

Abstract
Despite all available therapies, the rates of hospitalization and death from heart failure (HF) remain unacceptably high. The most common reasons for hospital admission are symptoms related to congestion. During hospitalization, most patients respond well to standard therapy and are discharged with significantly improved symptoms. Post-discharge, many patients receive diligent and frequent follow-up. However, rehospitalization rates remain high. One potential explanation is a persistent failure by clinicians to adequately manage congestion in the outpatient setting. The failure to successfully manage these patients post-discharge may represent an unmet need to improve the way congestion is both recognized and treated. A primary aim of future HF management may be to improve clinical surveillance to prevent and manage chronic fluid overload while simultaneously maximizing the use of evidence-based therapies with proven long-term benefit. Improvement in cardiac function is the ultimate goal and maintenance of a “dry” clinical profile is important to prevent hospital admission and improve prognosis. This paper focuses on methods for monitoring congestion, and strategies for water and sodium management in the context of the complex interplay between the cardiac and renal systems. A rationale for improving recognition and treatment of congestion is also proposed.
Keywords: Heart failure, Congestion, Post-discharge, Fluid intake, Sodium diet, Management, Outcome
Introduction
Despite all available therapies, there are over one million hospitalizations for heart failure (HF) annually in the USA alone [1], and a similar number in Europe. Symptoms responsible for hospitalization are typically related to pulmonary or systemic congestion that result in dyspnea, rales and edema [2]. Growing evidence suggests that congestion itself leads to HF progression [3]. Owing to exerts detrimental effects on the heart (altered ventricular geometry, functional mitral insufficiency, further increase in intra-cardiac pressures) and other organs (kidneys and liver) via increased venous pressures [4]. Congestion, ideally, should be prevented and its early detection, possibly with the help of new technologies, may allow for early intervention long before overt symptoms develop. Current therapies for congestion should be personalized according to congestion severity and renal function, and should be used to “bridge” patients through episodes of worsening congestion, while providing opportunities to add proven therapies that improve cardiac function and outcomes. In this respect, this paper will focus on (1) methods for monitoring congestion and related kidney injury; (2) strategies for fluid and sodium management; and (3) the rationale for improved recognition and treatment of congestion with the goal of improving HF outcomes.
Congestion in Heart Failure
Congestion is a manifestation of several concurrent processes both structural and functional including ventricular remodeling, progression of coronary artery disease, valvular abnormalities, neurohormonal and inflammatory activation, vascular adaptations and renal dysfunction [5]. It is often not recognized until it becomes severe enough to necessitate hospital admission or acute therapies in diverse settings. It can be divided into two general categories that represent a continuum; hemodynamic and clinical congestion [6]. Hemodynamic congestion refers to the state of increased intra-cardiac filling pressures accompanied by cardiopulmonary volume overload that can occur in the absence of clinically evident signs/symptoms. Clinical congestion refers to the presence of signs/symptoms related to elevated intra-cardiac filling pressures. These pressures may begin to rise days to three weeks prior to the development of symptoms or weight gain [7]. Some studies have suggested that in patients with pulmonary congestion, fluid overload is caused by fluid redistribution because of an increased vascular resistance/stiffness which may lead to both reduced capacitance in the large veins and increased arterial resistance with consequent endogenous fluid shift from splanchnic bed into effective circulating volume rather than on endogenous fluid gain. Fluid redistribution and fluid accumulation may be variably combined in such patients [8]. However, aside from this potential redistribution, true accumulation of fluid due to sodium and water retention secondary to adaptative neurohormonal changes is also at play. Congestion can increase LV wall stress, functional mitral regurgitation and neurohormonal/inflammatory activation, thus exacerbating myocardial remodeling (chamber dilatation, increased ventricular sphericity and aggravated ischemia), loss of myocardial cells, decreasing ventricular function and leading to worsening hemodynamics and progressive HF (Fig. 1). LV impairment often leads to right ventricular (RV) dysfunction either through ventricular interdependence or because of chronically elevated left-sided filling pressures that lead to an increase in pulmonary pressures which in turn affects RV afterload. This increase in RV afterload (pulmonary venous hypertension extends to pulmonary arteries) leads to RV dysfunction, tricuspid regurgitation, and subsequent further RV impairment and systemic congestion, reinforcing the vicious cycle of HF (Fig. 2). Conversely, systemic congestion increases RV preload that in long term leads to RV dysfunction, tricuspid incompetence and increased right-side filling pressure. The result is the increased central venous pressure with subsequent renal dysfunction and further congestion. Thus, the concept of hemodynamic congestion illustrates that hemodynamic derangements can substantially precede clinical manifestations and that careful detection of hemodynamic congestion allows a window for early preclinical intervention (Fig. 1).
Fig. 1. Pathophysiological course and vicious cycle of HF until clinical congestion, including renal dysfunction by cardio-renal interaction, subdivided in phases of preclinical medical interventions and hospitalization, suggesting a window for earlier intervention on hemodynamic congestion.
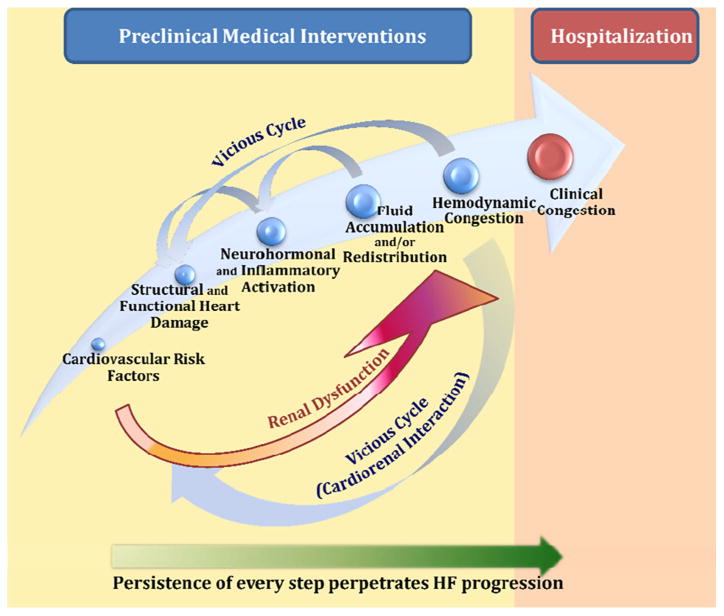
Fig. 2. The vicious cycle of HF progression with mutual involvement of left and right sides of the heart and the kidney: key role of congestion.
