded by two different genetic systems. The core circuit genes for mitochondrial energy generation are encoded by the maternally inherited, high-copy-number mtDNA, whereas all of the genes for mitochondrial structure, intermediate metabolism, and biogenesis are encoded by nuclear DNA (nDNA). The mtDNA has a very high sequence evolution rate. In the short term, this results in the generation of numerous deleterious mutations that have been found to cause all of the clinical problems associated with the “complex” diseases. In the long term, a subset of these mutations has proven to be advantageous in certain regional environments and these have been enriched to high frequencies by adaptive selection. Although they are optimal for their adaptive local environment, these same variants can be maladaptive when exposed to a different energetic environment.
The Mendelian component of the mitochondrial genome encompasses well over 1000 genes dispersed across the chromosomes. Although the sequence evolution rate of the nDNA genes is very low, deleterious mutations do arise that can affect the mitochondrion. Polymorphisms also occur in energy-regulating genes, but these are highly region-specific and thus are averaged out in large-scale GWAS.
Because the nDNA-encoded mitochondrial proteins must interact with the mtDNA-encoded proteins, the two genetic systems must be coordinately regulated. Indeed, the entire growth and reproduction system of the cell must be coordinated with the availability of the energy to fuel those processes. These systemic regulatory processes are coordinated via mitochondrially generated high-energy and redox intermediates. Therefore, the entire epigenome and all of the cellular signal transduction pathways are interfaced with mitochondrial energy flux.
Finally, the environment directly impinges on OXPHOS. The environment determines the amounts and types of calories available to the animal and imposes demands on the individual's energetic capacity to survive and reproduce. In our industrial society, an increasing number of chemicals are being added to our environment and food supply that are inhibitors of mitochondrial OXPHOS. These are eroding our cellular energetic capacity and increasing our predilection for chronic diseases.
Our mitochondria and mtDNAs replicate and are turned over by mitophagy throughout our lives. This means that mtDNA mutations accumulate in our somatic tissues as we age. This creates an aging clock that progressively erodes our energetic capacity until there is insufficient energy flow for optimal tissue function, at which point symptoms ensue.
Each person is born with a mitochondrial genotype with different degrees of robustness. Over their lifetime, they accumulate somatic mtDNA mutations that erode their energy reserves until the energy-generating capacity of their mitochondria falls below the minimum necessary for optimal function of one of the high-energy-demand organs. At this point, symptoms appear. As the severity of the energy defect increases, more systems become affected until death ensues.
Every tissue and organ in the body requires mitochondrial energy, but certain tissues have an especially high energy demand. These include the central nervous system, heart, muscle, kidney, and endocrine systems. As mitochondrial energy output declines, the most energetic organs are affected first, analogous to the sequential failure of electrical systems during a metropolitan brown-out. Hence, progressive mitochondrial decline generates clinical symptoms in the same organs that are affected by aging and the age-related diseases.
Because the mitochondria lie at the interface between the availability and use of environmental calories, mitochondrial dysfunction directly affects energy balance. Hence, mitochondrial defects are central to metabolic diseases such as diabetes, obesity, hypertension, and cardiovascular disease.
The mitochondria are symbiotic bacteria and mitochondrial biogenesis is bacteria-like. Hence, within each of our cells are hundreds of bacteria. In fact there are on the order of 1017 mitochondrial bacteria within our cells but only about 1014 bacteria within our gut. If the mitochondrial bacteria are released into our bloodstream, they will initiate an immune response, which suggests that one of the main reasons for having apoptosis is to degrade the intracellular bacteria before they escape from the cell. Unfortunately, apoptosis is an energy-driven process. Hence, chronic energy deficiency can impede apoptosis and increase the rate of cellular necrosis, facilitating mitochondrial release. This may provide the priming events for developing autoimmunity.
Lastly, whereas the cancer cell has escaped tissue and developmental regulation, it has not escaped the constraints of energy flow. Therefore, the successful cancer cell must develop strategies for adjusting its energy metabolism to accommodate its varying environmental conditions. This is accomplished by developing the capacity to switch between glycolytic and oxidative metabolism, use mitochondrial ROS to drive mitosis, and redirect energy resources into cellular biogenesis and reproduction.
BIOENERGETIC NEXUS BETWEEN ENVIRONMENT AND CELL
The origin of bioenergetics probably occurred when a spherical membrane trapped a charge differential on its two sides. This type of biological capacitor remains the source of the “vital force” in our cells today.
From its humble origins as free-floating nucleic acids, information growth produced prokaryotic cells with a variety of energetic systems. About 2 billion years ago, two prokaryotic organisms formed an association; one was an oxidative α-protobacterium and the other was probably a motile glycolytic cell. Ultimately, the motile cell engulfed the oxidative cell, creating the eukaryotic cell encompassing a nucleus–cytosol organism and a mitochondrial organism. The stage was then set for the evolution of multicellular plants and animals. During the next ~1.2 billion years, the two organisms within the eukaryotic cell underwent extensive metabolic consolidation and genetic exchange. The ultimate result was that the nucleus–cytosol became specialized in encoding and elaborating structural forms, whereas the mitochondrion became specialized in generating energy. Ultimately, the nucleus incorporated all of the structure and biogenesis genes of the mitochondrion and the mtDNA was reduced to encoding only the key elements of the circuit diagram for the mitochondrial power plants (Wallace 2007). As anatomical structures became more complex, more genes were added to the nucleus. However, it takes about one bacterium's worth of energy-generating capacity to sustain 2000–3000 genes. Hence, as the number of nuclear genes increased, the number of mitochondria also had to increase (Lane and Martin 2010), resulting in the human cell today, which contains about 20,000+ nDNA genes plus the hundreds of mitochondria and thousands of mtDNAs needed to sustain them.
Not long after achieving an optimal specialization and arrangement of the nDNA and mtDNA genes, one line of single-cellular eukaryotic organisms acquired a second symbiotic organism, a cyanobacterium. This bacterium ultimately became the chloroplast of plants.
The bacterial symbiotic events that generated the mitochondria and chloroplasts completed the energy system of higher plants and animals. The cyanobacterial chloroplasts absorbed the sun's photons, used the energy to split H2O into hydrogen and oxygen, released the oxygen (O2) into the atmosphere, and fixed the hydrogen to the carbon of CO2 to generate the six-carbon molecule, glucose, stored in starch. Animals eat the plants to collect the glucose, which is transported to the cells where it is split by glycolysis into two three-carbon pyruvates. The pyruvates enter the mitochondrion where they are converted by pyruvate dehydrogenase into NADH + H+ and acetyl-coenzyme A (CoA). Acetyl-CoA, NADH, and FADH2 are also generated in the mitochondrion by fatty acid β-oxidation. Acetyl-CoA feeds the mitochondrial tricarboylic acid cycle (TCA), the purpose of which is to remove the hydrogens from the hydrocarbons and transfer them onto the carrier NAD+ to generate NADH + H+. The hydrogens from NADH + H+ are then reacted with oxygen in a series of steps within the mitochondrion known as the electron transport chain (ETC) and the energy released is used to pump protons from the inside of the mitochondrial matrix, out through the mitochondrial inner membrane, into the intermembrane space. The resulting capacitor is approximately −180 mV and is used as a source of energy to drive a broad spectrum of processes, including conversion of electrochemical energy into chemical energy in the form of ATP. This integrated system for energy transformation is known as OXPHOS and the mitochondrial inner membrane potential is the “vital force” that the ancient Greeks recognized was present when a person was alive and breathing and lost when the person died and breathing stopped.
