

Abstract
We expanded flow-sorted Foxp3+ cynomolgus monkey regulatory T cells (Treg) >1000-fold after three rounds of stimulation with anti-CD3 mAb-loaded artificial antigen-presenting cells, rapamycin (first round only) and IL-2. The expanded Treg maintained their expression of Treg signature markers, CD25, CD27, CD39, Foxp3, Helios, and CTLA-4, as well as CXCR3, which plays an important role in T cell migration to sites of inflammation. In contrast to expanded effector T cells (Teff), expanded Treg produced minimal IFN-γ and IL-17 and no IL-2 and potently suppressed Teff proliferation. Following cryopreservation, thawed Treg were less viable than their freshly-expanded counterparts, although no significant changes in phenotype or suppressive ability were observed. Additional rounds of stimulation/expansion restored maximal viability. Furthermore, adoptively-transferred autologous Treg expanded from cryopreserved second round stocks and labeled with CFSE or VPD450 were detected in blood and secondary lymphoid tissues of normal or immunosuppressed recipients at least two months after their systemic infusion.
Keywords: regulatory T cells, ex vivo expansion, cynomolgus macaques, cryopreservation, in vivo monitoring
1. Introduction
Regulatory T cells (Treg) characterized by expression of the transcription factor Forkhead box P3 (Foxp3) function to maintain immune tolerance and prevent inflammatory diseases [1]. Thus, considerable effort has been expended to exploit their immunomodulatory properties. Adoptive transfer of Treg has been shown to prevent rejection of organ allografts in mice and humanized mouse models[2–4], and to prevent experimental graft-versus-host disease (GVHD) [5]. These preclinical studies support Treg as a highly promising potential therapeutic agent to suppress adverse immune-mediated inflammatory reactions and promote immune tolerance in transplantation and autoimmune disease [6–8].
A major challenge in the clinical application of Treg is their paucity in peripheral blood. Expansion of isolated, naturally-occurring Treg ex vivo may provide a solution via generation of massive numbers of these cells for therapeutic application [5, 9]. It has been reported that combination of high dose IL-2, rapamycin, anti-CD3 and (or) anti-CD28 beads or monoclonal antibodies and artificial APCs (aAPC) can expand Treg massively in mice and humans [10–12]. In contrast to these studies in mice and humans, few reports have addressed expansion of Treg from non-human primates (NHP), -important pre-clinical models in organ transplantation [13–18]. Given the challenge of manufacturing Treg on a large scale and the potential advantage of banking these cells, cryopreservation may facilitate their clinical application [19]. However, there is evidence that cryopreservation can affect Treg recovery, viability [11], expression of essential surface markers, cytokine secretion and function [20, 21]. Here, we optimized a protocol for expansion of NHP polyclonal Treg that retained their phenotype and suppressive capacity; after two rounds of expansion we cryopreserved the Treg, then resuscitated and restimulated them for an additional round of expansion to overcome the negative effect of cryopreservation.
An additional important issue concerns the stability, trafficking and fate of Treg after their infusion in vivo. Like other immune cells, Treg vary in their homing and chemokine receptor expression, that controls their in vivo tissue distribution [22]. In the setting of transplantation or autoimmune disease, immunosuppressive (IS) drug administration may affect the properties of infused Treg. For optimal function, adoptively-transferred Treg must be able to survive and migrate to host lymphoid tissues [23, 24] and/or sites of inflammation [25], even in the presence of IS therapy. We therefore also investigated the persistence of expanded autologous Treg in blood and lymphoid tissues following their infusion in cynomolgus monkey recipients, with or without IS treatment. The IS regimen used comprised a lymphocyte-depleting agent (anti-thymocyte globulin; ATG [26, 27]), tacrolimus and rapamycin. The latter agents inhibit T cell proliferative responses. Both ATG [28] and rapamycin [29] have been considered “Treg-sparing” IS agents.
2. Materials and methods
2.1 Animals and cell lines
Healthy male cynomolgus macaques (Macaca fascicularis) of Indonesian origin, weighing 3–5 kg, were obtained from specific pathogen-free colonies at Alpha Genesis, Inc, or the NIAID NHP colony (both Yamassee, SC). All animal procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and conducted under a University of Pittsburgh Institutional Animal Care and Use Committee-approved protocol. Specific environment enrichment was provided.
Artificial antigen-presenting cells (aAPC) (L-32) that stably express CD32, CD80 and CD58, were kindly provided by Dr. M. K. Levings, University of British Columbia, Vancouver, Canada.
2.2 PBMC isolation
Fresh peripheral blood mononuclear cells (PBMC) were isolated from blood by one of the three methods (Fig. 1A, i–iii). The first method was to dilute the blood with PBS at 1:1 ratio, overlay 8 ml diluted blood on 4ml Ficoll-Paque Plus (GE Healthcare Bio-Sciences AB), spin for 20 min at 1500 rpm, then collect the buffy coat after centrifugation. The second method was to overlay 4 ml blood, diluted as in the first method, on 3ml Lympholyte-mammal (Cedarlane, Burlington, NC) and collect the buffy coat post centrifugation. The third method was to mix 10 ml blood with 1.25ml OptiPrep (Axis-Shield PoC AS, Oslo, Norway) thoroughly, overlay with 1ml PBS, spin down, and then collect the PBMC layer between plasma and PBS. PBMC layers obtained using these three approaches were treated with red blood cell (RBC) lysis buffer to remove red blood cells. RBC lysis buffer-treated whole blood was used as a control. The yield and purity of the mononuclear cells was affected to a considerable extent by the efficiency of red blood cell lysis. PBMC composition was determined by flow cytometry. Absolute counts of each cell type were determined by CountBright™ absolute counting beads (Invitrogen), according to the manufacturer’s protocol. Percent recovery was calculated as: absolute number of cells of interest in PBMC isolated from 1 ml blood / absolute number of cells of interest in 1 ml whole blood X 100%.
Fig. 1. Optimization of cynomolgus PBMC isolation for subsequent Treg sorting.
