

Abstract
Immunologically immature neonates suffer the highest incidence of paediatric sepsis. Postnatal immunological maturation is characterized by a relatively hypo-inflammatory immune response. The mechanisms that differentiate the mature and immature immune responses resemble those that differentiate the hyper- and hypo-inflammatory responses in severe sepsis. Immunological maturational differences likely affect the neonate's ability to mount an appropriate hyper-inflammatory response, a counteractive hypo-inflammatory response, and subsequent return to immune system homeostasis. To better understand the role of the hypo-inflammatory response in paediatric sepsis, we will explore the maturation of the immune system and the effect it may have on the sepsis-induced hypo-inflammatory response.
Keywords: endotoxin tolerance, immune maturation, immune tolerance, neonatal sepsis, paediatric sepsis
Introduction
Severe sepsis is a systemic mixed, pro- and anti-inflammatory response to an infectious organism and severe tissue injury (Table1). Commonly, the exaggerated inflammatory response is associated with progression to organ dysfunction and failure.1 The prevailing model of the severe sepsis response recognizes a pro-inflammatory response and a temporally variable counter-regulatory anti-inflammatory response, characterized by a hypo-inflammatory innate immune response and an impaired adaptive immune response.1 In clinical studies, the presence and persistence of a hypo-inflammatory state in severe sepsis is associated with an increased risk of secondary infections and death.2 However, whether the hypo-inflammatory response is mechanistically associated with progression to and persistence of organ failure remains controversial.
Table 1.
Definitions
| Hyper-inflammatory response the net effect of the immunological response is characterized by the production of pro-inflammatory cytokines and activation of leucocytes1 |
| Hypo-inflammatory response the net effect of the immunological response is characterized by the production of anti-inflammatory cytokines, negative regulators of Toll-like receptor signalling, and decreased innate-adaptive immune system communication1 |
| Immune tolerance (IT) a transient state seen in cells of the innate immune system after repeated exposure to low concentrations of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), after which they are unable to respond normally to further PAMP or DAMP exposure1 |
| Endotoxin tolerance (ET) a transient state seen in cells of the innate immune system after repeated exposure to low concentrations of endotoxin after which they are unable to respond normally to endotoxin challenge34 |
| Cross-tolerance the transient state of IT to one PAMP or DAMP induced by low exposure to a different PAMP or DAMP34 |
Paediatric sepsis in the USA disproportionately affects the neonatal population with a prevalence of 9·7/1000 neonates. This contrasts with 2·25/1000 in non-newborn infants and 0·23–0·52/1000 in older children.3 Postnatally, the immune system undergoes rapid maturation, approaching adult maturity by 2 years of age, with continued maturation into the second decade of life4 (Fig.1). The normal neonatal immune response is relatively hypo-inflammatory compared with the typical adult response.5 Interestingly, the mechanisms that differentiate the mature adult immune response from the immature neonatal response recapitulate the hyper-inflammatory and hypo-inflammatory states in sepsis (Table2).
Figure 1.
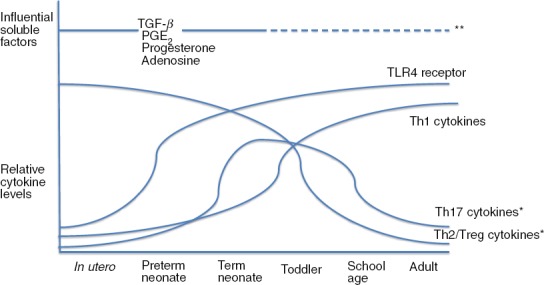
Age-dependent changes in endotoxin-induced T-cell responses. The T-cell response in utero and in preterm and term neonates is characterized by predominantly T helper type 2 (Th2) and regulatory T (Treg) responses. As the immune system matures early in life, these responses are replaced by the more mature Th1 response. Toll-like receptor 4 (TLR4) expression is relatively low in utero and in very premature neonates, increasing to adult levels later in utero. Maternal- and fetal-derived soluble factors [transforming growth factor-β (TGFβ), prostaglandin E2 (PGE2), progesterone and adenosine] influence the T-cell response in utero and in early postnatal life. *Conflicting data exist regarding the capacity of the neonatal Th17 and Treg responses. **Influences of these soluble factors in adulthood are unclear but may be important.4,5,9,10,12,13,16,18–20,27,28
Table 2.
Comparison of the immature, mature and tolerant responses to endotoxin stimulation
| Immature immune response relative to mature response | Relative effect of difference | Tolerance recapitulates immature immune response? |
|---|---|---|
| Cellular adhesion functions | ||
| ↓Leucocyte adhesion and extravasation5 | Impaired ability to fight infections5 | Yes20 |
| PRR/Signal transduction | ||
| ↓TLR4 receptor expression13,18,20,22 | Minimal effects13,18,20,22 | Yes8,21,37 |
| ↓Soluble TLR4 co-receptor expression18 | Minimal effects18 | Yes8,21,37 |
| ↓TLR/MyD88 pathway signalling14,23,24 | ↓ROS production24 | Yes2,8,21,37 |
| Regulation of the adaptive response | ||
| ↑Th2-skewing cytokine production4,12,14,18 | ↑Humoral immune response33 | Yes2,21 |
| ↓Th1-skewing cytokine production4,12,14,18 | ↓Pro-inflammatory immune response4,12,14,18 | Yes2,21 |
| ↓Stimulated T-cell proliferation4 | ↓Cytotoxic T-cell response and ↓Adaptive response4,24 | Yes44 |
| ↑Th17/Treg differentiationa13,16,20,22,23,27 | ↑Anti-inflammatory response13,16,20,22,23,27 | Unknown |
| Soluble factors | ||
| ↑Maternally produced factors (TGF-β, progesterone, PGE2)12 | ↑Th2-skewed response12 | Similar PGE2 (macrophage-produced)21 |
| ↑Adenosine12,18–20,31 | ↑Th2-skewed response12,18–20,31 | Unknown |
| ↓Antimicrobial protein peptides31 | Impaired ability to fight infections31 | Unknown |
| Cell–cell interactions | ||
| ↓CD8+ T-cell activation24 | ↓Cytotoxic response24 | Yes8 |
| ↓CD4+ stimulation by APCs33 | ↑Th2-skewed response33 | Yes2 |
| ↑Apoptosis of CD4+ Th1 cells24 | ↑Th2-skewed response24 | Yes2 |
| ↓Memory T cells9 | ↓Eradication of viral and intracellular pathogens5,13 | Unknown |
PGE2, prostaglandin E2; PRR, pattern recognition receptor; ROS, reactive oxygen species; TGF-β, transforming growth factor-β; Th2, T helper type 2; TLR, Toll-like receptor; Treg, regulatory T.
