

Abstract
Serotonin, a highly pro-inflammatory molecule released by activated platelets, is formed by tryptophan. Tryptophan is also needed in the production of kynurenine, a process mediated by the type I interferon (IFN)-regulated rate-limiting enzyme indoleamine 2,3-dioxygenase (IDO). The aim of this study was to investigate levels of serotonin in patients with the autoimmune disease systemic lupus erythematosus (SLE), association to clinical phenotype and possible involvement of IDO in regulation of serotonin synthesis. Serotonin levels were measured in serum and plasma from patients with SLE (n=148) and healthy volunteers (n=79) by liquid chromatography and ELISA, as well as intracellularly in platelets by flow cytometry. We found that SLE patients had decreased serotonin levels in serum (p=0.01) and platelets (p<0.0001) as compared to healthy individuals. SLE patients with ongoing type I IFN activity, as determined by an in-house reporter assay, had decreased serum levels of serotonin (p=0.0008) as well as increased IDO activity (p<0.0001), as determined by the kynurenine/tryptophan ratio measured by liquid chromatography. Furthermore, SLE sera induced IDO expression in WISH cells in a type I IFN-dependent manner (p=0.008). Also platelet activation contributed to reduce overall availability of serotonin levels in platelets and serum (p<0.05). Decreased serum serotonin levels were associated with severe SLE with presence of anti-dsDNA antibodies and nephritis. In all, reduced serum serotonin levels in SLE patients were related to severe disease phenotype, including nephritis, suggesting involvement of important immunopathological processes. Further, our data suggest that type I IFNs, present in SLE sera, are able to up-regulate IDO expression, which may lead to decreased serum serotonin levels.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune rheumatic disease characterized by systemic inflammation and involvement of multiple organ systems including skin, joints and kidneys [1]. The inflammation is mediated by tissue-deposited immune complexes (ICs) causing complement activation, infiltration of immune cells and tissue destruction. ICs are phagocytosed by plasmacytoid dendritic cells (pDCs) and may, if containing nucleic acids, activate toll-like receptors (TLR)7 or TLR9 and promote production of type I interferons (IFNs) [2, 3]. Type I IFNs, in particular IFN-alpha, are increased in SLE patients and related to disease activity. A type I IFN signature with several type I IFN-regulated genes and proteins highly up-regulated is often seen in SLE. Type I IFNs are considered key cytokines in SLE pathogenesis due to potent immunomodulatory effects [4–6].
Indoleamine 2,3-dioxygenase (IDO) is a type I IFN-regulated protein that is up-regulated in SLE patients [7–9]. IDO is the rate-limiting enzyme in the conversion of tryptophan into kynurenine. Kynurenine is an essential building block for several neuroactive metabolites, and skewing of the kynurenine pathways by inflammatory cytokines have been linked to central nervous system diseases [10–12]. By increasing the tryptophan conversion into kynurenine, IDO also limits synthesis of other tryptophan-dependent molecules, including serotonin [8, 13] (Fig 1). Serotonin is probably most known for its role as a signaling molecule in CNS synapses. However, most serotonin is produced in the periphery by intestinal enterochromaffin cells, where it is picked up by platelets and stored in dense granules [14, 15]. Upon platelet activation, serotonin is released locally to modulate hemostasis and inflammation [16–22]. Serotonin has been implicated as the driving force in establishing intestinal inflammation and serotonin receptor antagonists are able to block the inflammatory process in experimental animal models [23, 24]. Furthermore, serotonin has been shown to be involved in activation of monocytes and T cells, monocyte cytokine production, recruitment of neutrophils to inflammatory sites, extravasation of immune cells and regulation of type I IFN production through scavenging of ROS [18–22, 25], and those serotonin-mediated inflammatory effects have been implicated in rheumatic disorders [26–29]. Even though serotonin has been described to have profound inflammatory effects very little is known about its role in the pathogenesis of SLE and other chronic inflammatory diseases. To our knowledge very few attempts have been made to investigate serotonin in SLE and we found only four publications from the 80’s which all demonstrate decreased platelet serotonin levels in SLE [30–33]. The underlying mechanism behind the decreased levels of serotonin in SLE patients is however still unknown.
Fig 1. Summarizing figure of the main results and possible hypothesis.

Nucleic acid-containing immune complexes (IC) are phagocytosed by plasmacytoid dendritic cells (pDC) and induce large amounts of interferon (IFN)-alpha. IFN-alpha up-regulates the expression of indoleamine 2,3-dioxygenase (IDO), increasing the conversion of tryptophan to kynurenine and limiting the availability of tryptophan for serotonin synthesis. The produced serotonin is taken up by the platelet close to the enterochromaffin cells. Upon platelet activation, serotonin is released and consumed by immune cells to amplify the inflammatory environment together with ICs and IFN-alpha.
The aim of this study was to investigate peripheral levels of serotonin in SLE, association to clinical phenotype and the possible role of type I IFNs in regulation of serotonin synthesis. In brief we found that SLE patients had decreased platelet and serum levels of serotonin as compared to healthy individuals. The decreased serum levels of serotonin were highly associated with a severe SLE phenotype with involvement of nephritis and anti-dsDNA antibodies. Finally, a type I IFN-mediated skewing of the tryptophan metabolism and up-regulation of IDO, as well as ongoing platelet activation were identified as possible underlying mechanisms.