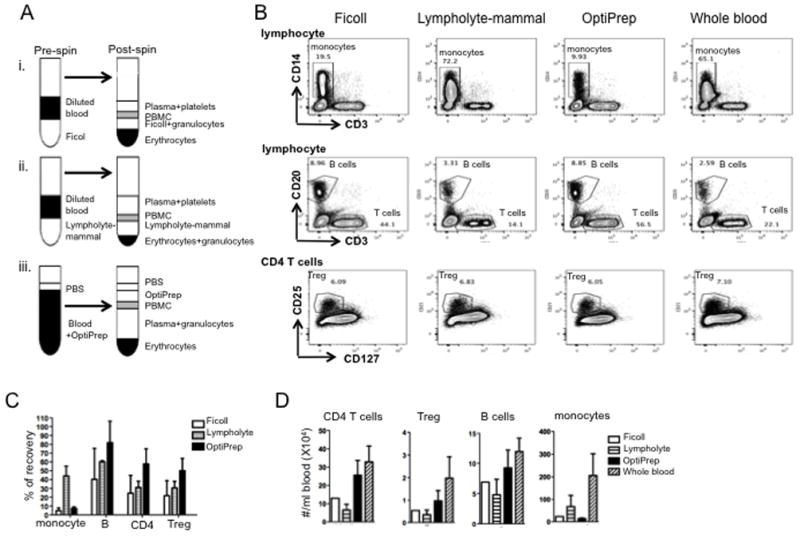
(A) Outlines of three methods tested to isolate cynomolgus PBMC. (B) Percentages of monocytes, B cells, and T cells in PBMC isolates and percentages of Treg in total CD4+ T cells. Lymphocytes were gated based on forward (FSC) and side-scatter (SSC) of singlets. Monocytes were gated on CD14+; B cells on CD3−CD20+; T cells on CD3+CD20−; and Treg on CD25+CD127−/loCD3+CD4+ T cells. Data are representative of 3 individual experiments. (C) Percent recovery of monocytes, B cells, CD4+ T cells and Treg from equivalent amounts of cynomolgus blood using different PBMC isolation methods. Absolute counts per ml blood of each type of cell were determined by counting beads as: number of cell events / number of bead events X total number of bead added / volume of sample. Percent recovery was defined as: absolute number of cells in PBMC isolated from 1ml blood / absolute number of cells in 1ml blood treated with red blood cell lysis buffer X 100%. Data are means + 1SD from 3 individual experiments. (D) Absolute number of indicated cells in PBMC isolated using the three methods. Whole blood was used as control. Data are means + 1SD from 3 individual experiments.
2.3 Treg isolation and ex-vivo expansion
PBMCs isolated by Ficoll-Paque Plus or OptiPrep were labeled with fluorochrome-labeled anti-CD4, anti-CD25 and anti-CD127 (BD PharMingen, Franklin Lakes, NJ, or BioLegend, San Diego, CA) antibodies, and then flow-sorted into populations of CD4+CD25+CD127− Treg and CD4+CD25−CD127+ effector T cells (Teff) (Fig. 2) on a BD FACS Aria II high-speed cell sorter (BD Biosciences, San Jose, CA). Irradiated (80 Gy) and anti-CD3 Ab-preloaded artificial antigen-presenting cells (aAPCs) L-32 were cultured with sorted Treg or Teff control at 1:1 T to aAPC ratio in complete RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine (Mediatech, Inc., Herndon, VA), 100U/mL penicillin-streptomycin (BioWhittaker), 10 mM HEPES (Mediatech) and 55 μM β-2 mercaptoethanol (Invitrogen) in the presence of 300 U/ml recombinant human IL-2 (R&D Systems, Minneapolis, MN) and 100 ng/ml rapamycin (LC Laboratories, Woburn, MA) for 3–4 days. Then the cells were split and transferred to larger vessels without L-32 cells or anti-CD3 mAb for an additional 4 days. At the end of the initial round of culture (7–8 days), non-adherent T cells were harvested and re-stimulated with L-32 cells as in the first round for an additional 2 rounds, except that no rapamycin was added. During each round, cultures were supplemented with fresh medium and 300U/ml IL-2 at intervals. The protocol used for Treg expansion and investigation is shown in Fig. 3. To cryopreserve cells at the end of each round, they were first suspended in 80% medium, 20% FCS at < 2 × 107 cells per ml, and then diluted 1:1 with 60% medium, 20% FCS, 20% dimethyl sulfoxide (DMSO, Fisher Scientific, Fairlawn, New Jersey) and stored in liquid nitrogen. In some experiments, cryopreserved expanded Treg were re-stimulated and expanded for an additional round, as described above.
Fig. 2. Purification of Treg from PBMC of normal cynomolgus macaques.
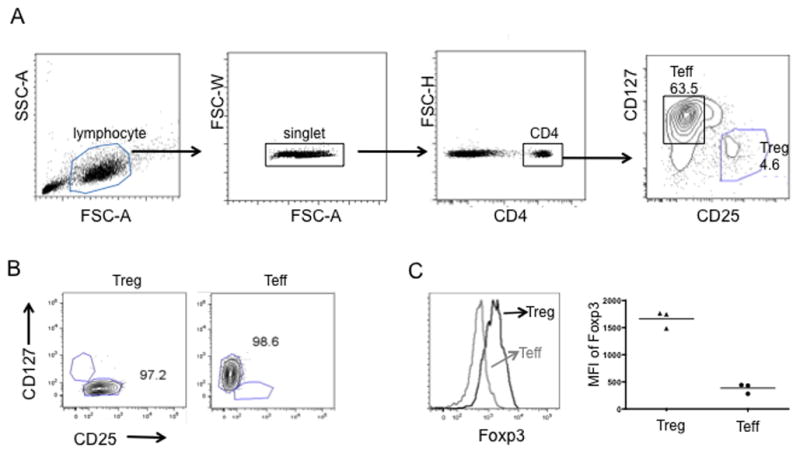
(A) Gating for flow sorting of Treg and Teff. Lymphocytes were gated based on forward (FSC) and side-scattered light (SSC), followed by single cell and CD4+ cell gating. Treg and Teff were gated on CD25+CD127−/low and CD25−CD127high, respectively, from CD4+ T lymphocytes. The percentages of putative Treg and Teff in CD4+ T cells are shown. (B) Representative flow cytometric plots of sorted Treg (left panel) and Teff (right panel). The numbers indicate the purity of Treg and Teff after sorting with a FACS Aria cell sorter. (C) Foxp3 expression by freshly-sorted putative Treg and Teff based on CD25 and CD127 gating. Histogram overlay from one representative experiment (left panel) and MFI from 3 separate experiments (right panel) are shown.
Fig. 3. Protocol for ex vivo-expansion of cynomolgus Treg using artificial APC and the assays performed.