Conflicting evidence exists regarding the impact of Th17 and Treg differentiation in the immature immune response.
The differing responses based on the immune system's maturational state are likely to play a role in a child's ability to mount an appropriate hyper-inflammatory response, use the hypo-inflammatory response to control and curtail ongoing harmful inflammation, and re-establish immune homeostasis. To more fully understand the potential role of the hypo-inflammatory responses, we explore the developmental differences between the mature and immature immune response to endotoxin and the immune regulatory effects in neonatal and paediatric sepsis. We also explore the role of developmental immune recapitulation in the mature immune responses that characterize sepsis.
The typical immune response to endotoxin
The canonical immune response to endotoxin has been thoroughly reviewed by Kawai and Akira6,7 and Medzhitov and Janeway.6,7 In brief, the cells of the innate immune system recognize foreign pathogens (pathogen-associated molecular patterns, PAMPS) and damage-associated molecules (DAMPS) through cell-surface expression of pattern-recognition receptors [PRR, e.g. Toll-like receptors (TLRs)]. The TLRs transduce the extracellular signal to intracellular domains through adapter protein signalling cascades (e.g. TRAM, TRIF, TIRAP, MyD88)8 (Fig.2). These signalling cascades form the initial response to endotoxin and create the environment in which T cells of the adaptive immune system are stimulated. Naive CD4+ and CD8+ T cells differentiate into one of several types of effector and memory T helper cells based on co-stimulatory molecules expressed on antigen-presenting cells (APCs) and the cytokine milieu within which they are stimulated5,9 (Table3; Fig.3). The differentiation into particular effector T-cell subtypes defines the quality and effectiveness of the adaptive response to a specific infection. CD4+ T cells include T helper type 1 (Th1) cells that produce a primarily pro-inflammatory response, Th2 cells that stimulate an antibody-mediated response in addition to combating parasitic infections and promoting allergic reactions, regulatory T (Treg) cells that are immune suppressive, and Th17 cells that fight extracellular bacteria10–17 (Table3). The interaction between the innate and adaptive responses depends on a coordinated sequence of developmental processes and is fundamental to the maturation-dependent functional difference between neonatal and adult immune responses.18
Figure 2.
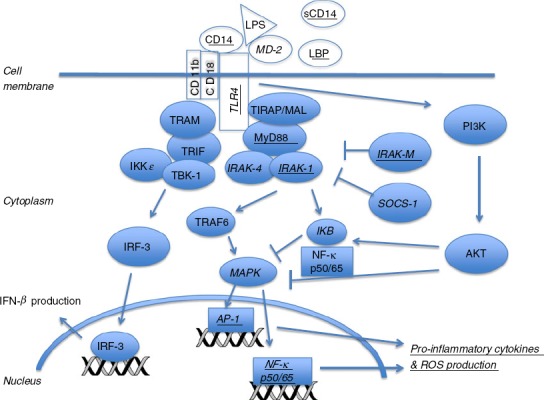
Toll-like receptor 4 (TLR4) signalling. Lipopolysaccharide (LPS) binding to TLR4 with co-stimulatory molecules, CD14, lipopolysaccharide binding protein (LBP), CD11b, CD18 and MD-2, recruits MyD88-dependent and -independent intracellular proteins. MyD88-independent pathway: signalling proteins, TRAM and TRIF, and kinases, IKKε and TBK-1, are recruited to the intracellular domain of TLR4. These are activated and phosphorylate interferon regulatory factor 3 (IRF-3). Phosphorylated IRF-3 translocates into the nucleus for transcription of interferon-β. MyD88-dependent pathway: MyD88 and TIRAP/MAL are recruited to the intracellular domain of the TLR4 receptor. IRAK-4 and IRAK-1 are activated via phosphorylation. IRAK-1 interacts with TRAF6, inducing activation of MAPK and transcription of AP-1 and NF-κB genes to produce pro-inflammatory cytokines and reactive oxygen species (ROS). Activation of the PI3K–AKT pathway results in impaired AP-1/NF-κB cytokine production. IRAK-M negatively regulates MyD88-pathway signalling.34 Underlined co-stimulatory molecules and signalling proteins represent those that are differentially expressed in the immature immune response. Italicized co-stimulatory molecules and signalling proteins represent those that are differentially expressed in the endotoxin and immune tolerant responses.
Table 3.