Because energy flow in the terrestrial environment is variable, multicellular animals have developed the capacity to adjust to the changing environments. During the plant growing season, glucose is abundant. Consequently, animals rely more on glycolysis and less on OXPHOS to store the excess calories as fat. In the nongrowing season, the animals mobilize the stored fat and up-regulate OXPHOS to burn the fat because it can only be oxidized by mitochondrial β-oxidation. These transitions in calorie availability are monitored by pancreatic islets, which signal the changes in available calories through insulin and glucagon. During the growing season, when blood sugars are high, pancreatic β cells secrete insulin, which signals the cells to reduce OXPHOS and shift energy production toward glycolysis. The insulin signal acts through the insulin receptor to activate Akt to phosphorylate the FOXO transcription factor, thus barring FOXO from the nucleus. This minimizes the transcription of peroxisome-proliferating-activated receptor γ (PPARγ) coactivator 1 (PGC-1α), which coordinately stimulates the transcription of an array of mitochondrial genes. FOXO can also be acetylated, which excludes it from the nucleus, and deacetylated by Sirt1 using NAD+ as a coreactant. Under highly glycolytic conditions, NAD+ is likely to be reduced to NADH and inaccessible to Sirt1, favoring inactivation of FOXO. During the non-growing season, when carbohydrates are limiting, the pancreatic islet α cells secrete glucagon. Glucagon binds to its receptors on the body's tissues, activating adenylcyclase to produce cAMP. cAMP activates protein kinase A to phosphorylate CREB, and phosphoCREB enters the nucleus and activates the transcription of PGC-1α. This up-regulates OXPHOS to burn fats to sustain ATP production during periods of privation.
The body is thus organized according to an energy anatomy including energy-utilizing tissues, the brain, heart, muscle, kidney, etc.; energy-storing tissues including white adipose tissue to store fat to generate ATP and brown adipose tissue to store fat to generate heat; an energy homeostasis tissue, the liver, that modulates blood glucose levels because glucose is our connection with plant energy fixation; and an energy-sensing tissue, the pancreatic islets, which senses glucose levels and modulates tissue energy metabolism accordingly. Thus, the primary factor in our “environment” is the availability of calories and the demands made on our caloric system for survival and reproduction (Wallace 2007).
The generation and utilization of the mitochondrial electrochemical gradient is a biophysical process involving the linked flow of both negative and positive charges (Fig. 3). The electrons borne by NADH H + H+ are transferred to mitochondrial inner membrane protein complex I or NADH dehydrogenase. Complex I oxidizes the NADH + H+ and reduces the lipid-soluble electron carrier coenzyme Q10 (CoQ). CoQ or ubiquinone incorporates two electrons successively, the first electron generating ubisemiquinone and the second ubiquinol. Electrons from other dehydrogenases are also transferred to CoQ. This includes complexII, succinate dehydrogenase, which collects electrons from the TCA cycle intermediate, succinate. From CoQ, the electrons are transferred to complex III (b–c1 complex), then to cytochrome c, then to complex IV (cytochrome c oxidase, COX), and finally to O2 to generate H2O. As the electrons traverse complexes I, III, and IV, the energy released is used to pump protons out across the mitochondrial inner membrane to create the electro-chemical gradient: ΔP = Δψ + ΔμH+ (Fig. 3). The accumulated power generated by 1000 mitochondria within each of 100 trillion cells or 1017 mitochondria provides the energy that powers our lives.
Figure 3.
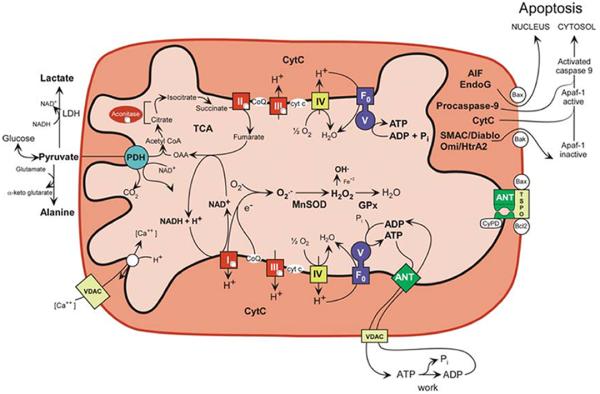
Mitochondrial energy production and its reaction to the pathophysiology of disease. Five features of mitochondrial metabolism are central to the pathophysiology of the common age-related diseases: (1) energy production by OXPHOS; (2) regulation of cellular oxidation–reduction (redox) state; (3) ROS generation as a by-product of OXPHOS; (4) buffering of the cytosolic and mitochondrial Ca2+ levels; and (5) regulation of apoptosis through activation of the mtPTP. ADP or ATP, adenosine di- or triphosphate, ANT, adenine nucleotide translocator; cytc, cytochrome c; GPx, glutathione peroxidase-1; LDH, lactate dehydrogenase; MnSOD, manganese superoxide dismutase or SOD2; NADH, reduced nicotinamide adenine dinucleotide; TCA, tricarboxylic acid cycle; VDAC, voltage-dependent anion channel; I, II, III, IV, and V, OXPHOS complexes I–V. Complex I is composed of 45 polypeptides, seven (ND1, 2, 3, 4L, 4, 5, 6) encoded by the mammalian mtDNA; complex II consists of four nDNA-encoded polypeptides; complex III consists of 11 polypeptides, one (cytb) encoded by the mtDNA; complex IV is composed of 13 polypeptides, three (COI, II, III) encoded by the mtDNA; and complex V is composed of ~15 polypeptides, two (ATP6, 8) encoded by the mtDNA. (Modified from Wallace 2005, 2007; Ruiz-Pesini et al. 2007.)
The mitochondrial capacitors provide energy to drive a wide range of biological processes, but to extend mitochondrial utility to fueling reactions and performing work, it needs to convert the capacitor's potential energy into the high-energy chemical intermediate ATP. This is accomplished by the H+-transporting ATP synthase, complex V, bound to the mitochondrial inner membrane and pointing into the matrix. Mitochondrial ATP is then exchanged by cytosolic ADP by the adenine nucleotide translocators (ANTs). Therefore, the proton-pumping complexes I, III, and IV of the ETC are coupled to ATP synthesis through the proton gradient. The efficiency by which complexes I, III, and IV pump protons out of the mitochondrion and by which the ATP synthase converts the proton gradient into ATP is known as the coupling efficiency. Because the calorie is a unit of heat, the more calories that are burned by the cell, the more heat that is generated, whether the energy goes through the high-energy intermediate ATP or not. As it turns out, people differ significantly in their coupling efficiencies. Therefore, those that are more tightly coupled generate the maximum ATP per calorie burned and the minimum heat, whereas individuals that are less tightly coupled have to burn more calories for the same amount of ATP and thus generate more internal heat. This provides one of the fundamental connections between our genes and our environment.
The biophysics of the linkage among calorie oxidation, the proton gradient, and ATP synthesis is now being elucidated by defining the composition and structure of the central OXPHOS complexes (Fig. 3). Complex I is one of the largest enzymes known, being composed of 45 polypeptides, seven (ND1, 2, 3, 4L, 4, 5, and 6) encoded by the mtDNA and the rest by the nDNA. For every two electrons that traverse complex I, up to four protons are pumped into the intermembrane spaces (Efremov et al. 2010; Brandt 2011; Efremov and Sazanov 2011). Complex II is composed of four polypeptides, all encoded by the nDNA, and does not pump protons. Complex III is composed of 11 polypeptides, cytochrome b being encoded by the mtDNA whereas the remainder of the polypeptides are encoded by the nDNA. The stoichometry of complex III is two protons pumped per two electrons that transit the complex (Iwata et al. 1998). Complex IV is composed of 13 polypeptides, three (COI, II, and III) encoded by the mtDNA (Tsukihara et al. 1996). This complex collects two pairs of electrons from two successive cytochrome cs. The four electrons reduce O2 to two H2Os coupled to the transport of four protons, giving a stoichometry of two protons pumped per two electrons oxidized. Complex V (ATP synthase) is the most dynamic of the OXPHOS complexes. It is composed of approximately 15 subunits, two (ATP6 and 8) encoded by the mtDNA, organized into a fixed element, the stator, and a rotating element, the rotor. The rotor is a wheel, with eight spokes that spins within the plan of the inner membrane. Each spoke has a negatively charged glutamate that picks up one proton from the intermembrance space, rotates around the circumference of the wheel, and releases the proton into the matrix. As the wheel spins, it drives an axel that protrudes into a fixed barrel structure of three pairs of alternating α and β subunits. As the axel contacts the β subunits, it successively binds ADP and Pi, fuses them into ATP, and releases the generated ATP. Therefore, the stoichometry of ATP synthesis is 2.7 protons per one ATP (Saraste 1999; Watt et al. 2010).
