

Abstract
Alzheimer's disease (AD) and familial Danish dementia (FDD) are degenerative neurological diseases characterized by amyloid pathology. Normal human sera contain IgG antibodies that specifically bind diverse preamyloid and amyloid proteins and have shown therapeutic potential in vitro and in vivo. We cloned one of these antibodies, 3H3, from memory B cells of a healthy individual using a hybridoma method. 3H3 is an affinity-matured IgG that binds a pan-amyloid epitope, recognizing both Aβ and λ Ig light chain (LC) amyloids, which are associated with AD and primary amyloidosis, respectively. The pan-amyloid-binding properties of 3H3 were demonstrated using ELISA, immunohistochemical studies, and competition binding assays. Functional studies showed that 3H3 inhibits both Aβ and LC amyloid formation in vitro and abrogates disruption of hippocampal synaptic plasticity by AD-patient-derived soluble Aβ in vivo. A 3H3 single-chain variable fragment (scFv) retained the binding specificity of the 3H3 IgG and, when expressed in the brains of transgenic mice using an adeno-associated virus (AAV) vector, decreased parenchymal Aβ amyloid deposition in TgCRND8 mice and ADan (Danish Amyloid) cerebral amyloid angiopathy in the mouse model of FDD. These data indicate that naturally occurring human IgGs can recognize a conformational, amyloid-specific epitope and have potent anti-amyloid activities, providing a rationale to test their potential as antibody therapeutics for diverse neurological and other amyloid diseases.
Keywords: Alzheimer's disease, amyloid, amyloid beta, animal study, antibody, IVIG
Introduction
Alzheimer's disease (AD) and Familial Danish dementia (FDD) are degenerative neurological diseases characterized by the deposition of pathological amyloids. Amyloid deposits and inclusions are characterized by the accumulation of normally soluble proteins in insoluble fibrillar forms. They adopt a characteristic amyloid conformation, which is highly enriched for β-pleated sheets and exhibits green birefringence under polarized light when stained with Congo red. Soluble preamyloid structures have also been identified, which include both oligomers with relatively little β-pleated sheet structure and protofibrils (PFs) that have a high degree of β-pleated sheet structure. In AD, Aβ oligomers may be more toxic than amyloid fibrils (Lambert et al., 1998; Walsh et al., 2002).
AD and FDD are examples of >30 human amyloid diseases that range from common disorders such as AD to rare autosomal disorders such as FDD (Chiti and Dobson, 2006). These diseases produce amyloid proteins with entirely different sequences that share conformational epitopes resulting from their β-pleated sheet structure (Kayed et al., 2003). Monoclonal antibodies (mAbs) have been isolated that recognize these pan-amyloid conformers and have anti-amyloid activities (Hrncic et al., 2000; O'Nuallain and Wetzel, 2002; O'Nuallain et al., 2011; Zhang et al., 2011; Zhao et al., 2014). Human intravenous Ig's (IVIGs) derived from pools of plasma from healthy donors contain IgGs that bind these epitopes (Du et al., 2001; Du et al., 2003; O'Nuallain et al., 2006). We reported that Aβ-reactive polyclonal IgGs in IVIGs cross-reacted with different types of amyloid fibrils and oligomers, including Ig light chain amyloid (AL) (O'Nuallain et al., 2006; O'Nuallain et al., 2008). These antibodies impaired Aβ and AL fibrillogenesis in vitro and dissociated AL amyloid in vivo. Experiments testing IVIG in transgenic AD mouse models suggested that short-term or long-term treatments can benefit cognitive function (Mengel et al., 2013; St-Amour et al., 2014). Results of clinical testing of IVIG in mild to moderate AD patients have been mixed, but a randomized trial suggested cognitive benefits for moderate AD patients and APOE-ε4 carriers (Relkin, 2014).
These data support the hypothesis that conformation-specific human IgGs exist and may have useful anti-amyloid properties. However, it is not clear whether the conformational amyloid binding and anti-amyloid capabilities observed with polyclonal IVIGs can be replicated by individual mAbs or if these may contribute to the potential utility of IVIG in AD (Relkin et al., 2009; Magga et al., 2010; Sudduth et al., 2013; Gu et al., 2014).