Inducing factors, cytokine production and functions of T-cell subtypes
| T-cell subtype | Effects of immune system immaturity on differentiation and/or activity of T-cell subtypes | Inducing factors | Cytokines produced | T-cell functions |
|---|---|---|---|---|
| CD8+ | ↓ | APC MHC Class I9 | IFN-γ, TNF-α11 | Memory & Effector Cytolysis, pro-inflammatory, intracellular bacteria clearance10,11 |
| CD4+ | APC MHC Class II11 | Memory & Effector | ||
| Th1 | ↓ | IL-12, HMGB1, IFN-γ12,15 | IFN-γ, TNF-α, IL-2, IL-1β11,15 | Pro-inflammatory, intracellular pathogen clearance, induces cell-mediated immune response15 |
| Th2 | ↑ | IL-4, IL-33, IL-25, or absence of Th1 promoting cytokines12,15 | IL-4, IL-10, IL-13, IL-511,12,15 | Enhance antibody-mediated responses, induce allergic reactions, fight parasitic infections11,15 |
| Treg | ↑1 | IL-10, TGF-β15 | IL-10, IL-35, TGF-β15 | Immune suppression, balance Th1/Th2 responses10,17 |
| Th17 | ↑1 | IL-6, TGF-β15 | IL-17, IL-21, IL-22, IL-2611,15 | Neutrophil recruitment and activation, extracellular bacteria clearance, enhance mucosal host defence15 |
APCs, antigen-presenting cells; HMGB1, high mobility group B1; IFN-γ, interferon-γ; IL-2, interleukin-2; TGF-β, transforming growth factor-β; Th2, T helper type 2; TNF-α, tumour necrosis factor-α; Treg, regulatory T.
Conflicting evidence exists regarding the impact of Th17 and Treg cell differentiation in the immature immune response.Bold indicate the general roles of the CD8+ and CD4+ Tcell subtypes.
Figure 3.
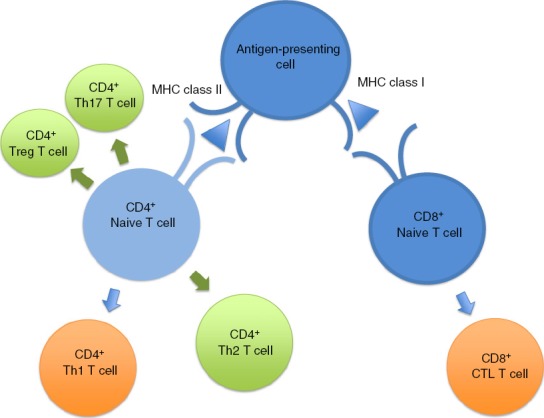
The innate immune response directs differentiation of T-cell subsets and the adaptive immune response: predominant immature versus mature responses. The antigen-presenting cells of the innate immune system stimulate CD4+ and CD8+ naive T cells via antigen (triangle) MHC class II and I interactions, respectively. The differentiated green T-cell subsets represent the predominant immature T-cell subtypes produced in response to stimulation [CD4+ T helper type 17 (Th17), regulatory T (Treg) and Th2 cells]. The differentiated orange T-cell subsets represent the predominant mature T-cell subtypes produced in response to stimulation [CD4+ Th1 and CD8+ cytotoxic T lymphocyte (CTL) T cells].4,5,9,10,12,13,16,18–20,27,28
The immature immune response
The immature immune response in preterm and term neonates places them at high risk of developing sepsis.5 Although some studies conclude that the neonatal inflammatory response is similar to an adult's after a highly toxic exposure, less potent PAMP activation consistently results in an attenuated pro-inflammatory response in neonates compared with an adult exposed to a similar stimulus.10,13,18–20 These data indicate that neonatal phagocytes are less responsive to PAMP stimulation, resulting in relatively deficient adhesion and extravasation. Sub-maximal cytokine proliferation, preferential Th2-skewed cytokine production, and decreased antigen-presenting capacity result in a globally hypo-inflammatory immune response.5 A comprehensive understanding of the functional differential endotoxin activation of the immature versus mature immune systems can be explored from four interacting perspectives: PAMP signal transduction, cytokine release, plasma components and cell–cell interactions.
Pathogen recognition and signal transduction
Transduction of the prototypical endotoxin immune signal is initiated by PAMP–PRR binding and is regulated at multiple levels including the monocyte cell surface receptors, intracellular signalling molecules, mRNA transcription, protein translation, cytokine secretion and protein ubiquitination pathways8,11,21 (Fig.2).
Signal transduction begins with the TLR4 receptor, which has lower cell surface density expression in very premature infants compared with neonates delivered after 30 weeks of gestation. From that latter time-point, TLR4 expression approximates that of adults.13,18,20,22 Initiation of the neonatal TLR4 response is impaired by lower soluble co-receptor expression and impaired up-regulation of the CD14 co-receptor expression after stimulation.18 However, these deficiencies probably play a minor role in attenuating the neonatal pro-inflammatory response.18,19,23
Downstream, activation of the neonatal TLR4/MyD88 pathway results in decreased production of reactive oxygen species (ROS) compared with the mature adult response. This difference significantly hinders the neonate's ability to clear infection.24 The clinical effects of this impairment are best exemplified by impaired ROS production in patients with chronic granulomatous disease who suffer recurrent, severe invasive bacterial and fungal infections.25
Further evaluation of the individual steps in the TLR4/MyD88 pathway reveals differential expression of signal transducing proteins based on immune maturation status. Reported in a systematic comparison of the TLR4 signalling pathways of adults and neonates, similar basal cell surface receptor and intracellular signalling protein expression Levy et al.14 Separately, Yan et al. demonstrated that differential tumour necrosis factor-α (TNF-α) production is due, in part, to decreased MyD88 expression in neonatal monocytes after low-dose endotoxin stimulation.23 However, high-dose endotoxin stimulation abolished this differential effect, suggesting a MyD88-independent pathway with receptor-saturating insults.23
Hence, the initial immature innate immune response is effectively inhibited at the level of intracellular signal transduction with evidence of some ability to overcome this inhibition with high-dose stimulation.