As a byproduct of OXPHOS, the mitochondrion generates much of the cellular ROS, initiated by the transfer of a single electron from complex I or III to O2. The resulting superoxide anion generated by complex I in the matrix is converted to hydrogen peroxide (H2O2) by Mn superoxide dismutase (MnSOD), whereas the produced in the intermembrane space by complex III is converted to H2O2 by Cu/ZnSOD. Mitochondrial H2O2 can be reduced to water by glutathione peroxidases.
The mitochondrial membrane potential can be used to import Ca2+ into the mitochondrion (Baughman et al. 2011; De Stefani et al. 2011). Hence, the mitochondrion is a major buffering system for cellular Ca2+ (Fig. 3).
The mitochondrion also contains a self-destruct system, the mtPTP, that spans the mitochondrial inner and outer membranes. The mtPTP interacts with cyclophilin D and the proapoptotic and antiapoptotic proteins of the Bax and Bcl-2 family. The pore is activated by reduced mitochondrial membrane potential, increased matrix Ca2+, and oxidative stress. When the levels of the factors exceed the activation threshold, the pore opens, short-circuiting the membrane potential. This is coordinated with the opening of a megachannel in the mitochondrial outer membrane, releasing cytochrome c and other proapoptotic factors, which results in the digestive destruction of the cell containing the faulty mitochondria (Fig. 3).
The bacterial origin of the mitochondrion is still apparent in its biogenesis systems. The mitochondria synthesize the 13 OXPHOS polypeptides encoded by the mtDNA on bacteria-like, mitochondria-specific, chloramphenicol-sensitive ribosomes. The ribosomes are assembled from 12S and 16S ribosomal RNAs (rRNAs) transcribed from the mtDNA plus an array of polypeptides encoded by the nDNA, the polypeptides assembled using 22 mtDNA-encoded transfer RNAs (tRNAs) that initiate mitochondrial translation with a formylmethionine. The mitochondrial formylmethionine polypeptides plus the mtDNA can be recognized by the innate immune system as endogenous “damage”-associated molecular patterns (Zhang et al. 2010). Thus, release of mitochondria into the extracellular space can initiate the inflammatory response and prime the immune system to initiate an autoimmune response.
MITOCHONDRIAL GENETICS IS QUANTITATIVE AND COMPLEX
The mtDNA of the mitochondrial genome is a 16,569-nucleotide closed circular molecule that encodes the 13 OXPHOS polypeptides, the 12S and 16S rRNA, 22 tRNAs, and a 1121-nucleotide control region that contains the promoters and one of the two origins of DNA replication. The two mtDNA strands are transcribed symmetrically from three control region promoters: two for the G-rich heavy (H) strand that encodes the rRNA genes and 12 of the 13 polypeptides, and one for the light (L) strand that encodes a single polypeptide, the ND6 complex I gene. Most of the genes are punctuated by tRNAs and, as the polycistronic RNAs are transcribed, the tRNAs fold in the transcript and are cleaved out, generating the mature rRNAs and messenger RNAs (mRNAs), which are posttranscriptionally polyadenylated. The L-strand promoter has been proposed to form the primer for H-strand replication, which then proceeds two-thirds around the mtDNA to the L-strand origin, at which point replication proceeds back in the opposite direction (Coskun et al. 2004; Wallace 2005).
In addition to the mtDNA, the mitochondrial genome encompasses > 1000 nDNA-encoded mitochondrial genes (Pagliarini et al. 2008). These include the remaining structural subunits of the OXPHOS complexes, the structural polypeptides to assemble the mitochondrion, the mitochondrial intermediate metabolism proteins, and the proteins for mitochondrial biogenesis proteins, including the mitochondrial-specific DNA, RNA polymerases, and the ribosomal proteins.
All of the mtDNA-encoded polypeptides are core electrical components of complexes I, III, IV, and V. These complexes share a common substrate and product, the inner membrane capacitance. Therefore, it is essential that the proton permeability of all four complexes be balanced to avoid one complex from becoming leaky for protons and shorting the capacitor. Hence, all 13 mtDNA protons must coevolve. This is accomplished by the exclusive maternal inheritance of the mtDNA, which requires that each new mtDNA mutation be tested for its fitness against the background of that mtDNA's previous variants. Furthermore, uniparental inheritance blocks the mixing and recombination between mtDNAs from two different maternal lineages, thus avoiding the combining of incompatible circuit elements (Wallace 2007).
Mitochondria are continuously replicating and the excess mitochondria turned over by mitophagy within cells. The replication of the mtDNA involves the mitochondria-specific DNA polymerase γ (POLG), a helicase (Twinkle), single-stranded binding protein, and an array of other proteins that are assembled into the mitochondrial nucleoid. Nucleoids may contain one mtDNA molecular complexed with Tfam (Bogenhagen et al. 2008; Gilkerson 2009; Prachar 2010).
The mtDNA has a very high sequence evolution rate, in part because it is exposed to ROS. When a new mtDNA mutation arises, it creates an intracellular mixture of mutant and normal mtDNAs, a state known as heteroplasmy. During cell division, the mutant and normal mtDNAs are randomly transmitted to the daughter cells. As a result, the percentage of mutant and normal mtDNAs can drift during either mitosis or meiosis (replicative segregation). As the percentage of mutant mtDNAs increases, energy output declines until it falls below the minimum required for a particular tissue, the bioenergetic threshold, at which cellular dysfunction occurs and symptoms ensue. The milder the mtDNA mutation, the higher the percentage of mutant mtDNAs that must be present before the individual manifests symptoms, with the mildest mutants becoming pure mutant (homoplasmy). The most severe mtDNA mutations are eliminated before ovulation by an ovarian selection system, thus reducing the mutational genetic load and permitting the introduction of numerous mild mtDNA variants into the population to foster rapid adaptation to regional environmental differences (Fan et al. 2008; Stewart et al. 2008).
Mitochondria within the cell are dynamic organisms, constantly undergoing fission and fusion. Fission is mediated by the Drp1 and hFis1 proteins and fusion by the Opa1 and Mfn1 and Mfn2 proteins. Fusion permits the complementation of defects between different mitochondria and mtDNAs (Schon and Gilkerson 2010). Fission is essential to separate mitochondria for transport throughout large cells such as neurons (Chen and Chan 2005, 2009; Chen et al. 2010). Fission is also modulated by the Bax–Bcl-2 family of proteins (Graef and Nunnari 2011a,b) and permits isolation of mutant nucleoids in mitochondrial vesicles where the mtDNA can be expressed and, if defective, result in a reduced membrane potential, signaling removal by mitophagy.
Mitophagy is initiated by the mitochondrial membrane protein PINK1, which is degraded in normal mitochondria but stabilized in the mitochondrial outer membrane in mitochondria with lowered membrane potential (Narendra et al. 2010). PINK1 phosphorylates the cytosolic protein parkin, attaching it to the mitochondrion (Narendra et al. 2008), where it ubiquinates mitofusins 1 and 2 (Gegg and Schapira 2011) and activates a mitochondrial ubiquitin–proteosome system (Chan et al. 2011). The ubiquinated mitofusins attract the adaptor protein p62/SQSTM1 (Ziviani and Whitworth 2010) that recruits the mitophagy apparatus including the microtubule-associated protein 1 light chain 3 (LC3) protein and Atg autophagy proteins to engulf the mitochondria in an autophagosome, preparatory to degradation (Burbulla et al. 2010; Guo 2010; Zhu and Chu 2010; Graef and Nunnari 2011a,b).
