

Abstract
Systemic lupus erythematosus is an autoimmune disorder characterized by increased levels of lymphocyte activation, antigen presentation by dendritic cells, and the formation of autoantibodies. This leads to immune complex-mediated glomerulonephritis. Toll-like receptor 7 (T7) and TLR9 localize to the endosomal compartment and play important roles in the generation of autoantibodies against nuclear components, as they recognize RNA and DNA, respectively. In contrast, very little is known about endogenous TLR8 activation in mice. We therefore tested whether TLR8 could affect autoimmune responses in a murine model of lupus. We introduced a Tlr8 null mutation into C57BL/6 mice congenic for the Nba2 (NZB autoimmunity 2) locus and bearing the Yaa (Y-linked autoimmune acceleration) mutation containing a tlr8 duplicated gene, and monitored disease development, autoantibody production, and glomerulonephritis-associated mortality. Cellular responses were investigated in female Nba2.TLR8−/− mice bearing no copy of tlr8. The TLR8 deficiency accelerated disease progression and mortality, increased the number of circulating antibodies and activated monocytes, and heightened cellular responses to TLR7 ligation. TLR8-deficient antigen-presenting cells exhibited increased levels of MHC class II expression. The ability of dendritic cells to present antigens to allogeneic T cells after TLR7 ligation was also improved by TLR8 deficiency. TLR8 deletion accelerated autoimmunity in lupus-prone mice in response to TLR7 activation. Antigen-presenting cell function seemed to play a key role in mediating the effects of TLR8 deficiency.
Keywords: monocytes, systemic lupus erythematosus, Toll-like receptors
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies and the subsequent development of immune complex-mediated lupus nephritis. In SLE, B-cell hyperactivity leads to the production of autoantibodies against components of the nucleus, such as chromatin, double-stranded and single-stranded DNA (dsDNA and ssDNA, respectively), and RNA-related antigens, such as small nuclear ribonucleoproteins (snRNP).1,2 Involvement of RNA-related antigens in the development of nephropathy has not been clearly established in models of lupus. It is also unclear how host nuclear compounds break tolerance and initiate autoimmune responses.
Discovery of the Toll-like receptor (TLR) gene family has revealed some of the mechanisms by which autoantibodies are produced. Recognition of microbial components by TLRs is an important step in host-cell responses to pathogens. The TLRs recognize conserved motifs presented by pathogenic bacteria, protozoa, fungi and viruses, including unmethylated CpG DNA (TLR9) and viral single-stranded RNA (ssRNA)3 (see Huyton et al. for a recent review, ref. 4). The finding that nucleic acids can serve as endogenous ligands for TLRs5 and that RNA-related autoantigens activate B cells by engaging B-cell receptors and TLR76 led researchers to investigate the role of endosomal TLR7 in regulating autoantibody production in murine models of lupus.7–11 Furthermore, one SLE model is the male BXSB mouse bearing the Y-linked autoimmune acceleration (Yaa) mutation. Yaa-bearing mice have a duplicated copy of the X-linked Tlr7 gene on the Y chromosome,12,13 which results in autoreactive B-cell responses to RNA-related antigens. Tlr7 duplication in lupus models involving the C57BL/6 (B6) background (B6.FcγRII−/− and the congenic B6.Sle1) results in high levels of nucleolar-related autoantibodies and splenomegaly. In addition, the B6.FcγRII−/− mouse exhibits proteinuria, whereas the B6.Sle1 mouse exhibits enhanced T-cell and B-cell activation.12–14 In contrast, when Tlr7 null mutations are introduced into models of lupus, autoantibodies and lupus manifestations are reduced.8,15,16 We have shown that TLR7 contributes to: (i) autoantibodies against DNA, RNA, snRNP and gp70, (ii) the development of monocytosis, (iii) the incidence of lupus nephritis and central nervous system autoimmunity.17–20 Hence, TLR7 mediates deleterious autoimmune responses.
Murine Tlr8, which is very close to Tlr7 on the X chromosome and is included in the Yaa mutation,12,13 has been considered non-functional but recent studies have revealed a function for this gene and shown that its induction can be independent of TLR7.21–23 Splenic B cells and myeloid cells (both murine and human) express Tlr8 mRNA24,25 and ssRNA is a natural ligand (both for TLR8 and TLR7). Hence, TLR8 could play a role in innate stimulations that precede autoimmune responses. Furthermore, we have shown that Yaa-mediated acceleration of SLE cannot be explained by duplication of Tlr7 alone, suggesting that others genes within the translocated X chromosome are involved in SLE pathogenesis.17 Indeed we cannot exclude the possibility that Tlr8 contributes to autoantibody production and disease manifestation. It is therefore important to determine whether TLR8 is involved in autoimmune processes observed in the Yaa-bearing lupus-prone mice.
To address this issue, we studied the effect of Tlr8 deletion on the development of lupus-related autoimmune traits in the congenic Nba2.Yaa mouse model. Nba2.TLR8−/− mice, with or without Yaa, developed a more severe disease. Surprisingly, TLR8 protected against the deleterious effects of TLR7. The Tlr8 null mutation affected primarily the myeloid compartment by increasing levels of MHC class II and TLR7 in monocytes and dendritic cells (DC). These results improve our understanding of the interplay between endosomal TLRs during lupus pathogenesis.
Materials and methods
Mice
B6.Nba2 and B6.Nba2.Yaa mice were generated as described.26–28 TLR8−/− mice were generously provided by Prof. S. Akira and backcrossed for seven generations into a B6 background using marker-assisted selection as described previously.17 The Tlr8 null mutation was introduced in B6.Nba2.Yaa mice by breeding. Resulting female and male mice were named Nba2 and Nba2.TLR8−/− or Nba2.Yaa and Nba2.TLR8−/Yaa, respectively. All animal experiments were approved by the Ethics Committee for Animal Experimentation of the Faculty of Medicine, University of Geneva.