Materials and Methods
Patients
Patients with SLE (n = 148) were recruited at the Department of Rheumatology, Skåne University Hospital, Lund, Sweden. Healthy volunteers (n = 79) were age- and sex-matched to the SLE patients (SLE median 47 years, range 18–81 years, and healthy volunteers median 48 years, range 20–82 years, respectively and 85% and 87% women, respectively). An overview of the clinical characteristics is presented in Table 1. Disease activity was assessed using SLEDAI-2K [34]. All but two individuals fulfilled at least four American College of Rheumatology (ACR) 1982 criteria for SLE [35]. These two patients fulfilled three ACR criteria, had a clinical SLE diagnosis with at least two organ manifestations characteristic of SLE, autoimmune phenomena, and no other diagnosis that better could explain the symptoms. The following treatments were used in the SLE cohort at the time of blood sampling: corticosteroids (n = 98), hydroxychloroquine (n = 105), azathioprine (n = 32), mycophenolatmofetil (n = 20), methotrexate (n = 13), intravenous immunoglobulins (n = 2), non-steroidal anti-inflammatory drugs (n = 12), acetylsalicylic acid (n = 44), Warfarin (n = 23) and selective serotonin re-uptake inhibitors (SSRI) or other serotonin transporter antagonists (n = 21).
Table 1. Distribution of the American College of Rheumatology (ACR) 1982 classification criteria and patient characteristics for SLE patients (n = 148) included in the study.
| Disease duration, median (range), years | 11 (0–46) |
| SLEDAI score, median (range) | 1.5 (0–18) |
| SLICC/ACR-DI score, median (range) | 0 (0–8) |
| ACR criteria, median (range) | 5 (3–10) |
| Malar rash % | 52 |
| Discoid rash % | 20 |
| Photosensitivity % | 56 |
| Oral ulcers % | 24 |
| Arthritis % | 78 |
| Serositis % | 39 |
| Renal disease % | 33 |
| Neurological disorder % | 6 |
| Hematological manifestations % | 55 |
| Leukopenia % | 37 |
| Lymphopenia % | 24 |
| Thrombocytopenia % | 14 |
| Immunology % | 69 |
| Anti-dsDNA antibodies % | 59 |
| ANA % | 98 |
Ethics statement
The study was approved by Lund University regional ethics board (LU-2010/544) and an informed written consent was obtained from all participants.
Flow cytometry
Intracellular levels of serotonin in platelets were only measured in a subgroup of the recruited SLE patients (40/148) and healthy volunteers (64/79) due to logistic difficulties with fresh blood samples. For these platelet intracellular analyses, platelet rich plasma (PRP) was isolated from sodium-citrate blood through centrifugation for 10 min at 280 g. Isolated PRP was incubated with 2% paraformaldehyde for 45 min at room temperature. The incubation continued for 30 min at 37°C with the addition of 0.2% Triton X-100 to permeabilize the platelets. Between each of the following incubations, the platelets were washed once at 2500 g for 3 min in phosphate-buffered saline pH 7.4 (PBS). Anti-human serotonin antibodies produced in mouse (Dako, Glostrup, Denmark) were added and incubated for 15 min at 37°C followed by incubation with a FITC-conjugated rabbit anti mouse IgG antibody for 30 min at room temperature. The platelets were analyzed by flow cytometry on a BD Accuri C6 (BD, Franklin Lakes, NJ, USA). The results are presented as the median fluorescence intensity (MFI) ratio of the FITC fluorescence of the sample divided by the fluorescence of the secondary FITC-conjugated antibody alone.
Interferon activity assay
Type I IFN activity was measured as previously described [36, 37]. Briefly, WISH cells (CCL-25; American Type Culture Collection, Manassas, VA, USA) were cultured for 6 h with patient serum after which lysis mixture (Panomics Inc., Fremont, CA, USA) was added. Cell lysates were analyzed on a Luminex 100 (Luminex Corporation, Austin, TX, USA) for mRNA expression of three house-keeping genes (GAPDH, PPIB, B2M) and six type I IFN-regulated genes (LY6E, MX1, OAS1, ISG15, IFIT1 and EIF2AK2) using the Quantigene Plex 2.0 assay as described by the manufacturer (Panomics Inc.). The type I IFN activity was calculated by the mean fold change of the type I IFN-regulated genes in WISH cells exposed to SLE serum as compared to unstimulated WISH cells.
Measurement of serotonin, tryptophan and kynurenine
Serum was diluted 1/2 in PBS at a total volume of 200 μL with 90 μM 3-nitro-L-tyrosine (Sigma Aldrich) as an internal standard. For protein precipitation, trichloroacetic acid (0.4 M final concentration) was added and the samples centrifuged at 6000 rpm for 5 minutes. The upper 100 μL of the samples were saved for analysis of serotonin, tryptophan and kynurenine by liquid chromatography with ultra violet fluorescence detection (Agilent, Santa Clara, CA, USA) at Red Glead Discovery, Lund, Sweden. Tryptophan and serotonin levels were measured by fluorescence with excitation wavelength at 286 nm and emission at 366 nm. Kynurenine and 3-nitro-L-tyrosine were both detected by UV at 360 nm. The levels of serotonin were also measured with a commercially available ELISA according to the manufacturer’s instructions (LDN, Nordhorn, Germany) with similar levels as for the liquid chromatography (p<0.0001, r = 0.75, Spearman’s correlation). The 10th percentile of the serotonin serum level in healthy individuals was used as a cut-off level for decreased serum levels of serotonin. Serotonin was also measured in EDTA plasma (isolated through centrifugation at 3000 g for 10 minutes at room temperature) with a commercially available ELISA according to the manufacturer’s instructions (LDN, Nordhorn, Germany). To estimate the relative amount of released serotonin, plasma levels (peripheral serotonin) were divided with serum levels (total serotonin). The 90th percentile of the relative amount of released serotonin was used as a cut-off for increased peripheral serotonin.