INHERITED MITOCHONDRIAL VARIATION
The discovery and analysis of diseases resulting from mutations in the mitochondrial genome have shown that bioenergetic defects result in the same clinical phenotypes as seen in the common complex diseases. Moreover, mutations in different mitochondrial genes as different as mtDNA encoded versus nDNA encoded can give the same phenotype.
Clinically relevant inherited mtDNA mutations fall into two major categories: recent pathogenic mutants and ancient adaptive polymorphisms. Additional clinical mutations occur in nDNA-encoded mitochondrial genes.
Recent Pathogenic mtDNA Mutations
Pathogenic mtDNA mutations can alter either the rRNA or tRNA mitochondrial protein synthesis genes or the OXPHOS polypeptide genes. Severe mutations can cause clinical phenotypes when they are heteroplasmic, whereas milder mutations cause symptoms when they are homoplasmic.
Pathogenic mitochondrial protein synthesis mutations are generally heteroplasmic and variation in the percentage of heteroplasmy can result in widely different clinical phenotypes. One common mtDNA protein synthesis mutation in the tRNALys gene at nucleotide A8344G causes myoclonic epilepsy and ragged red muscle fibers (MERRF) when present at a high percentage of heteroplasmy and sensory neural hearing loss when at lower heteroplasmy levels (Wallace et al. 1988b; Shoffner et al. 1990). The common tRNALeu(UUR) gene mutation at nucleotide A3243G (Goto et al. 1990) causes severe mitochondrial encephalomyelopathy, lactic acidosis, and stroke-like episodes (MELAS syndrome) at high heteroplasmy levels; mitochondrial myopathy with ragged red muscle fibers (RRFs); and ophthalmoplegia, ptosis, and cardiomyopathy at intermediate heteroplasmy levels, but it accounts for between 0.5% and 1.0% of all Type I and Type II diabetes throughout Euroasia at low levels of heteroplasmy (10%–30%) (van den Ouweland et al. 1992; Wallace 2005).
Pathogenic mtDNA missense mutations can be either heteroplasmic or homoplasmic depending on the severity of the biochemical defect associated with the mutation. In Europeans, the sudden-onset blindness, Leber hereditary optic neuropathy (LHON), can result from milder homoplasmic missense mutations such as the ND4 gene at nucleotide G11778A (R340H; Wallace et al. 1988a), ND1 nucleotide G3460A (A52T; Huoponen et al. 1991), and ND6 nucleotide T14484C (M64V; Johns et al. 1992) mutations. All three of these mutations predominantly affect males more than females and have milder complex I defects (Carelli et al. 1997, 1999; Brown et al. 2000, 2001; Baracca et al. 2005). A more severe missense mutation in the complex I ND6 gene at nucleotide G14459A (A72V) causes LHON when heteroplasmic and the movement disorder generalized dystonia when approaching homoplasmy (Jun et al. 1994, 1996).
Direct proof that mtDNA missense mutations are sufficient in themselves to cause degenerative disease has come from the introduction of an mtDNA COI gene missense mutation at nucleotide T6589C (V421A) into the female mouse germline. This resulted in a 50% reduction in complex IV activity in association with mitochondrial myopathy and cardiomyopathy (Fan et al. 2008).
mtDNA “Adaptive” Polypeptide Variants
Another factor that modulates the severity of mtDNA gene mutations is the mtDNA background. Soon after the discovery of the ND4 G11778A mtDNA mutation, a number of other mtDNA variants were found to be at increased frequency in ND4 G11778A, ND1 G3460A, and ND6 14484C LHON patients. A noteworthy example is the cytochrome b variant at nucleotide G15259A (D171N), which alters the Qo binding site of complex III and changes an amino acid that is conserved down to bacteria (Brown et al. 1995). It was subsequently discovered that the cytochrome b G15259A variant was one of several variants associated with European mtDNA lineage “J.”
Because mtDNA mutations accumulate sequentially along radiating maternal lines, nodal mtDNAs give rise to groups of descendant mtDNA haplotypes called haplogroups. By reconstructing the mutational history of the world's mtDNAs from indigenous peoples, we have been able to reconstruct the origin and ancient migrations of women (Denaro et al. 1981; Johnson et al. 1983). The African mtDNA tree coalesces back to a founding mtDNA at between 150,000 and 200,000 years before the present (Cann et al. 1987; Merriwether et al. 1991), which radiated in that continent to yield an array of haplogroups that constitute macrohaplogroup L (Chen et al. 1995). Out of all of the African mtDNAs, only two mtDNA types that arose in Ethiopia left Africa to found all of the mtDNAs of modern Eurasian and Native American populations, creating macrohaplogroups M and N (Wallace 1994; Wallace et al. 1999; Schurr et al. 2005). Macrohaplogroup N radiated into Europe, giving rise to eight haplogroups H, I, J, T, U and Uk, V, W, and X, whereas M and N radiated into Asia, giving rise to a plethora of mtDNA lineages. Of these haplogroups, A and B from N, and C and D from M from Asia, together with haplogroup X gave rise to all extant Native American mtDNAs (Wallace et al. 1999).
Frequently, regional mtDNA haplogroups were discovered to be founded by functionally significant amino acid substitution variants. Macrohaplogroup N, for example, was initiated by amino acid substitutions, an ND3 at nucleotide 10398 (A114T) and an ATP6 at nucleotide 8701 (A59T). This led to the hypothesis that mtDNA mutations that changed cellular energetics may have been adaptive in particular local energetic environments and enriched by adaptive selection (Wallace 1994; Mishmar et al. 2003; Wallace et al. 2003; Ruiz-Pesini et al. 2004; Ruiz-Pesini and Wallace 2006). Functional and biochemical analysis has confirmed that different haplogroups can be energetically different (Ruiz-Pesini et al. 2000; Kazuno et al. 2006; Montiel-Sosa et al. 2006; Gomez-Duran et al. 2010).
Association studies using mtDNA haplogroups have subsequently revealed a wide range of clinical associations. Haplogroup J was found to increase the penetrance of the milder LHON mutations (G11778A, G3460A, and T14484C) (Brown et al. 1995, 1997, 2002; Torroni et al. 1997). Other studies have found significant associations with neuropsychiatric disorders, including depression, AD, PD, macular degeneration, and stroke; metabolic diseases, including Type II diabetes, cardiovascular disease, and metabolic syndrome; immunological diseases, including sepsis, changes in IgE levels, asthma, osteoarthritis, AIDS progression, anti-AID HAART-induced lipodystrophy; various forms of cancer; longevity; and athletic performance (Wallace 2008; Gomez-Duran et al. 2010). Some mtDNA haplogroup associations have very high odds ratios. For example, mtDNA haplogroup U5a was found in one study to correlate perfectly with age-related macular degeneration (Udar et al. 2009).
Variation in nDNA-Encoded Mitochondrial Genes
Mutations in the nDNA-encoded mitochondrial genes can also cause a broad spectrum of neurological diseases in children and adults. These can include mutations in the structural genes of OXPHOS and in the nDNA genes for mitochondrial biogenesis such as ribosomal subunits (Smeitink et al. 2006; Wallace et al. 2007, 2010; Antonicka et al. 2010). Mutations in the nDNA-encoded genes for mtDNA replication can destabilize mtDNA, resulting in mtDNA multiple deletions and/or depletion (Zeviani et al. 1990; Nishino et al. 1999; Mandel et al. 2001; Tyynismaa et al. 2005; Copeland 2010; Fratter et al. 2010).