Here, we report on the cloning, characterization, and functional properties of an anti-amyloid IgG isolated from memory B cells of a healthy subject using a hybridoma method. This IgG mAb, 3H3, recognized diverse amyloids, inhibited elongation of Aβ and LC amyloids, and reduced the disruption of synaptic plasticity in the rat hippocampus by AD brain extract. Expression of a single-chain variable fragment (scFv) derived from 3H3, attenuated Aβ amyloid deposition in the brains of TgCRND8-transgenic mice and cerebral amyloid angiopathy (CAA) from ADan deposition in a mouse model of FDD. This study supports further investigation of the clinical utility of naturally occurring human mAbs that recognize conformational amyloid epitopes.
Materials and Methods
Proteins and polypeptides.
Human Aβ40 and S26C Aβ40, a mutant Aβ40 containing a cysteine in place of serine 26, which enables the formation of a disulfide cross-linked dimer, [S26C Aβ40]2 (O'Nuallain et al., 2010), were purchased from Quality Controlled Biochemicals. Wild-type (WT) Aβ40 monomers, monomeric S26C Aβ40, [S26C Aβ40]2, [S26CAβ]2 PFs, and Aβ40 fibrils were generated as below and were used immediately or frozen at −80°C. WT and S26C peptide concentrations were determined by absorbance at 275 nm using the molar extinction coefficient for tyrosine at 275 nm (e275 = 1400 M−1 cm−1).
Before use, the lyophilized Aβ40 was disaggregated by sequential exposure to trifluoroacetic acid and hexafluoroisopropanol (Thermo Fisher) followed by the addition of 2 mm NaOH and 2× PBS (1× final) and ultracentrifuged to give a final peptide concentration of ∼0.2 mg/ml (O'Nuallain et al., 2006). [S26C Aβ40]2 was prepared by solubilizing the S26C Aβ40 peptide to ∼0.2 mg/ml in MilliQ water (Millipore), diluted 1:1 with 20 mm ammonium bicarbonate, pH 8.2, to generate an ∼20 μm (with respect to monomer) peptide solution, and bubbled with oxygen for ∼5–10 min. The peptide was incubated at room temperature for ∼3 d and each day bubbled with oxygen.
WT and S26C Aβ40 monomers and [S26C Aβ40]2 were isolated from different sized Aβ40 assemblies using size exclusion chromatography. Peptides were incubated in 50 mm Tris-HCl containing 6 m guanidine HCl, pH 8.0, to dissociate preexisting aggregates and then characterized on a HiLoad 16/60 Superdex 75 column (GE Healthcare Bio-Sciences) eluted at 0.8 ml/min in 25 mm ammonium acetate, pH 8.5 (O'Nuallain et al., 2010). Fractions that contained Aβ40 monomers or [S26C Aβ40]2 were analyzed by SDS-PAGE using 16% polyacrylamide Tris-tricine gels and silver staining (Shevchenko et al., 1996).
Meta-stable thioflavin T (ThT)-positive PFs were generated by diluting freshly isolated [S26C Aβ40]2 to ∼0.1 mg/ml in 20 mm sodium phosphate, pH 7.4, and incubating the reaction mixture for ∼3 d at 37°C (O'Nuallain et al., 2010). The reaction was monitored by ThT fluorescence and was judged complete when the fluorescent signal reached a maximum plateau value (LeVine, 1999). Formation of PFs was confirmed by electron microscopy and by the presence of ThT-positive aggregates in reaction supernatants after centrifugation at 16,000 × g for 20 min.
JTO is a recombinant amyloidogenic λ6 LC variable region cloned from a multiple myeloma patient (Wall et al., 1999). JTO was produced in an E. coli expression system and purified as described previously (Wall et al., 1999). The soluble LC was sterile-filtered using a 0.22 μm polyvinylidene fluoride 25 mm Millex-GV syringe-driven filter unit (Millipore) and shown to be >90% pure, consisting of monomers and dimers using Sephadex G25 (GE Healthcare) gel filtration and SDS-PAGE. The protein concentration was determined by the MicroBCA assay (Thermo Fisher).
