

Abstract
Sepsis initially starts with a systemic inflammatory response (SIRS phase) and is followed by a compensatory anti-inflammatory response syndrome (CARS) that causes impaired adaptive T-cell immunity, immune paralysis and an increased susceptibility to secondary infections. In contrast, parasitic filariae release thousands of microfilariae into the peripheral blood without triggering inflammation, as they induce regulatory, anti-inflammatory host responses. Hence, we investigated the impact of chronic filarial infection on adaptive T-cell responses during the SIRS and CARS phases of a systemic bacterial infection and analysed the development of T-cell paralysis following a subsequent adenovirus challenge in BALB/c mice. Chronic filarial infection impaired adenovirus-specific CD8+ T-cell cytotoxicity and interferon-γ responses in the absence of a bacterial challenge and led to higher numbers of splenic CTLA-4+ CD4+ T cells, whereas splenic T-cell expression of CD69 and CD62 ligand, serum cytokine levels and regulatory T-cell frequencies were comparable to naive controls. Irrespective of filarial infection, the SIRS phase dominated 6–24 hr after intravenous Escherichia coli challenge with increased T-cell activation and pro-inflammatory cytokine production, whereas the CARS phase occurred 6 days post E. coli challenge and correlated with high levels of transforming growth factor-β and increased CD62 ligand T-cell expression. Escherichia coli-induced impairment of adenovirus-specific CD8+ T-cell cytotoxicity and interferon-γ production was not additionally impaired by chronic filarial infection. This suggests that filarial immunoregulation does not exacerbate E. coli-induced T-cell paralysis.
Keywords: compensatory anti-inflammatory response syndrome, helminth, immune paralysis, sepsis, T-cell cytotoxicity
Introduction
Sepsis is a complex dysregulation of the immune system and one of the worldwide leading causes of complications in intensive care units and of death.1 Indeed, the mortality rate of severe sepsis and septic shock amounts to 30–60%.2 The initial phase of a septic assault is known as systemic inflammatory response syndrome (SIRS), which is caused by an excessive pro-inflammatory immune response triggered by microorganisms.3–5 The SIRS phase is followed by a compensatory anti-inflammatory response syndrome (CARS),6,7 which is associated with a decreased production of pro-inflammatory cytokines like interferon-γ (IFN-γ) and interleukin-12 (IL-12),8,9 increased release of anti-inflammatory cytokines and apoptosis of activated immune cells.10–12 Although this process limits sepsis-induced inflammation and pathology, it also impairs cellular immunity and leads to paralysis of the adaptive immune system.13 As a result, CARS patients have an increased susceptibility to secondary or opportunistic infections because of their inability to control viral or bacterial infections.14
Due to their fundamental role in modulating innate and adaptive immune responses, T cells are essentially involved during sepsis. T-cell production of pro-inflammatory cytokines like IFN-γ activate macrophages, so facilitating bacterial clearance.15 However, in more severe cases of sepsis, activated T cells may have detrimental effects by triggering inflammation, morbidity and tissue injury.15 Hence, patients with severe sepsis have increased numbers of activated cytotoxic CD8+ T cells16 and depletion of CD8+ T cells in combination with natural killer cells improves sepsis outcome in caecal ligation and puncture-treated animals.17 Accordingly, mice lacking T cells, showed a significantly improved survival rate following bacterial challenge.18 Furthermore, substantial T-cell apoptosis following SIRS requires the removal of apoptotic cells by macrophages and results in an increased production of anti-inflammatory cytokines.15 Regulatory Foxp3+ T cells, cytotoxic T-lymphocyte antigen 4 (CTLA-4; CD152) expressing effector T cells and regulatory T cells,19,20 anti-inflammatory IL-10,21,22 and programmed death-1/ programmed death ligand -1 (PD1/PD-L1) interactions have all been shown to contribute to the development of CARS.23 Prevention of those immunosuppressive mechanisms to maintain CD8 T-cell cytotoxicity and IFN-γ responses is hereby essential to avoid immune paralysis and secondary infections.9,10,13,24–27 Hence, sepsis-associated mortality is not only the result of excessive pro-inflammatory immune responses, but also the development of regulatory, anti-inflammatory immune responses that lead to immune paralysis and impair both adaptive and innate immune responses.
Infections with parasitic helminths also induce regulatory, anti-inflammatory immune responses in their hosts, which allows their long-term survival. In addition, helminths trigger type 2 immune responses that are characterized by the production of IL-4, IL-5 and IL-13, elevated levels of IgE, as well as eosinophilia.28 During chronic infection, filariae establish a regulatory, anti-inflammatory milieu in their hosts by increasing the production of the anti-inflammatory cytokines IL-10 and transforming growth factor-β (TGF-β), expansion of alternatively activated macrophages, regulatory T and B cells and by increasing the expression of the inhibitory co-receptor CTLA-4.28–30 Filariae also modulate the function of antigen-presenting cells. Microfilariae induce apoptosis in dendritic cells31 and suppress their production of IL-12 in response to lipopolysaccharide (LPS).32 This helminth-induced immunomodulation impairs adaptive immune responses to vaccinations,33,34 but has benefits for the host. Accordingly, a multitude of studies have demonstrated that helminth infections ameliorate or prevent autoimmune diseases.35–37
Recent studies further suggest that exposure to helminths and their products may also reduce acute pro-inflammatory immune responses as they occur during LPS-induced endotoxaemia or bacterial infection.38 Fasciola hepatica defence molecule 1 directly binds to LPS and prevents LPS-induced immune responses,39 whereas the F. hepatica tegumental antigen reduces dendritic cell maturation and suppresses IL-12p70 and IFN-γ levels during SIRS.40 Filarial antigens like the glycoprotein chitohexaose increase endotoxaemia survival by reducing pro-inflammatory cytokine levels41 or, in the case of the recombinant anticoagulant protein c2, reduce IL-10 production and thrombin generation without exerting an influence on pro-inflammatory cytokines.42 Similarly, infections with the intestinal nematode Nippostrongylus brasiliensis leads to a reduced bacterial burden after Klebsiella pneumoniae injection and improves sepsis survival.43 Although those studies suggest a beneficial effect of helminths and helminth-derived products on the initial pro-inflammatory phase of sepsis, helminth-induced regulatory, anti-inflammatory immune responses may worsen sepsis-induced CARS and lead to a stronger immune paralysis.