Quantitative RT-PCR
RNA from spleen cells was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). The abundance of Tlr8 mRNA was determined using quantitative real-time RT-PCR. Tlr8 and Tlr7 cDNA was amplified using the following primers: Tlr8 forward (5′-TGGATGTTAAGAGAGAAACAAACG-3′), Tlr8 reverse (5′-GATATGGACGACCCAACGGAC-3′), Tlr7 forward (5′-GTACCAAGAGGCTGCAGATTAGAC-3′), and Tlr7 reverse (5′-AGCCTCAAGGCTCAGAAGATG-3′). PCR was performed using an iCycler iQ real-time PCR detection system (Bio-Rad, Hercules, CA) and iQ SYBR green supermix (Bio-Rad). Results were quantified relative to a standard curve generated with serial dilutions of a reference cDNA preparation from spleen and normalized using β-actin mRNA.
Histopathology and immunohistochemistry
Kidney samples were collected when mice were moribund or 14 months old. Histological sections were stained with periodic acid-Schiff reagent. The extent of glomerulonephritis (GN) was scored on a 0–4 scale based on histopathological changes, as described previously.27 GN with a grade ≥ 3 was considered a significant contributor to the clinical disease. For immunohistochemistry, kidneys were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek Europe) and snap-frozen in liquid nitrogen. Five-micrometre frozen sections were labelled with Texas Red-conjugated goat antibodies against mouse IgM (Southern Biotechnology, Birmingham, AL) and AlexaFluor 488-conjugated goat antibodies against mouse IgG (Invitrogen), or with phycoerythrin-conjugated anti-CD11b and FITC-conjugated CD4 (eBioscience, San Diego, CA) and then placed into mounting medium (Dako, Glostrup, Denmark).
Serological analysis
Enzyme-linked immunosorbent assays (ELISAs) were used to determine serum levels of: (i) total IgG, (ii) total IgM, (iii) IgG and IgG isotype autoantibodies against dsDNA, chromatin and RNA, and (iv) IgM autoantibodies against dsDNA and RNA. Chromatin, goat anti-IgM (Lomm9) and goat anti-mouse IgG (Sigma Aldrich, Saint-Louis, MO) were coated directly onto the ELISA plates. In contrast, dsDNA and yeast RNA were coated onto ELISA plates that were pre-coated with poly-l-lysine (Sigma Aldrich). Plates were incubated with diluted serum samples and the assays were developed with alkaline phosphatase-conjugated goat anti-mouse IgG, or IgG1, IgG2b, IgG2c, IgG3 (Southern Biotechnology), rat anti-mouse IgM monoclonal antibody (Lomm9), or rat anti-mouse κ chain monoclonal antibody. For total IgM and IgG, results are expressed in mg/ml. For autoantibodies, results are expressed in units per milliliter (U/ml) based on a standard curve derived from pooled sera from normal mice or MRL-Faslpr mice. The amounts of interleukin-12 p40 (IL-12p40) and tumour necrosis factor-α (TNF-α) from mixed lymphocyte reaction supernatant were also quantified using ELISA (eBioscience).
IgG autoantibodies directed against native dsDNA or antinuclear autoantibodies were titrated by immunofluorescence assay using dsDNA (Crithidia luciliae) slides or Hep-2 slides, respectively (INOVA Diagnostics, Inc., San Diego, CA).
Flow cytometric analysis
Flow cytometry was performed using four-colour staining of splenocytes or peripheral blood mononuclear cells and analysed with a FACSCalibur system (BD Biosciences, San Jose, CA). To analyse splenic DC subsets, spleen cells were prepared after spleen fragments were digested with liberase (Roche Diagnostics, Rotkreuz, Switzerland) and DNase I (Sigma-Aldrich), as described previously.29 We used antibodies against CD4 (GK1.5), CD8 (53-6.7), B220 (RA3-6B2), CD11c (N418), CD11b (M1/70), PDCA (eBio927), CD115 (AFS98, monocyte colony-stimulating factor receptor), Gr1 (RB6-8C5), FcγRIV (9E9), MHC class II I-A (Y3P), CD40 (HM40-3), CD86 (GL1), and CD69 (H1.2F3). For proliferation analyses, cells were labelled with antibodies against intracellular KI67 (BD Biosciences). Stainings were performed in the presence of saturating concentrations of monoclonal antibodies against FcγRII/III (2.4G2).
Mixed lymphocyte reaction
Differentiation of bone marrow-derived plasmacytoid dendritic cells (pDC) was performed by incubating, during 1 week, bone marrow cells in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 100 ng/ml fms-like tyrosine kinase 3 ligand (FLT3L; Amgen Inc., Thousand Oaks, CA). Mixed lymphocyte reactions were performed using pDC from Nba2.Yaa or Nba2.TLR8−/Yaa mice and CD4+ T lymphocytes from 6–8-week-old BALB/c mice. Cell-sorted purified pDC were plated in 24-well plates at a concentration of 6 × 105 cells/well and cultured overnight in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum with TLR agonists that included 1 μg of CpG2216, or 3 μg/ml imiquimod or resiquimod (all from InvivoGen). Isolated total CD4+ T cells were then added at a concentration of 2 × 106 cells/well. After 48 hr of co-culture, CD4+ T cells were labelled with antibodies against KI67 and proliferation was measured using flow cytometric analysis. Alternatively, similar cultures were performed on 96-well plates with 6 × 104 pDc per well and 2 × 105 CD4+ T cells. After 48 hr of co-culture, T cells were pulsed overnight with 1 μCi/well of [3H]thymidine, and [3H]thymidine incorporation was measured the next day.
Injection of TLR7 agonist
Toll-like receptor 7 ligand II (1V136; Calbiochem, EMD Millipore, Billerica, MA) was diluted in sterile PBS and injected intraperitoneally into Nba2 or Nba2.TLR8−/− female mice, as described previously.19 Controls were injected with PBS. Blood samples were collected 24 hr after the injection and cells were analysed using flow cytometry.