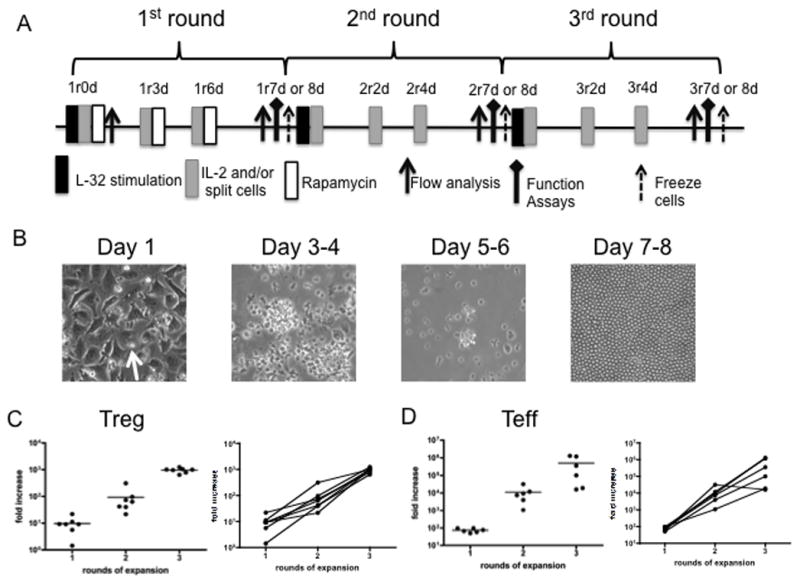
(A) Treg expansion protocol. Freshly-sorted Treg (CD4+CD25hiCD127−/lo) or conventional Teff (CD4+CD25−CD127hi) were stimulated with anti-CD3-loaded artificial APC (L-32 cells) in the presence of 300 U/ml recombinant human IL-2 for 7–8 days. Rapamycin (100 ng/ml) was also added to the cultures for the first round. The cells were harvested and expanded for an additional 2 rounds, as in the first round, except that no rapamycin was added. At the end of the 2nd round (2r7 or 8 days), a portion of the cells was frozen for future expansion. r: round of expansion; d: days in a round (B) Appearance of the cultures during the course of round 1. Treg (arrow) appear to attach to L-32 cells on day 1. Cultured cells were transferred to larger vessels after day 3–4. Homogeneous appearance of expanded Treg on days 7–8, with minimal/no L-32 cell contamination. (C, D) Fold expansion of Treg and Teff at the end of each of 3 rounds (results of 6 experiments).
2.4 Flow cytometry
Single cell suspensions of T cells were stained as described [17] at 4°C with fluorochrome-labeled anti-CD3, anti-CD4, anti-CD25, anti-CD27-PE, anti-CD39, anti-CD44, anti-CD45RA, anti-CD62L, anti-CD127, anti-CXCR3, anti-CCR7 (BD PharMingen, Franklin Lakes, NJ, or BioLegend, San Diego, CA). Intracellular Foxp3, cytotoxic T lymphocyte Ag-4 (CTLA-4), Helios, T-bet, IL-2, IL-17 and IFN-γ were stained using an eBioscience Foxp3 Staining kit, according to the manufacturer’s instructions. Data were acquired on a LSR II or LSR Fortessa (BD Bioscience) and analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
2.5 Cytokine detection
Ex-vivo-expanded Treg or for comparison, Teff, were stimulated with anti-CD3-preloaded L-32 cells at a Treg to L-32 ratio of 1:1 for 3 days. Supernatants were collected on day 1, 2 and 3, and IL-2, IL-17 and IFN-γ measured using a Cytometric Bead Assay (CBA) Flex set system following the manufacturer’s instructions. Data were acquired on a LSR Fortessa and analyzed using FCAP Array Software (BD Bioscience). For intracellular cytokine staining, Treg or Teff were further activated on d 1 for 4h with phorbol 12-myristate 13-acetate (PMA) (1μg/ml) and ionomycin (1μg/ml) in the presence of GolgiStop (BD Bioscience), followed by staining with LIVE/DEAD fixable dye (Molecular Probes, Invitrogen) and fluorescent-labelled anti-CD4 mAb. Intracellular expression of IL-2, IFN-γ or IL-17 was detected according to the eBioscience Intracellular Foxp3 Staining Protocol.
2.6 Apoptosis assay
Cells were stained using Annexin V Apoptosis Detection Kits (eBioscience) following the manufacturer’s instructions. Briefly, expanded Treg were resuspended in 50μl Annexin V binding buffer and incubated with 2.5 μl FITC or eFluor 405-labeled Annexin V at room temperature for 15 min. Two μl propidium iodide (PI) was added to each sample before flow cytometry was performed. Annexin-V+ PI− cells and Annexin V+ PI+ cells were considered early-apoptotic and late-apoptotic/necrotic, respectively.
2.7 In vitro suppression assay
PBMC were labeled with 4 μM carboxyfluorescein succinimidyl ester (CFSE; Invitrogen) or Violet Proliferation Dye 450 (VPD450, BD Biosciences) and stimulated with NHP-specific anti-CD3/CD28 microbeads (Miltenyi) at a cell: bead ratio of 10:1 for 4 days in the addition of indicated numbers of expanded Treg from each round. Proliferation was determined by dye dilution and Percent Divided Cells was analyzed by Flowjo software. Percent suppression was determined as: (percent divided T cells without addition of Treg – percent divided T cells with Treg)/percent divided T cells without addition of Treg X 100%. Cultures of responder cells with titrated numbers of Teff were used as controls.
2.8 In vivo detection of infused autologous Treg
Autologous Treg expanded from cryopreserved 2nd round stock were labeled with 2 μM CFSE or VPD450. Approximately 2×107/kg Treg were injected intravenously (i.v.) into normal untreated or IS monkeys (on day 0) that had received 2 consecutive intravenous (i.v.) infusions of rabbit ATG (Genzyme, Boston, MA) 2 days apart (day −20 and day −18) at doses of 10 and 5 mg/kg and Tacrolimus from day −20 to −16 (target whole blood trough levels between 10 and 15 ng/ml) followed by rapamycin (LC Laboratories) monotherapy from day −15 (target trough levels between 5 and 15 ng/ml) through the duration of the experiment. On day 66 post-infusion for one normal untreated monkey and d50 post-infusion for one IS monkey, PBMC (3ml), inguinal lymph node (LN) (2–3), mesenteric LN (2–3) and spleen cells were stained for CD3, CD4, CD25, CCR7, CD62L, and CXCR3. Data were analyzed with FlowJo software (Tree Star) and graphed with GraphPad Prism (Graph Pad Software, San Diego).
2.9 Statistical analysis
Differences between means were evaluated using Student’s “t” test or ANOVA, as appropriate. Statistical analysis was conducted using the standard formula in Prism Graphpad software.
3. Results
3.1. Use of OptiPrep achieves the best yield of cynomolgus macaque PBMC for Treg purification
In order to maximize the yield of Treg from cynomolgus macaque blood, we compared three PBMC isolation procedures using Ficoll-hypaque, Lympholyte-mammal or OptiPrep (Fig. 1A). PBMC were stained for CD3, CD14, CD20, CD25, and CD127. Counting beads were used to calculate the absolute numbers of B cells (CD20+CD3−), monocytes (CD14+CD3−CD20−), total CD4 T cells (CD3+CD4+CD20−CD14−) and Treg (CD3+CD4+CD25+CD127−). Cells isolated using Lympholyte-mammal formed a clear mononuclear cell layer. However, as shown in Fig. 1B, the majority of cells (>72%) in this layer were monocytes. OptiPrep yielded the highest absolute number of B, T, and Treg (Fig. 1C, D). Consistently, >50% of B, T and Treg were recovered by OptiPrep, whereas about 50% of monocytes were recovered using Lympholyte-mammal (Fig. 1C). Thus, we selected OptiPrep for subsequent Treg isolation.