The current study investigated whether chronic Litomosoides sigmodontis infection alters splenic T-cell responses during the SIRS and CARS phase of an intravenous (i.v.) Escherichia coli-induced sepsis. Litomosoides sigmodontis is an excellent murine model for studying human filarial infections, as it establishes patent infections in susceptible BALB/c mice and induces immune responses that are comparable to those arising during human filarial infections.44,45 Development of sepsis-induced immune paralysis was analysed in this study by investigating cytokine and T-cell responses over time and determining adenovirus-specific CD8+ T-cell cytotoxicity in vivo.
Materials and methods
Mice and filarial infection
Female BALB/c mice were obtained from Janvier Labs (Le Genest-Saint-Isle, France) and housed in the animal facility of the Institute of Medical Microbiology, Immunology and Parasitology of the University Hospital of Bonn, Germany with access to food and water ad libitum. Experimental procedures performed in this study were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz, Cologne, Germany (84-02.04.2011.A326).
Female BALB/c mice were infected with L. sigmodontis at 6–8 weeks of age by natural infection via the tropical rat mite Ornithonyssus bacoti as previously described.46 Sepsis experiments were performed 90 days post-infection (dpi). At the time of necropsy, the infection was confirmed by identification of adult worms within the thoracic cavity and presence of microfilariae, the progeny of adult worms, was checked in the peripheral blood as previously described.47 At the time of analysis one to five L. sigmodontis adult worms could be found in the pleural cavity of all mice in all of the experiments.
Sepsis induction
For sepsis induction, chronic L. sigmodontis-infected mice and age-matched controls were intravenously challenged with 1 × 108 to 4 × 108 colony-forming units of E. coli K12. At the indicated time-points, mice were killed with an overdose of isoflurane (AbbVie Deutschland GmbH & Co. KG, Wiesbaden, Germany), blood was taken for determination of plasma cytokine levels and spleens were removed as previously described.47 The bacterial count (colony-forming units) in the spleen was determined from single spleen cell suspensions that were plated as serial dilutions on Luria–Bertani agar plates and incubated overnight at 37°.
Spleen cell preparation
Spleens were homogenized using a 70-µm sieve to obtain single cell suspensions and washed with medium (RPMI-1640; PAA Laboratories, Pasching, Austria) including 100 U/ml penicillin, 100 µg/ml streptomycin) and centrifuged at 300 g for 10 min. The obtained cell pellet was re-suspended in 1× RBC lysis buffer (eBioscience, San Diego, CA) and incubated for 5 min at room temperature for red blood cell lysis. Following another washing step, spleen cells were counted using the CASY TT® (Roche, Pensberg, Germany).
Assessment of T-cell activation via flow cytometry and cytokine production by ELISA
For analysis of T-cell activation, spleen-derived lymphocyte preparations were isolated at different time-points post E. coli challenge, blocked with PBS/1% BSA and 0·1% Fc block (Sigma-Aldrich, St Louis, MO) and stained with CD4-Peridinin chlorophyll protein and CD8-allophycocyanin conjugated antibodies (eBioscience). For analysis of cell activation, T cells were additionally stained with CD62L-phycoerythrin-Cy7 and CD69-phycoerythrin conjugated antibodies (eBioscience). Regulatory T cells and their activation were analysed after overnight fixation and permeabilization (eBioscience) and thereafter stained with CD4-Peridinin chlorophyll protein, Foxp3-FITC, CD25-allophycocyanin and CTLA-4(CD152)-phycoerythrin conjugated antibodies (eBioscience).
Spleen cells from animals that were analysed for CD8+ T-cell cytotoxicity in vivo (see below) were left either unstimulated (medium) or activated with H9L (HYLSTQSAL) or 2·5 μg/ml concanavalin A for 72 hr. Thereafter, cytokine concentrations were determined from spleen cell culture supernatants or serum by ELISA [IFN-γ, IL-6, TGF-β, tumour necrosis factor-α (TNF-α)] or ProcartaPlex Multiplex Immunoassays (IL-7, IL-15/IL-15R, IL-10) according to kit protocols (eBioscience).
Measurement of in vivo CD8+ T-cell cytotoxicity
To assess in vivo CD8 T-cell cytotoxicity, chronic L. sigmodontis-infected BALB/c mice and uninfected controls were i.v. challenged 1 day post i.v. E. coli challenge with 2 × 107 plaque-forming units of an enhanced green-fluorescent-protein (eGFP), ovalbumin aa257_264 SIINFEKL and luciferase (CBG68Luc) expressing DNA-containing adenovirus (AdGOL).48 Five days following AdGOL injection, mice were injected with 5 × 106 spleen cells from naive donor mice that were primed with the H-2Kd peptide HYLSTQSAL (H9L, eGFP200–208) and the same number of non-primed spleen cells of the same donor mice. To differentiate H9L exposed and unexposed donor cells in the recipient mice, donor cells were stained with two concentrations of carboxyfluorescein diacetate succinimidyl ester (CFSE), i.e. 2 µm and 0·2 µm CFSE, respectively. Relations of primed to non-primed target cells were analysed by flow cytometry 4 hr after cell transfer according to the following quotation to assess CD8+ T-cell cytotoxicity: % specific cytotoxicity = {100 − [(CFSEhigh/CFSElow) sample/(CFSEhigh/CFSElow) control] × 100}.
Statistical analysis
The statistical analysis was performed using prism graphpad 5.01 (GraphPad Software, San Diego, CA). All experiments with multiple groups were tested by one-way analysis of variance followed by Tukey's multiple comparison test. Differences between two unpaired groups were tested for significance with the two-tailed Mann–Whitney U-test. Significance is defined as P < 0·05 and the error bars represent mean ± SEM.