Statistical analysis
Mortality rates were analysed using the Kaplan–Meier log-rank test. Analyses of serological parameters and the intensity of glomerular lesions were performed using the Mann–Whitney U-test. Unpaired comparisons involving cellular parameters or mRNA expression levels were performed using the Student's t-test. Probability values < 5% were considered significant.
Results
Tlr8 deletion from the X chromosome in Nba2.Yaa lupus-prone mice leads to accelerated lupus nephritis and splenomegaly
To determine the effect of the Yaa chromosome, i.e. Tlr8 duplication, on Tlr8 expression we used quantitative RT-PCR to analyse Tlr8 transcripts in males of different genotypes. Similar to what is observed with Tlr7,17 Tlr8 mRNA abundance was twofold higher in spleen cells from B6.Tlr8+/Yaa male mice, compared with cells from B6.Tlr8+/Y (non-Yaa) males. Cells from male B6.Tlr8−/Yaa mice, which bear the Tlr8 null mutation on the X chromosome, expressed levels of Tlr8 mRNA comparable to those of the male B6.Tlr8+/Y (non-Yaa mice), consistent with these mice having the same Tlr8 gene copy number (Fig.1a).
Figure 1.

Toll-like receptor 8 (TLR8) deletion accelerates autoimmune responses. (a) Quantitative RT-PCR was used to analyse Tlr8 expression in splenic B cells from 2-month-old male mice. Values (mean ± SD) were normalized to β-actin (b) Survival curves for Nba2 (n = 12), Nba2.Yaa (n = 15), and Nba2.TLR8−/Yaa (n = 15) male mice over a 14-month period. The difference in mortality between Nba2.Tlr8−/Yaa and Nba2.Yaa mice was highly significant (P < 0·001). (c) The severity of glomerular lesions was scored on a 0–4 scale. Tissue samples were obtained from moribund mice or at the end of the 14-month period. Incidence of severe glomerulonephritis (grade ≥ 3) in Nba2.Tlr8−/Yaa mice was increased compared with Nba2.Yaa mice. Mean values are indicated by horizontal lines. (d) Representative histological appearance of kidney glomerular lesions from Nba2.Yaa, Nba2.TLR8−/Yaa, and Nba2-control (Ct) male mice (n = 4 mice/group). Kidney sections were labelled with antibodies against IgG and IgM, or against CD11b. (e) Spleens from 4-month-old Nba2.Yaa or Nba2.TLR8−/Yaa male mice were compared. Nba2.TLR8−/Yaa spleens were heavier and had more cells than Nba2.Yaa spleens (n = 4 mice/group). Representative images of analysed spleens are shown. *P < 0·05; **P < 0·01; ***P < 0·001.
We have shown that Nba2.Yaa mice develop a lethal form of lupus nephritis that causes 50% mortality by 14 months of age.28 To test whether Tlr8 influences the autoimmune pathology of Nba2.Yaa mice we inserted a Tlr8 deletion into this genetic background, generating Nba2.TLR8−/Yaa mice. Nba2, Nba2.Yaa, and Nba2.TLR8−/Yaa male mice were followed for 14 months and survival curves were generated (Fig.1b,c). Nba2.TLR8−/Yaa male mice had a shorter lifespan compared with Nba2.Yaa males. None of the non-Yaa Nba2 control males succumbed or developed significant GN during the analysed period. In contrast, only 53·3% (8/15) of the Nba2.Yaa males lived for 14 months, whereas all (15/15) of the Nba2.TLR8−/Yaa mice succumbed before the end of the experiment. Kidneys from these mice were harvested and sectioned to generate GN scores. This revealed that 93·3% (14/15) of the Nba2.Tlr8−/Yaa mice developed fatal GN (i.e. had a GN score ≥ 3) compared with 53·3% (8/15) for Nba2.Yaa mice and none for Nba2 controls (Fig.1c). Frozen kidney sections were also stained for IgM and IgG deposits. Nba2.TLR8−/Yaa glomeruli exhibited more immunoglobulin deposits and macrophage infiltration (Mac1+ cells) than Nba2.Yaa glomeruli (Fig.1d). In both strains we barely detected CD4+ T cells staining in glomeruli (not shown).
Finally, at 4 months of age both Nba2.TLR8−/Yaa and Nba2.TLR8−/− mice developed splenomegaly, as spleens from these animals were larger, heavier and contained more cells than spleens from Nba2.Yaa males and Nba2 females, respectively (Fig.1e).
Tlr8 deletion from the X chromosome enhances the production of circulating antibodies
To examine whether Tlr8 deletion affects antibody and autoantibody production, serum was collected from 8-month-old mice and antibodies were detected using ELISA. Levels of IgM and IgG were significantly higher in mice deficient for Tlr8 on the X chromosome (both females and males) compared with controls (Table1). We did not detect differences in autoantibody levels between these mice, although levels of IgM and IgG against dsDNA or RNA were elevated in Nba2.TLR8−/Yaa male mice compared with Nba2.Yaa. In contrast, levels of IgG directed against chromatin were not affected by Tlr8 dosage. In females, autoantibody levels remained low because they did not carry the Yaa mutation.
Table 1.