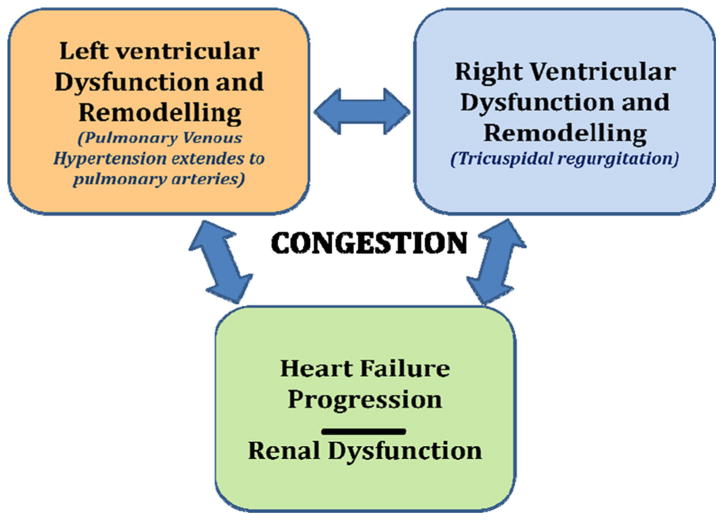
Clinicians are likely failing to recognize and treat congestion because of the insidious onset with which it develops. Furthermore, the clinical evaluation of volume overload is limited. In ambulatory non-edematous patients with HF, clinically unrecognized hypervolemia (as determined by blood volume analysis) is frequently present and associated with increased cardiac filling pressures and worse patient outcomes [9]. Once congestion is detected, it is an obvious target for therapy. Aggressive reduction of intra-cardiac filling pressures is beneficial by producing symptom relief with concomitant improvements in mitral regurgitation, RV function, neurohormonal activation and exercise tolerance. Often, however, congestion is inadequately treated and patients are often discharged with residual elevation in circulating volume and symptoms that are improved but not resolved. This contributes to instability and readmission early after discharge [10].
Congestion Assessment
The “gold standard” for evaluating congestion in hospitalized patients is the measurement of pulmonary capillary wedge pressure (PCWP) that closely approximates LV end-diastolic pressure. However, PCWP measurement involves invasive catheterization, limiting its clinical use. Body weight monitoring is readily available, but is often not representative of changes in filling pressures [10]. Clinical assessment of jugular venous pressure remains the most sensitive and specific test for detecting elevated LV filling pressures. Campbell et al. [11] provide reassurance that concordance of elevated right- and left-side filling pressures allows reliance upon jugular venous assessment in majority of patients with chronic HF. However, when therapy guided by right-sides assessment does not produce the desired responses, consideration should be given to invasive measurement of left-side filling pressures. Furthermore, other physical findings such as rales and peripheral edema can be absent in a large proportion of patients despite measured intravascular volume overload. Therefore, new technologies that supplement clinical evaluation are required for monitoring fluid overload in HF. Chest radiography can demonstrate chronically elevated filling pressures but does not change rapidly enough to guide acute evaluation and therapy. Efforts to improve clinical monitoring have included scoring systems. One such system, which contains the variables of pulmonary crackles, pathological jugular venous distention, peripheral edema and third heart sound, results in a 95 % negative predictive value for left atrial pressure <20 mmHg [12]. Plasma natriuretic peptides (NP) were first used with the promise of increasing the diagnostic accuracy of HF, diagnosing elevated LV filling pressures and defining “congestion”. However, NP should not be used alone to assess congestion but must be evaluated in the appropriate clinical context because there is no defined cut-point and their pattern of production and release is slow and variable. However, having a baseline NP concentration may help determine a patient's “target” level and may be helpful to monitor filling pressure and to optimize therapy.
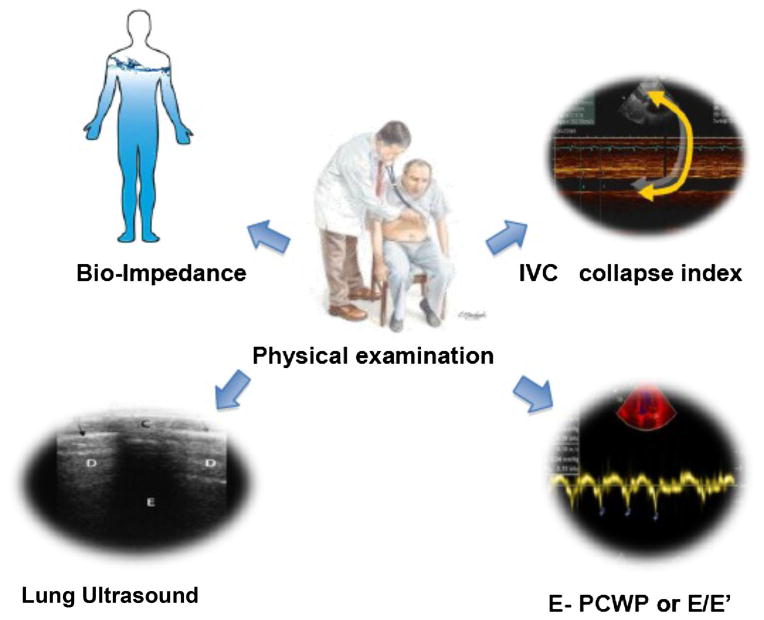
New noninvasive instrumental methods for congestion assessment include:
Ultrasonography of the inferior vena cava (IVC), a rapid method to estimate elevated right atrial pressure by measuring IVC diameter and its collapsibility;
Echocardiographic assessment of PCWP or LV filling pressure estimated by trans-mitral E/e1 the ratio of peak early mitral flow velocity (E) divided by mitral annular early diastolic velocity (e1), measured by Tissue Doppler Imaging;
Lung ultrasound for detecting B lines (also called ultrasound lung comet), which correlate with a radiographic score of pulmonary congestion and invasively measured extravascular lung water values [13] (Table 1);
Transthoracic bioimpedance or thoracic impedance cardiography which utilizes the principle that electrical impedance is specifically and inversely correlated with the content of tissue fluids. It provides an assessment of cardiac output and stroke volume, systemic vascular resistance and thoracic fluid content (Fig. 3).
Table 1. Main cutoff of noninvasive methods of congestion monitoring according to heart failure clinical profile.
| Parameters | Wet profile | Dry profile |
|---|---|---|
| IVC collapse index | <50 % → RAPs >10 mmHg [54] | ≥50 % → RAPs ≤10 mmHg |
| <45 % → RAPs >8 mmHg [55] | >45 % → RAPs ≤8 mmHg | |
| <40 % → RAPs >10 mmHg [56] | >40 % → RAPs ≤10 mmHg | |
| IVC max expiratory diameter | ≥2 cm | <2 cm |
| ≤1.2 cm are indicative of normal RAPs (≤10 mmHg) at 100 % [57] | ||
| Echocardiographic PCWP | >12 [58] | ≤12 |
| E/e1 (a) | ≥15 (Sep.); ≥ 12 (Lat); ≥ 13 (Av.) [59] | <15 (Sep.); <12 (Lat.); <13 (Av.) |
| ≥11 [60] | <11 | |
| ≤8 (sep, lat, or Av.) indicates very low LV filling pressure | ||
| Lung ultrasound | Multiple bilateral B lines assessed on the anterior and lateral chest: two or more positive regions bilaterally (a positive region is defined by the presence of ≥3 ultrasound B lines in a longitudinal plane between two ribs) [61] | ≤2 ultrasound B lines in any chest region |
Sep septal, Lat lateral, Av average, RAP right arterial pressure
E/e1 ratio ranging from 9 to 14 is a gray zone considered suggestive but non-diagnostic of diastolic LV dysfunction and needs to be implemented with other noninvasive investigations to confirm the diagnosis of HF
Fig. 3. Key points of congestion assessment.