Analysis of IDO expression
WISH cells (CCL-25 ATCC, Manassas, VA, USA), were cultured in RPMI-1640 medium supplemented with 2.05 mM L-Glutamine, 10% fetal calf serum, 1% non-essential amino acid (vol/vol), 1 mM sodium pyruvate (PAA Laboratories GmbH, Pasching, Austria) and 100 μg/mL gentamicin (Invitrogen, Carlsbad, CA, USA) in a volume of 200 μL using Lab-Tek chamber slide (Nalge Nunc International, Naperville, IL, USA) for 3 h. In some experiments, anti-human interferon alpha/beta receptor (IFNAR) chain 2 (Clone NMHAR-2, pbl interferonsource, Piscataway Township, NJ, USA) blocking antibodies were added at a concentration of 5 μg/mL 3 hours prior to addition of 100 μL sera from three healthy individuals or eight SLE patients selected to have anti-ribonucleoprotein (RNP) antibodies. As a positive control, 650 U/mL of recombinant IFN-alpha (IntronA, Schering-Plough Company, Innishannon, Ireland) was used. After four days of incubation, cells were detached by 0.5 mM EDTA in PBS. WISH cells were fixed in 2% paraformaldehyde (Alfa Aesar, Karlsruhe, Germany) for 10 min and permeabilized in 0.25% Triton X-100 for 15 min, followed by addition of a phycoerythrin-conjugated anti-human IDO antibody (R&D Systems Minneapolis, MN, USA) for another 30 min at room temperature. IDO expression was analyzed by flow cytometry (Accuri C6, BD Pharmingen).
Platelet activation
Platelet cell surface deposition of C4d was analyzed by flow cytometry as been described previously [38, 39]. For microparticle (MP) detection, flow cytometry was performed directly on EDTA plasma isolated through centrifugation at 3000 g for 10 minutes at room temperature [40, 41]. MPs were labeled with murine monoclonal anti-CD61-APC or the relevant isotype-matched control antibody. To reduce background noise, all buffers were filtered through 0.1 μm pore size filters (MiniSart HF, Sartorius Stedim Biotech S.A., Aubagne, France). Labeling was carried out by adding 5 μL heparin-sodium salt 10% w/v (Sigma-Aldrich 194 USP/mg dry basis) to 5 μL plasma followed by the addition of 5 μL pre-diluted anti-CD61-APC (IgG1k, final dilution 0.065 μg/mL, clone C7280, Dako, Glostrup, Denmark) or the isotype-matched control antibodies (IgG1k-APC, clone DAK-GO1, Dako). Then the suspension was diluted to 955 μL in low phosphate buffered saline with citrate (154 mM NaCl, 1.4 mM phosphate, 10.5 mM trisodium citrate, pH 7.4). The samples were analyzed using a FACSCalibur flow cytometer (BD, Franklin Lakes, NJ, USA) controlled by CellQuest software version 5.1.1 in the “high” flow rate mode. Flow rate was measured before each experiment. Both forward scatter (FSC) and side scatter (SSC), and the FL-4 fluorescence (anti-CD61) were recorded with logarithmic gain. μm Acquisition time was 60 seconds. MP gating was accomplished using 1 μm beads (Flow Cytometry Size Calibration Kit, Molecular Probes, Inc., Eugene, OR, USA) for setting upper limits in both FSC and SSC signals, and a lower limit was placed to exclude buffer noise as previously described [42]. Results from pilot studies clearly demonstrated that this gating strategy mainly captured MPs, whereas normal platelets were not included in the gate [40, 42]. Data analysis was performed using FlowJo software version 7.6.1 (Tree StarTreeStar, Inc., Ashland, OR, USA). Plasma concentrations (MPs/mL) were calculated on the basis of MP count per unit time, flow rate of the flow cytometer, and net dilution during sample preparation of the analyzed sample.
Statistics
Spearman’s correlation test was used to analyze correlations between platelet intracellular levels and serum levels of serotonin. Variables not assuming Gaussian distribution were normalized through logarithms. Mann-Whitney U-test and ANCOVA was used for group analyses. Logistic regression analysis was used for all dichotomous variables. All analyses were adjusted for differences in age and sex when applicable. GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) was used for the figures and IBM SPSS Statistics 20 (Armonk, NY, USA) was used for the statistical calculations. A p-value <0.05 was considered statistically significant. All data utilized in the statistical analyses are provided in the Supporting Information S1 Table.
Results
Platelet and serum levels of serotonin are decreased in SLE patients
The platelet levels of serotonin were markedly decreased in SLE patients as compared to healthy volunteers (p<0.0001, Fig 2A), and correlated with the serum concentration of serotonin in both healthy individuals (r = 0.45, p<0.0001) and in SLE patients (r = 0.48, p = 0.006, Fig 2B). A fairly large proportion of the SLE patients (n = 21/148, 14%) used selective serotonin re-uptake inhibitors (SSRI) which highly affect the concentration of serotonin in serum (p<0.0001, Fig 2C). Among the healthy volunteers, only 4/79 (5%) used SSRI treatment and they also had a decreased serotonin concentration in serum (p = 0.0009, Fig 2C). In concordance with the intracellular levels of serotonin in platelets, SLE patients had decreased levels of serotonin in serum as compared to healthy individuals (p = 0.01, Fig 2D). Even though the use of SSRI was evenly distributed among the SLE patients and not increased in any of the investigated clinical phenotypes, patients (and controls) on SSRI treatment were excluded from all serotonin analyses, including the results depicted in Fig 2. However, to verify the robustness of the statistical models all analyses were also performed with the SSRI-treated patients included, but adjusted for, with similar results. None of the immunosuppressive treatments used by the SLE patients affected serotonin levels (data not shown).
Fig 2. Serotonin levels in platelets and serum are dysregulated in SLE patients.

A) Platelet intracellular serotonin concentration was analyzed by flow cytometry in a subset of the SLE patients and healthy volunteers (HV). B) Correlation analysis for serum and platelet levels of serotonin in SLE patients. C) Treatment with selective serotonin re-uptake inhibitors was associated with decreased serum levels of serotonin in both SLE-patients and HV. D) Serum concentrations of serotonin in HV and SLE patients. The bars represent the median value.