Serum levels of IgM, IgG, IgG2b and autoantibodies
| Females | Males | |||
|---|---|---|---|---|
| Nba2 | Nba2.TLR8−/− | Nba2.Yaa | Nba2.TLR8−/Yaa | |
| IgM (μg/ml) | 699 ± 118 | 908 ± 65* | 1333 ± 497 | 4393 ± 1450** |
| IgM against dsDNA (U/ml) | 70 ± 9·5 | 70 ± 7·3 | 541 ± 240 | 1057 ± 413 |
| IgM against RNA (U/ml) | 55 ± 6·4 | 60 ± 6·4 | 7084 ± 2980 | 14 104 ± 4119 |
| IgG (mg/ml) | 13 ± 1·5 | 16 ± 0·6* | 11 ± 1 | 17 ± 1·6** |
| IgG2b (μg/ml) | 45·5 ± 12·9 | 69·3 ± 19·7 | 35·4 ± 16·6 | 121·7 ± 33·7** |
| IgG against dsDNA (U/ml) | 5·3 ± 0·7 | 6·5 ± 1·6 | 4·4 ± 1·6 | 15 ± 6·1 |
| IgG1 against dsDNA (U/ml) | 13·6 ± 4·1 | 34·9 ± 14·8 | 8·9 ± 1·7 | 43·7 ± 27·1 |
| IgG2b against dsDNA (U/ml) | 66·4 ± 14·9 | 57·8 ± 8·6 | 78·8 ± 13·5 | 259·8 ± 44·8*** |
| IgG2c against dsDNA (U/ml) | 3·9 ± 1·4 | 5·6 ± 1·8 | 15·8 ± 2·89 | 27·1 ± 3·7* |
| IgG3 against dsDNA (U/ml) | 0·9 ± 0·5 | 3·2 ± 1·2 | 0·9 ± 0·5 | 5·0 ± 2·7** |
| IgG against RNA (U/ml) | 107 ± 44 | 111 ± 26 | 504 ± 272 | 1087 ± 272 |
| IgG1 against RNA (U/ml) | 16·7 ± 3·8 | 70·1 ± 41·3 | 72·3 ± 41·1 | 227 ± 92 |
| IgG2b against RNA (U/ml) | 35·2 ± 8·0 | 228 ± 88 | 346 ± 114 | 881 ± 147** |
| IgG2c against RNA (U/ml) | 1·5 ± 1·1 | 8·9 ± 3·3 | 22·8 ± 7·5 | 36·0 ± 14·1 |
| IgG3 against RNA (U/ml) | 3·7 ± 1·2 | 4·2 ± 1·2 | 10·3 ± 4·5 | 252 ± 126* |
| IgG against Chromatin (U/ml) | 5·9 ± 1·1 | 9·4 ± 1·2 | 11 ± 1·2 | 12 ± 1·3 |
Serum was collected from 8-month-old Nba2 (n = 14), Nba2.TLR8−/− (n = 23), Nba2.Yaa (n = 9), and Nba2.TLR8−/Yaa (n = 12) mice. Levels were determined using ELISA. Values are the mean ± SEM.
P < 0·05;
P < 0·01
P < 0·001.
We also analysed autoantibodies with Crithidia luciliae slides and found that all groups of mice were positive for anti-dsDNA staining but no main differences between groups were observed, as already seen by ELISA for total IgG anti-dsDNA. However, analysis of autoantibodies with Hep-2 slides showed slightly increased staining for anti-nuclear antibodies with serum from Nba2.TLR8−/Yaa mice compared with WT (Fig.2).
Figure 2.
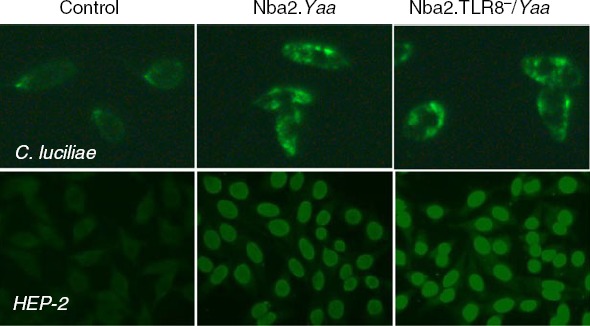
Anti-dsDNA and antinuclear autoantibodies are detected in male Nba2.Yaa and Nba2.TLR8−/Yaa. Staining of Crithidia luciliae or Hep2 cell coated slides for the presence of IgG anti-dsDNA or IgG antinuclear autoantibodies in the serum of Nba2.Yaa and Nba2.TLR8−/Yaa mice or from B6 mice as negative control. Pictures are representative of five animals analysed per group.
IgG isotypes were also quantified as total levels or directed against dsDNA and RNA. We showed that total IgG2b as well as IgG2b against dsDNA and RNA were significantly increased in Nba2.TLR8−/Yaa mice compared with controls. Interestingly, IgG2b was shown to be highly involved in pathogenic responses in murine SLE. In addition, levels of IgG3 antibodies against dsDNA and RNA were also significantly elevated in Nba2.TLR8−/Yaa mice as well as IgG2c anti-dsDNA, when compared with TLR8-sufficient mice (Table1). Notably, IgG2b, IgG2c and IgG3 are isotypes released upon T helper type 1-related immune responses, which are known to correlate to SLE immune profile.
Enhanced development of monocytosis in Nba2.TLR8−/Yaa mice
Yaa-bearing mice develop monocytosis, which is characterized by the selective accumulation of Gr1– monocytes.19,30 To determine whether the enhanced activated phenotype observed in Nba2.TLR8−/Yaa mice involved monocytosis, we analysed monocytosis development and circulating monocytes (Gr1− and Gr1+ subsets) in 6-month-old male Nba2.Yaa, Nba2.TLR8−/Yaa and female Nba2.Yaa, Nba2.TLR8−/− mice. We found increased monocytosis and a selective expansion of the Gr1– subset in mice deficient for Tlr8 on the X chromosome (both females and males) compared with controls (Table2).
Table 2.