Results
Increased CD4+ and CD8+ T-cell numbers in L. sigmodontis-infected animals
CD4+ and CD8+ T cells are essential players of the adaptive immune response during sepsis. To investigate whether filaria-induced regulatory immune responses during chronic L. sigmodontis infection mitigate T-cell responses during the SIRS and CARS phases, we examined splenic CD4+ and CD8+ T cells following i.v. E. coli challenge. After 24 hr, the bacterial loads in the spleens of L. sigmodontis-infected and uninfected mice were comparable and both groups cleared the bacteria within 6 days post bacterial challenge (Fig.1a). Total spleen cell numbers were increased in both L. sigmodontis-infected and uninfected mice 24 hr and 6 days post E. coli challenge (Fig.1b), reaching statistical significance at both time points between E. coli-only challenged mice and naive controls. Interestingly, L. sigmodontis-infected mice had continuously increased spleen cell numbers compared with controls, before as well as after E. coli challenge, although this difference was only statistically significant in the absence of E. coli challenge (Fig.1b). Accordingly, total numbers of splenic CD4+ and CD8+ T cells were significantly increased in L. sigmodontis-infected mice compared with naive controls in the absence of an E. coli challenge and remained at an elevated level after E. coli challenge (Fig.1c,d), whereas relative numbers of CD4+ and CD8+ T cells were not influenced by L. sigmodontis infection (Fig.1e,f).
Figure 1.

Increased CD4+ and CD8+ T-cell numbers in the spleens of Litomosoides sigmodontis-infected animals. (a) Splenic bacterial load in L. sigmodontis-infected and uninfected mice 24 hr and 6 days post Escherichia coli K12 injection. (b) Absolute spleen cell numbers, (c) CD8+ T-cell and (d) CD4+ T-cell numbers, as well as frequencies of (e) CD8+ and (f) CD4+ T cells of L. sigmodontis-infected and uninfected mice before, and 24 hr and 6 days post E. coli K12 injection. Data represent one of two independent experiments with at least six mice per group. Data are shown as mean ± SEM and were tested for statistical significance by one-way analysis of variance followed by Tukey's multiple comparison test (a, b) or Mann–Whitney U-test (c–f). *P < 0·05; **P < 0·01.
Chronic L. sigmodontis infection does not alter CD4+ and CD8+ T-cell activation
To examine if chronic filarial infection modulates E. coli-induced T-cell activation, CD69 and CD62L expression was measured on splenic CD4+ and CD8+ T cells (Fig.2a). CD69 expression on CD4+ T cells was not altered by L. sigmodontis infection, neither in the absence of nor 24 hr and 6 days post E. coli challenge in comparison to uninfected controls (Fig.2b). Hence, E. coli-induced CD4+ T-cell activation was comparable in L. sigmodontis-infected and uninfected controls with both groups reaching the highest frequencies of CD4+ CD69+ T cells and CD69 expression levels at 24 hr post E. coli challenge and demonstrating a similar decline in CD69 expression levels and CD4+ CD69+ T-cell frequencies 6 days post E. coli challenge (Fig.2b). Similar to the CD4+ T-cell activation, frequencies of CD8+ CD69+ T cells and CD69 mean fluorescence intensity of CD8+ T cells peaked at 24 hr post E. coli challenge and declined back to baseline levels at 6 days post E. coli challenge and were not influenced by L. sigmodontis infection (Fig.2c). Frequencies of CD4+ and CD8+ T cells that expressed the homing receptor CD62L reached their lowest rate 24 hr after E. coli challenge and were not affected by the L. sigmodontis infection (Fig.2d,e). Expression intensity of CD62L on CD4+ and CD8+ T cells inversely correlated with CD69 expression levels during E. coli challenge and peaked 6 days post E. coli challenge. L. sigmodontis infection significantly reduced CD62L expression levels on CD4+ and on CD8+ T cells 6 days post E. coli challenge (Fig.2d,e). Those results suggest that T-cell activation peaked 24 hr following i.v. E. coli challenge, whereas increased CD62L expression 6 days post E. coli challenge indicates a recruitment of naive T cells to the spleen that was less pronounced in L. sigmodontis-infected animals.
Figure 2.
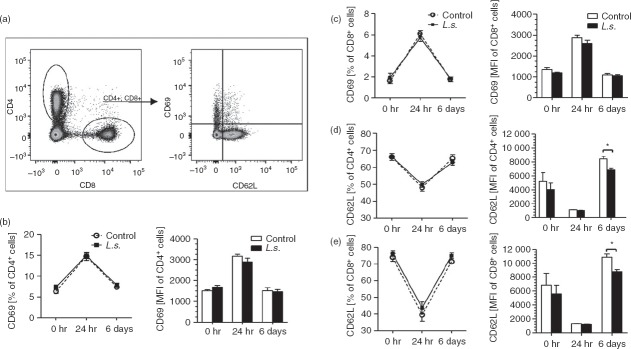
Litomosoides sigmodontis infection reduces CD62L expression on CD4+ and CD8+ T cells 6 days after Escherichia coli injection. (a) Gating strategy to identify splenic CD4+ and CD8+ T cells expressing the activation marker CD69 and the homing receptor CD62L. Frequency and mean fluorescence intensity (MFI) of CD4+ T cells and CD8+ T cells expressing CD69 (b, c) and CD62L (d, e). T cells were analysed from spleens of L. sigmodontis-infected (L.s.) and uninfected animals before, and 24 hr and 6 days post E. coli challenge (uninfected n = 6; L.s. n = 4; uninfected + E. coli n = 6; L.s. + E. coli n = 6 per point of time). One representative of two independent experiments is shown. Data are shown as mean ± SEM and were analysed by Mann–Whitney U-test. *P < 0·05.
Systemic cytokine production in the case of i.v. E. coli challenge is not modulated by L. sigmodontis infection
To assess if systemic cytokine production after E. coli challenge is modulated by L. sigmodontis infection, the pro-inflammatory cytokines TNF-α, IFN-γ and IL-6 as well as the anti-inflammatory cytokines IL-10 and TGF-β were measured at the time-points 0, 6, 24 hr and 6 days (Fig.3). In addition, concentrations of IL-7, because of their important role in T-cell survival and expansion, and levels of IL-15, which promotes effector T-cell differentiation, were analysed.26,27 Six hours after E. coli injection concentrations of all measured pro-inflammatory cytokines (TNF-α, IFN-γ, IL-6) were strongly increased in both L. sigmodontis-infected and non-infected animals and declined back to baseline levels 6 days after bacterial injection (Fig.3a–c). In contrast, IL-7 levels dropped in both groups 6 hr after E. coli injection and increased back to baseline levels in L. sigmodontis-infected mice 6 days post E. coli challenge (Fig.3d). At the peak of inflammation (6 hr post E. coli challenge), concentrations of IL-15/IL-15R and IL-10 levels were highest, whereas TGF-β cytokine concentrations only rose by 6 days post injection (Fig.3e–g). Besides the significant increase in IL-6 release 24 hr following E. coli challenge, chronic L. sigmodontis infection did not alter the aforementioned systemic cytokine levels either before or after E. coli challenge. Hence, the initial systemic pro-inflammatory cytokine release triggered by i.v. E. coli challenge and the consequential anti-inflammatory responses do not appear to be modulated by chronic filarial infection.