WT Aβ40 and JTO fibrils were generated by incubating 0.2 mg/ml concentrations of the amyloidogenic proteins in PBS containing 0.02% sodium azide, pH 7.4, at 37°C for 14 d [23]. Fibrillogenesis was judged complete when ThT fluorescence reached maximum plateau values. The reaction products were harvested by centrifugation at 20,200 × g for 30 min at room temperature and fibril morphology confirmed by negative contrast electron microscopy.
Amyloid fibrils derived from AL patient tissues and AD patient brain plaque cores have been described previously (O'Nuallain et al., 2007; O'Nuallain et al., 2008).
Cloning and initial characterization of 3H3 mAb.
The 3H3 mAb was cloned following previously described methods (Adekar et al., 2008b). Work with human peripheral blood cells was performed under a protocol approved by the Main Line Hospitals Institutional Review Board and consistent with the principles set out in the WMA Declaration of Helsinki and the National Institutes of Health Belmont Report. After receipt of informed consent, a blood sample from a healthy adult male was collected in heparin containing tubes. Peripheral blood mononuclear cells (PBMCs) were isolated by gradient density centrifugation with FicollPaque Plus (GE Healthcare). PBMCs were stored frozen in 90% heat-inactivated fetal calf serum (Life Technologies) and 10% DMSO (Sigma-Aldrich) under liquid nitrogen. Before cell fusion, CD27+ cells were isolated with anti-CD27 magnetic beads (Miltenyi Biotec) according to the manufacturer's instructions and cultured for 8 d on a monolayer of tCD40L cells (Urashima et al., 1995) (courtesy of Gordon Freeman, Dana Farber/Partners Cancer Care) in IMDM supplemented with 10% human AB serum, IL-4, IL-10, transferrin, gentamycin, insulin, and cyclosporine. On day 8, cultured CD27+ cells were electrofused to the B5-6T heteromyeloma cell line (Adekar et al., 2008a), plated in 96-well plates, and selected in HAT medium (Sigma-Aldrich).
Hybridoma supernatants were screened by ELISAs with Aβ40 or JTO fibrils bound to 96-well high binding (Corning) plates at 4°C overnight with 100 μl/well at 5 μg/ml in PBS. Specific IgG was detected with the HRP-conjugated anti-human IgG monoclonal antibody (9040-05) (Southern Biotechnology). Hybridoma supernatants that were positive in ELISA were selected for subcloning by limiting dilution. After 3 rounds of subcloning, stable hybridomas were adapted to serum-free medium (IS MAB-CD; Irvine Scientific), incubated for 5 d in a 500 ml roller bottle. Filtered supernatants were purified over protein G-Sepharose (GE Healthcare). Purity was assessed by SDS-PAGE (Life Technologies) and shown to be >90% pure. Protein concentrations were determined using the NanoDrop spectrophotometer (NanoDrop Technologies).
IgG heavy-chain (HC) and LC subtypes were assessed by capture of the antibodies with a polyclonal rabbit anti-human whole IgG (6145-01; Southern Biotech) and identification of reactivity with the HRP-conjugated mouse anti-human IgG3 monoclonal antibody (053620; Life Technologies) and the λ LC-specific HRP-conjugated goat anti-human polyclonal antibody (2070-05; Southern Biotech), respectively. For all ELISAs, o-phenylenediamine was used as the colorimetric substrate; optical density at 490 nm was measured using a BioTek ELISA reader.
Variable domain cDNA sequences of the 3H3 mAb were amplified with consensus primer sets specific for human Ig heavy chains (Campbell et al., 1992) and for λ light chains (Coronella et al., 2000), as previously described (Adekar et al., 2008b). DNA fragments were sequenced by the Kimmel Cancer Center Nucleic Acid Facility, Thomas Jefferson University (Philadelphia, Pennsylvania) and analyzed using the V-Quest program (Lefranc et al., 2005).
Direct and competitive assays of antibody binding to amyloids.