Regulation of the immature adaptive immune response
Cytokine regulation
Discrete cytokine profiles in response to TLR4 stimulation typify the neonatal versus adult responses to monocyte stimulation. While ex vivo stimulation of neonatal monocytes in isolation reveals a similar maximal cytokine production capacity to that of adults, endogenous cytokine production appears to be heavily dependent on soluble plasma-derived mediators.10,12,19,20,23,26 Neonatal monocytes suspended in autologous plasma produce less TNF-α and higher concentrations of cytokines that induce Th2-skewing following low-dose endotoxin stimulation.4,12,14,18 Furthermore, relatively deficient interferon-γ production results in further skewing towards a Th2 T-cell response4,12,14,18 (Fig.3). In association with maturational cytokine changes, the magnitude and nature of the T-cell response is similarly skewed towards a Th2 response and away from a Th1 response in the immature immune system4,5,9,10,12,13,16,18–20,27,28 (Fig.3).
Lavoie et al. reported clinically relevant Th1 and additional Th17 deficiencies in the most immature immune response of premature neonates. They showed significantly lower post-stimulation production of the p40 subunit common to interleukin-12 (IL-12) and IL-23 in extremely premature neonatal cord blood compared with term cord blood.29 This difference is observed despite similar numbers of monocyte-derived cells responsible for production of these cytokines. The reduced IL-12 production in these preterm neonates results in deficiency of the Th1 response. Additionally, the deficient IL-23 production results in an impaired Th17 response. More significantly, in a nested case–control cohort study of neonates matched for gestational age and birthweight, Lavoie et al.29 reported that those infants with lower production of the IL-12 and IL-23 p40 subunit had an increased risk of early onset neonatal sepsis despite higher levels of other inflammatory cytokines. These findings implicate a clinically relevant deficiency in Th17 and Th1 stimulating cytokines in the most immature immune response of preterm neonates.
Further evaluations in term neonates have yielded conflicting evidence regarding their ability to mount appropriate Th17 and Treg responses.13,22,23 Schaub et al.16 describe decreased Treg proliferation and less effective Treg anti-inflammatory effects. In contrast, Dijksta et al. demonstrate preferential Treg cell differentiation until 1 year of age.27 Further evidence describes differential Treg cell activity based on atopic phenotype wherein atopic children display delayed Treg cell maturation.30 Dijksta et al. provide further evidence that neonates have an inability for Th17 differentiation until 3 months of age after which Th17 differentiation is no longer inhibited.27 This contrasts with evidence suggesting that the Th17 response predominates at birth and decreases over the first year of life.20 While incompletely understood, the balance of Th1 and Th2 phenotypes and regulation of the pro-inflammatory and anti-inflammatory responses to pathogens that are dependent on the proliferation of Th17 and Treg cell types is an area that warrants further study.16,31
In addition to differential stimulation by the innate immune system, impaired T-cell cytokine production also attenuates the neonatal hyper-inflammatory response through limitation of T-cell proliferation. T-cell levels are high at birth, increase throughout the first year of life, and subsequently stabilize to adult levels by school age. However, relatively impaired IL-2 production results in sub-optimal T-cell function and proliferative capacity after stimulation.4
Over the first few years of life, the neonatal-predominant Th2 response shifts to a more mature Th1 response.20 Although incompletely elucidated, evidence suggests that soluble plasma factors, in addition to the cytokines described above, play a dominant role in directing the immature to mature transition.14
Other soluble factors
Neonatal haemocytes suspended in adult plasma demonstrate a significantly greater capacity to produce TNF-α compared with suspension in autologous plasma. Conversely, adult cells suspended in neonatal plasma yield reduced TNF-α production capacity.26 These observations suggest the presence of an agonist in adult plasma and/or an inhibitor in neonatal plasma. Further, both neonatal and adult monocytes display enhanced responses to low-dose endotoxin stimulation after adult plasma incubation compared with stimulation in media alone.23 Additionally, Kollmann et al.19 report greater maturity-based differences in TNF-α production after endotoxin stimulation of whole blood compared with monocytes suspended in medium alone. Of note, neonatal monocytes, particularly when stimulated in whole blood, elaborate a more constricted cytokine profile compared with adult cells.19 Taken together, these studies strongly suggest that soluble factors are a significant maturation-associated and independent determinant of differential monocyte-derived pro-inflammatory cytokine production after endotoxin stimulation.
Of the many putative soluble factors, extracellular adenosine, a purine metabolite, is established as a key factor in directing the character of the neonatal immune response. Adenosine's disruption of the cAMP–mitogen-activated protein kinase pathway impairs production of Th1 cytokines while enhancing production of Th2 and Th17 skewing cytokines.12,18–20 Additionally, adenosine inhibits neutrophil–endothelial adhesion and attenuates T-cell proliferation and effector functions.31 Neonatal blood has a fourfold higher concentration of adenosine and a 20-fold higher concentration of cAMP when compared with adult levels.18,20 Furthermore, neonatal monocytes are more sensitive to the inhibitory effects of adenosine.20,23
Additional, potentially influential factors that impair effective resistance to infection are antimicrobial protein peptides such as cathelicidin, lactoferrin, bactericidal/permeability-increasing protein and α- and β-defensins.31 Likewise, maternally produced factors such as transforming growth factor-β, prostaglandin E2 and progesterone further skew lymphocytes towards a Th2 response in utero and postnatally.12 The differential concentrations of these soluble factors probably play a major role in the differential immature and mature immune response.
Cell–cell interactions
The regulatory effects of soluble factors on the adaptive immune response are strongly augmented by immune cell–cell interactions.10,12,19 The APCs are the cellular bridge between the innate and adaptive immune systems and regulate lymphocyte responses through direct interaction with naive T cells and via cytokine production. Relative to adults, neonatal CD8+ T cells respond more rapidly to infection but are unable to differentiate into memory CD8+ T cells because they undergo terminal differentiation.32 Additionally, the neonatal cytokine environment is characterized by lower IL-12 and higher IL-10, resulting in suboptimal activation of CD8+ T cells and a weakened cytotoxic response.24 Clinically, the high prevalence of severe viral infections in children younger than 2 years is likely to be a manifestation of the deficient cytotoxic lymphocyte response.5 Conversely, neonatal CD8+ T cells are capable of producing strong cytotoxic responses when stimulated in the presence of Th1-promoting agents.10 Hence, when stimulated in the appropriate environment, neonatal CD8+ T cells can produce mature cytotoxic responses, again suggesting a significant influence of cytokines and other soluble factors.