Figure 3.

Litomosoides sigmodontis infection does not alter systemic cytokine production during the course of an Escherichia coli-induced sepsis. (a) Tumour necrosis factor-α (TNF-α), (b) interferon-γ (IFN-γ), (c) interleukin-6 (IL-6), (d) IL-7, (e) IL-15/IL-15R, (f) IL-10, and (g) transforming growth factor-β (TGF-β) cytokine concentrations were measured in serum of L. sigmodontis-infected (L.s.) and uninfected animals before, and 6 hr, 24 hr and 6 days post E. coli challenge (uninfected n = 6; L.s. n = 4; uninfected + E. coli n = 6; L.s. + E. coli n = 6 per point of time). One representative of two independent experiments is shown. Bars show mean + SEM and were analysed for statistical significance by one-way analysis of variance followed by Tukey's Multiple Comparison Test. *P < 0·05; **P < 0·01; ***P < 0·001.
Expansion of regulatory T cells after filarial infection and bacterial challenge
To investigate whether chronic filarial infection increases the risk for CARS by promoting regulatory cell types, CTLA-4 expression and regulatory T cells were analysed before, and 24 hr and 6 days after E. coli challenge via flow cytometry (Fig.4a). Absolute Foxp3+ CD4+ regulatory T-cell numbers, but not frequencies, tended to be increased before (P = 0·16) and 24 hr post E. coli injection (P = 0·06) in L. sigmodontis-infected mice (Fig.4b,c), but did not differ 6 days post E. coli challenge (P = 1·0). Interestingly, 24 hr after E. coli challenge the relative and absolute numbers of splenic regulatory T cells peaked in both L. sigmodontis-infected and non-infected mice (Fig.4b,c), which coincided with the highest activation of the conventional CD4+ and CD8+ T cells (Fig.2). Similarly, absolute numbers of CD4+ T cells expressing CTLA-4 were significantly increased in L. sigmodontis-infected animals before, and 24 hr and 6 days post E. coli challenge, peaking 24 hr after E. coli injection (Fig.4e). Frequencies of CTLA-4+ CD4+ T cells were consistently increased in L. sigmodontis-infected mice following E. coli challenge, although the differences did not reach statistical significance (24 hr P = 0·06; 6 days P = 0·13; Fig.4d). Mean fluorescence intensity of CTLA-4 on CD4+ T cells peaked at 24 hr post E. coli challenge and was not effected by L. sigmodontis infection (Fig.4f). Taken together, during L. sigmodontis infection regulatory cell types were more abundant and peaked 24 hr following i.v. E. coli challenge.
Figure 4.
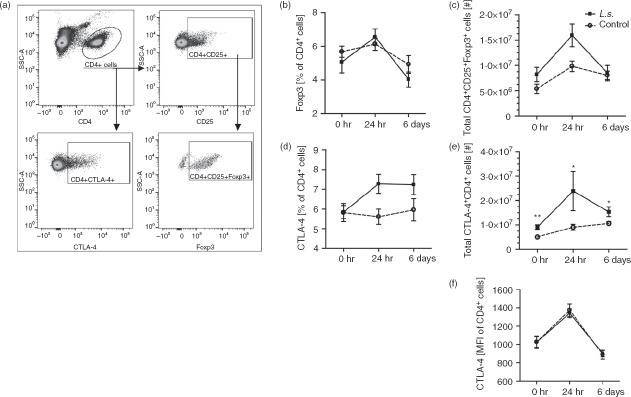
Litomosoides sigmodontis infection increases suppressive CTLA-4+ CD4+ cells during Escherichia coli challenge. (a) Gating strategy to identify splenic CD4+ CD25+ FoxP3+ regulatory T cells and CD4+ CTLA-4+ T cells by flow cytometry. (b) Frequency and (c) total numbers of CD4+ Foxp3+ T cells and (d) frequency and (e) total numbers of CTLA-4+ CD4+ T cells. (f) Mean fluorescence intensity of CTLA-4 on CD4+ T cells. Spleen cells were analysed from L. sigmodontis-infected (L.s.) and uninfected animals before, 24 hr and 6 days post E. coli challenge (uninfected n = 6; L.s. n = 4; uninfected + E. coli n = 6; L.s. + E. coli n = 6 per point of time). One representative of two independent experiments is shown. Data are shown as mean ± SEM and were analysed for statistical significance by Mann–Whitney U-test. *P < 0·05; **P < 0·01.
Sepsis impaired CD8 T-cell cytotoxicity is not further dampened by chronic filarial infection
During CARS CD8+ T-cell cytotoxicity is reduced, which increases the risk for opportunistic infections.13 To investigate whether specific CD8+ T-cell cytotoxicity after E. coli challenge is negatively influenced by chronic filarial infection, naive and chronic L. sigmodontis-infected mice were injected with a non-lethal dose of E. coli. The following day an eGFP, ovalbumin and luciferase expressing adenovirus (AdGOL) was injected intravenously. Six days after E. coli injection, AdGOL-specific CD8+ T-cell cytotoxicity was measured in vivo after an injection of CFSE-labelled and H-2Kd H9L-peptide (HYLSTQSAL)-pulsed donor spleen cells (Fig.5a). Subsequently, H9L-specific IFN-γ production was analysed after further ex vivo culture. As expected, the CD8+ T-cell cytotoxicity was reduced by ∼60% after i.v. E. coli challenge in non-infected mice (P < 0·001; Fig.5b). Accordingly, spleen cells from AdGOL- and E. coli-challenged mice produced significantly less IFN-γ in response to H9L compared with cells from AdGOL-only-treated mice (Fig.5c). In the absence of an E. coli challenge, L. sigmodontis-infected mice showed a significantly reduced AdGOL-specific CD8+ T-cell cytotoxicity compared with uninfected, AdGOL-treated controls (Fig.5b; see Supporting information, Fig. S1). Similarly, ex vivo H9L-induced IFN-γ responses were suppressed in L. sigmodontis-infected mice (Fig.5c; see Supporting information, Fig. S1).