Antibody reactivity with wild-type Aβ40 fibrils, S26C Aβ40 monomers, [S26C Aβ40]2, monomeric Aβ40, bovine elastin fibrils (Sigma-Aldrich), JTO, and patient-derived Aβ fibrils and LC amyloids was determined using a europium (Eu3+)-based fluoroimmunoassay (EuLISA) (Adekar et al., 2010). The EuLISA was performed with purified 3H3 in assay buffer (1% BSA in PBSA containing 0.05% Tween 20) in activated high-binding Costar microtiter plate wells coated with 400 ng of target protein and blocked with 1% BSA (Sigma-Aldrich) in PBS. A biotinylated goat anti-human IgG (γ-chain specific; Sigma-Aldrich) served as the secondary antibody. After the addition of a Eu3+ streptavidin conjugate, followed by the releasing enhancement solution, Eu3+ time-resolved fluorescence was measured using a Victor2 1420 Multilabel Counter (PerkinElmer). The amount (fM) of lanthanide released was calculated from a standard curve using known concentrations of Eu3+. All measurements were done in triplicate (error bars in the figures represent SD) and antibody half-maximal effective concentration (EC50) values were determined from the sigmoidally fit binding curves (SigmaPlot 2000 ver. 8; Systat Software).
For the competition binding studies, we used the Eu3+ time-resolved fluoroimmunoassay (Diamandis, 1988) incorporating Eu3+-streptavidin and time-resolved fluorometry (DELFIA system; PerkinElmer) as described previously (O'Nuallain et al., 2006). Ninety-six-well plates were coated with 400 ng of soluble or fibrillar protein or peptide and blocked with 1% BSA in PBSA. The concentration of the antibody remained constant in the presence up to a 100- to 50-fold molar excess of soluble protein or peptide or 0.5–0.1 mg/ml fibrillar competitors. Biotinylated goat anti-human IgG (Sigma-Aldrich) served as the secondary antibody. Time-resolved fluorescence was measured as described above. Half-maximal inhibitory concentration (IC50) values were determined by fitting each titration dataset to a sigmoid equation using the SigmaPlot 2000 version 8 software package (Systat Software).
Human tissue samples and immunohistochemistry.
Human brain samples, including pathologically confirmed AD cases, were obtained at autopsy, fixed in formalin, embedded in paraffin blocks, and sliced into 4- to 6-μm-thick slices and placed on poly-l-lysine coated microscope slides. Antigen retrieval was with Vecta stain antigen retrieval solution (Vector Labs), boiled in a pressure cooker for 30 min, blocked for 30 min at 37°C, developed with the Vector ABC kit (Vector Labs) and 3,3′-diaminobenzidene, followed by counterstaining with hematoxylin. Staining of human kidney, heart, and spleen samples from patients with amyloid deposition was performed on 4- to 6-μm-thick sections cut from formalin-fixed, paraffin-embedded blocks of amyloid-laden tissue and placed on poly-L-lysine coated microscope slides for Congo red and antibody staining. Antigen retrieval was performed by 30 min boiling in Glyca solution (BioGenex). Tissues were exposed overnight at 4°C to 0.1 μg/ml biotinylated 3H3 mAb in HEPES-buffered saline. Slides were developed using a Vectastain ABC kit (Vector Labs) and 3,3′-diaminobenzidene and examined with a Leica DM500 microscope.
Inhibition of fibril formation.
To assay the formation of amyloid fibrils in real time, we used ThT fluorescence to measure amyloid fibril formation in solution (Ban et al., 2003). Wells of an ultra-low-binding 96-well plate (Corning) were filled with 15 μm soluble Aβ40 or 50 μm JTO monomer, 30 μm ThT, and serial dilutions of the test antibodies (1.0 μm-10 nm) in PBS containing 10 μm ThT, pH 7.4 (Adekar et al., 2008b). The plates were incubated at 37°C and fluorescence intensity was measured hourly for 5 h with the FL600 microplate reader.
Measurement of 3H3 binding kinetics.
Off-rate screening and affinity determination were performed on an Octet RED384 instrument (Pall ForteBio). All measurements were performed at 30°C; 96-well microplates were agitated at 1000 rpm. Various 3H3 or pan-Aβ antibody dilutions in a range of 10 nm to 1 mm were prepared in a new 384-well microtiter plate for secondary screening. Monomeric Aβ40 or Aβ42 were biotinylated using EZ-Link sulfo-NHS-LC-biotin (Thermo Fisher) according to the manufacturer's protocol and immobilized at a concentration of 50 μg/ml on streptavidin sensors for 10 minutes. For off-rate screening, sensors were blocked with 1% bovine serum albumin (Sigma-Aldrich) for 5 min, transferred to the wells containing the antibody dilution for the association step (5 min), and transferred back to the blocking well for the measurement of the dissociation step (5 min). Dissociation rate constants (koff) for each antibody concentration were calculated applying a 1:1 interaction model (fitting local, full) using the ForteBio data analysis software 7.0.1.5. Curves that could not be reliably fitted with the software (mostly full R2 < 0.96), presumably due to heterogeneous binding, were excluded from the analysis.