Low serum levels of serotonin are associated with a severe SLE phenotype
After establishing that serotonin levels were decreased in SLE, the association between serotonin concentrations and clinical phenotype according to ACR classification criteria was analyzed. SLE patients with nephritis had decreased serotonin levels in serum as compared to patients without this manifestation (OR = 3.1 (1.3–7.2), p = 0.01, Table 2). Furthermore, presence of anti-dsDNA antibodies, which is often seen in SLE patients with nephritis, were associated with decreased serum levels of serotonin (OR = 2.7 (1.2–6.5), p = 0.02, Table 2). No associations were found between serotonin levels and clinical disease activity at the time-point of blood sampling, possibly due to very few or no signs of clinical disease activity in most of the recruited patients. However, decreased serotonin levels were associated with complement consumption at the time-point of blood sampling (OR = 3.5 (1.4–8.4), p = 0.006, Table 2). In summary, decreased serotonin levels were seen in patients with a severe clinical phenotype with autoantibodies, complement consumption and nephritis.
Table 2. Low serum levels of serotonin are associated with severe SLE.
| Manifestation | Positive (n = 127) | p-value a | Odds ratio (95% CI) a |
|---|---|---|---|
| Nephritis b | 39 | 0.01 | 3.1 (1.3–7.2) |
| Anti-dsDNA b | 76 | 0.02 | 2.7 (1.2–6.5) |
| Low C3/C4 levels c | 35 | 0.006 | 3.5 (1.4–8.4) |
aAll analyses were adjusted for differences in age and sex.
bAccording to the 1982 ACR classification criteria.
cAccording to the SLEDAI-2K definition.
The tryptophan metabolism is dysregulated in SLE patients
Since total serum serotonin levels were decreased in SLE we next explored whether it was related to skewing of the tryptophan metabolism via IDO, as has been previously suggested [7–9]. Indeed, the activity of IDO, measured indirectly as the ratio between kynurenine and tryptophan, was increased in SLE patients as compared to healthy volunteers (p = 0.007, Fig 3A). SLE patients had a modest reduction in serum levels of tryptophan as compared to healthy individuals (p = 0.03, Fig 3B) whereas no differences were seen for the kynurenine levels (p = 0.28, Fig 3C). Thus, these findings suggest that SLE patients have a skewed tryptophan metabolism due to increased IDO activity.
Fig 3. Dysregulated tryptophan metabolism in SLE patients.

A) The activity of the enzyme indoleamine 2,3-dioxygenase (IDO) was indirectly measured as the ratio between kynurenine and tryptophan. The levels of B) tryptophan and C) kynurenine were measured by liquid chromatography.
The IDO activity is regulated by type I IFNs in SLE patients
IDO activity has been described to be regulated by type I IFNs [13], a cytokine highly increased in SLE patients [43]. To investigate if type I IFNs were responsible for the increased IDO activity seen in SLE patients, sera were incubated with a type I IFN-sensitive cell line (WISH cells) and IDO expression was analyzed by flow cytometry. Physiological concentrations of recombinant IFN-alpha increased the IDO expression about 2-fold, and the IFN-alpha-mediated IDO expression could readily be inhibited by addition of an IFNAR-blocking antibody (p = 0.008, Fig 4A). SLE sera supported IDO expression to a higher extent than sera from healthy individuals (p = 0.01, Fig 4B), in a similar type I IFN-dependent manner as the positive control (p = 0.008, Fig 4B).
Fig 4. SLE sera increase IDO expression in a type I IFN-dependent manner.
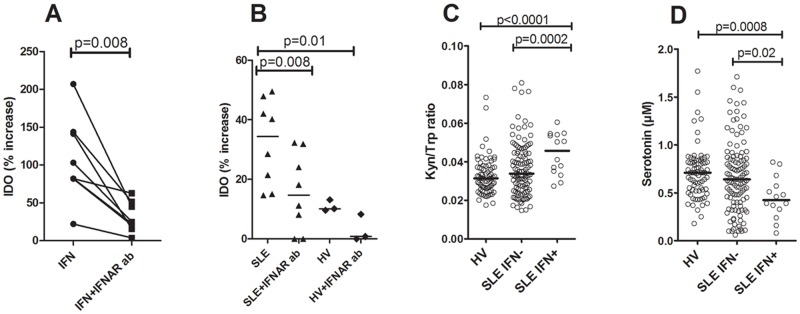
Endothelial cells (WISH cells) were incubated with A) recombinant IFN-alpha with or without pre-incubation with an IFN alpha/beta receptor blocking antibody (IFNAR ab) and IDO expression analyzed in permeabilized cells by flow cytometry. B) WISH cells were incubated with serum from SLE patients or healthy individuals with or without pre-incubation with an IFNAR ab. The results are presented as percentage increased IDO expression in median fluorescence intensity (MFI) as compared to non-stimulated cells. C) Patients with ongoing type I IFN activity (SLE IFN+) had increased IDO activity as compared to patients with no type I IFN activity (SLE IFN-) and healthy volunteers (HV). D) Patients with ongoing type I IFN activity had decreased serotonin levels as compared to patients with no type I IFN activity and healthy volunteers.