In addition, there is a maturation-dependent skewing in HLA class-restricted CD4+ T-cell subsets. Neonatal and adult immature APCs have similar basal expression of MHC Class II receptors and CD80 and CD28 co-stimulatory molecules12 (Fig.3). However, after endotoxin stimulation, adult APCs demonstrate a more pronounced up-regulation of receptors and co-stimulatory molecules compared with neonatal APCs.33 This relatively ineffective response in neonatal cells produces a weakened stimulus of CD4+ T cells to produce interferon-γ, resulting in the Th2-skewed response previously described33 (Table3). Additional evidence supports a Th2-skewed response due to apoptosis of CD4+ Th1 cells, but not Th2 cells.24 Furthermore, lack of antigen-driven memory T cells further impairs the neonatal Th1 response. Naive T cells have a higher stimulation threshold, are more reliant on co-stimulatory molecules, produce less IL-2, and proliferate poorly compared with memory T cells.2,9 This results in a deficient Th1, pro-inflammatory, cell-mediated response and a relatively impaired ability to eradicate viral infections and intracellular pathogens.5,13 Instead, the Th2-skewed response is functionally effective in developing immunological memory33 (Table3). As the neonate gains memory T cells over the first year of life, their lymphocyte activation threshold decreases and pro-inflammatory cytokine production increases.9
Perinatal immune response as an adaptation
The neonate's hypo-inflammatory response may appear teleologically counterproductive; however, a blunted pro-inflammatory response may be functionally protective.10,14,18,20,29 The dampened immune response in utero is thought to decrease the risk of alloimmune reactions between the mother and fetus.18 Furthermore, in the intrauterine environment, a hyper-inflammatory response is repressed unless a stimulus, such as a significant infection, is of considerable force to activate the fetus to speed lung maturity and remove itself from the infected environment by stimulating preterm labor.29 Unnecessarily eliciting this type of response would likely prove maladaptive.
After delivery, the neonate transitions from the sterile intrauterine environment to the microbe-rich world, newly exposed to a massive number of antigens, both intrinsic and extrinsic. It is suspected that the hypo-inflammatory response is beneficial as a full Th1 and/or Th17 response could induce detrimental hyper-inflammatory and/or autoimmune reactions.10,29 For example, neonatal intestinal epithelial cells have been shown to be particularly hypo-inflammatory after endotoxin stimulation. Mechanistically, these cells down-regulate the expression of interleukin receptor-associated kinase 1 (IRAK 1), a key mediator in TLR4 signalling, and increase expression of IRAK-M, a negative regulator of this pathway.14,20 This response may be protective in that it allows the neonate to tolerate colonization as it forms an intestinal microbiome. Further supporting this theory, the development of necrotizing enterocolitis in preterm neonates is characterized by a dysregulated, hyper-inflammatory response.18,20 Although both the fetus and neonate maintain a hypo-inflammatory environment with low-level stimuli, their respective immune systems are able to produce a more mature hyper-inflammatory response when stimulated aggressively.10 This maturational difference favours a safe perinatal transition but feasibly increases the risk that amplified immune activation or infection could have paradoxical pathological effects to a greater extent in neonates than adults.
Endotoxin tolerance as a recapitulation of immature immune function
Endotoxin tolerance (ET) in the mature immune system shares several features with the immature neonatal immune response (Table2). Endotoxin tolerance, the reduced pro-inflammatory response observed in monocytes after repeated endotoxin exposure, is a transient component of the normal immune response seen in humans and animals.34 Additionally, the phenomenon of cross-tolerance, the hypo-inflammatory response of endotoxin tolerized monocytes upon re-stimulation with Gram-positive PAMPs, demonstrates that this response is not specific to endotoxin exposure.34 As such, the concept of immune tolerance (IT) is defined even more broadly as a transient state seen in cells of the innate immune system after repeated exposure to low concentrations of PAMPs and/or DAMPs after which they are unable to respond normally to further PAMP and/or DAMP exposure1 (Table1). Endotoxin tolerance is a marker of innate immune exhaustion and is mechanistically discrete from adaptive T-cell tolerance characterized as a lack of reactivity to self-antigens or foreign tissue antigens.35
Endotoxin tolerance is highly recapitulative of the normal neonatal immune condition; tolerized phagocytes such as neutrophils, monocytes and dendritic cells in adults exhibit decreased adhesion and extravasation, produce fewer pro-inflammatory cytokines and have impaired antigen-presenting capacity.20 Additionally, the ET phenotype is associated with decreased neutrophil recruitment and reduced efficacy of endotoxin-induced ROS production similar to what is observed in neonates.2,36 Furthermore, tolerized innate immune cells produce an altered cytokine profile, shifting the T-cell response from a Th1 to a Th2 response.2 In the following sections, we will compare the mechanisms that induce the hypo-inflammatory state in neonates with that of experimental ET. Their similarities could provide insights regarding regulatory mechanisms and potential therapeutic targets.