3.2 Purity and Foxp3 expression by flow-sorted CD25+CD127−/lo Treg from cynomolgus peripheral blood
High expression of CD25 and no or low expression of CD127 are used to identify CD4+ Treg in mice and humans [30]. After labeling with the Abs indicated, cynomolgus PBMC were then flow sorted into populations of CD4+CD25+CD127−/low Treg and CD4+CD25−CD127+ Teff (Fig. 2A). Treg compromised approx. 4.7% (2–6.9%, n=35) of the total CD4 T cell compartment. Both sorted cell populations consistently exceeded 95% purity (Fig. 2B). The freshly-sorted Treg and Teff were further analyzed for their intracellular expression of Foxp3. As shown in Fig. 2C, putative Treg expressed significantly higher levels of Foxp3 (mean fluorescence intensity; MFI) than Teff.
3.3 Expansion of cynomolgus Treg using artificial APC
Previously, we have reported that polyclonal cynomolgus Treg can be expanded ex vivo using anti-CD3/CD28 mAb-coated beads [17]. To minimize any possible bead contamination of the Treg cultures in this study, we stimulated Treg expansion for 3 rounds (Fig. 3A) with adherent aAPC (mouse fibroblast L-32 cells) that express the high-affinity Fc receptor CD32 and the costimulatory molecule CD80 [31], in the presence of anti-CD3 mAb and IL-2. Rapamycin was added only in the first round to inhibit Teff proliferation. As shown in Fig. 3B, during each round of expansion, Treg attached to the L-32 cells, were observed to cluster and blast after stimulation (day 3 or 4), and continued to cluster until day 5–6. During the expansion, the cells were transferred to larger vessels after day 3 or 4 and thus adherent L-32 cells were removed. They appeared more rested by day 7–8, when they were re-stimulated. Very few, if any contaminating L-32 cells were detected at the end of each round (Fig. 3B). Treg were consistently expanded >1000-fold after 3 rounds (22–24 days) (Fig. 3 C). Their expansion is compared with that of Teff in Fig. 3D.
3.4 Ex-vivo expanded cynomolgus Treg maintain expression of Treg signature molecules and display migration feature of Th1-like cells
To determine whether stimulation/expansion of naturally-occurring, thymus-derived cynomolgus Treg (nTreg) affected their phenotype, we compared expanded Treg after each round for the expression of specific markers. Fresh cynomolgus PBMC (CD4+CD25+CD127− gate for fresh Treg and CD4+CD25−CD127+ gate for fresh Teff) and expanded 3rd round Teff were stained as controls. As shown in Fig. 4A, both expanded Treg and Teff were CD25hi and CD127lo, whereas the expanded Treg consistently expressed high levels of CD27 (a costimulatory molecule linked to Treg with increased suppressive ability), the ecto-nucleotidase CD39 (a marker of activated Treg [32–34]), CTLA-4, Foxp3, and Helios. There was no significant difference between Treg obtained after each round of expansion. Thus the ex vivo-expanded cynomolgus nTreg maintained their expression of Treg signature molecules and were phenotypically distinct from expanded Teff. Three rounds of stimulation/expansion with aAPCs did not significantly affect the phenotype of Treg.
Fig. 4. Phenotype and function of expanded cynomolgus Treg and Teff from each round.
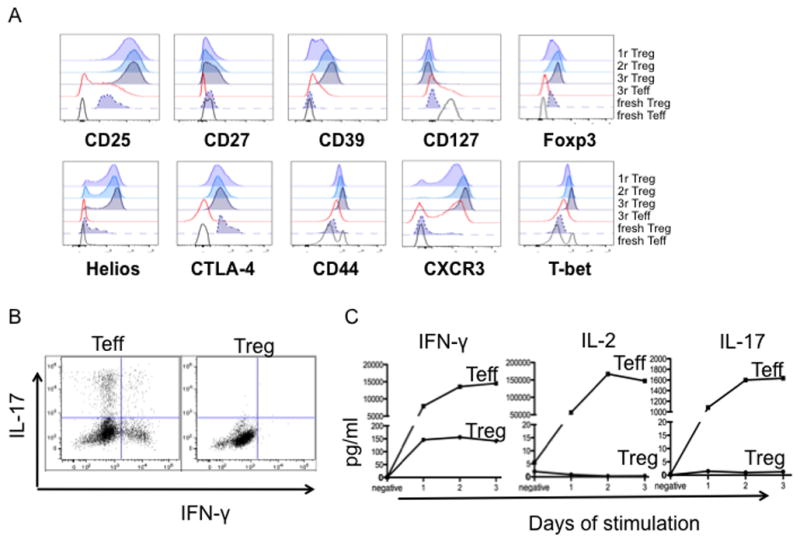
(A) Expanded Treg and Teff were stained for Foxp3, CD25, CD27, CD39, CD44, Helios, CTLA-4, CD127, CXCR3 and T-bet at the end of rounds 1, 2 and 3 of expansion. PBMC were used as controls: fresh Teff (CD4+CD25−CD127+); fresh Treg (CD4+CD25+CD127−). Data are representative of 3 experiments. (B, C) Expanded (3 rounds) Treg and Teff were stimulated with L-32 cells for 3 days. (B) On day 1, some cells were further activated for 4h with PMA and ionomycin in the presence of GolgiStop (BD Bioscience), followed by staining with LIVE/DEAD fixable dye (Molecular Probes: Invitrogen) and fluorescent-tagged Ab against CD4. Intracellular IFN-γ and IL-17 expression were detected according to the eBioscience Intracellular Foxp3 Staining Protocol. (C) Supernatants on days 1, 2, and 3 were collected and IFN-γ, IL-2 and IL-17 levels quantified. Data are representative of 2 experiments.
Homing receptor expression and activation status affect the in vivo behavior of Treg, while expression of the transcription factor T-bet, a Th1 cell-specifying transcription factor, is required for homeostasis and function of Treg during type-1 inflammation [35, 36]. Thus we next examined ex vivo-expanded cynomolgus Treg for the expression of activation markers, homing receptors and T-bet. Compared to fresh bulk CD4+ T cells and expanded Teff, expanded Treg expressed high levels of the activation marker CD44 and comparatively high levels of the inflammatory site migration molecule CXCR3. CCR7, CD62L and CD45RA expression was more variable from culture to culture and from monkey to monkey (data not shown). The expression of T-bet, a known Th1 cell-specifying transcription factor, remained high on the expanded Treg, but not Teff. In addition, unlike expanded Teff, the expanded cynomolgus Treg did not produce IFN-γ or IL-17 upon TCR or mitogen stimulation (Fig. 4B). Collectively, the expanded Treg have a Th-1 like phenotype without producing the inflammatory cytokine IFN-γ.
