

Abstract
While age-related macular degeneration (AMD) is a leading cause of central vision loss among the elderly, many inherited diseases that present earlier in life share features of AMD. These diseases of juvenile-onset macular degeneration include Stargardt disease, Best disease, retinitis pigmentosa, X-linked retinoschisis, and other allied disorders. In particular, they can be accompanied by the appearance of drusen, geographic atrophy, macular hyperpigmentation, choroidal neovascularization, and disciform scarring just as in AMD, and often may be confused for the adult form of the disease. Diagnosis based on funduscopic findings alone can be challenging. However, the use of diagnostic studies such as electroretinography, electrooculography, optical coherence tomography, and fundus autofluorescence in conjunction with genetic testing can lead to an accurate diagnosis.
Definitions, Basic Concepts, Classifications
Age-related macular degeneration (AMD) is a leading cause of central vision loss among the elderly population. There are two forms of AMD: the non-neovascular form, which is characterized by macular drusen and other abnormalities of the retinal pigment epithelium (RPE) such as geographic atrophy (GA) and macular hyperpigmentation, and the neovascular form, which features choroidal neovascularization (CNV) and subsequent disciform scarring.
In contrast to AMD, juvenile-onset macular degeneration can result from a number of inherited diseases and may present early in childhood or later in life. This chapter will review the pathologic and diagnostic characteristics of these inherited diseases that may be mistaken for AMD.
The diseases of juvenile-onset macular degeneration can be classified by their patterns of inheritance: autosomal recessive (Stargardt disease, STGD, and cone dystrophy); autosomal dominant (cone dystrophy, adult vitelliform dystrophy, pattern dystrophy, North Carolina macular dystrophy, Doyne honeycomb dystrophy, Sorsby macular dystrophy, and some cases of Stargardtlike macular dystrophy); X-linked (X-linked retinoschisis, XLRS), and mitochondrial (maternally inherited diabetes and deafness, MIDD). Retinitis pigmentosa (RP) results from mutations in mul tiple genes and can be inherited in an autosomaldominant, X-linked recessive or, most commonly, an autosomal recessive pattern.
Stargardt Disease
The autosomal recessive form of STGD, or fundus flavimaculatus, is caused by a mutation in the ABCA4 gene in photoreceptors [1]. Mutations in ABCA4 can lead to retinopathies other than STGD, including AMD in heterozygous carriers [2], bull’s eye maculopathy [3, 4], autosomal recessive (ar) cone-rod dystrophy, and arRP [5–9]. The underlying pathology of STGD results from accumulation of lipofuscin in the RPE through a process of photoreceptor outer segment disc shedding and phagocytosis [10, 11]. Mutations in ABCA4 lead to accumulation of all-trans retinal in the photoreceptor, which is toxic to the cell at certain concentrations. This accumulation can result in the formation of bis-pyridinium molecules such as N-retinyledeneN-retinyl-ethanolamine (A2E). A2E is a component of lipofuscin and causes complement activation and inhibition of 11-cis retinal regeneration [12]. Since pigmentary changes and RPE atrophy seen in STGD may be confused with features of AMD, it is particularly important to consider both diagnoses.
The true prevalence of STGD may be higher than the estimated 1 in 8,000–10,000 [13] since the carrier frequency for an ABCA4 defect may be as high as 1 in 20 [14, 15]. Furthermore, of the estimated 600 diseases associated with mutations in the ABCA4 gene, the three most common account for less than 10% of the disease phenotypes [16].
The age at onset of STGD varies widely, with typical presentation between 10 and 20 years of age. Earliest symptoms are consistent with slowly progressive central vision loss [17], whereas later ages at onset have been associated with a more favorable visual prognosis [18, 19]. In some cases, asymptomatic patients may be diagnosed before the onset of symptoms by the diagnosis of a symptomatic sibling [20].
STGD progresses through four stages (fig. 1) [21]. In stage I, both the electrooculogram (EOG) and dark adaption measured with the electroretinogram (ERG) are normal. Pigmentary changes and RPE atrophy are confined to the fovea or parafoveal macula, and a discontinuous ring of flecks often encircles the fovea. In stage II, these flecks become more widespread, and there is a subnormal cone and rod response with delayed dark adaption. Stage III sees resorption of the flecks and widespread atrophy of the choriocapillaris. There is further resorption of flecks in stage IV with extensive choriocapillaris and RPE atrophy.