Several studies have shown that decreasing thoracic impedance correlates with HF hospitalizations. Similarly, some studies indicate that new noninvasive methods that detect whole-body bioelectrical impedance are capable of rapidly assessing intra- and extra-cellular total body fluid content (overhydration or dehydration) and the effectiveness of diuresis [14]. These methods, however, require large trials to confirm clinical utility.
Cardio-renal Interaction in Heart Failure
The ADHERE registry revealed high prevalence of renal dysfunction in acute HF patients. In particular, moderate (GFR 30–59 ml/min/1.73 m2) and severe renal dysfunction (GFR 15–29 ml/min/1.73 m2) and kidney failure (GFR < 15 ml/min/1.73 m2) occurs in 43.5 %, 13.1 % and 7.0 % respectively [15]. Additionally, in patients with HF, an acute or chronic reduction in glomerular filtration rate (GFR) has been independently associated with poor outcomes [16, 17]. Cardio-renal interactions can be divided into five categories based on the classification of Ronco et al. [18]. The first two include the forms where the heart is the primary failing organ: type 1 occurs when acute HF leads to acute kidney injury and type 2 refers to chronic HF causing progressive and potentially permanent chronic kidney disease (CKD). This is much more useful than the initial use of the term cardio-renal syndrome which referred only to WRF which occurs during therapy to relieve congestive HF symptoms. The pathophysiology of HF-related renal dysfunction remains complex. Multiple mechanisms are likely involved [19], including: (1) reduced cardiac output (CO) and renal perfusion; (2) elevated central venous pressure (CVP); (3) elevated intra-abdominal pressure (defined as >8 mmHg); (4) activation of inflammatory and neurohormonal systems and oxidative stress; (5) preexisting chronic renal disease; and (6) drug mediated diuretics, antibiotics, NSAIDs. A failing heart is unable to generate adequate “forward” CO, leading to prerenal hypoperfusion and arterial underfilling with compensatory neurohormonal activation including the renin–angiotensin–aldosterone system (RAAS), sympathetic nervous system and arginine vasopressin (AVP) expression [19]. In the presence of diminished CO and low systemic pressures, the kidney itself can maintain adequate renal perfusion by means of autoregulatory mechanisms [20]. Neurohumoral activation may be useful during acute stress to help restore CO and preserve renal perfusion and filtration fraction by increasing circulating volume. Chronic volume retention, however, can lead to a vicious cycle in which increased preload and afterload can further diminish CO. In parallel, persistent renal hypoperfusion may lead to chronic renal hypoxia, inflammation and oxidative stress causing progressive renal dysfunction. Improvements in cardiac index (CI) alone may not, however, result in improved renal function, as supported by the ESCAPE trial where, of the hemodynamic parameters measured, only right atrial pressure (a surrogate for venous congestion) was correlated with baseline renal dysfunction [21]. Similarly, Mullens et al. [22] found WRF during hospitalization (serum creatinine increase >0.3 mg/ dL) to be associated with higher central venous pressure on admission and discharge and they failed to demonstrate an association with lower cardiac index. RV dysfunction and tricuspid regurgitation have an important role in this process, as reflected by the ability of CVP to stratify risk across various levels of cardiac index. This is also supported by improved renal outcome after relief of venous congestion. These findings are consistent with the hypothesis that elevated CVP can be transmitted back on the renal veins with subsequent increased renal interstitial pressure. This may lead to impaired GFR and hypoxic damage similar to congestive liver dysfunction in HF. An increase in hydrostatic pressure in Bowman's capsule and afferent arteriolar vasoconstriction can result in reduction in GFR independent of CO [23]. Intrarenal vasoconstriction may result from sympathetic and neurohormonal stimulation. Moreover, venous congestion, through stretch of endothelial cells, can modulate the synthetic and endocrine phenotype of the vascular endothelium from a quiescent state to an activated one, leading to a pro-oxidant, pro-inflammatory and vasoconstrictive state. This may contribute to the development and progression of functional or structural changes in the kidneys (in particular in the tubulo-interstitium) with subsequent sodium and water retention [24].
Pharmacotherapies used in the management of HF may worsen renal function: diuresis associated hypovolemia, early introduction of RAAS blockade, and drug-induced hypotension have all been suggested as contributing factors. In particular, hemoconcentration in subjects aggressively treated with diuretics is significantly associated with deterioration in renal function, but 180-day mortality was reduced in these subjects, when compared to subjects treated more conservatively. WRF may therefore be acceptable upon start of therapy with diuretics [25] as well as with angiotensin-converting enzyme inhibitors. It should be noted that impairment of renal function is as likely to occur as improvement during diuresis for hypervolemia in chronic HF.