Clinically relevant regional polymorphisms have also been observed in nDNA-encoded mitochondrial genes that predispose to common clinical problems. A Pro 121 Ala polymorphism in the PPARα has been associated with Type II diabetes (Altshuler et al. 2000), and a Gly482Ser polymorphism in the PGC-1α gene has been associated with Type II diabetes in Danish populations (Ek et al. 2001) and in Pima Indians (Muller et al. 2003). Polymorphisms in mitochondrial uncoupling proteins (UCP1, 2, 3) have also been correlated with diabetes and obesity in a number of regional studies (Jun et al. 2009; Jia et al. 2010; Srivastava et al. 2010; Dalgaard 2011).
Finally, the expression of nDNA genes is regulated via mitochondrially generated high-energy intermediates through modification of the histones. Presumably, in the early single-cell eukaryote, growth and reproduction had to be directly linked to the availability of calories. Therefore, the structure of the chromatin had to be keyed to energy flow. This was accomplished by packaging the DNA in nucleosomes composed of histones with positively charged amino acid tails. When unmodified, the positive tails bind electrostatically to the negatively charged DNA, shutting down transcription. However, when calories become available, they are rapidly converted into ATP, acetyl-CoA, and S-adenosylmethionine by glycolysis and the mitochondrion. These substrates then permit the modification of histone trails, neutralizing the negative charges. This opens the chromatin, permitting transcription, cell growth, and cellular replication. These high-energy intermediates also contribute to the modification of proteins in the signal transduction pathways, permitting modulation of metabolism based on energy flow (Wallace and Fan 2010). The availability of reducing equivalents from NADH/NAD+ and NADPH/NADP+ also modulated the cellular redox state, which controlled the equilibrium for thiol-disulfide systems, both soluble within the cell (reduced versus oxidized glutathione) and incorporated into enzymes and transcriptions factors, thus further coupling the metabolic and transcriptional control to the cell's energetic environment (Wallace and Fan 2010; Wallace et al. 2010).
SOMATIC mtDNA MUTATIONS AND THE AGING CLOCK
The continuous turnover of mtDNAs in our cells and tissues means that our mtDNAs replicate throughout our lives and thus are prone to accumulating de novo mutations. Both deletions and base substitution mutations have been found to accumulate with age. These mutations can occur either early in development and become widely disseminated throughout our tissues or later in development and become confined to one or a few tissues. In the former case, a single mtDNA mutation will come to predominate in all tissues, whereas in the latter case each tissue will have a different array of mutations. Deletions that arise in oocytes or very early in development may be origin deletion syndromes such as chronic progressive ophthalmoplegia, Kearns–Sayre syndrome, or Pearson marrow pancreas syndrome (Holt et al. 1988; Poulton et al. 1995; Wallace et al. 2007), the pathology of which has been confirmed by the introduction of deleted mtDNA into the female mouse germline (Inoue et al. 2000).
Somatic deletion mutations accumulate throughout life but may accrue at different rates in different tissues. In the brain, for example, the highest deletion levels are found in the basal ganglion, followed by the cortex, and then the cerebellum (Corral-Debrinski et al. 1992; Cortopassi et al. 1992).
Base substitution mutations also accumulate with age. These mutations have been most thoroughly studied in the mtDNA control region, with specific mutations being found in particular tissues (Michikawa et al. 1999; Murdock et al. 2000; Wang et al. 2001; Zhang et al. 2003).
The accumulation of “somatic mtDNA mutations” is thought to erode cellular energetics, ultimately traversing bioenergetic thresholds and resulting in symptoms. This conclusion has been supported by the observation that mice harboring a mutant mitochondrial polymerase γ with a defective proofreading function have increased mtDNA somatic mutation rates and exhibit evidence of premature aging (Trifunovic et al. 2004; Kujoth et al. 2005), whereas transgenic mice in which the peroxisomal antioxidant enzyme catalase was introduced into the mitochondrial matrix have reduced mtDNA mutation rates and increased life span (Schriner et al. 2005).
MITOCHONDRIAL GENETICS OF AGE-RELATED DISEASES
From these observations, we envision that each individual inherits a mitochondrial genome that defines his initial bioenergetic state. However, as he ages, he accumulates somatic mtDNA mutations that progressively erode mitochondrial function until mitochondrial energy output falls below the expression threshold of one or more tissues, at which point age-related symptoms appear (Wallace 1992a,b; 2005). These principles now provide a framework for understanding diseases such as PD and AD.
PD is a late-life-onset movement disorder associated with basal ganglia dysfunction and the accumulation of Lewy bodies in affected dopaminergic neurons (Guzman et al. 2010). Mitochondrial dysfunction is demonstrated because PD can be induced in humans and rats by exposure to the complex I inhibitors MPTP (Langston et al. 1983) and rotenone (Betarbet et al. 2000; Cannon and Greenamyre 2010).
Subsets of PD patients are members of Mendelian pedigrees. A number of these genes have been cloned and their functions examined. Most have been found to affect mitochondrial biology. PARK1 (α-synuclein) is involved in mitochondrial membrane integrity (Kamp et al. 2010), PARK8 (LRRK2) deficiency results in reduced OXPHOS function (Greggio et al. 2011), PARK13 (Omi/HtrA2) is a mitochondrial apoptosis regulatory protein, PARK7 (DJ-1) affects mitochondria function and oxidative stress (Krebiehl et al. 2010; McCoy and Cookson 2011), and PARK2 (parkin) and PARK6 (PTEN-induced kinase 1, PINK1) regulate mitophagy in response to mitochondrial dysfunction and mtDNA heteroplasmy (Narendra et al. 2008; Burbulla et al. 2010; Guo 2010; Zhu and Chu 2010; Ziviani and Whitworth 2010; Gegg and Schapira 2011). The PINK and parkin association with PD and thus mitophagy dysfunction strongly implicates the accumulation of somatic mtDNA mutations in the genetics and pathophysiology of PD.
An mtDNA role in PD was also shown by our discovery that an mtDNA tRNAGln polymorphism at nucleotide A4336G found on haplogroup H5a is present in about 5% of PD patients and 7% of PD + AD patients versus 0.4% of controls (Shoffner et al. 1993). Subsequent studies have confirmed that haplogroup H increases the risk of PD and that haplogroups J, U, and Uk decrease the risk (Khusnutdinova et al. 2008).
Similarly, mtDNA deletions (Bender et al. 2006; Kraytsberg et al. 2006) and base substitution (Coskun et al. 2011) mutations have also been found to be elevated in PD brains. Furthermore, when mtDNAs from PD patients are transferred into cultured cell cybrids, mitochondrial defects have been observed (Borland et al. 2009; Keeney et al. 2009; Trimmer and Bennett 2009; Coskun et al. 2011).
AD is a progressive dementia associated with cortical atrophy, amyloid plaques, and neurofibrillary tangles. Most AD cases have a late-onset, spontaneous appearance, but 1% of cases have an early-onset course and Mendelian inheritance (Alzheimer's Association 2011). These early-onset cases have been linked to mutations in the amyloid precursor protein and presenilin 1 and 2 complex genes. These mutations increase the generation of amyloid peptides Aβ1 – 40 and Aβ1 – 42. Oligomeric aggregates of Aβ peptides have been shown to be the toxic form of Aβ (Deshpande et al. 2006; Glabe and Kayed 2006), and Aβ has been reported to inhibit complex IV, cyclophilin D, ATP synthase, Aβ alcohol dehydrogenase (ABAD), mitochondrial fission and fusion, and mitophagy (Coskun et al. 2011).
The risk for developing late-onset AD is increased by the inheritance of the ApoE ε4 allele and an allele of the mitochondrial outer membrane protein import gene TOMM40 (Potkin et al. 2009; Roses 2009). Several studies have indicated that the ApoE ε4 allele is toxic to mitochondria (Coskun et al. 2011).