Fig. 1.

STGD. a Wide-view fundus photograph of an STGD patient. b, d Disease progression in a second STGD patient. b Fundus photograph demonstrating pigmentary changes and RPE atrophy of the fovea. FAF (c) and fundus photograph (d) after a 3-year interval showing widespread accumulation of lipofuscin and increased areas of RPE atrophy in a classic Bull’s eye shape, but with peripapillary sparing (arrows).
Funduscopic examination alone has made stratification into groups and counseling on prognosis of these patients challenging. Use of functional testing, autofluorescence imaging and optical coherence tomography (OCT) can assist with classification and prediction of future visual function. Full-field ERG testing can be used to divide patients into three groups, ranging from normal rod and cone function in group 1 and relative loss of generalized cone function in group 2 to abnormal rod and cone function in group 3. Progression of visual field changes is expected with marked abnormality of both cone and rod systems, and thus group 3 patients represent the worst prognosis for retention of peripheral vision. Abnormal cone-rod physiology or observation of loss-of-function ABCA4 alleles at the initial visit is likely a reliable predictor of disease severity [18].
The presence of a dark or silent choroid on fluorescein angiography (FA) has assisted in making the clinical diagnosis of STGD and is seen in 85.9% of cases [22]. Sometimes, the area of blocked choroidal fluorescence is only evident in the peripapillary region. Background choroidal fluorescence is masked due to a build-up of lipofuscin in the RPE that causes absorption of short-wavelength light. Importantly, the absence of this sign does not rule out a diagnosis of ABCA4 disease.
In general, macular autofluorescence is abnormally high in STGD patients. Fundus autofluorescence (FAF) provides a qualitative assessment of the build-up and distribution of lipofuscin and can be used to detect changes in the RPE before these can be appreciated on fundus biomicroscopy [23, 24]. Flecks of focal hyperfluorescence and areas of hypofluorescence that correspond to RPE atrophy can be seen on FAF (fig. 1). These regions of atrophy, as seen on both FA and FAF, also correlate with the loss of the inner segment-outer segment (ISOS) junction appreciated with OCT [25]. As the retinal disease progresses, total loss of the ISOS junction is seen in the macula and is associated with widespread thinning of the inner and outer retina as well as the RPE.
Best Disease
Best disease, the early-onset form of vitelliform macular dystrophy, is an autosomal dominant retinal disease with incomplete penetrance and wide phenotypic variability that was initially described by Franz Best is 1905. The mutation is in the BEST1 gene, previously known as VMD2, which encodes bestrophin-1, a calcium-dependent chloride channel found in the RPE [26]. Mutations lead to accumulation of lipofuscin within and beneath the RPE and result in the characteristic bilateral egg yolk appearance of the macula (fig. 2). In particular, OCT has demonstrated that the material accumulates on the outer retina, which is suggestive of photoreceptor shedding from the outer segment [27].
Fig. 2.

Best disease. Fundus photograph (a) and FAF (b) demonstrating central areas of atrophy that correspond to areas of serous detachment. b Lipofuscin accumulates inferiorly due to gravity.
Best disease usually presents in childhood and has been characterized as progressing through five stages, although some controversy exists as to the chronological order of these stages: the previtelliform, vitelliform, pseudohypopyon, vitelliruptive, and atrophic stages [28]. The atrophic stage may be followed by the development of CNV in a subset of cases and often represents the worst visual prognosis. Best disease is characterized by a clinically normal ERG and an abnormal EOG, with a reduced or nonexistent light to dark ratio. Additionally, as both FAF and OCT can reveal abnormalities even when fundoscopy is normal, these techniques should be included in the diagnostic workup whenever possible [29].
The yellow macular deposits seen in Best disease may be confused with adult foveomacular vitelliform dystrophy, which usually develops in the fourth to sixth decades of life [30]. The same gene, BEST1, is implicated in the adult form of the disorder, but it has not been found to significantly predispose the patient to AMD as was previously considered [31]. Despite the similarities of the yellow, yolk-like macular deposits, the younger age at onset and characteristically abnormal EOG help differentiate Best vitelliform dystrophy from the adult form. Further complicating the distinction, the macular lesions may eventually resolve, leaving areas of RPE atrophy that can be confused for AMD later in life.