Kidney Injury and Biomarkers
Acute kidney injury (AKI) is a common complication in patients hospitalized for acute HF and has been associated with longer hospitalization and increased morbidity and mortality. It occurs as a consequence of new onset kidney injury or acute deterioration of preexisting chronic kidney disease (CKD) (acute-on-chronic kidney injury). AKI was defined by the Acute Kidney Injury Network (AKIN) criteria as an abrupt (within 48 h) reduction in kidney function with as an absolute increase in serum creatinine >0.3 mg/dl (≥26.4 μmol/L), a percentage increase in serum creatinine >50 % or a reduction in urine output <0.5 ml/kg per hour for more than 6 hours [26]. More recently, the Kidney Disease Improving Global Outcomes (KDIGO) group has refined this definition to be an increase in serum creatinine >50 % within 7 days, or an increase in serum creatinine >0.3 mg/dl (26.5 μmol/L) within 2 days, or oliguria. Damman et al. [27] have reported a 61 % increase in the risk of death and a 30 % increase in the risk of all-cause readmissions, when there was AKI after 2–6 months of follow-up. Serum creatinine is not always a reliable indicator of early kidney injury. Serum creatinine varies with age, gender, ethnicity, muscle mass and volume status. Furthermore, changes in serum creatinine may reflect hemodynamic factors without any associated tubular, vascular or interstitial injury. Traditionally, GFR remains the gold standard for assessing renal function. However, measuring accurate real time GFR remains difficult in the clinical setting. Formulas estimating GFR have been validated when serum creatinine is in a steady state and thus are not accurate during acute changes in renal function. Conversely, blood urea nitrogen, has recently been emerged as a stronger predictor of outcome than creatinine and estimated GFR represents an emerging surrogate marker for the “renal response” to neurohormonal activation and congestion. Of the many new biomarkers available, serum and/or urine neutrophil gelatinase-associated lipocaptin (NGAL), serum cystatin C, kidney injury molecule 1 (KIM-1) and N-acetyl-beta-d-glucosaminidase (NAG) appear to be the most promising panel of biomarkers for renal tubular injury and/or functional assessment. A recent study demonstrated that tubulo-interstitial damage detected by measuring urinary NAG, KIM-1 NGAL, was associated with an adverse prognosis in HF patients even when GFR was normal [28]. Another study found that KIM-1 and NAG were predictive of all-cause mortality and the composite of all-cause mortality and rehospitalization for HF, whereas NGAL was not associated with either outcome [29]. Furthermore, Damman et al. [30] showed that in CHF patients, urinary NAG, but not NGAL or KIM-1 correlated with GFR (r = −0.34, p = 0.001) and effective renal plasma flow (r = −0.29, p = 0.006). Both NAG (r = 0.21, p = 0.048) and KIM-1 (r = 0.23, p = 0.033) correlated with plasma N-terminal probrain natriuretic peptide levels. Furthermore, both urinary NAG (HR = 1.42, p = 0.039) and KIM-1 (HR = 1.15, p = 0.025) were associated with an increased risk of death or HF hospitalizations, independent of GFR. Importantly, a recent study on subclinical modulation of volume status found that diuretic withdrawal resulted in significant increases in urinary KIM-1, and NAG while NGAL and serum creatinine were unaffected [31]. After reinstitution of furosemide treatment, both urinary KIM-1 and NAG concentrations returned to baseline, but NGAL was unaffected. These results suggest that subclinical alterations in volume status in HF patients are associated with changes in markers of renal tubular dysfunction and that diuretic therapy may favorably affect renal tubular function by decreasing congestion. All these findings suggest an important future role for markers of renal tubular damage to monitor cardio-renal interaction in HF.
From Low-Dose to High-Dose Loop Diuretics: Flexible Titration
Diuretics remain the mainstay of treatment in 90 % of HF patients hospitalized with worsening HF in the USA and Europe [32]. The relationship between diuretic delivery and response is characterized by a sigmoidal dose response curve where efficacy (maximal effect) is the same for all loop diuretics. Several features of this pharmacodynamic relationship are clinically important. First, there is a threshold drug concentration which must be achieved at the active site to elicit a response, and this threshold differs from patient to patient. Clinically, this means that patients should have doses tailored to their individual needs. Second, a maximal response can be identified, allowing a definition of the ceiling dose of a diuretic, namely, the smallest dose of a diuretic eliciting a maximal response and, therefore, the dose that should not be exceeded. In patients with renal insufficiency, the plasma half-life of furosemide is prolonged because both urinary excretion and renal conjugation are decreased. When sufficient doses are administered to attain effective amounts of loop diuretic in the urine, the diuretic response in functional nephrons is the same in patients with renal insufficiency as in healthy volunteers. However, a response in terms of total urinary sodium excretion never reaches that of a healthy volunteer because the decrease in renal function limits filtered sodium. Clinically, this means that a maximally effective dose of a loop diuretic in a patient with renal insufficiency may not result in the required overall diuresis and that other measures, including high doses, frequent dosing, combining diuretics, may also need to be employed.
In HF patients, the quantity of furosemide absorbed is the same of healthy subjects but the absorbtion is slowed. The sigmoidal dose–response curve is shifted downward and rightward [33], resulting in a natriuretic response that is one-fourth to one-third of what occurs normally with maximally effective doses of loop diuretics. In HF, chronic treatment with loop diuretic is associated with intrarenal resistance that may be initially overcome by larger doses of furosemide. Alternative, therapeutic strategy is to administer modest doses more frequently, combine different class of loop diuretic or add a thiazide. This last strategy may have a synergistic response with profound diuresis.
In summary, a patient who has renal insufficiency should be given increasing doses of a loop diuretic until an effective dose is found or the ceiling dose relative to the individual patient's renal function is reached. In patients with congestive HF and preserved renal function, delivery of loop diuretics to the tubular fluid is normal [34]. Given that the pharmacokinetics of loop diuretics are essentially normal in patients with HF, it can be said that pharmacodynamic mechanisms associated with enhanced proximal sodium reabsorption in the nephron account for a diminished response. Felker et al. [35] in a prospective randomized trial in ADHF, observed that global assessment of symptoms or changes in renal function not differ significantly when diuretic therapy was administered by bolus as compared with continuous infusion or at high dose as compared with a low dose. The high-dose strategy was associated with a greater diuresis and more favorable outcomes in some secondary measures but also with transient worsening of renal function [35].
Regarding diuretic titration, several sets of HF consensus/guideline statements support the use of a flexible diuretic dosing regimen for outpatient management of fluid overloadrelated signs and symptoms. The rationale is to titrate the diuretic increasing or reducing doses according to the state of congestion, the symptoms or the possible risk of excessive volume depletion. Today, only five randomized studies have evaluated this issue in HF. Three randomized trials included flexible diuretic titration as part of a broader multifaceted disease management program, and 2 were designed to specifically evaluate the sole contribution of flexible diuretic titration. Collectively, data from these studies supported the idea that flexible and individualized diuretic dosing is potentially associated with reduced emergency room visits, reduced rehospitalization, and improved quality of life in HF patients with reduced ejection fraction [36].
Improving Post-Discharge Outcomes
Despite multiple trials aimed at improving outcomes, patients hospitalized with HF face mortality and rehospitalization rates as high as 15 and 30 % within 60–90 days post-discharge, respectively [37]. Results from the ESCAPE trial demonstrate associations between congestion and clinical course with patients who continue to have a “wet” profile after acute HF treatment, showing a significantly increased risk of adverse outcomes at 6 months compared with those who are “dry” at discharge [10].