Subsets of circulating monocyte and splenic B cells, T cells and dendritic cells
| Cell type | Females | Males | ||
|---|---|---|---|---|
| Nba2 | Nba2.TLR8−/− | Nba2.Yaa | Nba2.TLR8−/Yaa | |
| Circulating monocytes | ||||
| Gr1+ | 5·5 ± 2·5 | 6·2 ± 1·7 | 8·6 ± 1·6 | 10·8 ± 1·8 |
| Gr1− | 4·3 ± 0·6 | 7·3 ± 3·2* | 11·9 ± 1·4 | 20·4 ± 2·8* |
| Total | 9·8 ± 3·0 | 13·5 ± 4·3* | 20·5 ± 2·9 | 31·1 ± 4·2* |
| Splenic populations | ||||
| T CD4+ | 22·7 ± 0·9 | 22·0 ± 1·0 | 16·3 ± 2·2 | 18·0 ± 0·4 |
| T CD8+ | 11·5 ± 0·6 | 8·9 ± 1·1 | 9·2 ± 1·7 | 10·9 ± 0·6 |
| B220+ | 51·7 ± 2·1 | 53·5 ± 2·6 | 43·0 ± 3·8 | 51·4 ± 2·1 |
| cDC | 1·7 ± 0·1 | 1·9 ± 0·1 | 2·4 ± 0·8 | 1·8 ± 0·1 |
| pDC | 0·29 ± 0·05 | 0·36 ± 0·05 | 0·21 ± 0·01 | 0·26 ± 0·03 |
Percentages of circulating monocyte subtypes (CD115+) were determined in 6-month-old Nba2 (n = 5), Nba2.TLR8−/− (n = 8) females, Nba2.Yaa (n = 14) and Nba2.TLR8−/Yaa (n = 12) male mice. Values are the mean ± SEM.
Percentages of splenic T cells (CD3+ gated), B cells and dendritic cells were determined in 4-month-old Nba2, Nba2.TLR8−/− females and Nba2.Yaa and Nba2.TLR8−/Yaa male mice (n = 3/group). Results are from one representative experiment. Three independent experiments were performed. Values are the mean ± SEM.
P < 0·05.
cDC, conventional dendritic cells; pDC, plasmacytoid dendritic cells.
We also analysed the cellular composition of spleens from 4-month-old mice using flow cytometry. We found no significant differences in the percentages of B cells, CD4+ T cells, CD8+ T cells, pDC, and conventional DC between Nba2.TLR8−/Yaa and Nba2.Yaa mice nor between Nba2.TLR8−/− and Nba2 females (Table2). However, Nba2.TLR8−/Yaa mice had twofold more total spleen cells than controls (Fig.1e), suggesting that the absolute number of each splenic population was increased in Nba2.TLR8−/Yaa mice compared with controls.
Elevated autoantibody levels and monocytosis are therefore also observed in TLR8-deficient females not bearing the Yaa mutation. Because of the expression of a ‘Yaa copy’ of Tlr7 and Tlr8 genes as the result of a translocation from the telomeric end of the X chromosome onto the Y chromosome in Yaa-bearing mice,12,13 the analysis of these mice did not allow us to conclusively investigate the increased autoimmune responses observed in Nba2.TLR8−/− mice. Therefore, we assessed the following cellular responses in female TLR8-deficient Nba2 mice in comparison with TLR8-sufficient Nba2 female mice.
MHC class II expression is up-regulated in Nba2.TLR8−/− antigen-presenting cells
Because TLR8 had a limited effect on autoantibody production, we asked whether TLR8 contributed to the activation of antigen-presenting cells by using TLR8-deficient Nba2 female mice. We used flow cytometry to analyse the expression of various activation molecules on antigen-presenting cells. B cells gated on B220+ CD11c–, pDC gated on CD11clo PDCA+ B220+, and conventional DC gated on CD11chi had higher levels of MHC class II on their surface when isolated from Nba2.TLR8−/− mice than when isolated from Nba2 controls (Fig.3a). For all examined cell types, CD86 and CD40 were expressed at similar levels in the two mouse strains.
Figure 3.

Enhanced antigen-presenting cell function in TLR8−/− mice. (a) Splenocytes from 4-month-old Nba2 and Nba2.TLR8−/− mice were analysed by flow cytometry for MHC class II expression. B cells were gated ‘B220+ CD11c–’, plasmacytoid dendritic cells (pDC) were gated ‘B220+ CD11clow PDCA+’, and conventional DC were gated ‘B220– CD11chi’. Representative flow cytometric profiles and mean MHC class II fluorescence intensities (MFI, four mice per group) are shown. Compared with Nba2 controls, TLR8 deletion elevated MHC class II expression, but did not affect CD86 or CD40. (b) Mixed lymphocyte reactions using bone marrow-derived pDC. Immature pDC were incubated with Toll-like receptor 7-ligand (TLR7-L), CpG, or control medium for 48 hr, then analysed for CD86 and MHC class II expression by flow cytometry to verify maturation (left panel) and then proliferation of CD4+ T cells was measured using [3H]thymidine incorporation or KI67 staining (right panel). One representative flow cytometric profile is shown (n = 3 independent experiments). (c) TLR7-L-dependent releases of interleukin-12 (IL-12) from pDC and tumour necrosis factor-α (TNF-α) from T cells were measured using ELISA. NS: non-stimulated. *P < 0.05; **P < 0.01; ***P < 0.001.
Because an activated pDC phenotype correlates with lupus pathogenesis,31 we investigated whether pDC from Tlr8-deficient animals were functionally activated by conducting a mixed lymphocyte reaction in vitro. TLR7 is expressed at high levels in pDC, and TLR7 ligation was used to activate immature BM-derived pDC, as seen by the resulting elevated surface expression of CD86 and MHC class II (Fig.3b, left panel). Mature pDC were then co-cultured with allogeneic CD4+ T cells and CD4+ T-cell proliferation was analysed 48 hr later using [3H]thymidine incorporation or KI67-specific labelling. Plasmacytoid DC from Nba2.TLR8−/− mice induced more allogeneic T-cell proliferation than Nba2 pDC (Fig.3b), after TLR7 but not TLR9 (CPG) ligation. In addition, because pDC release cytokines as part of the T-cell activation process,32 we measured levels of IL-12 in the supernatant of the pDC-CD4+ T-cell co-culture. We also measured TNF-α released by activated T cells. Nba2.TLR8−/− pDC released significantly more IL-12 than Nba2 controls and Nba2.TLR8−/− T cells released more TNF-α than Nba2 controls (Fig.3c).