Allied Disorders
Several other inherited retinal dystrophies share features of AMD, including RP, cone-rod dystrophy, and Bull’s eye maculopathy. Autosomal dominant retinal dystrophies include cone dystrophy, pattern dystrophy, North Carolina macular dystrophy, Doyne honeycomb dystrophy, and Sorsby macular dystrophy. XLRS and MIDD, inherited via defects in mitochondrial genes, are other allied disorders.
RP is a retinal degenerative disease characterized by progressive vision loss. Unlike Stargardt and Best diseases, where accumulation of lipofuscin leads to pathology (e.g. secondary photoreceptor degeneration), the symptoms of RP are caused by primary photoreceptor death throughout the retina. RP can be inherited in an autosomal dominant, autosomal recessive, or X-linked recessive pattern, though autosomal recessive is the most common and is associated with 25 different genes. It is estimated that the aggregate carrier frequency for arRP alleles may be as high as 10% [32].
In contrast to AMD, in which photoreceptors in the macula may be lost, RP is characterized by loss of both rods and cones throughout the retina, including in the periphery. Though disease progression in RP can vary, patients typically present initially with night blindness due to loss of rods followed by central vision loss as a result of cone degeneration and, eventually, total blindness [33].
Several methods can be used to clinically assess disease progression in RP patients. For instance, ERG wave amplitudes correlate with visual field size and can be used to accurately diagnose the disease [34]. OCT can be used to measure retinal thickness in patients with RP: both retinal thinning due to cell loss and thickening due to edema can be demonstrated and are associated with lower visual acuity [35]. FAF in RP shows a hyperautofluorescent ring that constricts progressively throughout the course of the disease in conjunction with a shortening of width of the ellipsoid zone (fig. 3) [36]. In fact, this ring appears to delineate the functional and dysfunctional areas of the retina [37].
Fig. 3.
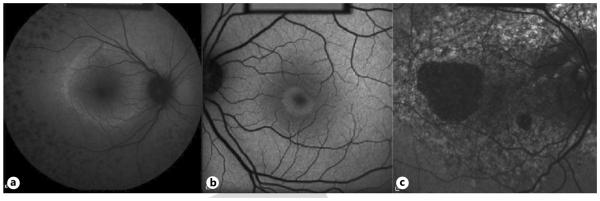
Allied disorders. a RP FAF with high-density autofluorescent ring and peripheral drop-off of photoreceptors. This high-density ring corresponds to visual field loss. b Early bull’s eye maculopathy FAF showing lesion in the classic shape with central foveal atrophy. c Pattern dystrophy FAF with diffuse accumulation of lipofuscin and profound central atrophy without peripapillary sparing.
Variation in the RP 1-like protein 1 gene RP1L1 is associated with a spectrum of diseases, including RP and occult macular dystrophy (OCMD) [38]. OCMD is characterized by central retinal cone degeneration. The age at onset is variable, between 6 and 50 years, and is typically followed by progressive decline of visual acuity. Despite significant loss of vision in these patients, funduscopic, FA, and full-field ERG findings are all normal. However, multifocal ERG findings demonstrate depressed responses from the central retina with preserved peripheral responses [39]. OCT shows reduced foveal thickness with thinning of the outer nuclear layer and is predictive of the degree of vision loss [40].
Cone dystrophies without rod involvement affect the central retina while sparing the periphery [41]. Typical cases of autosomal dominant cone dystrophy show a bull’s-eye maculopathy that can be seen on FAF, while others may demonstrate varying degrees of macular atrophy similar to AMD (fig. 3). In addition, temporal optic nerve pallor may be present. Patients typically present in their teens or early adulthood with photophobia and varying degrees of color vision loss. ERG findings are consistent with cone involvement, specifically a reduced 30-Hz flicker amplitude and increased implicit time, while rod responses are normal.
Autosomal dominant pattern dystrophy has been linked to RDS/peripherin gene mutations [42]. Affected individuals often present in midlife and may be asymptomatic. FAF of patients with pattern dystrophy demonstrates a wide variety of RPE pigment deposits in the macula (fig. 3). Some patients may eventually develop areas of macular GA with a small subset developing CNV. These features mimic AMD, and thus the two diagnoses may be confused.