In vivo electrophysiology measurements of long-term potentiation.
Experiments were performed on groups of five to seven urethane-anesthetized adult male Lister hooded rats, and were licensed by the Department of Health and Children, Ireland. Single pathway recordings of field EPSPs from the stratum radiatum in the CA1 area of the hippocampus were evoked by stimulation of the ipsilateral Schaffer collateral-commissural pathway (Klyubin et al., 2004). Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity adjusted to trigger an EPSP amplitude of 50% of maximum. The high-frequency stimulation (HFS) protocol for inducing long-term potentiation (LTP) consisted of 10 trains of 20 stimuli, interstimulus interval of 5 ms (200 Hz), intertrain interval of 2 s. The stimulation intensity during the HFS remained unchanged. To inject samples, a cannula was implanted in the lateral cerebral ventricle (coordinates 1 mm lateral to the midline and 4 mm below the surface of the dura) just before electrode implantation. The preparation of the soluble AD brain extract and AD brain extract immunodepleted of Aβ used in the present experiments has been described previously (Hu et al., 2014) and was obtained and used in accordance with local institutional review board guidelines. For the intracerebral coinjection paradigm, either 3H3 or the control antibody was coincubated with the human AD brain extract for 10 min at room temperature before the mixture was injected.
LTP is expressed as the mean ± SEM percent baseline field EPSP amplitude recorded over a 45 min baseline period. Statistical analysis (GraphPad Prism version 6 for PC) comparing LTP values at 3 h post-HFS was performed using one-way ANOVA followed by Holm–Sidak's multiple-comparisons post hoc tests.
Construction of a 3H3 scFv and expression in adeno-associated virus (AAV).
3H3 scFv cDNA was synthesized at Blue Heron Biotechnology based on mAb HC and LC variable domain sequences in VH-linker-VL orientation and separated by a Gly4Ser3 linker. The scFv was cloned into the pSecTag plasmid under control of the cytomegalovirus enhancer and chicken β-actin (CBA) promoter with an Ig κ secretion leader at the N terminus and c-Myc and 6xHIS tags at the C terminus, a Woodchuck hepatitis virus posttranscriptional regulatory element, and the bovine growth hormone polyA sequence (Levites et al., 2006a). Nonspecific scFv (scFv ns) was obtained from a phage library (Medical Research Council) and showed no affinity to Aβ.
rAAV2/1 was prepared by standard methods. Briefly, pAAVs expressing the scFv were generated by transfection of the vector plasmid with helper plasmids for serotype 1 in HEK293T cells (Levites et al., 2006a). Forty-eight hours after transfection, cells were harvested and lysed in the presence of 0.5% sodium deoxycholate and 50 U/ml benzonase (Sigma-Aldrich) by freeze thawing; the virus was isolated using a discontinuous iodixanol gradient and affinity purified on a HiTrap HQ column (GE Healthcare). The genomic titer of each virus was determined by quantitative PCR.
HEK cells were transfected with pSecTag scFv or pAAV scFv. Conditioned media were collected 48 h later and cells were lysed in 1% Triton X-100 with protease inhibitors. Media and cells were then subjected to SDS-PAGE Western blotting analysis with chicken anti-c-myc (1:1000; Invitrogen) as primary antibody. When expression was confirmed, media from transfected cells was analyzed by direct ELISA method with Aβ fibrils (100 ng/well) as capture and anti-c-myc-HRP (1:2000) as detection.
Animal studies of rAAV2/1 expression.