Increased TLR7-dependent activation of monocytes in Nba2.TLR8−/− mice
In response to TLR7 ligation, TLR8-deficient DC elicited a more potent allogeneic response than TLR8-expressing cells. We hypothesized that this resulted from an increase in TLR7-dependent activation, as we have previously observed in TLR9−/− cells.29 To test this hypothesis we investigated the in vivo response of monocyte activation to TLR7 ligation. We have previously shown that injecting B6 mice with the TLR7 agonist 1V136 increases monocytes within 24 hr.19 Monocytosis is associated with the induction of Gr1int monocytes and an increase in FcγRIV-expressing Gr1− monocytes. To verify that the in vivo activation of TLR7 increased the response of TLR8−/− mice, we injected 1V136 into Nba2 and Nba2.TLR8−/− mice and then checked for monocytosis and the expression of Gr1 and FcγRIV by CD115+ blood monocytes. TLR7 ligation generated more Gr1− monocytes and Gr1int monocytes in Nba2.TLR8−/− mice than in Nba2 controls. Hence, TLR8−/− monocytes are more activated in vivo upon TLR7 ligation compared with TLR8-expressing cells (Fig.4a).
Figure 4.
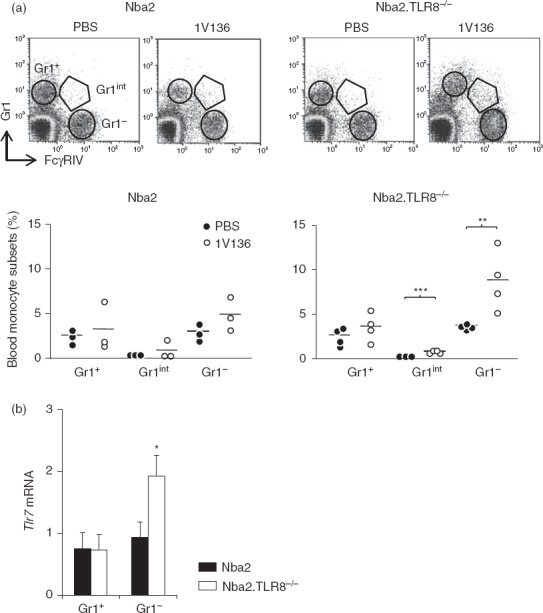
Toll-like receptor 7 (TLR7)-dependent monocytes activation in TLR8-deficient mice. (a) Five-month-old Nba2 or Nba2.TLR8−/− female mice were intraperitoneally injected with the TLR7-L 1V136 or PBS. Twenty-four hours later peripheral blood mononuclear cells were labelled for CD115, Gr1 and FcγRIV. Representative flow cytometric profiles (three or four mice per group) are shown. Gr1+, Gr1int and Gr1lo monocytes are highlighted by gating. Percentages of monocyte subsets are shown in the graphs below. Mean values are indicated by horizontal lines. (b) Quantitative RT-PCR analysis was used to assess Tlr7 expression in purified Gr1+ and Gr1– monocytes from 5-month-old Nba2 or Nba2.TLR8−/− female mice (three mice per group). Levels of Tlr7 mRNA (mean ± SEM) were normalized to β-actin. *P < 0·05; **P < 0.01; ***P < 0.001.
TLR7 expression is up-regulated in activated Nba2.TLR8−/− Gr− monocytes
We next tested whether the increased response of TLR8−/− monocytes to TLR7 ligation was caused by the up-regulation of TLR7 expression. Gr1+ and Gr1− blood monocytes were purified from 4-month-old Nba2.TLR8−/− and Nba2 mice and quantitative RT-PCR was used to quantify Tlr7 mRNA abundance. Tlr7 mRNA in Gr1– monocytes was up-regulated by 48% in Nba2.TLR8−/− mice compared with Nba2 controls (P < 0·01, Fig.4b). TLR7 appeared to be selectively up-regulated in activated monocytes, because Tlr7 mRNA levels were not affected in Gr1+ TLR8-deficient cells.
Discussion
We investigated the role of TLR8 in systemic autoimmunity by studying the effects of Tlr8 deletion on the development of autoimmune disease in the Nba2.Yaa lupus-prone mouse. Reducing TLR8 levels did not protect mice from the disease, but accelerated the spontaneous development of lupus-like GN, splenomegaly and mortality. We show that disease acceleration was associated with changes in total IgG and IgM levels in both males and females, with elevated IgG2b, IgG2c, IgG3 isotypes of autoantibodies against DNA and RNA but not with elevated levels of autoantibodies against chromatin. Loss of TLR8 also modulated the activation status of antigen-presenting cells, as levels of MHC class II were elevated in B cells and splenic DC (both plasmacytoid and conventional). Hence, TLR8−/− pDC presented allogeneic antigens more effectively. In addition, TLR8-deficient mice developed monocytosis, suggesting causal links between TLR8, activation of myeloid cells in autoimmune responses, and worsening of the SLE. Cell activations observed in TLR8-deficient mice depended on TLR7 signalling, and Tlr7 expression was elevated in TLR8-deficient myeloid cells. This agrees with a previous study that reported increased levels of TLR7 signalling and TLR7 expression in bone marrow-derived DC from TLR8-deficient mice (a B6 non-autoimmune background compared with normal B6 mice).33
Total IgM, IgG levels were increased in Nba2.TLR8-deficient mice, which is consistent with broad B-cell activation. Furthermore, TLR8 did not affect the total production of autoantibodies against dsDNA or RNA in this model system, but we observed isotype modulation of autoantibodies toward a T helper type 1 profile response (i.e. IgG2b, IgG2c and IgG3), which is consistent with the T helper type 1-associated autoantibody production observed in lupus-prone mice.34 We have previously demonstrated that high levels of antibodies against dsDNA and RNA-related antigens (i.e. snRNP) depend on TLR7, whereas IgG against chromatin depends on TLR9.17,29 As TLR8 deficiency did not modulate anti-chromatin levels, antibodies against dsDNA and RNA-related antigens are probably regulated by the TLR7 pathway, which is up-regulated in this model. This does not exclude the possibility that TLR8 acts as a receptor for self dsDNA and/or RNA in endocytosed immune complexes.