North Carolina macular dystrophy arises from mutations in the MCDR1 gene on chromosome 6. The disease was first described in families living in mountainous regions of North Carolina, but has since been found in unrelated families living elsewhere. Its onset is typically in infancy with stabilization of the dystrophy by the teenage years [43]. Affected patients may share clinical features of AMD, including drusen-like deposits in the macula and areas of severe atrophy that can appear staphylomatous or colobomatous (fig. 4).
Fig. 4.
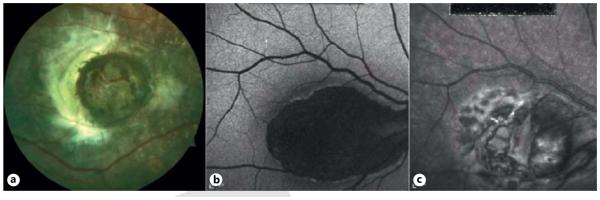
North Carolina macular dystrophy. a Fundus photograph of a child with NCMD. b FAF of a 55-year-old man showing severe central hypoautofluorescence. c Infrared image of same adult.
Doyne honeycomb dystrophy, also known as malattia leventinese, is caused by mutations in the EFEMP1 gene on chromosome 2 [44]. Affected individuals typically develop drusen in the macula and on the nasal side of the optic disc in the third decade of life. These patients typically have normal ERG and EOG findings, while FAF may help to highlight the abnormal deposits. These deposits may fade with age, and the subsequent development of peripapillary and/or macular atrophy and CNV may simulate AMD, making it difficult to elicit an accurate diagnosis.
Sorsby macular dystrophy is caused by a mutation in the gene encoding the tissue inhibitor of metalloproteinases-3. The typical age at onset is 40 years, and patients often present with difficulty transitioning between light and dark environments [45]. Central vision abnormalities arise and are followed by loss of peripheral vision late in the course of the disease. Drusen-like deposits may form early on with areas of GA, followed by development of bilateral CNV and subsequent disciform scars – features that mimic AMD, although the age at onset is much earlier than in cases of AMD. Symptoms of nyctalopia can be ameliorated with high-dose vitamin A supplementation.
XLRS is caused by a defect in the XLRS1 gene encoding retinoschisin, a protein thought to be involved in cell adhesion [46]. Over 100 distinct gene mutations causing similar phenotypes exist, with an estimated prevalence of 1 in 5,000–25,000. XLRS often presents with early vision loss in affected males, while clinical findings in carrier females are typically normal. The main clinical finding in affected males is a radial pattern of folds emanating from the fovea, which contains schisis cavities. OCT can provide detailed histopathologic images of these cavities. In roughly 50% of cases, peripheral schisis also develops, often in the nerve fiber layer. Areas of schisis may resolve in older patients, giving way to pigmentary changes and RPE atrophy that mimic AMD. Electronegative ERG findings, with a normal awave and a reduced b-wave, indicate relative sparing of the photoreceptors and involvement of the inner retina in XLRS.
MIDD is caused by mitochondrial gene defects, specifically a 3243 mitochondrial DNA mutation, involved in the oxidative production of energy [47]. This ensuing insulin secretion defect leads to diabetes, hearing loss, and a macular pattern dystrophy. Macular findings typically cause symptoms in the fifth decade of life and may present as a spectrum ranging from small, pigmented lesions to large areas of macular atrophy. Though macular findings in these patients may suggest AMD, the history of maternally inherited diabetes, sensorineural hearing loss, and kidney failure related to mitochondrial renal disease suggests a diagnosis of MIDD.
Conclusion
This chapter has reviewed a set of inherited retinal diseases that lead to juvenile-onset macular degeneration. Accurate diagnosis may be difficult based on fundus appearance alone, especially if the patient presents later in life. However, by careful review of a patient’s pedigree, as well as the judicious use of diagnostic studies such as the ERG and FAF, in conjunction with genetic testing, accurate diagnoses can be elicited.