Figure 5.
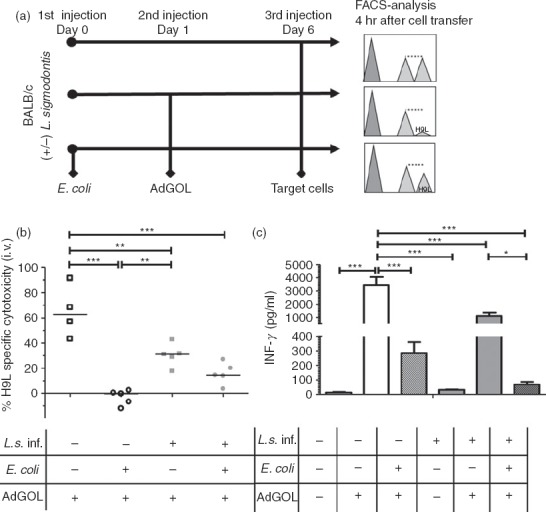
Adenovirus-specific CD8+ T-cell cytotoxicity is not exacerbated by Litomosoides sigmodontis infection after Escherichia coli-induced immune paralysis. (a) Experimental setup to assess CD8+ T-cell cytotoxicity and eGFP, ovalbumin and luciferase expressing adenovirus (AdGOL) -derived H9L-specific cytokine production in chronic L. sigmodontis-infected and uninfected mice. Two sets of naive spleen cells from donor mice were stained with different concentrations of CFSE and the cells stained with the higher CFSE concentration were subsequently pulsed with HYLSTQSAL-peptide (H9L). (b) In vivo CD8+ T-cell cytotoxicity and (c) ex vivo H9L-specific IFN-γ production of spleen cells from L. sigmodontis-infected (L.s.) and uninfected controls without (L.s. n = 5; uninf. n = 4) and 6 days after intravenous E. coli injection (L.s. + E. coli n = 5; uninf. + E. coli n = 5), 5 days after AdGOL was administered. Data are shown as mean + SEM and were tested for statistical significance by one-way analysis of variance followed by Tukey's multiple comparison test. *P < 0·05; **P < 0·01; ***P < 0·001.
Interestingly, following i.v. E. coli challenge, in vivo AdGOL-specific CD8+ T-cell cytotoxicity showed a trend to an improved H9L-specific cytotoxicity in L. sigmodontis-infected mice compared with E. coli-challenged controls (Fig.5b; (average 15·8 versus 0%; P > 0·05), whereas ex vivo H9L-specific IFN-γ production tended to be lower (Fig.5c; P > 0·05). The data presented in Fig.5 stemmed from observations made in animals that harboured one to five living adult worms at the time of analysis and were all microfilaraemic [median 28 (range 2–75) microfilariae/50 µl blood]. In a second experiment, although all five mice had one to five living adult worms, three were amicrofilaraemic and two had only two microfilariae/50 µl peripheral blood. In this experiment, in vivo AdGOL-specific CD8+ T-cell cytotoxicity following i.v. E. coli challenge was comparable in L. sigmodontis-infected and non-infected groups (see Supporting information, Fig. S1a). Ex vivo H9L-induced IFN-γ responses were also equal and significantly reduced when compared with the control group (see Supporting information, Fig. S1b). The results suggest therefore that chronic filarial infection impairs cytotoxic T-cell functions in the absence of a bacterial challenge, but does not further promote sepsis-induced cytotoxic T-cell impairment.
Discussion
Previous work demonstrated that immunomodulation by L. sigmodontis infection prevents the onset of diabetes in non-obese diabetic mice,37 modulates basophil functions,49 prevents cerebral malaria,50 and ameliorates allergies,51 highlighting the influence of a chronic filarial infection on bystander immune responses. Importantly, filarial infection does not seem to increase susceptibility to additional bacterial infections, as chronic L. sigmodontis-infected animals had no exacerbated Mycobacterium tuberculosis infection52 and had an improved sepsis survival after intraperitoneal E. coli challenge.53
In the current study we investigated the impact of a chronic filarial infection on T-cell responses following i.v. E. coli-induced sepsis. Litomosoides sigmodontis adult worms live within the thoracic cavity, where they induce alternatively activated macrophages, regulatory T cells and CTLA-4 expression on T cells.54 In the scenario studied here, systemic modulation of immune responses was less evident in chronic L. sigmodontis-infected mice, in the absence of E. coli challenge, as we observed no differences in expression levels of CD62L and CD69 on splenic CD4+ and CD8+ T cells, Foxp3+ regulatory T cell frequencies, or changes in pro-inflammatory and anti-inflammatory cytokine levels in sera compared with naive mice. However, total CD4+ and CD8+ T-cell numbers in the spleens of chronic L. sigmodontis-infected mice were significantly increased, including significantly higher numbers of splenic CD4+ T cells expressing CTLA-4. Furthermore, in the absence of E. coli challenge, in vivo adenovirus-specific CD8+ T-cell cytotoxicity was significantly impaired in L. sigmodontis-infected mice and ex vivo spleen cell re-stimulation revealed impaired H9L-specific IFN-γ responses in L. sigmodontis-infected mice, suggesting that chronic filarial infection induces splenic CD8+ T-cell anergy. Our new findings are in line with previous studies investigating CD8+ T-cell responses during helminth infection. For example, acute L. sigmodontis-infected mice showed impaired CD8+ T-cell responses after a single vaccination with Plasmodium berghei circumsporozoite protein (CSP), and presented reduced CSP-specific cytokine production, CSP-specific in vivo CD8+ T-cell cytotoxicity and impaired protection against P. berghei infection.55 Similarly, Schistosoma mansoni infection significantly down-regulated virus-specific CD8+ T-cell responses and impaired virus-specific CTL immunity leading to an increased virus titre and a delayed viral clearance.56 Recently, it was further shown using Trichinella spiralis and murine norovirus co-infected animals that the resulting impairment of virus-specific CD4+ and CD8+ T-cell responses was due to the induction of alternatively activated macrophages and their production of YM1.57