Toll-like receptor 8 is expressed in mouse and human myeloid cells, including monocytes and neutrophils.25 Hence, we expected that the Tlr8 null mutation affected the myeloid compartment. Indeed, we found major phenotypic and functional changes to DC and monocytes. Tlr8 deletion increased responsiveness to a TLR7 agonist in pDC and monocytes. This is in accordance with recent reports showing that TLR8 on DC restrain TLR7-mediated autoreactivity in non-lupus-prone mice.35 Increased levels of MHC class II on the cell surface of B cells and DC explained the heightened effectiveness of TLR8−/− pDC to stimulate allogeneic responses. Up-regulation of Tlr7 mRNA levels enhanced the response to TLR7 ligation. Moreover, increased levels of MHC class II on splenic DC in B6.TLR8−/− mice33 and Nba2.TLR8−/− mice (the present study) are a consequence of enhanced TLR7 activity, and TLR7 ligation makes TLR8-deficient pDC more effective antigen-presenting cells than TLR8-expressing pDC. Notably, pDC secrete more type I interferon in response to TLR7 and TLR9 ligation36,37 and it is clear that pDC and type I interferon contribute to SLE development.31,38–40
Circulating monocytosis can be divided into two major populations based on the expression of cell-membrane markers: Gr1hi FcγRIV– inflammatory monocytes and Gr1lo FcγRIV+ resting monocytes. We have shown that lupus-prone mice develop monocytosis and that this phenotype is associated with a selective expansion of Gr1hi FcγRIV+ monocytes.19,30,41 We showed that the in vivo ligation of TLR7 or TLR9 expanded the population of resting monocytes and generated transitional Gr1int monocytes.19 Here we demonstrated that the in vivo ligation of TLR7 in Nba2.TLR8−/− monocytes generated intermediate monocytes (Gr1int FcγRIVint) and expanded Gr1lo FcγRIV+ resting monocytes. TLR8−/− monocytes exhibited more extreme responses to TLR7-L because they expressed higher levels of Tlr7. It is interesting to note that TLR8-deficient B6.Nba2 female mice developed TLR7-mediated monocytosis, a cellular abnormality caused by the Yaa mutation.41–43 Duplication of Tlr7 is critical for Nba2.Yaa male mice to develop Yaa-associated monocytosis17 and our current study showed that monocytosis in Nba2.TLR8−/− females also seemed to depend on enhanced TLR7 activity. Only a fraction of Gr1hi monocytes become Gr1lo upon TLR activation,19 those that give rise to tissue-resident macrophages and DC.44 These cells are presumably further activated by circulating IgG immune complexes that contain self-nucleic acids, providing a link between stimulatory FcγRIV and endosomal TLRs.
In humans, TLR8 is thought to affect autoimmune responses because: (i) TLR8 is expressed primarily by myeloid cells,25 (ii) TLR8 contributes to DC differentiation and maturation in response to TLR8 agonists,45 and (iii) a Tlr8 polymorphism is genetically associated with SLE in a cohort of females with lupus.46 In addition, within human monocytes and pDC, autoantibodies induced the translocation of TLR7 and TLR8 to the endosomal compartment, a process that requires the endoplasmic reticulum membrane protein UNC93B. Interestingly, the expression of UNC93B in blood cells was higher for patients with active lupus than for healthy controls.47
In conclusion, we have demonstrated TLR7-dependent activation of B cells, DC and monocytes, as well as increased levels of Tlr7 expression in activated monocytes. These data provide a mechanistic link between TLR8-deficiency and accelerated pathogenesis in lupus-prone mice. Our study also demonstrated that TLR8 regulates TLR7-mediated autoimmune responses. Further studies are necessary to determine whether TLR8-based strategies can be used to treat immune complex-mediated human diseases such as SLE.
Acknowledgments
We thank Prof. S. Akira who generously provided TLR8-deficient mice, Prof. S. Izui for GN scoring of periodic acid-Schiff-stained kidney sections, Mr G. Brighouse and Ms M. Alvarez for technical assistance. This project was supported by the Swiss National Science Foundation. M-LS-R is supported by the Swiss National Science Foundation (310030 138338).
Author Contribution
NLT performed the experiments and analysed the data. CM-L performed the experiments. M-LS-R designed and performed the experiments, analysed the data and wrote the manuscript.
Disclosures
The authors declare no financial or commercial conflict of interest.