To assess if type I IFN-mediated regulation of IDO activity was seen in SLE patients, type I IFN activity was analyzed with an in-house cell reporter assay. SLE patients with high type I IFN production had increased IDO activity (kynurenine/tryptophan ratio) as compared to patients without type I IFN activity and healthy individuals (p = 0.0002, and p<0.0001, respectively, Fig 4C). Furthermore, SLE patients with high type I IFN production had a decreased serotonin concentration in serum as compared to SLE patients without ongoing type I IFN production as well as compared to healthy individuals (p = 0.02 and p = 0.0008, respectively, Fig 4D). Since type I IFNs clearly influenced serotonin levels through up-regulation of IDO, and have been associated with disease activity and severity in previous investigations [4], we re-analyzed the associations between disease manifestations and serotonin levels also taking into account differences in type I IFN activity. However, the association between decreased serotonin levels and disease severity, including development of nephritis, remained statistically significant even after adjusting for ongoing type I IFN activity, suggesting that decreased serotonin levels were associated with disease severity independently of type I IFN activity (data not shown). In all, our data suggest that type I IFNs, present in SLE sera, are able to up-regulate IDO expression, which may lead to decreased serotonin levels.
Platelet activation contributes to reduce total serotonin levels
Although type I IFNs clearly were able to reduce serotonin synthesis through up-regulation of IDO, several mechanisms may operate in SLE to decrease the overall availability of serotonin in platelets, including platelet activation as depicted in Fig 1. Previous studies have demonstrated inverse correlations between low intracellular levels of serotonin in platelets and heightened levels of peripheral serotonin in plasma samples from SLE patients [33], suggesting that release of serotonin by activated platelets and subsequent consumption by surrounding immune or tissue cells may also reduce overall availability of serotonin.
In contrast to serum (total/platelet) levels of serotonin, plasma (peripheral) levels of serotonin were increased in SLE, although it did not reach statistical significance (p = 0.14, Fig 5A). However, taking into account the reduced overall availability of serotonin, the percentage of peripheral serotonin was highly increased in SLE patients (p = 0.003, Fig 5B). Compatible with previous study [33], high levels of serotonin in the periphery (plasma) were associated with decreased levels of serotonin both in platelets and serum (p = 0.03 and p = 0.003, respectively, Fig 5C and 5D). Furthermore, high levels of peripheral serotonin were associated with ongoing platelet activation as assessed by platelet microparticle formation (p = 0.03, Fig 5E) and platelet C4d deposition (p = 0.005, Fig 5F). In all, those data clearly demonstrate that also platelet activation participates in regulating platelet serotonin levels through releasing it into the periphery where it could induce inflammation in neighboring cells. However, the role of platelet activation in association to clinical manifestations is outside of the scope of the current investigation and will be studied in more details in future investigations.
Fig 5. Decreased platelet serotonin levels are associated with platelet activation.

A) Serotonin levels were analyzed in EDTA plasma samples and compared between healthy volunteers (HV) and SLE patients. B) To adjust for differences in total serotonin levels, the relative amount of peripheral versus total serotonin was calculated and compared between HV and SLE patients. SLE patients with a high relative amount of peripheral serotonin (high) was compared to SLE patients with low relative amount of peripheral serotonin levels (low) with regard to C) platelet intracellular serotonin levels, D) serum levels of serotonin, E) platelet microparticle (PMP) count in plasma and F) platelet activation assessed by platelet C4d deposition analyzed by flow cytometry.
Discussion
Even though mainly thought of as an important neurotransmitter, the majority of serotonin is produced locally in the intestine supplying platelets with their serotonin stores enabling them to efficiently induce pro-inflammatory and pro-thrombotic signals upon activation and release of serotonin unto neighboring cells. In SLE, a disease characterized by systemic inflammation as well as markedly increased risk of developing thrombosis, decreased levels of serotonin in platelets have been described, but the underlying mechanism(s) was still lacking. In the current study, using a large cohort of SLE patients, we found decreased levels of serotonin in platelets from SLE patients as compared to healthy controls, also reflected by decreased levels of serotonin in serum. As illustrated in Fig 1, we hypothesized that the decreased serotonin levels found in SLE patients were dependent on deprivation of tryptophan, the building block of serotonin by rate-limiting enzyme IDO. Indeed, we were able to clearly demonstrate that SLE patients had increased IDO activity as measured by the shift in tryptophan and kynurenine, compatible with previous studies [8, 9]. Interestingly, even though IDO mainly catalyzes the conversion of tryptophan into kynurenine, several other IDO substrates have been described, including serotonin itself [44]. Thus, increased IDO activity may decrease serotonin content by increasing both tryptophan and serotonin turnover.
Next we investigated the cause of the increased IDO activity in SLE patients. IDO is well-known to be up-regulated in response to type I and type II IFNs, which are increased in SLE patients. Utilizing a reporter cell (WISH) we observed that SLE sera supported up-regulation of IDO expression to a higher extent than sera from healthy individuals. Furthermore, by using an IFNAR blocking antibody, we concluded that IDO was up-regulated in a type I IFN-dependent manner. This particular cell line is commonly used to analyze type I IFN activity in SLE sera due to its inability to respond to TLR7 and TLR9 ligands as well as low/absent expression of FcγRs [36, 45]. Not only did SLE sera support up-regulation of IDO in a type I IFN-dependent manner, but also SLE patients with ongoing type I IFN activity in vivo had increased IDO activity as well as decreased serotonin levels. Thus, we suggest that one explanation for the decreased levels of serotonin seen in SLE patients may be a type I IFN-mediated up-regulation of IDO (Fig 1). Up-regulation of IDO and depletion of serotonin is not specific for SLE, but has also been seen in patients with hepatitis C viral infections upon administration with recombinant IFN-alpha [13].
SLE is a very heterogeneous disease ranging from mild manifestations with rash to more severe inflammation in several organ systems including the kidneys and central nervous system. In our cohort, decreased serum levels of serotonin were strongly associated with a severe SLE phenotype, characterized by nephritis and anti-dsDNA antibodies, similar to previous studies in SLE [30, 31, 33, 46]. Importantly, the association was independent on type I IFN activity indicating a role of serotonin in the immunopathogenesis of SLE. The invert correlation with disease severity may at first seem contradicting considering the ample of literature supporting a role of serotonin in induction of inflammation, including activation and extravasation of immune cells, chemotaxis of neutrophils as well as regulation of type I IFN production [18–22, 25]. Furthermore, serotonin receptor antagonists are able to block the inflammatory process in several experimental animal models [19, 23, 24, 47]. Thus, serotonin has ample pro-inflammatory effects and therefore decreased serotonin levels would be expected to be associated with milder disease and not more severe disease as observed here.