3.5. Ex-vivo expanded cynomolgus Treg suppress T cell proliferation in vitro
The suppressive capacity of ex-vivo-expanded Treg obtained after each round of expansion for autologous and non-autologous Teff responders was assessed using a CD3/CD28 bead-stimulated T cell proliferation assay. Proliferation was determined by dilution of CFSE-labeled CD3+CD4+ or CD3+CD8+ responder T cells. In some experiments, Treg or control cells were labeled with VPD450 to distinguish Treg from extensively divided responder cells. As shown in Fig 5, addition of expanded Treg inhibited the proliferation of both non-autologous CD4+ and CD8+ responder T cells, as described for murine [37] and rhesus monkey Treg [16]. The suppression was dose-dependent (Fig 5A). No significant difference was observed between Treg obtained after 1, 2, or 3 rounds of expansion (Fig 5B, C). Similar degrees of suppression of autologous T cell proliferative responses were observed (data not shown).
Fig. 5. Expanded cynomolgus Treg strongly suppress CD4+ and CD8+ T cell proliferation.
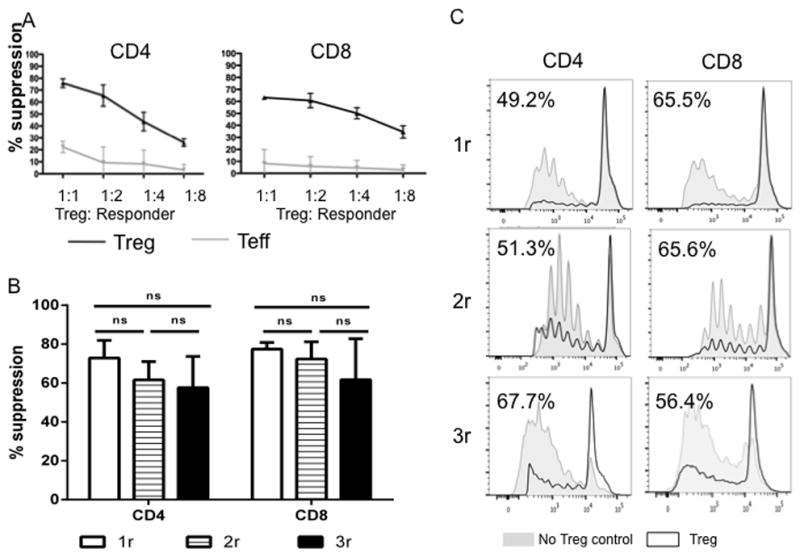
(A) Cynomolgus Treg expanded for 2 rounds were tested for their suppressive activity in CFSE proliferation assays using various responders (CD2+ T cells): Treg ratios, as described in the Materials and Methods. Percent Divided were calculated using FlowJo software. Treg function was expressed as percent suppression of autologous CD4+ or CD8+ T cell proliferation calculated using the formula: (percent divided T cells without addition of Treg or Teff – percent divided T cells with Treg or Teff)/percent divided T cells without addition of Treg or Teff X 100%. Expanded Treg (black lines) showed strong suppressive activity when added to bead-stimulated T cells, whereas expanded Teff (gray lines) did not. Data are representative of 2 independent experiments. (B, C) Cynomolgus Treg expanded for 1, 2 or 3 rounds were tested for their suppressive activity in CFSE proliferation assays using responder (PBMC): Treg ratio of 2:1. (B) Percent suppression from 4 independent experiments was plotted as bar graph. (C) Representative histogram overlay of proliferation with (black line) and without (grey shading) addition of Treg. All three types of Treg showed comparable suppressive capacity when added to bead-stimulated PBMCs.
3.6. Cryopreservation of expanded cynomolgus Treg impairs viability, which can be restored by additional stimulation/expansion
Cryopreservation of expanded Treg could offer several advantages from a clinical perspective. Current knowledge about how cryopreservation may affect expanded Treg is very limited [19]. We did not observe significant changes in the phenotype or suppressive function of expanded Treg after cryopreservation (Fig. 6A, B). However, as shown in Fig. 6C and D, Treg cryopreserved following 1 or 2 rounds of expansion exhibited more apoptosis than their fresh counterparts. This is consistent with finding that cryopreserved expanded Treg do not appear to persist as long as fresh Treg in vivo [11] (data not shown). It has been reported that expansion of frozen primary Treg restored their suppressive capacity impaired by the process of cryopreservation [38]. We next tested whether additional rounds of stimulation/expansion could improve the viability of cryopreserved expanded cynomolgus Treg. We found that, after expansion for a further round, the incidence of apoptotic cells was reduced to the level of their fresh counterparts (Fig. 6D). Taken together, these findings suggest that stimulation/expansion of cryopreserved Treg for a further round can overcome the negative impact of cryopreservation on their viability.
Fig. 6. Cryopreservation of expanded cynomolgus Treg does not affect their phenotype and suppressive capacity, but compromises their viability that can be restored by an additional round(s) of stimulation/expansion.
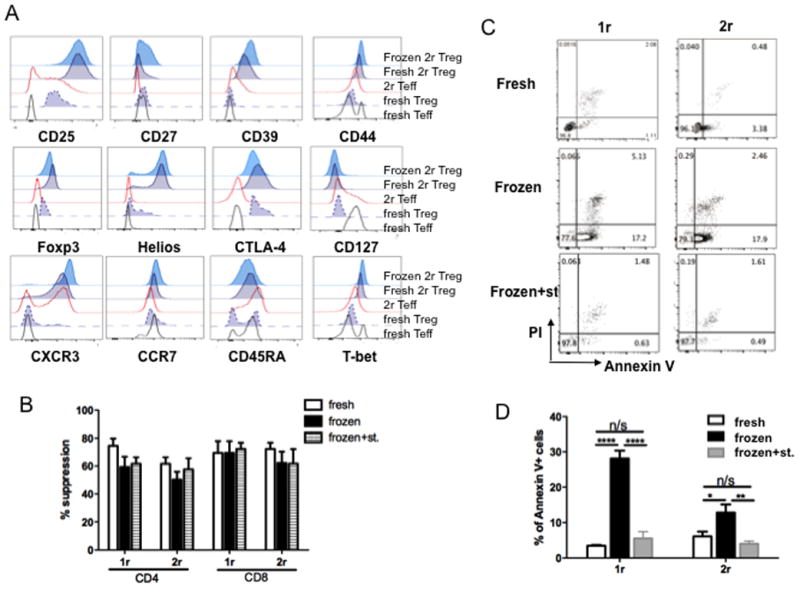
Cynomolgus Treg were expanded for 1 or 2 rounds. At the end of each round, a portion of the cells was frozen. (A, B, C) Frozen expanded Treg were compared with their fresh counterparts in phenotype (A), tested as in Fig. 4, suppressive capacity (B), tested as in Fig. 5, and viability (C) tested by staining for Annexin V and PI. Data are representative of 3 experiments. (D) Cryopreserved expanded Treg were stimulated/expanded for an additional round (Frozen+st.) and compared to their fresh and frozen counterpart in viability. Data were graphed as % Annexin V+ cells, that include early apoptotic cells (Annexin V+ PI−) and late apoptotic cells (Annexin V+ PI+) cells. n>3. ****, p<0.0001;**, p<0.001; *, P<0.01.