Following i.v. bacterial challenge, both CD8+ and CD4+ T-cell numbers expanded in the spleen of L. sigmodontis-infected and uninfected controls to a similar degree and showed a marked increase in CD69 expression and low expression levels of CD62L 24 hr after E. coli challenge, demonstrating their increased activation. In correlation with the development of anergy,58 baseline expression levels of CD69 expression returned on CD4+ and CD8+ T cells 6 days post E. coli challenge, whereas expression levels of CD62L rose, suggesting a loss of activated T cells and a replenishment with naive T cells. Our observations are therefore in line with the literature, reporting a decrease of naive T cells 24 hr following sepsis and subsequent apoptosis of effector cells and new recruitment of naive T cells to the spleen during the later phase of sepsis.15,18 CD8+ T-cell anergy was clearly demonstrated in our experiments 6 days post E. coli challenge, as in vivo CD8+ T-cell cytotoxicity and ex vivo H9L-specific IFN-γ production were significantly reduced compared with animals without E. coli challenge, being in line with the observations of Schwandt et al.13 In agreement with T-cell activation and the known function of IL-7 to prevent apoptosis and induce T-cell proliferation,26 and the ability of IL-15/IL-15R, to promote effector T cells,27 IL-7 levels were reduced 6–24 hr after E. coli challenge, whereas IL-15/IL-15R concentrations peaked at that time point. According to the kinetics of T-cell activation, concentrations of pro-inflammatory serum cytokines peaked 6 hr following E. coli challenge and declined back to baseline concentrations within 1–6 days post E. coli challenge. Similar to the study by Osuchowski et al.,59 serum IL-10 levels in our study showed kinetics comparable to pro-inflammatory cytokines following E. coli challenge, while TGF-β levels were significantly increased 6 days post E. coli. Those results suggest that the SIRS phase dominated 6–24 hr after E. coli challenge, whereas the CARS phase was dominant at 6 days post i.v. E. coli challenge, in accordance with previous studies.13,58,59
Although there were no differences in splenic T-cell activation as well as serum cytokine concentrations between L. sigmodontis-infected and uninfected mice during the SIRS phase following i.v. E. coli challenge, CD62L expression on T cells was reduced in L. sigmodontis-infected animals during the CARS phase following E. coli challenge, suggesting that fewer naive T cells were present in the spleen of L. sigmodontis-infected mice. However, CTLA-4+ CD4+ T cells remained increased in L. sigmodontis-infected mice following E. coli challenge, which may impair T-cell activation and proliferation.24 Despite the increased number of CTLA-4+ CD4+ T cells in L. sigmodontis-infected mice, no differences in CTLA-4 mean fluorescence intensity were observed and two independent experiments revealed no additional impairment of in vivo AdGOL-specific CD8+ T-cell cytotoxicity and ex vivo AdGOL-specific IFN-γ responses by L. sigmodontis infection during the CARS phase. Interestingly, the presence of microfilaraemia in L. sigmodontis-infected animals even resulted in an improved CD8+ T-cell cytotoxicity after E. coli challenge. A similar beneficial role of microfilariae-positive L. sigmodontis infections on protective CD8 T-cell responses has been previously suggested in the context of P. berghei co-infections because the effects were not observed in microfilariae-negative L. sigmodontis-infected mice.60 Those observations suggest that chronic filarial infection does not further impair specific CD8+ T-cell cytotoxicity during the CARS phase of a bacterial sepsis, although future studies will have to clarify whether this also improves sepsis survival during the CARS phase.
Importantly, we used i.v. E. coli challenges in this study, to allow the analysis of a sufficient immune paralysis within the spleen, which was previously shown to be essential in the initiation of CARS.13 Although current data from our group demonstrate that chronic L. sigmodontis infection has a beneficial effect on E. coli-induced sepsis after intraperitoneal bacterial challenge, including reduced serum levels of pro-inflammatory cytokines, an improved bacterial clearance and milder hypothermia,53 those beneficial effects were not observed when bacteria were administered intravenously. Hence, in a setting where chronic filarial infection improves bacterial clearance during sepsis, the impact on the spleen is less prominent and consequently T-cell paralysis is less pronounced.
Helminth-derived products are currently investigated for their potential to reduce acute pro-inflammatory immune responses,36,38 e.g. during SIRS, and may present new treatment strategies for sepsis. Based on our findings, chronic filarial infection does not further exacerbate the CARS phase following SIRS, which is an essential requirement for a helminth-based therapy.
Acknowledgments
This work was funded by the German Research Foundation (HU 2144/1-1); intramural funding by the University Hospital of Bonn (BONFOR, 2010-1-16 and 2011-1-10); and the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013 under Research Executive Agency Grant GA 276704. BB was supported by the Jürgen Manchot Stiftung, Düsseldorf. AH is a member of the German Centre for Infection Research (DZIF), and of the Excellence Cluster Immunosensation (DFG, EXC 1023). We would like to thank Jesuthas Ajendra, Afiat Berbudi, Anna-Lena Neumann and David Schmidt for their valuable technical assistance during the experiments and Laura E. Layland for critical reading of the manuscript.
Glossary
- AdGOL
eGFP, ovalbumin and luciferase expressing adenovirus
- CARS
compensatory anti-inflammatory response syndrome
- CSP
circumsporozoite protein
- CTLA-4
cytotoxic T-lymphocyte antigen-4
- eGFP
enhanced green fluorescent protein
- IFN-γ
interferon-γ
- IL-12
interleukin-12
- i.v.
intravenous
- LPS
lipopolysaccharide
- PD1
programmed death 1
- PD-L1
programmed death ligand 1
- SIRS
systemic inflammatory response syndrome
- TGF-β
transforming growth factor-β
- TNF-α
tumour necrosis factor-α
Disclosures
The authors declare no financial and commercial conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Adenovirus-specific CD8+ T-cell cytotoxicity is not exacerbated by Litomosoides sigmodontis infection after Escherichia coli-induced immune paralysis.