However, even though serotonin has been ascribed some anti-inflammatory properties [48–50] possibly able to explain the inverse association with disease severity, the localization of serotonin is also important. Whereas the serum levels, e.g. platelet-stored serotonin, were decreased in SLE patients, the bioactive released serotonin found in plasma samples was increased. Importantly, we observed an inverse correlation between increased peripheral levels of serotonin and decreased platelet-stored serotonin, highly related to ongoing platelet activation. Thus, decreased levels of serotonin in serum, e.g. in the form of decreased platelet-stored serotonin, may reflect increased release and pro-inflammatory actions of serotonin, and likely cause a more severe phenotype.
In conclusion, SLE patients had a reduced serotonin level in serum and platelets related to severe disease phenotype, including nephritis, driven by immune complexes and type I IFN activation, but also representing other immunopathological processes which are central in SLE. As depicted in Fig 1, our data support the hypothesis of a type I IFN-mediated increase in IDO activity and subsequent deprivation of available tryptophan for serotonin synthesis in favor of kynurenine. However, other mechanisms, including platelet activation, may also participate to reduce peripheral serotonin levels, and this will be the subject of further studies.
Supporting Information
(XLSX)
Acknowledgments
The valuable advices from Prof. Lars Rönnblom, Uppsala University, Sweden, in how to proceed with this project is gratefully acknowledged.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The study was supported by grants from the Medical Faculty at Lund University, The Crafoord Foundation, Greta and Johan Kock’s Foundation, King Gustaf V’s 80th Birthday Foundation, Lund University Hospital, the Swedish Rheumatism Association, Swedish Society of Medicine, the Foundation of the National Board of Health and Welfare and Österlund’s Foundation. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. CKL, CWE contributed to the design of the study and to the preparation of the manuscript. The collaboration was purely scientific and focused on basic science related to disease mechanisms. AnaMar did not provide support in the form of salaries for authors others then its employees (CKL and CWE). The specific roles of these authors are articulated in the ‘author contributions’ section. There were no AnaMar compounds involved in the study and AnaMar has no current interest in SLE.
References
- 1. Crispín JC, Liossis SN, Kis-Toth K, Lieberman LA, Kyttaris VC, Juang YT, et al. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med. 2010;16(2):47–57. Epub 2010/02/09. doi: S1471-4914(10)00006-7 [pii] 10.1016/j.molmed.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rönnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci. 2011;116(4):227–37. Epub 2011/11/10. 10.3109/03009734.2011.624649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17(5):394–9. Epub 2008/05/21. doi: 17/5/394 [pii] 10.1177/0961203308090020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000;9(9):664–71. Epub 2001/02/24. . [DOI] [PubMed] [Google Scholar]
- 5. Lood C, Amisten S, Gullstrand B, Jönsen A, Allhorn M, Truedsson L, et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood. 2010;116(11):1951–7. Epub 2010/06/12. doi: blood-2010-03-274605 [pii] 10.1182/blood-2010-03-274605 . [DOI] [PubMed] [Google Scholar]
- 6. Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5(6):279–87. Epub 2003/12/19. doi: 10.1186/ar1015ar1015 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furuzawa-Carballeda J, Lima G, Jakez-Ocampo J, Llorente L. Indoleamine 2,3-dioxygenase-expressing peripheral cells in rheumatoid arthritis and systemic lupus erythematosus: a cross-sectional study. Eur J Clin Invest. 2011;41(10):1037–46. Epub 2011/03/04. 10.1111/j.1365-2362.2011.02491.x . [DOI] [PubMed] [Google Scholar]
- 8. Widner B, Sepp N, Kowald E, Ortner U, Wirleitner B, Fritsch P, et al. Enhanced tryptophan degradation in systemic lupus erythematosus. Immunobiology. 2000;201(5):621–30. Epub 2000/06/02. doi: S0171-2985(00)80079-0 [pii] 10.1016/S0171-2985(00)80079-0 . [DOI] [PubMed] [Google Scholar]
- 9. Pertovaara M, Hasan T, Raitala A, Oja SS, Yli-Kerttula U, Korpela M, et al. Indoleamine 2,3-dioxygenase activity is increased in patients with systemic lupus erythematosus and predicts disease activation in the sunny season. Clin Exp Immunol. 2007;150(2):274–8. Epub 2007/08/23. doi: CEI3480 [pii] 10.1111/j.1365-2249.2007.03480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell BM, Charych E, Lee AW, Moller T. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci. 2014;8:12 Epub 2014/02/26. 10.3389/fnins.2014.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thevandavakkam MA, Schwarcz R, Muchowski PJ, Giorgini F. Targeting kynurenine 3-monooxygenase (KMO): implications for therapy in Huntington's disease. CNS Neurol Disord Drug Targets. 2010;9(6):791–800. Epub 2010/10/15. doi: BSP/CDTCNSND/E-Pub/00086 [pii]. . [DOI] [PubMed] [Google Scholar]