3.7 Ex-vivo expanded autologous cynomolgus Treg persist in peripheral blood, inguinal LN, mesenteric LN and spleen >50 days post-infusion
To achieve long-term regulation of immune reactivity, adoptively-transferred Treg may need to survive long enough to migrate to appropriate tissue sites and therein exert their function. To assess the persistence and distribution of transferred Treg expanded from frozen 2nd round Treg, we infused 2×107/kg CFSE- or VPD450-labeled autologous Treg into untreated (normal) and IS monkeys. Peripheral blood was tested up to 87 and 71 days post infusion in the control normal and IS monkeys, respectively. On day 66 (normal) and 50 (IS), inguinal (ing.) LN, mesenteric (mes.) LN and spleen were tested. Monkeys that did not receive fluorochrome-labeled autologous Treg were used as negative controls. As shown in Fig. 7 A, B, in both monkeys, labeled Treg could be detected in the peripheral blood, ing. LN, mes. LN, and spleen. Under the non-inflammatory conditions of this experiment, the expression of Foxp3 by the infused expanded Treg was high during the early period post-transplant, but then decreased over time to a level similar to that of endogenous Treg or slightly higher than that of endogenous Teff (Fig. 7A).
Fig. 7. Persistence of ex-vivo expanded Treg in peripheral blood, LN and spleen after systemic infusion.
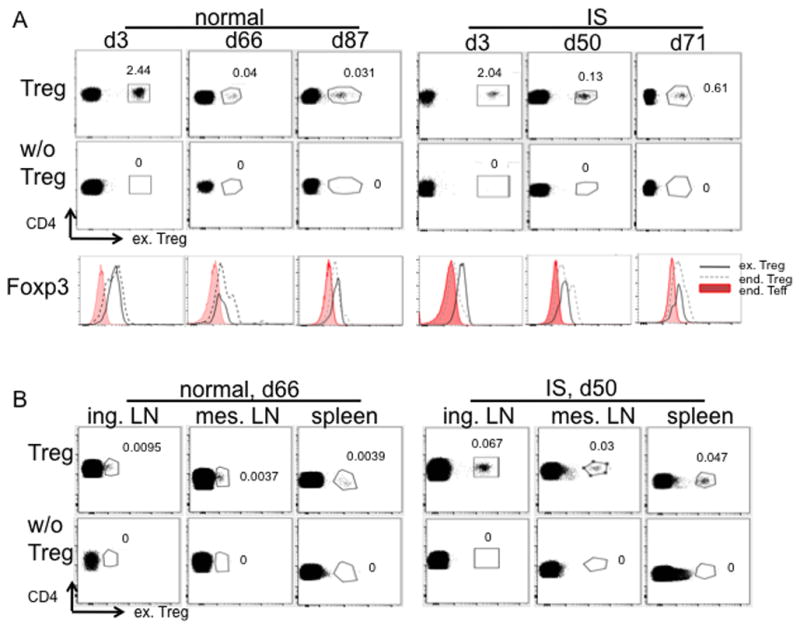
Autologous Treg expanded from frozen 2nd round Treg were labeled with CFSE or VPD450 dye and infused into normal or immunosuppressed (IS) monkeys. On the indicated days post infusion, 1ml blood (A, n=2) and/or lymphoid tissues (B, n=1) were assessed for the presence of infused cells (ex. Treg) by flow cytometry. Samples from monkeys without receiving cells labeled with the same dye were used as negative controls. Blood samples were further tested for Foxp3 expression in infused cells (CD3+CD4+CD25+CD127−Dye+, black solid line), endogenous (end.) Treg (CD3+CD4+CD25+CD127− Dye−, black dashed line) and end. Teff (CD3+CD4+CD25−CD127+, shaded profiles). Histogram overlay of the three populations is shown (A).
4. Discussion
The potential of Treg for therapy of autoimmunity [39–42], allograft rejection [43] and graft-versus-host disease (GVHD) [9, 44–46] has been well-documented in rodent models. Although three phase I clinical trials have confirmed the safety and potential efficacy of ex vivo- expanded Treg for therapy of GVHD after hematopoietic stem cell transplantation [11, 47, 48], generation of sufficient numbers of Treg ex vivo, and development of efficacious protocols in vivo remain significant challenges. Due to the similarity of their immune system to that of humans, NHP represent valuable models to address these issues during transition of regulatory immune cell therapy to the clinic. However, only limited studies of NHP Treg expansion, characterization and in vivo monitoring have been reported especially in cynomolgus macaques [13–18] that are important in studies of cell and organ transplant tolerance. In this study, we were able to expand flow-sorted cynomolgus Treg up to 1000-fold by repetitive stimulation with aAPC and to improve the viability of cryopreserved expanded Treg via restimulation. We were also able to use fluorochrome labeling to assess the in vivo persistence, location and stability of the expanded Treg following their adoptive transfer to normal or IS autologous recipients.
A key observation made in this study is that proliferation dye (CFSE or VPD450)-labeled, ex vivo-expanded cynomolgus autologous Treg can be detected in the blood and host lymphoid tissue for up to at least 2–3 months, the duration of the experiment. There are however, limitations to the use of proliferation dye for in vivo tracking. First, dye-labeled cells eventually become undetectable following extensive proliferation. We observed that the MFI of labeled Treg gradually diminished within the first 8 days after their infusion (data not shown), but remained relatively stable thereafter (Fig. 7). This correlates with results of Ki67 staining of these cells (in press) [49]. More specifically, during the first few days post-infusion, the cells exhibit strong Ki67 staining, which decreases over time to a negative level. Detection of a significant number of dye-labeled infused Treg for >87 days in vivo indicates that, in the absence of inflammation, at least a portion of the cells do not actively proliferate at the later time point and survive >87 days. However, we cannot rule out the possibility that some cells proliferate in the tissues and re-enter the blood when the MFI has decreased to an undetectable level. Another challenge is the detection of rare, dye-labeled cells in tissues against background fluorescence. Autofluorescence is a major contributor to background fluorescence, especially in tissues. To improve sensitivity and thus our ability to detect rare, labeled Treg (Fig. 7B, d66 normal monkeys), we corrected for autofluorescence by gating tightly on lymphocytes to eliminate cells with comparatively high autofluorescence, followed by subtracting signals detected by a detector dedicated to measure autofluorescence (e.g. FITC detector for VPD450-labeled cells) from the detector measuring the labeling dye (e.g. VPD450). To confirm the gate for labeled cells, we used monkeys that had not received dye-labeled cells as negative controls to establish the level of background fluorescence. As shown in Fig. 7B, with appropriate flow analysis to correct background fluorescence, and negative controls to verify gating, it is feasible to detect weak, dye-labeled Treg as low as <0.01% of CD4+ T cells. Our future research will determine the ability of these labeled cells to migrate to inflamed tissues, e.g. organ allografts.