List of Contributors
Petr Baranov Schepens Eye Research Institute, An affiliate of Harvard Medical School, Boston, MA 02114 (USA), E-Mail Petr_Baranov@meei.harvard.edu
Gerhard Bauer University of California Davis School of Medicine, Institute for Regenerative Cures, Sacramento, CA 95817 (USA), E-Mail gerhard.bauer@ucdmc.ucdavis.edu
Amanda-Jayne F. Carr The London Project to Cure Blindness, Division of ORBIT, Institute of Ophthalmology, University College London, 11–43 Bath Street, London EC1V 9EL (UK), E-Mail a.carr@ucl.ac.uk
RebeccaL.Carrier Chemical Engineering, Northeastern University, 360 Huntington Avenue, Boston, MA 02115 (USA), E-Mail r.carrier@neu.edu
Ricardo P. Casaroli-Marano Department of Surgery, School of Medicine, University of Barcelona, and Transplant Services Foundation (TSF), Hospital Clínic de Barcelona, ES-08028 Barcelona (Spain), E-Mail rcasaroli@ub.edu
Dennis O. Clegg Center for Stem Cell Biology and Engineering, Neuroscience Research Institute, University of California, Santa Barbara, CA 93106 (USA), E-Mail clegg@lifesci.ucsb.edu
Peter J. Coffey The London Project to Cure Blindness, Division of ORBIT, Institute of Ophthalmology, University College London, 11–43 Bath Street, London EC1V 9EL (UK), E-Mail p.coffey@ucl.ac.uk
Roxanne H. Croze Center for Stem Cell Biology and Engineering, Neuroscience Research Institute, University of California, Santa Barbara, CA 93106 (USA), E-Mail rhcroze@gmail.com
Lyndon da Cruz NIHR Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, London EC1V 2PD UK, E-Mail Lyndon.daCruz@moorfields.nhs.uk
Jacque L. Duncan 10 Koret Way, K129, San Francisco, CA 94143-0730 (USA), E-Mail duncanj@vision.ucsf.edu
Rony Gelman Department of Ophthalmology, Jules Stein Eye Institute, University of California Los Angeles, 100 Stein Plaza UCLA, Los Angeles, CA 90095 (USA), E-Mail rony_gelman@yahoo.com
Mark S. Humayun Doheny Retina Institute, University of Southern California, 1355 San Pablo St., DVRC 119, Los Angeles, CA 90033 (USA), E-Mail •••
Henry Klassen University of California Irvine School of Medicine, Gavin Herbert Eye Institute and, Stem Cell Research Center, Irvine, CA 92697 (USA), E-Mail hklassen@uci.edu
Joydip Kundu, Chemical Engineering, Northeastern University, 360 Huntington Avenue, Boston, MA 02115 (USA), E-Mail j.kundu@neu.edu
Jonathan H. Lin Shiley Eye Center, Department of Pathology, University of California San Diego, La Jolla, CA 92093 (USA), E-Mail JLin@ucsd.edu
Bo Lu Berkeley Lights, Inc., 5885 Hollis St., Suite 370, Emeryville, CA 94608 (USA), E-Mail lubomems@gmail.com
Christopher Malcuit Department of Biological Sciences, Kent State University, Kent, OH 44242 (USA), E-Mail cmalcuit@kent.edu
Moreno Menghini Department of Ophthalmology, University of California, San Francisco, 400 Parnassus, San Francisco, CA 94143 (USA), E-Mail moreno.menghini@me.com
Andrew Michaelson Chemical Engineering, Northeastern University, 360 Huntington Avenue, Boston, MA 02115 (USA), E-Mail michaelson.a@husky.neu.edu
Victoria North College of Physicians and Surgeons, Columbia University, 630 West 168th Street, New York, NY 10032 (USA), E-Mail vsn2104@mail.cumc.columbia.edu
Michael B. Powner The London Project to Cure Blindness, Division of ORBIT, Institute of Ophthalmology, University College London, 11–43 Bath Street, London EC1V 9EL (UK), E-Mail m.powner@ucl.ac.uk
Conor M. Ramsden The London Project to Cure Blindness, Division of ORBIT, Institute of Ophthalmology, University College London, 11–43 Bath Street, London EC1V 9EL (UK), E-Mail conor.ramsden.09@ucl.ac.uk
Rajesh C. Rao Department of Ophthalmology & Visual Sciences, W.K. Kellogg Eye Center, Department of Pathology, University of Michigan Medical School, 1000 Wall St., Ann Arbor, MI 48105 (USA), E-Mail rajeshr@med.umich.edu
Philip J. Rosenfeld Bascom Palmer Eye Institute, 900 NW 17th St., Miami, 33136 (USA), E-Mail prosenfeld@med.miami.edu