Contributing factors to this unacceptably high post-discharge event rate include the incomplete relief of fluid overload, insufficient patient education, the lack of implementation of evidence-based therapies and poor post-discharge follow-up [38]. The transition from the hospital to the outpatient setting involves not only changes in the physicians providing care, but also modifications in diet, self-dependence in the administration of new and complex drug therapies, demands for more physical activity, and confrontation with familial and social stresses [39]. Moreover, patients are often discharged not only before optimal volume status is achieved, but also without adequate control of their blood pressure or with an inadequate ventricular response to atrial fibrillation. All of these factors make the early post-discharge period a vulnerable phase which require, for all these considerations, clinical surveillance [37]. Most patients leave the hospital with relative symptomatic improvement but without complete optimization of filling pressures and with an optimistic, but perhaps unrealistic plan for physicians to “continue diuresis at home”. The recent COMPASS-HF study [40], enabling continuous monitoring of RV and estimated pulmonary artery diastolic pressures in patients with recent HF hospitalizations has shown how high the daily filling pressures remained elevated despite apparently intensive management, how slowly the filling pressures rose prior to HF decompensation events, and how poorly body weight reflected changes in filling pressures during extended outpatient follow-up [10]. Excluding the periods around events, the risk of subsequent HF events was clearly and continuously related to the level of chronic median filling pressures, with no threshold or shoulder level once the median daily pressure exceeded 14 mm Hg.
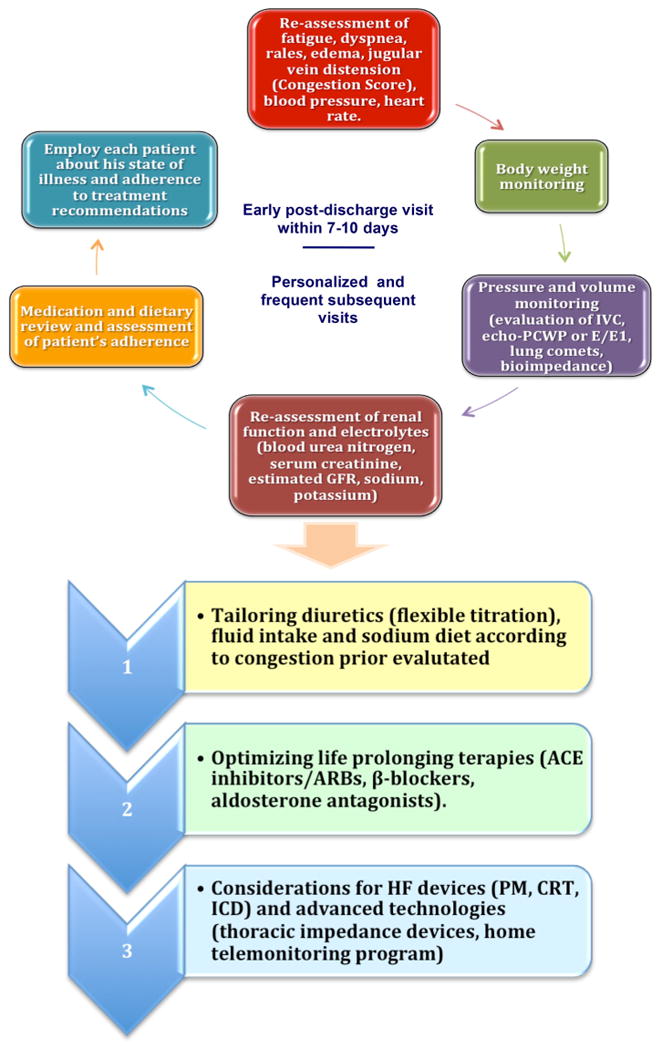
Post-discharge assessment is now deemed an essential component of the treatment of the patients hospitalized for HF to promote recovery and good health. The aim is to maintain lower filling pressures, relieve symptoms, improve exercise tolerance, decrease neurohormonal activation and reduce morbidity and mortality [10]. Simply decreasing body weight cannot be used as an indiscriminate target for reducing hospitalization events [41]. In a small observational study, physician-directed patient self-management of HF with direct left atrial pressure (LAP) monitoring was associated with improved LAP control, reduced symptoms, more optimal neurohormonal antagonist and diuretic dosing, hemodynamic remodeling and a reduction of early clinical events [42]. These data indicate that outpatient hemodynamic monitoring linked to a self-management therapeutic strategy could change current management of advanced HF and potentially facilitate more optimal therapy and improve outcomes. The self-management strategy is analogous to diabetes care in patients who regulate prescribed therapy using objective daily measurements of therapeutic efficacy by the use of a glucometer. The modern strategy of congestion management includes: treat filling pressures that go up (hit the peaks) and treat to reduce chronically elevated filling pressures even without acute change (hit the plateaus) seeking to adjust diuretics up and down (during dry spells) and empower the patient to make daily changes. Thus, a lower risk of rehospitalization will depend on early clinic follow-up post-discharge (within 7–10 days) and on personalized and frequent subsequent visits. At each follow-up visit, fluid status and body weight should be monitored by physical examination, bioimpedance and/or clinical scores (reassessment of signs/symptoms). In addition, when possible, IVC, PCWP or E/e1 (echocardiographic evaluation) and ultrasound B lines should be performed too. The steps of this personalized and congestion-oriented approach are summarized in Fig. 4 and include new technological advances such as the home telemonitoring and electronic assessment of weight, HF symptoms and thoracic impedance by devices and pressure sensors.
Fig. 4. Congestion-guided clinical approach and decisionmaking during post-discharge follow-up of patients with HF.
Dietary Sodium and Water intake in Heart Failure
All HF management guidelines recommend sodium restriction as a key factor in optimizing fluid balance; however, there are insufficient data to endorse any specific level of sodium intake with certainty, and differences among the various HF subpopulations are not known. The Heart Failure Society of America recommends 2,000–3,000 mg daily sodium intake for patients with the clinical syndrome of HF and preserved or depressed ejection fraction, with further restriction (<2,000 mg) for moderate to severe HF and patients with recurrent or refractory volume overload. European Guidelines indicate restriction of sodium intake to <2,000 mg/day in symptomatic patients. However, the level of restriction is controversial [43]. An observational cohort study in the Second and Third National Health and Nutrition Examination Survey (NHANES) showed an association between low sodium intake and cardiovascular mortality [44]. O'Donnell et al. [45] found a J-shaped association between estimated sodium excretion and CV events with the possibility of increased risk of CVD morbidity and mortality at both extremes of sodium intake. Compared with baseline sodium excretion of 4–5.99 g per day, sodium excretion of more than 7 g per day was associated with an increased risk of all CV events while a sodium excretion of <3 g per day was associated with increased risk of CV mortality and hospitalization for HF. Thus, there is some evidence for a “J” curve fit, with a safe zone of about 2.5–6.0 g/day [43].
In compensated HF patients receiving high-dose oral furosemide, it has been found that sodium restriction to 80 mmol/day (1,840 mg/day) was associated long term with significantly higher rates of hospitalization and increased levels of BNP, aldosterone, plasma renin activity and cytokines (TNF-α, IL-6) compared with those patients receiving less restricted sodium intake of 120 mmol/day (2,760 mg/day) who showed improvement in clinical compensation, neurohormonal and inflammatory activation, and outcome [46]. These findings were recently supported by a Cochrane review which showed that sodium reduction resulted in a significant increase in plasma renin, plasma aldosterone, plasma adrenaline and plasma noradrenaline [47]. Recently, Lennie et al. [48] interestingly showed <3,000 mg/d sodium was associated with better outcomes in NYHA class III to IV patients, whereas it was associated with significant increase in hospital visits, readmissions, and mortality in NYHA class I–II patients. All these data, in addition to recent data concerning the utility of hypertonic saline in decompensated HF, do not support universal strict sodium restriction in HF patients and indicates the need to define a safe range of sodium intake in this setting, recalling the experience with β-blockers which were previously contraindicated in HF.