- 12. Perez-De La Cruz V, Konigsberg M, Santamaria A. Kynurenine pathway and disease: an overview. CNS Neurol Disord Drug Targets. 2007;6(6):398–410. Epub 2008/01/29. . [DOI] [PubMed] [Google Scholar]
- 13. Schäfer A, Scheurlen M, Seufert J, Keicher C, Weissbrich B, Rieger P, et al. Platelet serotonin (5-HT) levels in interferon-treated patients with hepatitis C and its possible association with interferon-induced depression. J Hepatol. 2010;52(1):10–5. Epub 2009/11/10. doi: S0168-8278(09)00653-9 [pii] 10.1016/j.jhep.2009.10.007 . [DOI] [PubMed] [Google Scholar]
- 14. Fukami MH, Bauer JS, Stewart GJ, Salganicoff L. An improved method for the isolation of dense storage granules from human platelets. J Cell Biol. 1978;77(2):389–99. Epub 1978/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ni W, Watts SW. 5-hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol. 2006;33(7):575–83. Epub 2006/06/23. doi: CEP [pii] 10.1111/j.1440-1681.2006.04410.x . [DOI] [PubMed] [Google Scholar]
- 16. Li N, Wallen NH, Ladjevardi M, Hjemdahl P. Effects of serotonin on platelet activation in whole blood. Blood Coagul Fibrinolysis. 1997;8(8):517–23. Epub 1998/03/10. . [DOI] [PubMed] [Google Scholar]
- 17. Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf). 2009;196(2):193–222. Epub 2009/02/18. doi: APS1964 [pii] 10.1111/j.1748-1716.2009.01964.x . [DOI] [PubMed] [Google Scholar]
- 18. Leon-Ponte M, Ahern GP, O'Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. 2007;109(8):3139–46. Epub 2006/12/13. doi: blood-2006-10-052787 [pii] 10.1182/blood-2006-10-052787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durk T, Panther E, Muller T, Sorichter S, Ferrari D, Pizzirani C, et al. 5-Hydroxytryptamine modulates cytokine and chemokine production in LPS-primed human monocytes via stimulation of different 5-HTR subtypes. Int Immunol. 2005;17(5):599–606. Epub 2005/04/02. doi: dxh242 [pii] 10.1093/intimm/dxh242 . [DOI] [PubMed] [Google Scholar]
- 20. Soga F, Katoh N, Inoue T, Kishimoto S. Serotonin activates human monocytes and prevents apoptosis. J Invest Dermatol. 2007;127(8):1947–55. Epub 2007/04/13. doi: 5700824 [pii] 10.1038/sj.jid.5700824 . [DOI] [PubMed] [Google Scholar]
- 21. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2013;121(6):1008–15. Epub 2012/12/18. doi: blood-2012-06-437392 [pii] 10.1182/blood-2012-06-437392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cloutier N, Pare A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, et al. Platelets can enhance vascular permeability. Blood. 2012;120(6):1334–43. Epub 2012/05/01. doi: blood-2012-02-413047 [pii] 10.1182/blood-2012-02-413047 . [DOI] [PubMed] [Google Scholar]
- 23. Shajib MS, Wang H, Kim JJ, Sunjic I, Ghia JE, Denou E, et al. Interleukin 13 and serotonin: linking the immune and endocrine systems in murine models of intestinal inflammation. PLoS One. 2013;8(8):e72774. Epub 2013/09/10. doi: 10.1371/journal.pone.0072774 PONE-D-13-14207 [pii]. 10.1371/journal.pone.0072774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim JJ, Bridle BW, Ghia JE, Wang H, Syed SN, Manocha MM, et al. Targeted inhibition of serotonin type 7 (5-HT7) receptor function modulates immune responses and reduces the severity of intestinal inflammation. J Immunol. 2013;190(9):4795–804. Epub 2013/04/05. doi: jimmunol.1201887 [pii] 10.4049/jimmunol.1201887 . [DOI] [PubMed] [Google Scholar]
- 25. Eloranta ML, Lovgren T, Finke D, Mathsson L, Ronnelid J, Kastner B, et al. Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheum. 2009;60(8):2418–27. Epub 2009/08/01. 10.1002/art.24686. 10.1002/art.24686 [DOI] [PubMed] [Google Scholar]
- 26. Shajib MS, Khan WI. The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol (Oxf). 2014. Epub 2014/12/03. 10.1111/apha.12430. . [DOI] [PubMed] [Google Scholar]
- 27. Sarac H, Markeljevic J, Mokrovic G, Erdeljic V, Bozina N, Cicin-Sain L. Platelet serotonin in primary Sjogren's syndrome: level and relation with disease activity. J Neuroimmunol. 2012;251(1–2):87–9. Epub 2012/07/24. doi: S0165-5728(12)00212-3 [pii] 10.1016/j.jneuroim.2012.06.011 . [DOI] [PubMed] [Google Scholar]
- 28. Krishnadas R, Cavanagh J. Sustained remission of rheumatoid arthritis with a specific serotonin reuptake inhibitor antidepressant: a case report and review of the literature. J Med Case Rep. 2011;5:112. Epub 2011/03/23. doi: 1752-1947-5-112 [pii] 10.1186/1752-1947-5-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boilard E, Blanco P, Nigrovic PA. Platelets: active players in the pathogenesis of arthritis and SLE. Nat Rev Rheumatol. 2012;8(9):534–42. Epub 2012/08/08. doi: nrrheum.2012.118 [pii] 10.1038/nrrheum.2012.118 . [DOI] [PubMed] [Google Scholar]
- 30. Kanai H, Tsuchida A, Yano S, Naruse T. Intraplatelet and urinary serotonin concentrations in systemic lupus erythematosus with reference to its clinical manifestations. J Med. 1989;20(5–6):371–90. Epub 1989/01/01. . [PubMed] [Google Scholar]