References
- Egner W. The use of laboratory tests in the diagnosis of SLE. J Clin Pathol. 2000;53:424–32. doi: 10.1136/jcp.53.6.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–6. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Akira S. Manifold mechanisms of toll-like receptor-ligand recognition. J Clin Immunol. 2005;25:511–21. doi: 10.1007/s10875-005-7829-1. [DOI] [PubMed] [Google Scholar]
- Huyton T, Rossjohn J, Wilce M. Toll-like receptors: structural pieces of a curve-shaped puzzle. Immunol Cell Biol. 2007;85:406–10. doi: 10.1038/sj.icb.7100089. [DOI] [PubMed] [Google Scholar]
- Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter HH, Warnatz K. Molecules involved in T-B co-stimulation and B cell homeostasis: possible targets for an immunological intervention in autoimmunity. Expert Opin Biol Ther. 2005;5(Suppl 1):S61–71. doi: 10.1517/14712598.5.1.s61. [DOI] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–31. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Lartigue A, Courville P, Auquit I, et al. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006;177:1349–54. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- Pawar RD, Patole PS, Ellwart A, et al. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J Am Soc Nephrol. 2006;17:3365–73. doi: 10.1681/ASN.2006030263. [DOI] [PubMed] [Google Scholar]
- Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 2006;54:336–42. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. 2006;103:9970–5. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolland S, Yim YS, Tus K, et al. Genetic modifiers of systemic lupus erythematosus in FcγRIIB–/– mice. J Exp Med. 2002;195:1167–74. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland R, Fernandez L, Kari E, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–40. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Savarese E, Steinberg C, Pawar RD, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–15. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber ML, Kikuchi S, Borel P, et al. Evidence for genes in addition to Tlr7 in the Yaa translocation linked with acceleration of systemic lupus erythematosus. J Immunol. 2008;181:1556–62. doi: 10.4049/jimmunol.181.2.1556. [DOI] [PubMed] [Google Scholar]
- Yoshinobu K, Baudino L, Santiago-Raber ML, et al. Selective up-regulation of intact, but not defective env RNAs of endogenous modified polytropic retrovirus by the Sgp3 locus of lupus-prone mice. J Immunol. 2009;182:8094–103. doi: 10.4049/jimmunol.0900263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Raber ML, Baudino L, Alvarez M, et al. TLR7/9-mediated monocytosis and maturation of Gr-1hi inflammatory monocytes towards Gr-1lo resting monocytes implicated in murine lupus. J Autoimmun. 2011;37:171–9. doi: 10.1016/j.jaut.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Lalive PH, Benkhoucha M, Tran NL, et al. TLR7 signaling exacerbates CNS autoimmunity through downregulation of Foxp3+ Treg cells. Eur J Immunol. 2014;44:46–57. doi: 10.1002/eji.201242985. [DOI] [PubMed] [Google Scholar]
- Gorden KK, Qiu X, Battiste JJ, et al. Oligodeoxynucleotides differentially modulate activation of TLR7 and TLR8 by imidazoquinolines. J Immunol. 2006;177:8164–70. doi: 10.4049/jimmunol.177.11.8164. [DOI] [PubMed] [Google Scholar]
- Gorden KK, Qiu XX, Binsfeld CC, et al. Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol. 2006;177:6584–7. doi: 10.4049/jimmunol.177.10.6584. [DOI] [PubMed] [Google Scholar]
- Bauer S, Pigisch S, Hangel D, et al. Recognition of nucleic acid and nucleic acid analogs by Toll-like receptors 7, 8 and 9. Immunobiology. 2008;213:315–28. doi: 10.1016/j.imbio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Genestier L, Taillardet M, Mondiere P, et al. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol. 2007;178:7779–86. doi: 10.4049/jimmunol.178.12.7779. [DOI] [PubMed] [Google Scholar]
- Crozat K, Vivier E, Dalod M. Crosstalk between components of the innate immune system: promoting anti-microbial defenses and avoiding immunopathologies. Immunol Rev. 2009;227:129–49. doi: 10.1111/j.1600-065X.2008.00736.x. [DOI] [PubMed] [Google Scholar]
- Rozzo SJ, Allard JD, Choubey D, et al. Evidence for an interferon-inducible gene, Ifi202, in the susceptibility to systemic lupus. Immunity. 2001;15:435–43. doi: 10.1016/s1074-7613(01)00196-0. [DOI] [PubMed] [Google Scholar]
- Izui S, Higaki M, Morrow D, et al. The Y chromosome from autoimmune BXSB/MpJ mice induces a lupus-like syndrome in (NZW × C57BL/6)F1 male mice, but not in C57BL/6 male mice. Eur J Immunol. 1988;18:911–5. doi: 10.1002/eji.1830180612. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Fossati-Jimack L, Moll T, et al. Differential role of three major New Zealand Black-derived loci linked with Yaa-induced murine lupus nephritis. J Immunol. 2005;174:1111–7. doi: 10.4049/jimmunol.174.2.1111. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber ML, Dunand-Sauthier I, Wu T, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34:339–48. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber ML, Amano H, Amano E, et al. Fcγ receptor-dependent expansion of a hyperactive monocyte subset in lupus-prone mice. Arthritis Rheum. 2009;60:2408–17. doi: 10.1002/art.24787. [DOI] [PubMed] [Google Scholar]
- Ronnblom L, Alm GV. A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J Exp Med. 2001;194:F59–63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O, Pagni PP, Traub S, et al. TLR8 deficiency leads to autoimmunity in mice. J Clin Invest. 2010;120:3651–62. doi: 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA. 2002;99:5545–50. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnues B, Macedo AB, Roussel-Queval A, et al. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci USA. 2014;111:1497–502. doi: 10.1073/pnas.1314121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–23. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN, Baccala R, Beutler B, et al. Type I interferons (αβ) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. 2012;24:499–505. doi: 10.1097/BOR.0b013e3283562c3e. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Santiago-Raber ML, Amano H, et al. Contribution of NZB autoimmunity 2 to Y-linked autoimmune acceleration-induced monocytosis in association with murine systemic lupus. J Immunol. 2006;176:3240–7. doi: 10.4049/jimmunol.176.5.3240. [DOI] [PubMed] [Google Scholar]
- Wofsy D, Kerger CE, Seaman WE. Monocytosis in the BXSB model for systemic lupus erythematosus. J Exp Med. 1984;159:629–34. doi: 10.1084/jem.159.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano H, Amano E, Santiago-Raber ML, et al. Selective expansion of a monocyte subset expressing the CD11c dendritic cell marker in the Yaa model of systemic lupus erythematosus. Arthritis Rheum. 2005;52:2790–8. doi: 10.1002/art.21365. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Hackstein H, Knoche A, Nockher A, et al. The TLR7/8 ligand resiquimod targets monocyte-derived dendritic cell differentiation via TLR8 and augments functional dendritic cell generation. Cell Immunol. 2011;271:401–12. doi: 10.1016/j.cellimm.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Laska MJ, Troldborg A, Hansen B, et al. Polymorphisms within Toll-like receptors are associated with systemic lupus erythematosus in a cohort of Danish females. Rheumatology. 2014;53:48–55. doi: 10.1093/rheumatology/ket316. [DOI] [PubMed] [Google Scholar]
- Nakano S, Morimoto S, Suzuki S, et al. Up-regulation of the endoplasmic reticulum transmembrane protein UNC93B in the B cells of patients with active systemic lupus erythematosus. Rheumatology. 2010;49:876–81. doi: 10.1093/rheumatology/keq001. [DOI] [PubMed] [Google Scholar]