PAMP recognition and signal transduction alterations in endotoxin tolerance
Similar to the immature immune response, decreased expression of TLR4 and co-receptors (MD-2) have been described in ET, however, these alterations do not appear to be solely responsible for the induction of ET.8,21,37 Instead, downstream alterations in the MyD88/TLR4 signalling cascade described in tolerized murine models and humans are probably more significant.8 In human and mouse cell lines, several steps in the TLR4/MyD88 pathway are shown to be inhibited in ET. Examples include decreased IRAK adapter protein levels, impaired p38 MAPK activation, suppressed degradation of inhibitor of κB (IκB), and increased presence of p50 nuclear factor-κB (NF-κB) homodimers, all resulting in impaired transcription of pro-inflammatory cytokines in the NF-κB and activator protein-1 pathways37 (Fig.2). Additionally, IRAK-M, a negative regulator of the MyD88/TLR4 pathway, is expressed only after the initial endotoxin exposure.2 IRAK-M inhibits signal propagation down the MyD88/TLR4 pathway by impairing degradation of IκB, an enzyme complex that binds NF-κB and inhibits intra-nuclear translocation, resulting in decreased production of pro-inflammatory cytokines and ROS21 (Fig.2). The inability of IRAK-M-deficient mice to display ET further supports its role as a primary mediator of ET.2 Similarly, murine evidence suggests that SOCS-1 acts as a negative regulator of lipopolysaccharide signalling through interaction with the adapter protein, IRAK.21
Alterations in cytokines and other soluble factors in endotoxin tolerance
Similar to the neonatal immune response, soluble factors play a significant role in the induction of ET. Stimulated tolerized monocytes preferentially produce Th2 skewing cytokines and fail to produce pro-inflammatory cytokines.2 As discussed previously, this affects the innate immune response as well as the quality of the adaptive immune response, creating weakened cytotoxic and pro-inflammatory responses and skewing towards a more humoral and relatively anti-inflammatory adaptive response. One particular cytokine, IL-10, has been of particular interest in ET because of its anti-inflammatory properties. It has been evaluated as a sole inducer of the ET response. However, despite its capacity to induce down-regulation of pro-inflammatory cytokines, IL-10-deficient mice can develop ET, suggesting that IL-10 is not the sole mediator of the ET response.21
Prostaglandin E2, an arachidonic acid metabolite, is produced by macrophages after re-stimulation with endotoxin and suppresses pro-inflammatory cytokine production in macrophages and lymphocytes.21 Similar to the perinatal response, prostaglandin E2 appears to play a role in induction of the ET response. Further evaluation of soluble factors such as adenosine may reveal more similarities between the neonatal and ET responses that have not yet been studied.
Cell–cell interactions
A well-characterized phenomenon in studies of human tolerized monocytes is the reduced levels of MHC class II expression, functionally impairing their capacity for antigen presentation.8 This impaired ability for antigen presentation hinders the ability of the innate immune response to activate the adaptive response.
Clinical relevance: immune tolerance in sepsis
The regulation of the TLR4/MyD88 signalling cascade, impaired antigen presentation, and differential production of cytokines attenuate the pro-inflammatory Th1 response in the innate and adaptive immune systems similarly in neonates and ET models. In well-controlled murine models, induction of an ET response yields a survival advantage.2,36 This is attributed to significant repression of pro-inflammatory cytokine release that is causatively associated with organ injury. However, clinical evidence in humans points to an increased mortality and secondary infections in patients with evidence of the more generalized phenomenon, IT. This association has been seen in patients suffering from acute inflammatory illnesses such as myocardial infarctions, trauma, sepsis and cardiac surgery.38–43 Taken together, currently available evidence describing the adaptive or maladaptive nature of IT in neonatal and adult humans remains inconclusive. The aggregate biological effect appears to depend on the degree, timing and duration. However, the bulk of the evidence points to the tolerance response as being an essential adaptive component in early immune maturation.
The IT phenomenon has been described extensively in patients with sepsis.1,2,39–41,43,44 The prevailing model posits that the initial hyper-inflammatory response is temporally related with a counter-regulatory hypo-inflammatory IT response. Hence, the patient's immunological state is determined by the phenotype of the net response.1 The IT response is thought to be teleologically adaptive, resulting in abrogated propagation of the injurious hyper-inflammatory response. However, just as the hypo-inflammatory neonatal response although adaptive, may, in some circumstances, place the neonate at greater risk of severe infection, the IT response in severe sepsis may have a threshold above which it also becomes maladaptive.5 Described mechanisms of IT induction in sepsis patients include alterations in inflammatory signal transduction, cytokine production, and apoptosis of lymphocytes.2,44
In sepsis, the IT phenotype is pleiotropic and efforts to evaluate immune capacity rely on surrogate markers including monocyte HLA-DR expression and exogenous cytokine responses to PAMP stimulation such as quantification of TNF-α production after ex vivo endotoxin stimulation.39 These factors are fundamental for cell–cell communication and down-regulated expression represents attenuation of the hyper-inflammatory response, making them commonly used surrogate markers in clinical studies.
A translational study evaluated the signalling apparatus of APCs in post-mortem sepsis patients and revealed decreased expression of T-cell-activating co-stimulatory molecules, CD80 and CD86. Further hampering the communication between the innate to adaptive immune systems is the lack of T-cell expression of the co-stimulatory molecule, CD28, described in post-mortem sepsis patients compared with controls.44 The consequent altered signalling results in decreased IL-2 production and increased T-cell anergy and apoptosis.44 Furthermore, ex vivo studies have demonstrated significant inflammation-induced apoptosis of CD4+ and CD8+ T cells in sepsis patients.44 Additionally, IL-10 production suppresses cytotoxic CD8+ T cells and the Th1 hyper-inflammatory responses, favouring a Th2 skewed response.2,44 However, similar to murine ET models, the IL-10 response in humans with sepsis is variable and probably insufficient to induce IT.34
An important limitation of using ex vivo TNF-α production as a surrogate for integrated immune responses is highlighted by a study of 148 patients with severe sepsis in which 28-day mortality was not associated with ex vivo-stimulated TNF-α production.45 These findings contrast many studies that describe an association between worse outcomes including higher mortality, increased incidence of secondary infections and increased length of stay in patients who display a lower stimulated TNF-α production capacity.39–41 This is feasibly explained by the isolated cell-stimulation method used in this study, which contrasts with the whole blood endotoxin stimulation method used in the other studies.39–41 Given the influence of soluble factors and cell–cell interactions on the character of the inflammatory response, it is imperative that ex vivo immune stimulation closely approximates the in vivo environment for complete and valid interpretation.