David H. Sachs Transplantation Biology Research Center, Harvard Medical School and, Massachusetts General Hospital, 149 13th Street, Suite 9019, Boston, MA 02129 (USA), E-Mail david.sachs@tbrc.mgh.harvard.edu
Jonathan Sheu University of California Davis School of Medicine, Institute for Regenerative Cures, Sacramento, CA 95817 (USA), E-Mail jonathan.sheu@ucdmc.ucdavis.edu
Valentin M. Sluch Biochemistry, Cellular and Molecular Biology Graduate Program, Johns Hopkins University School of Medicine, Baltimore, MD 21287 (USA), E-Mail vsluch1@jhmi.edu
Matthew J.K. Smart The London Project to Cure Blindness, Division of ORBIT, Institute of Ophthalmology, University College London, 11–43 Bath Street, London EC1V 9EL (UK), E-Mail matthew.smart@ucl.ac.uk
Jeffrey Stern Neural Stem Cell Institute, One Discovery Drive, Rensselaer, NY 12144 (USA), E-Mail retina@nycap.rr.com
Ilene K. Sugino Institute of Ophthalmology and Visual Science, Rutgers-New Jersey Medical School, Rutgers University, Room 6155, Doctors Office Center, 90 Bergen Street, Newark, NJ 07103 (USA), E-Mail suginoik@njms.rutgers.edu
Jaime Tabera Transplant Services Foundation (TSF), Hospital Clínic de Barcelona, ES-08036 Barcelona (Spain), E-Mail JTABERA@clinic.ub.es
Yu-Chong Tai Electrical Engineering, California Institute of Technology, 1200 E. California Blvd, Pasadena, MC 136-93, CA 91125 (USA), E-Mail yctai@caltech.edu
Sally Temple Neural Stem Cell Institute, One Discovery Drive, Rensselaer, NY 12144 (USA), E-Mail sallytemple@neuralsci.org
Aseda Tena Transplantation Biology Research Center, Harvard Medical School and Massachusetts General Hospital, 149 13th Street, Suite 9019, Boston, MA 02129 (USA), E-Mail Aseda.Tena@tbrc.mgh.harvard.edu
Esteve Trias Transplant Services Foundation (TSF), Hospital Clínic de Barcelona, ES-08036 Barcelona (Spain), E-Mail ETRIAS@clinic.ub.es
Stephen H. Tsang Columbia University Departments of Pathology and Ophthalmology, 630 West 168th Street, New York, NY 10032 (USA), E-Mail sht2@columbia.edu
Anna Vilarrodona Transplant Services Foundation (TSF), Hospital Clínic de Barcelona, ES-08036 Barcelona (Spain), E-Mail AVILARRO@clinic.ub.es
Katherine J. Wert The Whitehead Institute for Biomedical Research, Jaenisch Laboratory, Nine Cambridge Center, Cambridge, MA 02142 (USA), E-Mail wert.katherine@gmail.com
Michael J. Young Schepens Eye Research Institute, An affiliate of Harvard Medical School, Boston, MA 02114 (USA), E-Mail michael_young@meei.harvard.edu
Donald J. Zack Ophthalmology, Wilmer Eye Institute, Johns Hopkins University School of Medicine, 400 N. Broadway, Smith Building 3029, Baltimore, MD 21287 (USA), E-Mail dzack@jhmi.edu
David N. Zacks Department of Ophthalmology & Visual Sciences, W.K. Kellogg Eye Center, University of Michigan Medical School, 1000 Wall St., Ann Arbor, MI 48105 (USA), E-Mail davzacks@umich.edu
Marco A. Zarbin Institute of Ophthalmology and Visual Science, Rutgers-New Jersey Medical School, Rutgers University, Room 6155, Doctors Office Center, 90 Bergen Street, Newark, NJ 07103 (USA), E-Mail zarbin@earthlink.net
Qian Sun Institute of Ophthalmology and Visual Science, Rutgers-New Jersey Medical School, Rutgers University, Room 6155, Doctors Office Center, 90 Bergen Street, Newark, NJ 07103 (USA), E-Mail •••
Noounanong Cheewatrakoolpong Institute of Ophthalmology and Visual Science, Rutgers-New Jersey Medical School, Rutgers University, Room 6155, Doctors Office Center, 90 Bergen Street, Newark, NJ 07103 (USA), E-Mail •••
References
- 1.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATPbinding transporter gene (ABCR) is mutated in recessive Stargardt’s macular dystrophy. Nat Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 3.Michaelides M, Chen LL, Brantley MA, Jr, Andorf JL, Isaak EM, Jenkins SA, et al. ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull’s-eye maculopathy. Br J Ophthalmol. 2007;91:1650–1655. doi: 10.1136/bjo.2007.118356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Koyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 5.Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 6.Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4 (ABCR) gene. Exp Eye Res. 2001;73:877–886. doi: 10.1006/exer.2001.1093. [DOI] [PubMed] [Google Scholar]