We observed the tRNAGln at nucleotide A4336G in 3% of late-onset AD (Shoffner et al. 1993) and this association has been confirmed multiple times (Hutchin and Cortopassi 1995; Wallace 2005; Santoro et al. 2010). Other mtDNA haplogroups have also been reported to influence AD risk (Chagnon et al. 1999; Carrieri et al. 2001; van der Walt et al. 2004; Lakatos et al. 2010).
Finally, somatic mtDNA deletions (Corral-Debrinski et al. 1994) and mtDNA control region base substitution mutations (Coskun et al. 2003, 2010) are elevated in AD brains as well as in the brains of Down syndrome patients with dementia (DSAD) (Coskun et al. 2010). Increased mtDNA control region mutation levels are also seen in blood cell DNAs from AD patients and in lymphoblastoid cell lines from AD and DSAD patients. Hence, the increased mtDNA mutation levels are due to systemic defects. AD and DSAD brains with increased somatic mtDNA mutations also exhibit an approximately 50% reduction in mtDNA ND6 transcript levels and in mtDNA copy number (Coskun et al. 2003, 2010). Finally, in the triple transgenic mouse model of AD (3XTg-AD) (Oddo et al. 2003), females were found to have higher mitochondrial respiration and complex I activity than controls when young. These levels dropped below that of control mice at about the time when the levels of Aβ might result in aggregation (Coskun et al. 2010). This raises the intriguing possibility that Aβ is induced to protect brain mitochondria from functional decline. However, when mitochondrial function becomes too severe, Aβ is designed to oligomorize and destroy the defective synapses and neurons.
ENERGY AND THE ENVIRONMENT
The bioenergetic paradigm can now begin to define the important parameters of the environmental influence on human health and disease. Organisms within the biosphere interface with energy flux at three levels: (1) the source of and requirement for the energy available to a species within its niche, the energy reservoir; (2) the regional differences in energy requirements and availability to subpopulations within a species, the energy environment; and (3) the short-term fluctuations in energy availability and demands made on the individual during life by biological and environmental cycles, the energy fluctuations (Fig. 4).
Figure 4.
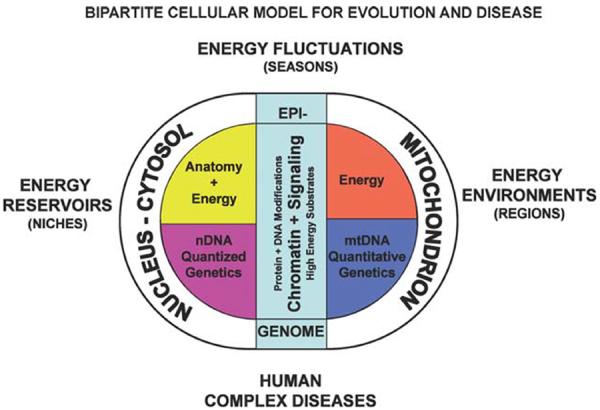
Three hypothesized levels of animal adaptation to energy resources and demands. The primary contributor to the biological environment is the flux of energy through the biosphere. The dichotomy between structure and energy in eukaryotes results from the symbiotic origin of the eukaryotic cell involving the proto-mitochondrion and the proto-nucleus–cytosol. The mitochondrion is specialized in energy production and retains the core genes for controlling energy production within the mtDNA. The nucleus–cytosol is specialized in encoding structure. Because growth and reproduction must be coordinated with the availability of energy, the status of the energetic flux through cellular bioenergetic systems, particularly the mitochondrion, is communicated to the nucleus–cytosol by alterations in nDNA chromatin, the epigenome, and cytosolic signal transduction systems, based on the production and availability of high-energy intermediates, reducing equivalents, and ROS produced primarily by the mitochondrion. As a consequence, biological systems interface with the energy environment at three levels: the species level, at which nDNA gene variation alters anatomical forms to exploit different environmental energy reservoirs; the species subpopulation level, at which primarily mtDNA bioenergetic genetic variation permits adaptation to long-term regional differences in the energetic environment of the species' niche; and the individual level, at which the epigenome and signal transduction pathways are modulated by the availability of high-energy intermediates in response to cyclic fluctuations in the individual's energy environment (Wallace 2009, 2010). (Reprinted, with permission, from Cold Spring Harbor Laboratory and Proceedings of the National Academy of Sciences.)
Adaptation to a new energy reservoir requires anatomical changes to exploit adjacent niches—Darwin's finches. This requires mutations in nDNA-encoded structural genes. However, the mutation rate of the nDNA is constrained because anatomical changes are only expressed in the adult organism. Because most mutations are deleterious, a high nDNA mutation rate would destroy many of the adults, imposing a severe genetic load on the species. Hence, the nDNA mutation rate must remain low, in the range of 10−6–10−7. Masked by heterozygosity, this will still permit the accumulation of sufficient variation to allow anatomical evolution within the time frame of the hundreds of thousands to millions of years required for speciation.
Adaptation to different energy environments encountered by regional populations of a species requires more rapid adjustments in a time frame of thousands of years. This rate of genetic change can only be achieved by the high-sequence evolution rate of the mtDNA. Whereas such a high mutation rate might be expected to cause severe genetic loads on the species, this problem has been resolved for the mtDNA by intraovarian selection against proto-oocytes that harbor highly deleterious mtDNA mutations. This is feasible because mitochondrial dysfunction can be selected at the cellular level and thus does not require the full life cycle of the individual to be manifested. Removal of the most deleterious mutations before fertilization then permits the mtDNA to have a high mutation rate and thus introduce into the species a wide range of modest bioenergetic alterations that provide the raw material for rapid genetic adaptation to novel regional energetic environments.
Finally, an individual's environment changes repeatedly throughout daily, monthly, yearly, and life cycles. These cyclic changes are reversible and thus cannot be mutational. Animals adjust to these energetic changes by modulating nuclear gene expression through modification of the epigenome and cellular signal transduction. These changes are mediated by mitochondrially regulated energetic intermediates, redox state, Ca2+, and ROS signaling.
Therefore, the search for common nDNA variants that predisposed to the common “complex” diseases may have been unproductive because most of the intraspecific genetic variation of relevance to the environment and to diseases is localized in the mtDNA and the epigenome, not in the nDNA.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants NS21328, AG24373, NS41850, AG13154, and DK73691.