References
- Martin G, Brunkhorst F, Janes JM, Reinhart K, Sundin DP, Garnett K, Beale R. Promoting Global Research Excellence in Severe Sepsis (PROGRESS): lessons from an international sepsis registry. Infection. 2009;37:222–32. doi: 10.1007/s15010-008-8203-z. [DOI] [PubMed] [Google Scholar]
- Morrell MR, Micek ST, Kollef MH. The management of severe sepsis and septic shock. Infect Dis Clin North Am. 2009;23:485–501. doi: 10.1016/j.idc.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Levy M, Fink M, Marshall J, et al. SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM Consensus Conference on Sepsis and Organ Failure. Chest. 1992;101:1481–3. doi: 10.1378/chest.101.6.1481. [DOI] [PubMed] [Google Scholar]
- Bosmann M, Ward P. The inflammatory response in sepsis. Trends Immunol. 2013;3:129–36. doi: 10.1016/j.it.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–8. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Adib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb Haemost. 2009;101:36–47. [PubMed] [Google Scholar]
- Ono S, Ueno C, Aosasa S, Tsujimoto H, Seki S, Mochizuki H. Severe sepsis induces deficient interferon-γ and interleukin-12 production, but interleukin-12 therapy improves survival in peritonitis. Am J Surg. 2001;182:491–7. doi: 10.1016/s0002-9610(01)00754-1. [DOI] [PubMed] [Google Scholar]
- Döcke WD, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-γ treatment. Nat Med. 1997;3:678–81. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- Hotchkiss R, Opal S. Immunotherapy for sepsis – a new approach against an ancient foe. N Engl J Med. 2010;1:87–9. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger PM, Hyvernat H, Breittmayer J, et al. Enhanced T-cell apoptosis in human septic shock is associated with alteration of the costimulatory pathway. Eur J Clin Microbiol Infect Dis. 2009;28:575–84. doi: 10.1007/s10096-008-0673-5. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- Schwandt T, Schumak B, Gielen G, et al. Expression of type I interferon by splenic macrophages suppresses adaptive immunity during sepsis. EMBO J. 2012;31:201–13. doi: 10.1038/emboj.2011.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–74. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten KR, Tschöp J, Goetzman H, et al. T-cell activation differentially mediates the host response to sepsis. Shock. 2010;34:377–83. doi: 10.1097/SHK.0b013e3181dc0845. [DOI] [PubMed] [Google Scholar]
- Napoli AM, Fast LD, Gardiner F, Nevola M, Machan J. Increased granzyme levels in cytotoxic T lymphocytes are associated with disease severity in emergency department patients with severe sepsis. Shock. 2012;37:257–62. doi: 10.1097/SHK.0b013e31823fca44. [DOI] [PubMed] [Google Scholar]
- Sherwood ER, Enoh VT, Murphey ED, Lin C. Mice depleted of CD8+ T and NK cells are resistant to injury caused by cecal ligation and puncture. Lab Invest. 2004;84:1655–65. doi: 10.1038/labinvest.3700184. [DOI] [PubMed] [Google Scholar]
- Van Schaik SM, Abbas AK. Role of T cells in a murine model of Escherichia coli sepsis. Eur J Immunol. 2007;37:3101–10. doi: 10.1002/eji.200737295. [DOI] [PubMed] [Google Scholar]
- Monneret G, Debard A, Venet F, Bohe J, Hequet O, Bienvenu J, Lepape A. Marked elevation of human circulating CD4+ CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. 2003;31:2068–71. doi: 10.1097/01.CCM.0000069345.78884.0F. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- O'Garra A, Murphy KM. From IL-10 to IL-12: how pathogens and their products stimulate APCs to induce TH1 development. Nat Immunol. 2009;10:929–32. doi: 10.1038/ni0909-929. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhou Y, Lou J, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. 2010;14:R220. doi: 10.1186/cc9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock. 2011;36:38–44. doi: 10.1097/SHK.0b013e3182168cce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmamdam P, Inoue S, Unsinger J, Chang K, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. 2010;88:233–40. doi: 10.1189/jlb.0110037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsinger J, McGlynn M, Kasten KR, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 2010;184:3768–79. doi: 10.4049/jimmunol.0903151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Unsinger J, Davis CG, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J. Immunol. 2010;184:1401–9. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11:375–88. doi: 10.1038/nri2992. [DOI] [PubMed] [Google Scholar]
- Hoerauf A, Satoguina J, Saeftel M, Specht S. Immunomodulation by filarial nematodes. Parasite Immunol. 2005;27:417–29. doi: 10.1111/j.1365-3024.2005.00792.x. [DOI] [PubMed] [Google Scholar]
- Maizels RM, Balic A, Gomez-Escobar N, Nair M, Taylor MD, Allen JE. Helminth parasites – masters of regulation. Immunol Rev. 2004;201:89–116. doi: 10.1111/j.0105-2896.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- Semnani R, Liu A, Sabzevari H, Kubofcik J, Zhou J, Gilden J, Nutman TB. Brugia malayi microfilariae induce cell death in human dendritic cells, inhibit their ability to make IL-12 and IL-10, and reduce their capacity to activate CD4+ T cells. J Immunol. 2003;171:1950–60. doi: 10.4049/jimmunol.171.4.1950. [DOI] [PubMed] [Google Scholar]
- Semnani R, Venugopal P, Leifer C, Mostböck S, Sabzevari H, Nutman TB. Inhibition of TLR3 and TLR4 function and expression in human dendritic cells by helminth parasites. Blood. 2008;112:1290–8. doi: 10.1182/blood-2008-04-149856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PJ, Espinel I, Paredes W, Nutman TB. Impaired tetanus-specific cellular and humoral responses following tetanus vaccination in human onchocerciasis: a possible role for interleukin-10. J Infect Dis. 1998;178:1133–8. doi: 10.1086/515661. [DOI] [PubMed] [Google Scholar]
- Cooper PJ, Chico M, Losonsky G, et al. Albendazole treatment of children with ascariasis enhances the vibriocidal antibody response to the live attenuated oral cholera vaccine CVD 103-HgR. J Infect Dis. 2000;182:1199–206. doi: 10.1086/315837. [DOI] [PubMed] [Google Scholar]