Regarding fluid restriction, international guidelines recommend fluid restriction of 1.5–2 liters/day during the initial management of an acute episode of HF associated with volume overload in symptomatic patients with severe hyponatremia <130 mEq/L and in all symptomatic subjects demonstrating fluid retention that is difficult to control despite high doses of diuretic and sodium restriction. More strict fluid restriction is recommended in patients with more severe hyponatremia (serum sodium <125 mEq/L) although the data are not conclusive. Many practices have found it impractical and unpleasant to restrict fluid to <2 L daily. Aliti et al. [49] observed in systolic ADHF patients with normal serum sodium that aggressive fluid (<800 ml/day) and sodium restriction (800 mg/day) when compared with diet without restriction have no differences on weight loss, clinical stability and 30-day readmission rate. Conversely, some new evidences suggest that fluid restriction to 1 L or less in addition to near-normal sodium diet may be useful in during intermediate term follow-up in recently decompensated HF patients without hyponatremia and might be considered by highly motivated patients who undergo frequent or persistent fluid overload despite optimized flexible diuretic regimens [46, 49–53]. Probably, the discrepancy in these results may depend on the different sodium diet strategy rather than fluid regimen.
Conclusions
The major goal in patients hospitalized with HF is to decrease the burdens of symptoms that limit daily life and lead to rehospitalization. A primary aim of initial and serial evaluation of patients with HF remains the identification of congestion and increased intra-cardiac pressures. The mandatory next step is to prevent or relieve chronic fluid overload, to preserve or achieve a dry clinical profile, and to maintain low BNP levels and intra-cardiac pressures without significant worsening of renal function. However, we are still failing to recognize and treat congestion largely because elevations of intra-cardiac pressures can occur well before obvious clinical signs/symptoms develop or because clinical signs/symptoms are underestimated. Fortunately, new and accurate strategies for monitoring congestion are now available. Every patient with suspected or evident congestion should undergo careful individualized assessment with serial evaluation that includes medical history, physical examination for congestion which may be supplemented by serial measurements of BNP, echocardiographic assessments of filling pressures and pulmonary interstitial edema, and measurements of ventilatory flows in order to unmask central fluid overload or to better monitor congestion before clinical signs/symptoms become evident (Fig. 3). Clinical surveillance after hospitalization with an optimized post-discharge follow-up planning is mandatory in HF management. The critical elements in this setting include frequent and personalized ambulatory visits including telephone monitoring and “telemedicine”, a tailored congestion-guided treatment regimen (dynamic diuretic titration beginning from low to high doses and eventual controlled dose reduction when a clinical steady state is reached), a controlled fluid and salt intake plan, renal function monitoring with traditional and novel biomarkers, and an organized network between the primary care provider, cardiologist, hospitalist and nurses. When clinical and noninvasive assessments fail to explain symptoms or lead to therapy that is poorly tolerated, consideration should be given to the possibility of R-L mismatch and other contributions to symptoms such as intrinsic pulmonary disease and the possible need for invasive hemodynamic measurement for clarification [11]. Further clinical studies are needed to recognize the submerged iceberg of congestion and its pathophysiological mechanisms at improving HF management and outcome.
Footnotes
Conflict of interest None of the authors have any potential conflict of interest in the submitted manuscript.
Contributor Information
Gaspare Parrinello, Email: gaspare.parrinello@unipa.it, Biomedical Department of Internal and Specialty Medicine (Di.Bi.M.I.S.), A.O.U.P “Paolo Giaccone”, University of Palermo, Piazza delle Cliniche 2, 90127 Palermo, Italy.
Stephen J. Greene, Center for Cardiovascular Innovation, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA
Daniele Torres, Biomedical Department of Internal and Specialty Medicine (Di.Bi.M.I.S.), A.O.U.P “Paolo Giaccone”, University of Palermo, Piazza delle Cliniche 2, 90127 Palermo, Italy.
Michael Alderman, Department of Epidemiology and Population Health, Albert Einstein College of Medicine, Yeshiva University, New York, USA.
Joseph Vincent Bonventre, Renal Division, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA.
Pietro Di Pasquale, Division of Cardiology, GF Ingrassia Hospital, Palermo, Italy.
Luna Gargani, Institute of Clinical Physiology, National Council of Research, Pisa, Italy.
Anju Nohria, Cardiovascular Division, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA.
Gregg C. Fonarow, Ronald Reagan-UCLA Medical Center, Los Angeles, CA, USA
Muthiah Vaduganathan, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Javed Butler, Division of Cardiology, Emory University, Atlanta, GA, USA.
Salvatore Paterna, Biomedical Department of Internal and Specialty Medicine (Di.Bi.M.I.S.), A.O.U.P “Paolo Giaccone”, University of Palermo, Piazza delle Cliniche 2, 90127 Palermo, Italy.
Lynne Warner Stevenson, Cardiovascular Division, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA.