Animal husbandry procedures were performed as approved by University of Florida Institutional Animal Care and Use Committee in an AAALAC-approved facility in accordance with National Institutes of Health guidelines. To generate TgCRND8 mice, male TgCRND8 mice containing double mutation in human APP gene (KM670/671NL and V717F; Chishti et al., 2001) were mated with female B6C3F1/Tac that were obtained from Taconic. Genotyping was performed by PCR as described previously (Levites et al., 2006b). Tg-ADan mice (TgFDD) and genotyping have been described previously (Vidal et al., 2009). All animals were housed three to five to a cage and maintained on ad libitum food and water with a 12 h light/dark cycle.
Injection of neonatal mice was performed based on Chakrabarty et al. (2013). Briefly, P0 pups were cryoanesthetized on ice for 5 min. 2 μl of rAAV2/1-scFv were injected intracerebroventricularly (ICV) into the both hemispheres using a 10 μl Hamilton syringe with a 30 g needle. The pups were then placed on a heating pad with their original nesting material for 3–5 min and returned to their mother for further recovery. For adult injections, mAb 3H3 (500 μg, 1 mg/ml) was administered though intraperitoneal injection to TgCRND8 mice.
Analysis of Aβ or ADan in the brain.
Biochemical and immunohistochemical analyses of Aβ in the brains of transgenic TgCRND8 and ADan mice were performed as described previously (Levites et al., 2006a; Levites et al., 2006c). The following mAbs against Aβ were used in the sandwich capture ELISA for brain Aβ40: monoclonal pan-Aβ antibody mAb5 for capture and Aβ40-specific mAb 40.1-HRP for detection (Levites et al., 2006b). For brain Aβ42, we used Aβ42-specific mAb42.2 for capture and mAb5-HRP for detection (Levites et al., 2006b). At the time of killing, the brains of mice were divided by midsagittal dissection and the first hemibrain was used for biochemical analysis. Each hemibrain was sequentially extracted in a 3-step procedure with RIPA followed by SDS followed by formic acid (FA) (Levites et al., 2006b).
Immunohistochemical staining was done using pan-Aβ antibody 33.1.1 (1:1500) (Kim et al., 2007), glial fibrillary acidic protein (GFAP; 1:500; Millipore), anti-c-Myc (1;500; Life Technologies). The second hemibrains were fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.6, and then dissected and stained for visualization of Aβ plaques (pan-Aβ mAb 33.1.1, 1:500; Todd Golde), ADan deposits (rabbit polyclonal antibody 1700, 1:500; Ruben Vidal), GFAP (1:500; Millipore), Iba-1 (1:1000; Wako Chemicals). For cerebrovascular amyloid detection, paraffin sections were stained with 1% Thio-S (Sigma-Aldrich).
Quantitation of Aβ plaque burden and intensity of astrogliosis-associated staining were calculated using the Positive Pixel Count program (Aperio Technologies). At least three sections per sample, 30 μm apart, were averaged by a blinded observer to calculate plaque burden. For Thio-S quantitation, three sections per sample were used to count the plaques or to calculate total positive pixel count using respective algorithms. All of the above analyses were performed in a blinded fashion. One-way ANOVA, followed by Dunnett's multiple-comparison test, were performed using the scientific statistic software Prism (version 3; GraphPad Software). p-values <0.05 were considered significant.
Results
Binding of 3H3 to oligomeric Aβ and LC fibrils
We performed a cell fusion using peripheral blood B cells obtained from a healthy male in his 30s. Hybridomas secreting human IgG were screened for binding to Aβ42 amyloid and an amyloid formed from a recombinant LC variable domain λ6 (JTO) (Wall et al., 1999). The IgG3λ mAb 3H3 was cloned and bound both antigens (data not shown). DNA sequence analysis of the variable domains revealed a HC complementarity determining region 3 (CDR3) length of 22 aa with 3.9% aa changes from germline and an LC CDR3 length of 10 aa with 4.0% aa changes. These features are typical of the antibodies produced by CD27+ memory B cells circulating in peripheral blood (Klein et al., 1998).