From a practical perspective, cryopreservation of Treg could facilitate the clinical application of these cells. Even though cryopreservation does not seem to significantly affect their phenotype and in vitro suppressive function [38, 50] (Fig. 6), frozen/thawed expanded Treg do not appear to persist as long as freshly-expanded Treg after infusion into the body [11] (data not shown). In this study, we observed a significant increase in apoptotic cells in cryopreserved expanded Treg. This could contribute to their relatively poor persistence in vivo. To restore their viability, we restimulated frozen/thawed expanded Treg for another round and observed that apoptotic cells (Annexin V+) in this restimulated population decreased to the pre-cryopreservation level. In addition, Treg expanded from cryopreserved 2nd round stock were detected in monkey blood up to 87 days post-infusion (Fig. 7). This supports the feasibility of cryopreservation-plus-restimulation of expanded Treg for therapeutic application.
The yield and purity of ex vivo –expanded Treg is affected by numerous factors. One of these is the efficiency of PBMC isolation, which varies from species to species when using standard Ficoll-hypaque density gradients. With this standard protocol, the mononuclear cell “buffy coat” from cynomolgus macaque blood is typically contaminated with RBC and granulocytes. In this study, we found that OptiPrep consistently yielded PBMC with a higher percentage of T cells, and thus a higher yield of Treg. It is noteworthy that using Lympholyte-mammal, a clear mononuclear cell layer with a significantly enhanced percentage of monocytes was obtained. Thus Lympholyte-Mammal might be a better choice for PBMC isolation for the generation of monocyte-derived dendritic cells. A second factor is the efficiency of Treg purification from PBMC. MACS-based Treg isolation has been used widely due to its convenience. However, the purity of CD4+CD25+CD127− Treg isolated by MACS is low [38, 51]. Infusion of contaminating non-regulatory T cells into patients suffering from chronic immune-mediated inflammatory disorders might potentially exacerbate the disease process. In this study, we flow-sorted CD4+CD25+CD127lo/− cells with >95% purity. To further limit contamination by non-Treg, in the first round of expansion, we added rapamycin, which suppresses proliferation of conventional T cells with low/no effect on Treg growth [52]. A further factor is stimulation of the Treg. Anti-CD3/CD28 beads have been used widely to expand Treg for in vivo infusion [12]. However, potential contamination with these potent stimulatory agents would be detrimental. Thus we took advantage of an adherent aAPC (L-32) [53] and generated Treg with no detectable L32 contamination by transferring the former to new vessels after the first 3–4 days of stimulation. Using this approach, we could expand cynomolgus Treg >1000 fold in 22–24 days, without compromising their Treg phenotype and suppressive capacity (Fig. 4, 5).
In conclusion, we have shown that it is possible to generate large numbers of suppressive NHP Treg by ex vivo expansion of flow-sorted nTreg using aAPC, and that restimulation of cryopreserved expanded Treg restores their viability. Furthermore, we have demonstrated that proliferation dye can be used to track these cells for at least 2–3 months after their infusion in blood and lymphoid tissue of normal or IS monkeys, in the absence of inflammation. Given the complexity of Treg function in vivo, it will be essential for the development of safe and efficacious protocols of Treg therapy to track their in vivo behavior, i.e. their lymphoid tissue- versus inflamed tissue-homing ability, their long-term stability and their suppressive capacity for regulating Teff functions at various differentiation stages. Our future research will apply this technique to investigate the in vivo behavior of expanded Treg in monkeys with organ allografts.
Highlights.
Rhesus monkey Treg expand massively with anti-CD3 Ab-pulsed artificial APC and IL-2
Rhesus Treg retain expression of signature markers after each round of expansion
Expanded Treg produce minimal IFNγ and IL-17 and no IL-2 and are potent suppressors
Re-stimulation/expansion of cryopreserved rhesus Treg restores maximal viability
Autologous Treg are evident in blood and lymphoid tissues 3 months after infusion
Acknowledgments
The work was supported by National Institutes of Health (NIH) grant U01 AI91197, part of the NIH NHP Transplantation Tolerance Cooperative Study group and sponsored by the NIAID and NIDDK. HG is supported by Tsinghua-Pittsburgh Joint Education Program and China Scholarship Council. HZ is the recipient of a Basic Science Fellowship from The American Society of Transplantation and a non-concurrent NIH T32 AI 74490 Fellowship.