Mihai Gheorghiade, Center for Cardiovascular Innovation, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Nohria A, Tsang SW, Fang JC, et al. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003;41:1797–1804. doi: 10.1016/s0735-1097(03)00309-7. [DOI] [PubMed] [Google Scholar]
- 3.Gheorghiade M, Zannad F, Sopko G, et al. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112:3958–3968. doi: 10.1161/CIRCULATIONAHA.105.590091. [DOI] [PubMed] [Google Scholar]
- 4.Gelman S. Venous function and central venous pressure: a physiologic story. Anesthesiology. 2008;108:735–748. doi: 10.1097/ALN.0b013e3181672607. [DOI] [PubMed] [Google Scholar]
- 5.Gheorghiade M, De Luca L, Fonarow GC, et al. Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol. 2005;96:11G–17G. doi: 10.1016/j.amjcard.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Gheorghiade M, Filippatos G, De Luca L, et al. Congestion in acute heart failure syndromes: an essential target of evaluation and treatment. Am J Med. 2006;119:S3–S10. doi: 10.1016/j.amjmed.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Adamson PB, Magalski A, Braunschweig F, et al. Ongoing right ventricular hemodynamics in heart failure: clinical value of measurements derived from an implantable monitoring system. J Am Coll Cardiol. 2003;41:565–571. doi: 10.1016/s0735-1097(02)02896-6. [DOI] [PubMed] [Google Scholar]
- 8.Cotter G, Metra M, Milo-Cotter O, et al. Fluid overload in acute heart failure–re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail. 2008;10:165–169. doi: 10.1016/j.ejheart.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Androne AS, Hryniewicz K, Hudaihed A, et al. Relation of unrecognized hypervolemia in chronic heart failure to clinical status, hemodynamics, and patient outcomes. Am J Cardiol. 2004;93:1254–1259. doi: 10.1016/j.amjcard.2004.01.070. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson LW, Theodore E. Woodward Award: coming in out of the rain. Relieving congestion in heart failure. Trans Am Clin Climatol Assoc. 2009;120:177–187. [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell P, Drazner MH, Kato M, et al. Mismatch of right- and left-sided filling pressures in chronic heart failure. J Card Fail. 2011;17:561–568. doi: 10.1016/j.cardfail.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Rohde LE, Beck-da-Silva L, Goldraich L, et al. Reliability and prognostic value of traditional signs and symptoms in outpatients with congestive heart failure. Can J Cardiol. 2004;20:697–702. [PubMed] [Google Scholar]
- 13.Volpicelli G, Elbarbary M, Blaivas M, et al. International Liaison Committee on Lung Ultrasound (ILC-LUS) for International Consensus Conference on Lung Ultrasound (ICC LUS). International evidence -based recommendations for point-of-care lung ultrasound. Intensive Care Med. 2012;38(4):577–591. doi: 10.1007/s00134-012-2513-4. [DOI] [PubMed] [Google Scholar]
- 14.Celik G, Kara I, Yilmaz M, Apiliogullari S. The relationship between bioimpedance analysis, haemodynamic parameters of haemodialysis, biochemical parameters and dry weight. J Int Med Res. 2011;39(6):2421–2428. doi: 10.1177/147323001103900643. [DOI] [PubMed] [Google Scholar]
- 15.Adams KF, Jr, Fonarow GC, Emerman CL, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE) Am Heart J. 2005;149:209–216. doi: 10.1016/j.ahj.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Mahon NG, Blackstone EH, Francis GS, et al. The prognostic value of estimated creatinine clearance alongside functional capacity in ambulatory patients with chronic congestive heart failure. J Am Coll Cardiol. 2002;40:1106–1113. doi: 10.1016/s0735-1097(02)02125-3. [DOI] [PubMed] [Google Scholar]
- 17.Akhter MW, Aronson D, Bitar F, et al. Effect of elevated admission serum creatinine and its worsening on outcome in hospitalized patients with decompensated heart failure. Am J Cardiol. 2004;94:957–960. doi: 10.1016/j.amjcard.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 18.Ronco C, Haapio M, House AA, et al. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–1539. doi: 10.1016/j.jacc.2008.07.051. [DOI] [PubMed] [Google Scholar]
- 19.Triposkiadis F, Starling RC, Boudoulas H, et al. The cardiorenal syndrome in heart failure: cardiac? renal? syndrome? Heart Fail Rev. 2012;17(3):355–366. doi: 10.1007/s10741-011-9291-x. [DOI] [PubMed] [Google Scholar]
- 20.Topalian S, Ginsberg F, Parrillo JE. Cardiogenic shock. Crit Care Med. 2008;36(1 Suppl):S66–S74. doi: 10.1097/01.CCM.0000296268.57993.90. [DOI] [PubMed] [Google Scholar]
- 21.Nohria A, Hasselblad V, Stebbins A, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51:1268–1274. doi: 10.1016/j.jacc.2007.08.072. [DOI] [PubMed] [Google Scholar]
- 22.Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53:589–596. doi: 10.1016/j.jacc.2008.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872–878. doi: 10.1016/j.ejheart.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Ganda A, Onat D, Demmer RT, et al. Venous congestion and endothelial cell activation in acute decompensated heart failure. Curr Heart Fail Rep. 2010;7(66–74):25. doi: 10.1007/s11897-010-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Testani JM, Chen J, McCauley BD, et al. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122:265–272. doi: 10.1161/CIRCULATIONAHA.109.933275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney Injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damman K, Navis G, Voors AA, et al. Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J Card Fail. 2007;13:599–608. doi: 10.1016/j.cardfail.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Damman K, Masson S, Hillege HL, et al. Clinical outcome of renal tubular damage in chronic heart failure. Eur Heart J. 2011;32:2705–2712. doi: 10.1093/eurheartj/ehr190. [DOI] [PubMed] [Google Scholar]
- 29.Jungbauer CG, Birner C, Jung B, et al. Kidney injury molecule-1 and N-acetyl-beta-d glucosaminidase in chronic heart failure: possible biomarkers of cardiorenal syndrome. Eur J Heart Fail. 2011;13:1104–1110. doi: 10.1093/eurjhf/hfr102. [DOI] [PubMed] [Google Scholar]
- 30.Damman K, Van Veldhuisen DJ, Navis G, et al. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96:1297–1302. doi: 10.1136/hrt.2010.194878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Damman K, Chuen MJNK, MacFadyen RJ, et al. Volume status and diuretic therapy in systolic heart failure and the detection of early abnormalities in renal and tubular function. J Am Coll Cardiol. 2011;57:2233–2241. doi: 10.1016/j.jacc.2010.10.065. [DOI] [PubMed] [Google Scholar]
- 32.Emerman CL, Marco TD, Costanzo MR, Peacock WFt, for the ASAC Impact of intravenous diuretics on the outcomes of patients hospitalized with acute decompensated heart failure: insights from the ADHERE(R) Registry. J Card Fail. 2004;10:S116–S117. doi: 10.1159/000164149. [DOI] [PubMed] [Google Scholar]