- 31. Zeller J, Weissbarth E, Baruth B, Mielke H, Deicher H. Serotonin content of platelets in inflammatory rheumatic diseases. Correlation with clinical activity. Arthritis Rheum. 1983;26(4):532–40. Epub 1983/04/01. . [DOI] [PubMed] [Google Scholar]
- 32. Meyerhoff J, Dorsch CA. Decreased platelet serotonin levels in systemic lupus erythematosus. Arthritis Rheum. 1981;24(12):1495–500. Epub 1981/12/01. . [DOI] [PubMed] [Google Scholar]
- 33. Parbtani A, Frampton G, Yewdall V, Kasai N, Cameron JS. Platelet and plasma serotonin in glomerulonephritis. III: The nephritis of systemic lupus erythematosus. Clin Nephrol. 1980;14(4):164–72. Epub 1980/10/01. . [PubMed] [Google Scholar]
- 34. Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–91. Epub 2002/02/13. . [PubMed] [Google Scholar]
- 35. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–7. Epub 1982/11/01. . [DOI] [PubMed] [Google Scholar]
- 36. Bengtsson AA, Sturfelt G, Lood C, Rönnblom L, van Vollenhoven RF, Axelsson B, et al. Pharmacokinetics, tolerability, and preliminary efficacy of paquinimod (ABR-215757), a new quinoline-3-carboxamide derivative: studies in lupus-prone mice and a multicenter, randomized, double-blind, placebo-controlled, repeat-dose, dose-ranging study in patients with systemic lupus erythematosus. Arthritis Rheum. 2012;64(5):1579–88. Epub 2011/12/02. 10.1002/art.33493 . [DOI] [PubMed] [Google Scholar]
- 37. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188(7):3522–31. Epub 2012/02/22. doi: jimmunol.1102404 [pii] 10.4049/jimmunol.1102404 . [DOI] [PubMed] [Google Scholar]
- 38. Lood C, Eriksson S, Gullstrand B, Jonsen A, Sturfelt G, Truedsson L, et al. Increased C1q, C4 and C3 deposition on platelets in patients with systemic lupus erythematosus—a possible link to venous thrombosis? Lupus. 2012;21(13):1423–32. Epub 2012/08/11. doi: 0961203312457210 [pii] 10.1177/0961203312457210 . [DOI] [PubMed] [Google Scholar]
- 39. Lood C, Tyden H, Gullstrand B, Sturfelt G, Jonsen A, Truedsson L, et al. Platelet activation and anti-phospholipid antibodies collaborate in the activation of the complement system on platelets in systemic lupus erythematosus. PLoS One. 2014;9(6):e99386. Epub 2014/06/13. doi: 10.1371/journal.pone.0099386 PONE-D-14-02789 [pii]. 10.1371/journal.pone.0099386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iversen LV, Ostergaard O, Nielsen CT, Jacobsen S, Heegaard NH. A heparin-based method for flow cytometric analysis of microparticles directly from platelet-poor plasma in calcium containing buffer. J Immunol Methods. 2013;388(1–2):49–59. Epub 2012/12/19. doi: S0022-1759(12)00347-X [pii] 10.1016/j.jim.2012.12.001 . [DOI] [PubMed] [Google Scholar]
- 41. Ostergaard O, Nielsen CT, Iversen LV, Jacobsen S, Tanassi JT, Heegaard NH. Quantitative proteome profiling of normal human circulating microparticles. J Proteome Res. 2012;11(4):2154–63. Epub 2012/02/15. 10.1021/pr200901p . [DOI] [PubMed] [Google Scholar]
- 42. Nielsen CT, Ostergaard O, Johnsen C, Jacobsen S, Heegaard NH. Distinct features of circulating microparticles and their relationship to clinical manifestations in systemic lupus erythematosus. Arthritis Rheum. 2011;63(10):3067–77. Epub 2011/06/28. 10.1002/art.30499 . [DOI] [PubMed] [Google Scholar]
- 43. Rönnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. 2013;25(2):248–53. Epub 2012/12/20. 10.1097/BOR.0b013e32835c7e32 . [DOI] [PubMed] [Google Scholar]
- 44. Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Biol Chem. 1978;253(13):4700–6. Epub 1978/07/10. . [PubMed] [Google Scholar]
- 45. Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54(6):1906–16. Epub 2006/06/01. 10.1002/art.21890 . [DOI] [PubMed] [Google Scholar]
- 46. Parbtani A, Frampton G, Cameron JS. Platelet and plasma serotonin concentrations in glomerulonephritis, II. Clin Nephrol. 1980;14(3):112–23. Epub 1980/09/01. . [PubMed] [Google Scholar]
- 47. Ito T, Ikeda U, Shimpo M, Yamamoto K, Shimada K. Serotonin increases interleukin-6 synthesis in human vascular smooth muscle cells. Circulation. 2000;102(20):2522–7. Epub 2000/11/15. . [DOI] [PubMed] [Google Scholar]
- 48. de las Casas-Engel M, Dominguez-Soto A, Sierra-Filardi E, Bragado R, Nieto C, Puig-Kroger A, et al. Serotonin skews human macrophage polarization through HTR2B and HTR7. J Immunol. 2013;190(5):2301–10. Epub 2013/01/29. doi: jimmunol.1201133 [pii] 10.4049/jimmunol.1201133 . [DOI] [PubMed] [Google Scholar]
- 49. Yu B, Becnel J, Zerfaoui M, Rohatgi R, Boulares AH, Nichols CD. Serotonin 5-hydroxytryptamine(2A) receptor activation suppresses tumor necrosis factor-alpha-induced inflammation with extraordinary potency. J Pharmacol Exp Ther. 2008;327(2):316–23. Epub 2008/08/19. doi: jpet.108.143461 [pii] 10.1124/jpet.108.143461 . [DOI] [PubMed] [Google Scholar]
- 50. Nau F Jr., Yu B, Martin D, Nichols CD. Serotonin 5-HT2A Receptor Activation Blocks TNF-alpha Mediated Inflammation In Vivo. PLoS One. 2013;8(10):e75426. Epub 2013/10/08. doi: 10.1371/journal.pone.0075426 PONE-D-12-30869 [pii]. 10.1371/journal.pone.0075426 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.