- Hübner MP, Stocker JT, Mitre E. Inhibition of type 1 diabetes in filaria-infected non-obese diabetic mice is associated with a T helper type 2 shift and induction of FoxP3+ regulatory T cells. Immunology. 2009;127:512–22. doi: 10.1111/j.1365-2567.2008.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccone P, Cooke A. Vaccine against autoimmune disease: can helminths or their products provide a therapy? Curr Opin Immunol. 2013;3:418–23. doi: 10.1016/j.coi.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Hübner MP, Shi Y, Torrero M, et al. Helminth protection against autoimmune diabetes in nonobese diabetic mice is independent of a type 2 immune shift and requires TGF-β. J Immunol. 2012;188:559–68. doi: 10.4049/jimmunol.1100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner MP, Layland LE, Hoerauf A. Helminths and their implication in sepsis – a new branch of their immunomodulatory behaviour. Pathog Dis. 2013;2:127–41. doi: 10.1111/2049-632X.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MW, Donnelly S, Hutchinson A, To J, Taylor NL, Norton RS, Perugini MA, Dalton JP. A family of helminth molecules that modulate innate cell responses via molecular mimicry of host antimicrobial peptides. PLoS Pathog. 2011;7:e1002042. doi: 10.1371/journal.ppat.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CM, Dowling DJ, Loscher CE, Morphew RM, Brophy PM, O'Neill SM. The Fasciola hepatica tegumental antigen suppresses dendritic cell maturation and function. Infect Immun. 2009;77:2488–98. doi: 10.1128/IAI.00919-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda SK, Kumar S, Tupperwar N, Vaidya T, George A, Rath S, Bal V, Ravindran B. Chitohexaose activates macrophages by alternate pathway through TLR4 and blocks endotoxemia. PLoS Pathog. 2012;8:e1002717. doi: 10.1371/journal.ppat.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pont A, Moons A, de Jonge E, et al. Recombinant nematode anticoagulant protein c2, an inhibitor of tissue factor/factor VIIa, attenuates coagulation and the interleukin-10 response in human endotoxemia. J Thromb Haemost. 2004;2:65–70. doi: 10.1111/j.1538-7836.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- Sutherland RE, Xu X, Kim S, Seeley EJ, Caughey GH, Wolters PJ. Parasitic infection improves survival from septic peritonitis by enhancing mast cell responses to bacteria in mice. PLoS One. 2011;6:e27564. doi: 10.1371/journal.pone.0027564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann WH, Petit G, Schulz-Key H, Taylor DW, Bain O, Le Goff L. Litomosoides sigmodontis in mice: reappraisal of an old model for filarial research. Parasitol Today. 2000;16:387–9. doi: 10.1016/s0169-4758(00)01738-5. [DOI] [PubMed] [Google Scholar]
- Allen JE, Adjei O, Bain O, et al. Of mice, cattle, and humans: the immunology and treatment of river blindness. PLoS Negl Trop Dis. 2008;2:e217. doi: 10.1371/journal.pntd.0000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann L, Bain O, Saeftel M, et al. Murine filariasis: interleukin 4 and interleukin 5 lead to containment of different worm developmental stages. Med Microbiol Immunol. 2003;192:23–31. doi: 10.1007/s00430-002-0155-9. [DOI] [PubMed] [Google Scholar]
- Ajendra J, Specht S, Neumann A, et al. ST2 deficiency does not impair type 2 immune responses during chronic filarial infection but leads to an increased microfilaremia due to an impaired splenic microfilarial clearance. PLoS One. 2014;9:e93072. doi: 10.1371/journal.pone.0093072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlleber D, Kashkar H, Gärtner K, et al. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep. 2012;2:478–87. doi: 10.1016/j.celrep.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Larson D, Hübner MP, Torrero M, et al. Chronic helminth infection reduces basophil responsiveness in an IL-10-dependent manner. J Immunol. 2012;188:4188–99. doi: 10.4049/jimmunol.1101859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht S, Ruiz DF, Dubben B, Deininger S, Hoerauf A. Filaria-induced IL-10 suppresses murine cerebral malaria. Microbes Infect. 2010;12:635–42. doi: 10.1016/j.micinf.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Dittrich A, Erbacher A, Specht S, et al. Helminth infection with Litomosoides sigmodontis induces regulatory T cells and inhibits allergic sensitization, airway inflammation, and hyperreactivity in a murine asthma model. J Immunol. 2008;180:1792–9. doi: 10.4049/jimmunol.180.3.1792. [DOI] [PubMed] [Google Scholar]
- Hübner MP, Killoran K, Rajnik M, et al. Chronic helminth infection does not exacerbate Mycobacterium tuberculosis infection. PLoS Negl Trop Dis. 2012;12:e1970. doi: 10.1371/journal.pntd.0001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondorf F, Berbudi A, Buerfent BC, et al. Chronic filarial infection provides protection against bacterial sepsis by functionally reprogramming macrophages. PLoS Pathog. 2015 doi: 10.1371/journal.ppat.1004616. ; accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Harris A, Babayan SA, Bain O, Culshaw A, Allen JE, Maizels RM. CTLA-4 and CD4+ CD25+ regulatory T cells inhibit protective immunity to filarial parasites in vivo. J Immunol. 2007;179:4626–34. doi: 10.4049/jimmunol.179.7.4626. [DOI] [PubMed] [Google Scholar]
- Kolbaum J, Tartz S, Hartmann W, et al. Nematode-induced interference with the anti-Plasmodium CD8+ T-cell response can be overcome by optimizing antigen administration. Eur J Immunol. 2012;42:890–900. doi: 10.1002/eji.201141955. [DOI] [PubMed] [Google Scholar]
- Actor JK, Shirai M, Kullberg MC, Buller RM, Sher A, Berzofsky JA. Helminth infection results in decreased virus-specific CD8+ cytotoxic T-cell and Th1 cytokine responses as well as delayed virus clearance. Proc Natl Acad Sci USA. 1993;90:948–52. doi: 10.1073/pnas.90.3.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LC, Monticelli L, Nice T, et al. Virus–helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science. 2014;345:578–82. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/Inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–74. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- Ruiz DF, Dubben B, Saeftel M, Endl E, Deininger S, Hoerauf A, Specht S. Filarial infection induces protection against P. berghei liver stages in mice. Microbes Infect. 2009;11:172–80. doi: 10.1016/j.micinf.2008.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Adenovirus-specific CD8+ T-cell cytotoxicity is not exacerbated by Litomosoides sigmodontis infection after Escherichia coli-induced immune paralysis.