- 7.Fishman GA, Stone EM, Eliason DA, Taylor CM, Lindeman M, Derlacki DJ. ABCA4 gene sequence variations in patients with autosomal recessive conerod dystrophy. Arch Ophthalmol. 2003;121:851–855. doi: 10.1001/archopht.121.6.851. [DOI] [PubMed] [Google Scholar]
- 8.Klevering BJ, van Driel M, van de Pol DJ, Pinckers AJ, Cremers FP, Hoyng CB. Phenotypic variations in a family with retinal dystrophy as a result of different mutations in the ABCR gene. Br J Ophthalmol. 1999;83:914–918. doi: 10.1136/bjo.83.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 10.Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, et al. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005;14:2769–2778. doi: 10.1093/hmg/ddi310. [DOI] [PubMed] [Google Scholar]
- 11.Koenekoop RK. The gene for Stargardt disease, ABCA4, is a major retinal gene: a mini-review. Ophthalmic Genet. 2003;24:75–80. doi: 10.1076/opge.24.2.75.13996. [DOI] [PubMed] [Google Scholar]
- 12.Moiseyev G, Nikolaeva O, Chen Y, Farjo K, Takahashi Y, Ma JX. Inhibition of the visual cycle by A2E though direct interaction with RPE65 and implications in Stargardt disease. Proc Nat Acad Sci. 2010;107:17551–17556. doi: 10.1073/pnas.1008769107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blacharski PA. Fundus flavimaculatus. In: Newsome DA, editor. Retinal Dystrophies and Degenerations. Raven Press; New York: 1988. pp. 135–159. [Google Scholar]
- 14.Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 15.Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4) Hum Genet. 2001;108:346–355. doi: 10.1007/s004390100493. [DOI] [PubMed] [Google Scholar]
- 16.Allikmets R. Stargardt disease: from gene discovery to therapy. In: Tombran-Tink J, Barnstable CJ, editors. Retinal Degenerations: Biology, Diagnostics and Therapeutics. Humana Press; Totowa: 2007. pp. 105–118. [Google Scholar]
- 17.Fishman GA. The electroretinogram. In: Fishman GA, editor. Electrophysiologic Testing in Disorders of the Retina, Optic Nerve and Visual Pathway. 2 The Foundation of the American Academy of Ophthalmology; Singapore: 2001. pp. 54–56. [Google Scholar]
- 18.Lois N, Holder GE, Bunce C, Fizke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119:359–369. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- 19.Rotenstreich Y, Fishman, GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003;110:1151–1158. doi: 10.1016/S0161-6420(03)00333-6. [DOI] [PubMed] [Google Scholar]
- 20.Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R, et al. Genotypephenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005;37:159–167. doi: 10.1159/000086073. [DOI] [PubMed] [Google Scholar]
- 21.Fishman GA. Fundus flavimaculatus: a clinical classification. Arch Ophthalmol. 1976;94:2061–2067. doi: 10.1001/archopht.1976.03910040721003. [DOI] [PubMed] [Google Scholar]
- 22.Fishman GA, Farber M, Patel BS, Derlacki DJ. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. 1987;94:809–814. doi: 10.1016/s0161-6420(87)33533-x. [DOI] [PubMed] [Google Scholar]
- 23.Boon CJ, Jeroen Klevering B, Keunen JE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vis Res. 2008;48:2569–2577. doi: 10.1016/j.visres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 24.von Rückmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997;115:609–615. doi: 10.1001/archopht.1997.01100150611006. [DOI] [PubMed] [Google Scholar]
- 25.Ergun E, Hermann B, Wirtitsch M, Unterhuber A, Ko TH, Sattmann H, et al. Assessment of central visual function in Stargardt’s disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest Ophthalmol Vis Sci. 2005;46:310–316. doi: 10.1167/iovs.04-0212. [DOI] [PubMed] [Google Scholar]