REFERENCES
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. Thies W, Bleiler L. Alzheimer's Association report: 2011 Alzheimer's disease facts and figures. Alzheimer's Dement. 2011;7:208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Pedersen AM, Rodenburg RJ, van der Knaap MS, Smeitink JA, Chrzanowska-Lightowlers ZM, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracca A, Solaini G, Sgarbi G, Lenaz G, Baruzzi A, Schapira AH, Martinuzzi A, Carelli V. Severe impairment of complex I-driven adenosine triphosphate synthesis in Leber hereditary optic neuropathy cybrids. Arch Neurol. 2005;62:730–736. doi: 10.1001/archneur.62.5.730. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF, Rousseau D, Burke S. The layered structure of human mitochondrial DNA nucleoids. J Biol Chem. 2008;283:3665–3675. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]
- Borland MK, Mohanakumar KP, Rubinstein JD, Keeney PM, Xie J, Capaldi R, Dunham LD, Trimmer PA, Bennett JP., Jr Relationships among molecular genetic and respiratory properties of Parkinson's disease cybrid cells show similarities to Parkinson's brain tissues. Biochim Biophys Acta. 2009;1792:68–74. doi: 10.1016/j.bbadis.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt U. A two-state stabilization-change mechanism for proton-pumping complex I. Biochim Biophys Acta. 2011;1807:1364–1369. doi: 10.1016/j.bbabio.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat. 1995;6:311–325. doi: 10.1002/humu.1380060405. [DOI] [PubMed] [Google Scholar]
- Brown MD, Sun F, Wallace DC. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet. 1997;60:381–387. [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Trounce IA, Jun AS, Allen JC, Wallace DC. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's Hereditary Optic Neuropathy mtDNA mutation. J Biol Chem. 2000;275:39831–39836. doi: 10.1074/jbc.M006476200. [DOI] [PubMed] [Google Scholar]
- Brown MD, Allen JC, Van Stavern GP, Newman NJ, Wallace DC. Clinical, genetic, and biochemical characterization of a Leber Hereditary Optic Neuropathy family containing both the 11778 and 14484 primary mutations. Am J Med Genet. 2001;104:331–338. [PubMed] [Google Scholar]
- Brown MD, Starikovskaya E, Derbeneva O, Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI, Wallace DC. The role of mtDNA background in disease expression: A new primary LHON mutation associated with Western Eurasian haplogroup J. Hum Genet. 2002;110:130–138. doi: 10.1007/s00439-001-0660-8. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Krebiehl G, Kruger R. Balance is the challenge—The impact of mitochondrial dynamics in Parkinson's disease. Eur J Clin Invest. 2010;40:1048–1060. doi: 10.1111/j.1365-2362.2010.02354.x. [DOI] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson's disease recent advances. Prog Brain Res. 2010;184:17–33. doi: 10.1016/S0079-6123(10)84002-6. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ghelli A, Ratta M, Bacchilega E, Sangiorgi S, Mancini R, Leuzzi V, Cortelli P, Montagna P, Lugaresi E, et al. Leber's hereditary optic neuropathy: Biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology. 1997;48:1623–1632. doi: 10.1212/wnl.48.6.1623. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ghelli A, Bucchi L, Montagna P, De Negri A, Leuzzi V, Carducci C, Lenaz G, Lugaresi E, Degli Esposti M. Biochemical features of mtDNA 14484 (ND6/M64V) point mutation associated with Leber's hereditary optic neuropathy. Ann Neurol. 1999;45:320–328. [PubMed] [Google Scholar]
- Carrieri G, Bonafe M, De Luca M, Rose G, Varcasia O, Bruni A, Maletta R, Nacmias B, Sorbi S, Corsonello F, et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer's disease. Hum Genet. 2001;108:194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- Chagnon P, Gee M, Filion M, Robitaille Y, Belouchi M, Gauvreau D. Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder population. Am J Med Genet. 1999;85:20–30. doi: 10.1002/(sici)1096-8628(19990702)85:1<20::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14(suppl 2):R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YS, Torroni A, Excoffier L, Santachiara-Benerecetti AS, Wallace DC. Analysis of mtDNA variation in African populations reveals the most ancient of all human continent-specific haplogroups. Am J Hum Genet. 1995;57:133–149. [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WC. The mitochondrial DNA polymerase in health and disease. Subcell Biochem. 2010;50:211–222. doi: 10.1007/978-90-481-3471-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- Cortopassi GA, Shibata D, Soong NW, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Ruiz-Pesini EE, Wallace DC. Control region mtDNA variants: Longevity, climatic adaptation and a forensic conundrum. Proc Natl Acad Sci. 2003;100:2174–2176. doi: 10.1073/pnas.0630589100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, Wallace DC. Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and down syndrome dementia. J Alzheimer's Dis. 2010;20(suppl. 2):S293–S310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner S, Chen HW, Marciniack C, LaFerla F, Wallace DC. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbagen.2011.08.008. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgaard LT. Genetic variance in uncoupling protein 2 in relation to obesity, type 2 diabetes, and related metabolic traits: Focus on the functional −866G>A promoter variant (rs659366) J Obes. 2011;2011:1–12. doi: 10.1155/2011/340241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaro M, Blanc H, Johnson MJ, Chen KH, Wilmsen E, Cavalli Sforza LL, Wallace DC. Ethnic variation in Hpa 1 endonuclease cleavage patterns of human mitochondrial DNA. Proc Natl Acad Sci. 1981;78:5768–5772. doi: 10.1073/pnas.78.9.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature. 2011;476:414–420. doi: 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Ek J, Andersen G, Urhammer SA, Gaede PH, Drivsholm T, Borch-Johnsen K, Hansen T, Pedersen O. Mutation analysis of peroxisome proliferator-activated receptor-γ coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia. 2001;44:2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire K, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, MacGregor GR, Wallace DC. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratter C, Gorman GS, Stewart JD, Buddles M, Smith C, Evans J, Seller A, Poulton J, Roberts M, Hanna MG, et al. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology. 2010;74:1619–1626. doi: 10.1212/WNL.0b013e3181df099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Schapira AH. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy. 2011;7:243–245. doi: 10.4161/auto.7.2.14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. Int J Biochem Cell Biol. 2009;41:1899–1906. doi: 10.1016/j.biocel.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Gomez-Duran A, Pacheu-Grau D, Lopez-Gallardo E, Diez-Sanchez C, Montoya J, Lopez-Perez MJ, Ruiz-Pesini E. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet. 2010;19:3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. EMBO J. 2011a;30:2101–2114. doi: 10.1038/emboj.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef M, Nunnari J. A role for mitochondria in autophagy regulation. Autophagy. 2011b;7:1245–1246. doi: 10.4161/auto.7.10.16508. [DOI] [PubMed] [Google Scholar]
- Greggio E, Bisaglia M, Civiero L, Bubacco L. Leucinerich repeat kinase 2 and α-synuclein: Intersecting pathways in the pathogenesis of Parkinson's disease? Mol Neurodegener. 2011;6:6. doi: 10.1186/1750-1326-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. What have we learned from Drosophila models of Parkinson's disease? Prog Brain Res. 2010;184:3–16. doi: 10.1016/S0079-6123(10)84001-4. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet. 1991;48:1147–1153. [PMC free article] [PubMed] [Google Scholar]
- Hutchin T, Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc Natl Acad Sci. 1995;92:6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, Hayashi J-I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- Jia JJ, Tian YB, Cao ZH, Tao LL, Zhang X, Gao SZ, Ge CR, Lin QY, Jois M. The polymorphisms of UCP1 genes associated with fat metabolism, obesity and diabetes. Mol Biol Rep. 2010;37:1513–1522. doi: 10.1007/s11033-009-9550-2. [DOI] [PubMed] [Google Scholar]
- Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- Johnson MJ, Wallace DC, Ferris SD, Rattazzi MC, Cavalli-Sforza LL. Radiation of human mitochondria DNA types analyzed by restriction endonuclease cleavage patterns. J Mol Evol. 1983;19:255–271. doi: 10.1007/BF02099973. [DOI] [PubMed] [Google Scholar]
- Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at np 14459 of the ND6 gene associated with maternally inherited Leber's hereditary optic neuropathy and dystonia. Proc Natl Acad Sci. 1994;91:6206–6210. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol Cell Biol. 1996;16:771–777. doi: 10.1128/mcb.16.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HS, Kim IK, Lee HJ, Lee HJ, Kang JH, Kim JR, Shin HD, Song J. Effects of UCP2 and UCP3 variants on the manifestation of overweight in Korean children. Obesity. 2009;17:355–362. doi: 10.1038/oby.2008.531. [DOI] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, Kato N, Miyawaki A, Kato T. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2006;2:e128. doi: 10.1371/journal.pgen.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney PM, Dunham LD, Quigley CK, Morton SL, Bergquist KE, Bennett JP., Jr Cybrid models of Parkinson's disease show variable mitochondrial biogenesis and genotype–respiration relationships. Exp Neurol. 2009;220:374–382. doi: 10.1016/j.expneurol.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, Magzhanov R, Wallace DC. A mitochondrial etiology of neurodegenerative diseases: Evidence from Parkinson's disease. Ann NY Acad Sci. 2008;1147:1–20. doi: 10.1196/annals.1427.001. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, Wolburg H, Gizatullina Z, Gellerich FN, Woitalla D, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One. 2010;5:e9367. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TS. The structure of scientific revolutions. University of Chicago Press; Chicago, IL: 1996. [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lakatos A, Derbeneva O, Younes D, Keator D, Bakken T, Lvova M, Brandon M, Guffanti G, Reglodi D, Saykin A, et al. Association between mitochondrial DNA variations and Alzheimer's disease in the ADNI cohort. Neurobiol Aging. 2010;31:1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7:531–532. doi: 10.4161/auto.7.5.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriwether DA, Clark AG, Ballinger SW, Schurr TG, Soodyall H, Jenkins T, Sherry ST, Wallace DC. The structure of human mitochondrial DNA variation. J Mol Evol. 1991;33:543–555. doi: 10.1007/BF02102807. [DOI] [PubMed] [Google Scholar]
- Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini EE, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, et al. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Sosa F, Ruiz-Pesini E, Enriquez JA, Marcuello A, Diez-Sanchez C, Montoya J, Wallace DC, Lopez-Perez MJ. Differences of sperm motility in mitochondrial DNA haplogroup U sublineages. Gene. 2006;368C:21–27. doi: 10.1016/j.gene.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Morowitz HJ. Energy flow in biology, biological organization as a problem in thermal physics. Academic Press; New York: 1968. [Google Scholar]
- Muller YL, Bogardus C, Pedersen O, Baier L. A Gly482Ser missense mutation in the peroxisome proliferator-activated receptor γ coactivator-1 is associated with altered lipid oxidation and early insulin secretion in Pima Indians. Diabetes. 2003;52:895–898. doi: 10.2337/diabetes.52.3.895. [DOI] [PubMed] [Google Scholar]
- Murdock DG, Christacos NC, Wallace DC. The age-related accumulation of a mitochondrial DNA control region mutation in muscle, but not brain, detected by a sensitive PNA-directed PCR clamping based method. Nucl Acids Res. 2000;28:4350–4355. doi: 10.1093/nar/28.21.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, Saykin AJ, Orro A, Lupoli S, Salvi E, et al. Hippo-campal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer's disease. PLoS One. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Morten KJ, Marchington D, Weber K, Brown GK, Rotig A, Bindoff L. Duplications of mitochondrial DNA in Kearns-Sayre syndrome. Muscle Nerve. 1995;3:S154–S158. doi: 10.1002/mus.880181430. [DOI] [PubMed] [Google Scholar]
- Prachar J. Mouse and human mitochondrial nucleoid—Detailed structure in relation to function. Gen Physiol Biophys. 2010;29:160–174. [PubMed] [Google Scholar]
- Ricardo A, Szostak JW. Origin of life on earth. Sci Am. 2009;301:54–61. doi: 10.1038/scientificamerican0909-54. [DOI] [PubMed] [Google Scholar]
- Roses AD. The medical and economic roles of pipeline pharmacogenetics: Alzheimer's disease as a model of efficacy and HLA-B(*)5701 as a model of safety. Neuropsycho-pharmacology. 2009;34:6–17. doi: 10.1038/npp.2008.153. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Wallace DC. Evidence for adaptive selection acting on the tRNA and rRNA genes of the human mitochondrial DNA. Hum Mutat. 2006;27:1072–1081. doi: 10.1002/humu.20378. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Lapena AC, Diez-Sanchez C, Perez-Martos A, Montoya J, Alvarez E, Diaz M, Urries A, Montoro L, Lopez-Perez MJ, et al. Human mtDNA haplogroups associated with high or reduced spermatozoa motility. Am J Hum Genet. 2000;67:682–696. doi: 10.1086/303040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Lott MT, Procaccio V, Poole J, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucl Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A, Balbi V, Balducci E, Pirazzini C, Rosini F, Tavano F, Achilli A, Siviero P, Minicuci N, Bellavista E, et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer's disease. PLoS One. 2010;5:e12037. doi: 10.1371/journal.pone.0012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: Mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta. 2010;1800:245–249. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Schurr TG, Donham BP, Morreale SC, Panter-Brick C, Donham DL, Armelagos GJ, Wallace DC. Genetic diversity in modern African populations and its use for reconstructing ancient and modern population movements. In: Reed DM, editor. Biomolecular archeology, genetic approaches to the past. Center for Archaeological Investigations, Southern Illinois University; Carbondale, IL: 2005. pp. 169–207. occasional paper no. 31. [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Brown MD, Torroni A, Lott MT, Cabell MR, Mirra SS, Beal MF, Yang C, Gearing M, Salvo R, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171–184. doi: 10.1006/geno.1993.1299. [DOI] [PubMed] [Google Scholar]
- Smeitink JA, Elpeleg O, Antonicka H, Diepstra H, Saada A, Smits P, Sasarman F, Vriend G, Jacob-Hirsch J, Shaag A, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N, Prakash J, Lakhan R, Agarwal CG, Pant DC, Mittal B. A common polymorphism in the promoter of UCP2 is associated with obesity and hyperinsulenemia in northern Indians. Mol Cell Biochem. 2010;337:293–298. doi: 10.1007/s11010-009-0311-2. [DOI] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni A, Petrozzi M, D'Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997;60:1107–1121. [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Bennett JP., Jr The cybrid model of sporadic Parkinson's disease. Exp Neurol. 2009;218:320–325. doi: 10.1016/j.expneurol.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cyto-chrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udar N, Atilano SR, Memarzadeh M, Boyer DS, Chwa M, Lu S, Maguen B, Langberg J, Coskun P, Wallace DC, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- van den Ouweland JM, Bruining GJ, Lindhout D, Wit JM, Veldhuyzen BF, Maassen JA. Mutations in mitochondrial tRNA genes: Non-linkage with syndromes of Wolfram and chronic progressive external ophthalmoplegia. Nucl Acids Res. 1992;20:679–682. doi: 10.1093/nar/20.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992a;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science. 1992b;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA sequence variation in human evolution and disease. Proc Natl Acad Sci. 1994;91:8739–8746. doi: 10.1073/pnas.91.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria as chi. Genetics. 2008;179:727–735. doi: 10.1534/genetics.104.91769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution. Cold Spring Harb Symp Quant Biol. 2009;74:383–393. doi: 10.1101/sqb.2009.74.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Colloquium paper: Bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci. 2010;107(suppl 2):8947–8953. doi: 10.1073/pnas.0914635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988a;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell. 1988b;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–230. doi: 10.1016/s0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Ruiz-Pesini E, Mishmar D. MtDNA variation, climatic adaptation, degenerative diseases, and longevity. Cold Spring Harb Symp Quant Biol. 2003;68:479–486. doi: 10.1101/sqb.2003.68.471. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Lott MT, Procaccio V. Mitochondrial genes in degenerative diseases, cancer and aging. In: Rimoin DL, et al., editors. Emery and Rimoin's principles and practice of medical genetics. 5th ed. Churchill Livingstone/Elsevier; Philadelphia: 2007. pp. 194–298. [Google Scholar]
- Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Michikawa Y, Mallidis C, Bai Y, Woodhouse L, Yarasheski KE, Miller CA, Askanas V, Engel WK, Bhasin S, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci. 2001;98:4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, Amati P, Moggio M, Servidei S, Scarlato G, DiDonato S. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: A new autosomal dominant disease. Am J Hum Genet. 1990;47:904–914. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Asin-Cayuela J, Fish J, Michikawa Y, Bonafe M, Olivieri F, Passarino G, De Benedictis G, Franceschi C, Attardi G. Strikingly higher frequency in centenarians and twins of mtDNA mutation causing remodeling of replication origin in leukocytes. Proc Natl Acad Sci. 2003;100:1116–1121. doi: 10.1073/pnas.242719399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chu CT. Mitochondrial dysfunction in Parkinson's disease. J Alzheimer's Dis. 2010;20(suppl 2):S325–S334. doi: 10.3233/JAD-2010-100363. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Whitworth AJ. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy. 2010;6:660–662. doi: 10.4161/auto.6.5.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]