Other studies in adults consistently show an association between a prolonged IT response and an increased risk of death and secondary infections.39,46,47
Immune maturation as a regulator of immune tolerance in severe sepsis
Similar to the neonatal immune response, IT in adult sepsis is Th2-skewed.34 Evaluations of IT in paediatric sepsis patients have shown an association between a severely suppressed pro-inflammatory response at admission and an increased risk of mortality and longer hospital stays.40,41,43 It is unknown, however, if the effects of a persistent IT response during the duration of a sepsis episode causatively affects outcomes. Additionally, it remains unknown if this association is present in neonates, the most immunologically immature demographic. Nor is it established if low-dose endotoxin exposure, shown to have tolerizing effects in adults, has similar, dampening or amplifying effects in neonates and whether the maturational-based immunological differences could alter the amplitude and/or direction of the immune response to an infection and clinical course of severe sepsis. Future research in neonatal and paediatric sepsis should focus on evaluation for a causative association between the inherently tolerant, immature immune response and poor outcomes. Further testing should focus on determining the specific cell–cell interactions and/or soluble factors most appropriate for trials of therapeutic modulation in the septic child. The care of profoundly immune-immature patients would be enriched by the ability to characterize and beneficially enhance their immunological response.
Conclusions
The overall characteristics of the immature immune response differ from the mature immune response in ways similar to the differences between the ET and normal endotoxin responses. A complete understanding of the specific mechanisms underlying these differences remains to be elucidated. To improve the care of adult, paediatric and neonatal sepsis patients, better characterization of the initial and sustained immune responses in neonatal versus paediatric and adult sepsis is imperative. More importantly, determining correlations between alterations in immune responses with intermediate and definitive outcomes from severe sepsis in neonates, children and adults will provide great insight into their specific immunological adaptations and deficiencies. A better understanding of the mechanisms that mediate these immune response impairments will allow targeting of future therapies to improve patient outcomes.
Disclosures
The authors listed on this manuscript do not have any conflicts of interest to report.
Glossary
- APC
antigen-presenting cell
- DAMP
damage-associated molecular pattern
- ET
endotoxin tolerance
- IL
interleukin
- IRAK
interleukin receptor-associated kinase
- IT
immune tolerance
- NF-κB
nuclear factor-κB
- PAMP
pathogen-associated molecular pattern
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- Th1
T helper type 1
- TLR
Toll-like receptor
- TNF
tumour necrosis factor
- Treg
regulatory T
References
- Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5:36–44. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Collazo E, Del Fresno C. Pathophysiology of endotoxin tolerance: mechanisms and clinical consequences. Crit Care. 2013;17:242. doi: 10.1186/cc13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ME, Linde-Zwirble WT, Angus DC, Watson RS. Trends in the epidemiology of pediatric severe sepsis*. Pediatr Crit Care Med. 2013;14:686–93. doi: 10.1097/PCC.0b013e3182917fad. [DOI] [PubMed] [Google Scholar]
- Ygberg S, Nilsson A. The developing immune system – from foetus to toddler. Acta Paediatr. 2012;101:120–7. doi: 10.1111/j.1651-2227.2011.02494.x. [DOI] [PubMed] [Google Scholar]
- Randolph AG, McCulloh RJ. Pediatric sepsis: important considerations for diagnosing and managing severe infections in infants, children, and adolescents. Virulence. 2014;5:179–89. doi: 10.4161/viru.27045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway C., Jr Innate immunity. N Engl J Med. 2000;343:338–44. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133–50. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- Remington J, Klein JO, Wilson CB, Baker CJ. Developmental Immunology and Role of Host Defenses in Fetal and Neonatal Susceptibility to Infection. Infectious Diseases of the Fetus and Newborn Infant. 6th edn. Philadelphia, PA: Elsevier Saunders; 2006. [Google Scholar]
- Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–64. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- Christaki E, Giamarellos-Bourboulis EJ. The complex pathogenesis of bacteremia: from antimicrobial clearance mechanisms to the genetic background of the host. Virulence. 2014;5:57–65. doi: 10.4161/viru.26514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diesner SC, Forster-Waldl E, Olivera A, Pollak A, Jensen-Jarolim E, Untersmayr E. Perspectives on immunomodulation early in life. Pediatr Allergy Immunol. 2012;23:210–23. doi: 10.1111/j.1399-3038.2011.01259.x. [DOI] [PubMed] [Google Scholar]
- Levy O. Innate immunity of the human newborn: distinct cytokine responses to LPS and other Toll-like receptor agonists. J Endotoxin Res. 2005;11:113–6. doi: 10.1179/096805105X37376. [DOI] [PubMed] [Google Scholar]
- Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR. Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-α induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol. 2004;173:4627–34. doi: 10.4049/jimmunol.173.7.4627. [DOI] [PubMed] [Google Scholar]
- Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014;5:4–11. doi: 10.4161/viru.27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub B, Liu J, Schleich I, Hoppler S, Sattler C, von Mutius E. Impairment of T helper and T regulatory cell responses at birth. Allergy. 2008;63:1438–47. doi: 10.1111/j.1398-9995.2008.01685.x. [DOI] [PubMed] [Google Scholar]
- Striz I, Brabcova E, Kolesar L, Sekerkova A. Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond) 2014;126:593–612. doi: 10.1042/CS20130497. [DOI] [PubMed] [Google Scholar]
- Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol. 2007;7:379–90. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- Kollmann TR, Crabtree J, Rein-Weston A, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol. 2009;183:7150–60. doi: 10.4049/jimmunol.0901481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37:771–83. doi: 10.1016/j.immuni.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res. 2004;10:71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- Sadeghi K, Berger A, Langgartner M, et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis. 2007;195:296–302. doi: 10.1086/509892. [DOI] [PubMed] [Google Scholar]
- Yan SR, Qing G, Byers DM, Stadnyk AW, Al-Hertani W, Bortolussi R. Role of MyD88 in diminished tumor necrosis factor α production by newborn mononuclear cells in response to lipopolysaccharide. Infect Immun. 2004;72:1223–9. doi: 10.1128/IAI.72.3.1223-1229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrid AM, Kollmann TR. Age-dependent differences in systemic and cell-autonomous immunity to L. monocytogenes. Clin Dev Immunol. 2013;2013:917198. doi: 10.1155/2013/917198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol. 2008;30:209–35. doi: 10.1007/s00281-008-0121-8. [DOI] [PubMed] [Google Scholar]
- Levy O, Coughlin M, Cronstein BN, Roy RM, Desai A, Wessels MR. The adenosine system selectively inhibits TLR-mediated TNF-α production in the human newborn. J Immunol. 2006;177:1956–66. doi: 10.4049/jimmunol.177.3.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Hoeks SB, Prakken BJ, de Roock S. TH17 differentiation capacity develops within the first 3 months of life. J Allergy Clin Immunol. 2014;133:891–4. doi: 10.1016/j.jaci.2013.09.022. e5. [DOI] [PubMed] [Google Scholar]
- Shah BA, Padbury JF. Neonatal sepsis: an old problem with new insights. Virulence. 2014;5:170–8. doi: 10.4161/viru.26906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie PM, Huang Q, Jolette E, et al. Profound lack of interleukin (IL)-12/IL-23p40 in neonates born early in gestation is associated with an increased risk of sepsis. J Infect Dis. 2010;202:1754–63. doi: 10.1086/657143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulic MK, Andrews D, Crook ML, Charles A, Tourigny MR, Moqbel R, Prescott SL, et al. Changes in thymic regulatory T-cell maturation from birth to puberty: differences in atopic children. J Allergy Clin Immunol. 2012;129:199–206. doi: 10.1016/j.jaci.2011.10.016. e1–4. [DOI] [PubMed] [Google Scholar]
- Pettengill MA, van Haren SD, Levy O. Soluble mediators regulating immunity in early life. Front Immunol. 2014;5:457. doi: 10.3389/fimmu.2014.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NL, Wissink E, Wang J, Pinello JF, Davenport MP, Grimson A, Rudd BD. Rapid proliferation and differentiation impairs the development of memory CD8+ T cells in early life. J Immunol. 2014;193:177–84. doi: 10.4049/jimmunol.1400553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D. Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clin Exp Immunol. 2002;128:118–23. doi: 10.1046/j.1365-2249.2002.01817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA. Mechanisms of tolerance. Immunol Rev. 2011;241:5–19. doi: 10.1111/j.1600-065X.2011.01019.x. [DOI] [PubMed] [Google Scholar]
- Ariga SK, Abatepaulo FB, Melo ES, Velasco IT, Pinheiro da Silva F, de Lima TM, Soriano FG. Endotoxin tolerance drives neutrophil to infectious site. Shock. 2014;42:168–73. doi: 10.1097/SHK.0000000000000175. [DOI] [PubMed] [Google Scholar]
- West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30(Suppl):S64–73. [PubMed] [Google Scholar]
- Cornell TT, Sun L, Hall MW, Gurney JG, Ashbrook MJ, Ohye RG, Shanley TP. Clinical implications and molecular mechanisms of immunoparalysis after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2012;143:1160–6. doi: 10.1016/j.jtcvs.2011.09.011. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier WJ, Hall MW. Immunoparalysis and adverse outcomes from critical illness. Pediatr Clin North Am. 2008;55:647–68. doi: 10.1016/j.pcl.2008.02.009. , xi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MW, Geyer SM, Guo CY, et al. Innate immune function and mortality in critically ill children with influenza: a multicenter study. Crit Care Med. 2013;41:224–36. doi: 10.1097/CCM.0b013e318267633c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mella C, Suarez-Arrabal MC, Lopez S, Stephens J, Fernandez S, Hall MW, Mejias A. Innate immune dysfunction is associated with enhanced disease severity in infants with severe respiratory syncytial virus bronchiolitis. J Infect Dis. 2013;207:564–73. doi: 10.1093/infdis/jis721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszynski JA, Nofziger R, Greathouse K, et al. Innate immune function predicts the development of nosocomial infection in critically injured children. Shock. 2014;42:313–21. doi: 10.1097/SHK.0000000000000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszynski JA, Nofziger R, Greathouse K, et al. Early adaptive immune suppression in children with septic shock: a prospective observational study. Crit Care. 2014b;18:R145. doi: 10.1186/cc13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence. 2014;5:45–56. doi: 10.4161/viru.26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez HG, Gonzalez SM, Londono JM, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit Care Med. 2014;42:771–80. doi: 10.1097/CCM.0000000000000100. [DOI] [PubMed] [Google Scholar]
- Hynninen M, Pettila V, Takkunen O, Orko R, Jansson SE, Kuusela P, Renkonen R, Valtonen M. Predictive value of monocyte histocompatibility leukocyte antigen-DR expression and plasma interleukin-4 and -10 levels in critically ill patients with sepsis. Shock. 2003;20:1–4. doi: 10.1097/01.shk.0000068322.08268.b4. [DOI] [PubMed] [Google Scholar]
- Landelle C, Lepape A, Voirin N, Tognet E, Venet F, Bohe J. Vanhems P, Monneret G. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. 2010;36:1859–66. doi: 10.1007/s00134-010-1962-x. [DOI] [PubMed] [Google Scholar]