- 33.Felker GM, O'Connor CM, Braunwald E. Loop diuretics in acute decompensated heart failure: necessary? Evil? A necessary evil? Circ Heart Fail. 2009;2:56–62. doi: 10.1161/CIRCHEARTFAILURE.108.821785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greither A, Goldman S, Edelen JS, et al. Pharmacokinetics of furosemide in patients with congestive heart failure. Pharmacology. 1979;19:121–131. doi: 10.1159/000137299. [DOI] [PubMed] [Google Scholar]
- 35.Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797–805. doi: 10.1056/NEJMoa1005419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piano MR, Prasun MA, Stamos T, Groo V. Flexible diuretic titration in chronic heart failure: where is the evidence? J Card Fail. 2011;17:944–954. doi: 10.1016/j.cardfail.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Metra M, Gheorghiade M, Bonow RO, Dei Cas L. Postdischarge assessment after a heart failure hospitalization: the next step forward. Circulation. 2010;122:1782–1785. doi: 10.1161/CIRCULATIONAHA.110.982207. [DOI] [PubMed] [Google Scholar]
- 38.Wilcox JE, Fonarow GC, Yancy CW, et al. Factors associated with improvement in ejection fraction in clinical practice among patients with heart failure: findings from IMPROVE HF. Am Heart J. 2012;163(49–56):e2. doi: 10.1016/j.ahj.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Lee DS, Stukel TA, Austin PC, et al. Improved outcomes with early collaborative care of ambulatory heart failure patients discharged from the emergency department. Circulation. 2010;122:1806–1814. doi: 10.1161/CIRCULATIONAHA.110.940262. [DOI] [PubMed] [Google Scholar]
- 40.Zile MR, Bennett TD, Sutton MSJ, et al. Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circulation. 2008;118:1433–1441. doi: 10.1161/CIRCULATIONAHA.108.783910. [DOI] [PubMed] [Google Scholar]
- 41.Blair JE, Khan S, Konstam MA, et al. Weight changes after hospitalization for worsening heart failure and subsequent rehospitalization and mortality in the EVEREST trial. Eur Heart J. 2009;30:1666–1673. doi: 10.1093/eurheartj/ehp144. [DOI] [PubMed] [Google Scholar]
- 42.Ritzema J, Troughton R, Melton I, et al. Hemodynamically Guided Home Self-Therapy in Severe Heart Failure Patients (HOMEOSTASIS) Study Group Physician-directed patient self-management of left atrial pressure in advanced chronic heart failure. Circulation. 2010;121:1086–1095. doi: 10.1161/CIRCULATIONAHA.108.800490. [DOI] [PubMed] [Google Scholar]
- 43.Alderman MH, Cohen HW. Dietary sodium intake and cardiovascular mortality: controversy resolved? Curr Hypertens Rep. 2012;14:193–201. doi: 10.1007/s11906-012-0275-6. [DOI] [PubMed] [Google Scholar]
- 44.Cohen HW, Hailpern SM, Alderman MH. Sodium intake and mortality follow-up in the Third National Health and Nutrition Examination Survey (NHANES III) J Gen Intern Med. 2008;23:1297–1302. doi: 10.1007/s11606-008-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Donnell MJ, Yusuf S, Mente A, et al. Urinary sodium and potassium excretion and risk of cardiovascular events. JAMA. 2011;306:2229–2238. doi: 10.1001/jama.2011.1729. [DOI] [PubMed] [Google Scholar]
- 46.Parrinello G, Di Pasquale P, Licata G, et al. Long-term effects of dietary sodium intake on cytokines and neurohormonal activation in patients with recently compensated congestive heart failure. J Card Fail. 2009;15:864–873. doi: 10.1016/j.cardfail.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Graudal NA, Hubeck-Graudal T, Jürgens G. Effects of low-sodium diet vs. high-sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride (Cochrane Review) Am J Hypertens. 2012;25:1–15. doi: 10.1038/ajh.2011.210. [DOI] [PubMed] [Google Scholar]
- 48.Lennie TA, Song EK, Wu JR, et al. Three gram sodium intake is associated with longer event-free survival only in patients with advanced heart failure. J Card Fail. 2011;17:325–330. doi: 10.1016/j.cardfail.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aliti GB, Rabelo ER, Clausell N, et al. Aggressive Fluid and Sodium Restriction in Acute Decompensated Heart Failure: a Randomized Clinical Trial. JAMA Intern Med. 2013;20:1–7. doi: 10.1001/jamainternmed.2013.552. [DOI] [PubMed] [Google Scholar]
- 50.Paterna S, Parrinello G, Cannizzaro S, et al. Medium term effects of different dosage of diuretic, sodium, and fluid administration on neurohormonal and clinical outcome in patients with recently compensated heart failure. Am J Cardiol. 2009;103:93–102. doi: 10.1016/j.amjcard.2008.08.043. [DOI] [PubMed] [Google Scholar]
- 51.Paterna S, Gaspare P, Fasullo S, et al. Normal-sodium diet compared with low-sodium diet in compensated congestive heart failure: is sodium an old enemy or a new friend? Clin Sci. 2008;114:221–230. doi: 10.1042/CS20070193. [DOI] [PubMed] [Google Scholar]
- 52.Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day read-mission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303:1716–1722. doi: 10.1001/jama.2010.533. [DOI] [PubMed] [Google Scholar]
- 53.Parrinello G, Torres D, Paterna S, et al. Early and personalized ambulatory follow-up to tailor furosemide and fluid intake according to congestion in post-discharge heart failure. Intern Emerg Med. 2013;8(3):221–228. doi: 10.1007/s11739-011-0602-y. [DOI] [PubMed] [Google Scholar]
- 54.Kircher BJ, Himelman RB, Schiller NB. Noninvasive estimation of right atrial pressure from the inspiratory collapse of the inferior vena cava. Am J Cardiol. 1990;66:493–496. doi: 10.1016/0002-9149(90)90711-9. [DOI] [PubMed] [Google Scholar]
- 55.Pepi M, Tamborini G, Galli C, et al. A new formula for echo-Doppler estimation of right ventricular systolic pressure. J Am Soc Echocardiogr. 1994;7:20–26. doi: 10.1016/s0894-7317(14)80414-8. [DOI] [PubMed] [Google Scholar]
- 56.Brennsn JM, Blair JE, Goonewardena S, et al. Reappraisal of the use of inferior vena cava for estimating right atrial pressure. J Am Soc Echocardiogr. 2007;20:857–861. doi: 10.1016/j.echo.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Jue J, Chung W, Schiller NB. Does inferior vena cava size predict right atrial pressures in patients receiving mechanical ventilation? J Am Soc Echocardiogr. 1992;5:613–619. doi: 10.1016/s0894-7317(14)80327-1. [DOI] [PubMed] [Google Scholar]
- 58.Traversi E, Cobelli F, Pozzoli M. Doppler echocardiography reliably predicts pulmonary artery wedge pressure in patients with chronic heart failure even when atrial fibrillation is present. Eur J Heart Fail. 2001;3:173–181. doi: 10.1016/s1388-9842(00)00140-9. [DOI] [PubMed] [Google Scholar]
- 59.Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur J Echocardiogr. 2009;10:165–193. doi: 10.1093/ejechocard/jep007. [DOI] [PubMed] [Google Scholar]
- 60.Kusunose K, Yamada H, Nishio S, et al. Clinical utility of single-beat E/e′ obtained by simultaneous recording of flow and tissue Doppler velocities in atrial fibrillation with preserved systolic function. JACC Cardiovasc Imaging. 2009;2:1147–1156. doi: 10.1016/j.jcmg.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 61.Cibinel GA, Casoli G, Elia F, et al. Diagnostic accuracy and reproducibility of pleural and lung ultrasound in discriminating cardiogenic causes of acute dyspnea in the emergency department. Intern Emerg Med. 2012;7:65–70. doi: 10.1007/s11739-011-0709-1. [DOI] [PubMed] [Google Scholar]