Abbreviations
- Treg
regulatory T cells
- Teff
effector T cells
- PBMC
peripheral blood mononuclear cells
- Foxp3
forkhead box P3
- APC(s)
antigen-presenting cell(s)
- NHP
non-human primate
- r
round(s) of expansion
- d
days in a round of expansion
- CFSE
carboxyfluorescein succinimidyl ester
- VPD450
violet proliferation dye 450
- MFI
mean fluorescence intensity
- mAb
monoclonal antibody
- IS
immunosuppressed
- PI
propidium iodide
- LN
lymph node(s)
- ing
inguinal
- mes
mesenteric
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. 2008;14:88–92. doi: 10.1038/nm1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, et al. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J Immunol. 2010;184:624–36. doi: 10.4049/jimmunol.0900936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–30. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 5.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493–9. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 6.Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7:585–98. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- 7.Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol. 2012;4:11–21. doi: 10.1093/jmcb/mjr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.June CH, Blazar BR. Clinical application of expanded CD4+25+ cells. Semin Immunol. 2006;18:78–88. doi: 10.1016/j.smim.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389–99. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. 2007;109:827–35. doi: 10.1182/blood-2006-05-025460. [DOI] [PubMed] [Google Scholar]
- 11.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–70. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- 13.Kean LS, Gangappa S, Pearson TC, Larsen CP. Transplant tolerance in non-human primates: progress, current challenges and unmet needs. Am J Transplant. 2006;6:884–93. doi: 10.1111/j.1600-6143.2006.01260.x. [DOI] [PubMed] [Google Scholar]
- 14.Torrealba JR, Katayama M, Fechner JH, Jr, Jankowska-Gan E, Kusaka S, Xu Q, et al. Metastable tolerance to rhesus monkey renal transplants is correlated with allograft TGF-beta 1+CD4+ T regulatory cell infiltrates. J Immunol. 2004;172:5753–64. doi: 10.4049/jimmunol.172.9.5753. [DOI] [PubMed] [Google Scholar]
- 15.Bashuda H, Kimikawa M, Seino K, Kato Y, Ono F, Shimizu A, et al. Renal allograft rejection is prevented by adoptive transfer of anergic T cells in nonhuman primates. J Clin Invest. 2005;115:1896–902. doi: 10.1172/JCI23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson A, Martens CL, Hendrix R, Stempora LL, Miller WP, Hamby K, et al. Expanded nonhuman primate Tregs exhibit a unique gene expression signature and potently downregulate alloimmune responses. Am J Transplant. 2008;8:2252–64. doi: 10.1111/j.1600-6143.2008.02376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dons EM, Raimondi G, Zhang H, Zahorchak AF, Bhama JK, Lu L, et al. Ex vivo-expanded cynomolgus macaque regulatory T cells are resistant to alemtuzumab-mediated cytotoxicity. Am J Transplant. 2013;13:2169–78. doi: 10.1111/ajt.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh K, Kozyr N, Stempora L, Kirk AD, Larsen CP, Blazar BR, et al. Regulatory T cells exhibit decreased proliferation but enhanced suppression after pulsing with sirolimus. Am J Transplant. 2012;12:1441–57. doi: 10.1111/j.1600-6143.2011.03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golab K, Leveson-Gower D, Wang XJ, Grzanka J, Marek-Trzonkowska N, Krzystyniak A, et al. Challenges in cryopreservation of regulatory T cells (Tregs) for clinical therapeutic applications. International immunopharmacology. 2013;16:371–5. doi: 10.1016/j.intimp.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg A, Song LY, Wilkening C, Sevin A, Blais B, Louzao R, et al. Optimization and limitations of use of cryopreserved peripheral blood mononuclear cells for functional and phenotypic T-cell characterization. Clinical and vaccine immunology : CVI. 2009;16:1176–86. doi: 10.1128/CVI.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costantini A, Mancini S, Giuliodoro S, Butini L, Regnery CM, Silvestri G, et al. Effects of cryopreservation on lymphocyte immunophenotype and function. Journal of immunological methods. 2003;278:145–55. doi: 10.1016/s0022-1759(03)00202-3. [DOI] [PubMed] [Google Scholar]
- 22.Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11:119–30. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MKt, Moore DJ, Jarrett BP, Lian MM, Deng S, Huang X, et al. Promotion of allograft survival by CD4+CD25+ regulatory T cells: evidence for in vivo inhibition of effector cell proliferation. J Immunol. 2004;172:6539–44. doi: 10.4049/jimmunol.172.11.6539. [DOI] [PubMed] [Google Scholar]
- 24.Vodanovic-Jankovic S, Hari P, Jacobs P, Komorowski R, Drobyski WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. 2006;107:827–34. doi: 10.1182/blood-2005-05-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–69. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beiras-Fernandez A, Thein E, Hammer C. Induction of immunosuppression with polyclonal antithymocyte globulins: an overview. Experimental and clinical transplantation : official journal of the Middle East Society for Organ Transplantation. 2003;1:79–84. [PubMed] [Google Scholar]
- 27.Gaber AO, Monaco AP, Russell JA, Lebranchu Y, Mohty M. Rabbit antithymocyte globulin (thymoglobulin): 25 years and new frontiers in solid organ transplantation and haematology. Drugs. 2010;70:691–732. doi: 10.2165/11315940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Tang Q, Leung J, Melli K, Lay K, Chuu EL, Liu W, et al. Altered balance between effector T cells and FOXP3+ HELIOS+ regulatory T cells after thymoglobulin induction in kidney transplant recipients. Transpl Int. 2012;25:1257–67. doi: 10.1111/j.1432-2277.2012.01565.x. [DOI] [PubMed] [Google Scholar]
- 29.Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–23. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 30.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol. 2013;190:2001–8. doi: 10.4049/jimmunol.1201379. [DOI] [PubMed] [Google Scholar]
- 32.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–32. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 33.Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant. 2010;10:2410–20. doi: 10.1111/j.1600-6143.2010.03291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J, Liu Y, Qin G, Lam KT, Guan J, Xiang Z, et al. Generation of human Th1-like regulatory CD4+ T cells by an intrinsic IFN-gamma- and T-bet-dependent pathway. Eur J Immunol. 2011;41:128–39. doi: 10.1002/eji.201040724. [DOI] [PubMed] [Google Scholar]
- 36.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steiner D, Brunicki N, Blazar BR, Bachar-Lustig E, Reisner Y. Tolerance induction by third-party “off-the-shelf” CD4+CD25+ Treg cells. Exp Hematol. 2006;34:66–71. doi: 10.1016/j.exphem.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJ, Joosten I. Clinical grade Treg: GMP isolation, improvement of purity by CD127 Depletion, Treg expansion, and Treg cryopreservation. PloS one. 2008;3:e3161. doi: 10.1371/journal.pone.0003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–77. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–6. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 41.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–43. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 42.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–65. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma A, Qi S, Song L, Hu Y, Dun H, Massicotte E, et al. Adoptive transfer of CD4+CD25+ regulatory cells combined with low-dose sirolimus and anti-thymocyte globulin delays acute rejection of renal allografts in Cynomolgus monkeys. International immunopharmacology. 2011;11:618–29. doi: 10.1016/j.intimp.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–50. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 45.Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, et al. Only the CD62L(+) subpopulation of CD4(+)CD25(+) regulatory T cells protects from lethal acute GVHD. Blood. 2005;105:2220–6. doi: 10.1182/blood-2004-05-2044. [DOI] [PubMed] [Google Scholar]
- 46.Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003;112:1688–96. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol. 2009;133:22–6. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–8. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 49.Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of rapamycin over tacrolimus in preserving nonhuman primate treg half-life and phenotype after adoptive transfer. Am J Transplant. 2014;14:2691–703. doi: 10.1111/ajt.12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41. doi: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peters JH, Hilbrands LB, Koenen HJ, Joosten I. Ex vivo generation of human alloantigen-specific regulatory T cells from CD4(pos)CD25(high) T cells for immunotherapy. PLoS One. 2008;3:e2233. doi: 10.1371/journal.pone.0002233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–8. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 53.Hippen KL, Harker-Murray P, Porter SB, Merkel SC, Londer A, Taylor DK, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood. 2008;112:2847–57. doi: 10.1182/blood-2008-01-132951. [DOI] [PMC free article] [PubMed] [Google Scholar]