- 26.Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- 27.Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmology. 2006;113:1392–1400. doi: 10.1016/j.ophtha.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 28.Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88:688–692. doi: 10.1016/s0161-6420(81)34965-3. [DOI] [PubMed] [Google Scholar]
- 29.Boon CJ, Theelen T, Hoefsloot EH, van Schooneveld MJ, Keunen JE, Cremers FP, et al. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina. 2009;29:835–847. doi: 10.1097/IAE.0b013e31819d4fda. [DOI] [PubMed] [Google Scholar]
- 30.Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S, et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet. 1999;104:449–453. doi: 10.1007/s004390050986. [DOI] [PubMed] [Google Scholar]
- 32.Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- 33.Tsui I, Song B, Lin C, Tsang SH. A practical approach to retinal dystrophy. Retinal Physician. 2007;4:18–26. [Google Scholar]
- 34.Sandberg MA, Weigel-DiFranco C, Rosner B, Berson EL. The relationship between visual field size and electroretinogram amplitude in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1996;37:1693–1698. [PubMed] [Google Scholar]
- 35.Sandberg MA, Brockhurst RJ, Gaudio AR, Berson EL. The association between visual acuity and central retinal thickness in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2005;46:3349–3354. doi: 10.1167/iovs.04-1383. [DOI] [PubMed] [Google Scholar]
- 36.Lima L, Burke T, Greenstein V, Chou C, Cella W, Yannuzi L, Tsang SH. Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am J Ophthalmol. 2012;153:718–727. doi: 10.1016/j.ajo.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popovic P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2005;243:1018–1027. doi: 10.1007/s00417-005-1186-x. [DOI] [PubMed] [Google Scholar]
- 38.Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–514. doi: 10.1002/humu.22264. [DOI] [PubMed] [Google Scholar]
- 39.Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet. 2010;87:424–429. doi: 10.1016/j.ajhg.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brockhurst RJ, Sandberg MA. Optical coherence tomography findings in occult macular dystrophy. Am J Ophthalmol. 2007;143:516–518. doi: 10.1016/j.ajo.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 41.Michaelides M, Hardcastle AJ, Hunt DM, Moore AT. Progressive cone and cone-rod dystrophies: phenotypes and underlying genetic basis. Surv Ophthalmol. 2006;51:232–258. doi: 10.1016/j.survophthal.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Marmor MF. The pattern dystophies. In: Heckenlively JR, Arden GB, editors. Principles and Practice of Clinical Electrophysiology of Vision. 2 MIT Press; Cambridge: 2006. pp. 757–761. [Google Scholar]
- 43.Small KW. North Carolina macular dystrophy: clinical features, genealogy, and genetic linkage analysis. Trans Am Ophthalmol Soc. 1998;96:925–961. [PMC free article] [PubMed] [Google Scholar]
- 44.Evans K, Gregory CY, Wijesuriya SD, Kermani S, Jay MR, Plant C, et al. Assessment of the phenotypic range seen in Doyne honeycomb retinal dystrophy. Arch Ophthalmol. 1997;115:904–910. doi: 10.1001/archopht.1997.01100160074012. [DOI] [PubMed] [Google Scholar]
- 45.Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–356. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- 46.Tantri A, Vrabec TR, Cu-Unjieng A, Frost A, Annesley WH, Jr, Donoso LA. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49:214–230. doi: 10.1016/j.survophthal.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 47.Guillausseau PJ, Massin P, Dubois-La-Forgue D, Timsit J, Virally M, Gin H, et al. Maternally inherited diabetes and deafness: a multicenter study. Ann Intern Med. 2001;134:721–728. doi: 10.7326/0003-4819-134-9_part_1-200105010-00008. [DOI] [PubMed] [Google Scholar]