We tested the binding of the 3H3 to Aβ40 conformers using an europium ELISA with plate-immobilized wild-type Aβ40 fibrils, Aβ40 monomers, S26C Aβ40 dimers ([S26C Aβ40]2), and S26C Aβ40 monomers, generated by β-mercaptoethanol reduction of the dimeric peptide (Fig. 1a). S26C Aβ40 is a synthetic Aβ peptide (1–40) containing cysteine in place of serine 26, which can form a disulfide cross-linked dimer (O'Nuallain et al., 2010). The binding curves demonstrated that the 3H3 mAb has nanomolar reactivity with plate-immobilized Aβ conformers and ∼2-fold weaker binding to the monomeric peptide. Calculated EC50 values were as follows: WT fibrils, 6.0 ± 0.02 nm, WT monomers, 17 ± 0.05 nm, [S26C Aβ40]2, 6.0 ± 0.04 nm, and S26C Aβ40 monomers, 9.1 ± 0.06 nm. A control, nonbinding mAb (30B), which binds serotype B botulinum neurotoxin (Adekar et al., 2008b), showed essentially no reactivity with [S26C Aβ40]2 (EC50 > 300 nm).
Figure 1.

mAb 3H3 is pan-reactive against immobilized Aβ conformers but in solution only recognizes aggregated peptide. A, Antibody binding curves for 3H3 demonstrate the antibody's low nm reactivity with plate-immobilized Aβ conformers, albeit ∼2-fold weaker binding to the monomeric peptide. A control mAb, 30B, had essentially no reactivity with plate-immobilized Aβ. S26C Aβ40 monomers were generated by β-mercaptoethanol reduction of the dimeric peptide. Filled triangles, WT Aβ40 fibrils; open squares, WT Aβ40 monomers; open circles, S26C Aβ40 dimers ([S26C Aβ40]2) filled circles, S26C Aβ40 monomers; all were tested with the 3H3 mAb. Filled diamonds, [S26C Aβ40]2, tested for binding of the 30B control mAb. B, Representative competition ELISA curves are shown for solution-phase Aβ conformer's inhibition of mAb 3H3 binding to plate-immobilized recombinant [S26C Aβ40]2. Filled triangles, WT Aβ40 fibrils; open circles, [S26C Aβ40]2; filled diamonds, S26C Aβ40 PFs; open squares, WT Aβ40 monomers. C, Antibody binding curves for 3H3 demonstrate the antibody's similar low nm reactivity against plate-immobilized Aβ (filled circles) and recombinant JTO LC amyloid fibrils (open circles), and inability to recognize nonamyloid elastin fibrils (open triangles). The calculated EC50 values were Aβ40 fibrils: 15 ± 0.05 nm, LC fibrils: 47 ± 0.50 nm, and elastin fibrils: >300 nm. D, 3H3 binding curves show the antibody's nm reactivity with plate-immobilized patient-derived Aβ (filled circles), κ1 LC (open triangles) and λ3 LC fibrils (filled triangles), with EC50 values of 14 ± 0.05 nm, 14 ± 0.15 nm and 123 ± 10 nm, respectively.
Because Aβ40 conformations may be altered by binding to the ELISA plate (O'Nuallain et al., 2007), we compared 3H3 binding to soluble Aβ40 aggregates using a competition binding ELISA (Fig. 1b). We bound [S26C Aβ40]2 to the plate and added 3H3 mAb in the presence of each of the following competitors: Aβ40 fibrils, Aβ40 monomers, [S26C Aβ40]2, or S26C Aβ40 PFs. Binding of the 3H3 to the plate-immobilized [S26C Aβ40]2 was inhibited only by aggregated Aβ and not by Aβ monomers, with the strongest inhibition provided by Aβ40 fibrils and PFs. The calculated IC50 values were Aβ40 fibrils: 0.62 ± 0.03 μg/ml, [S26C Aβ40]2: 8.4 ± 3.6 μg/ml, S26C Aβ40 PFs: 3.9 ± 0.28 μg/ml, and Aβ40 monomers: >40 μg/ml. We assessed whether 3H3 bound to elastin fibrils, which do not adopt an amyloid configuration (Fig. 1c), comparing their binding to plate-immobilized Aβ40 and JTO fibrils. The calculated EC50 values were Aβ40 fibrils: 15 ± 0.05 nm, LC fibrils: 47 ± 0.50 nm, and elastin fibrils: >300 nm. These data indicate that 3H3 preferentially binds amyloid fibrils and aggregated Aβ forms. We measured the avidity of 3H3 antibody to biotinylated fibrillar Aβ40 and Aβ42, loaded onto streptavidin sensors using the ForteBio Octet system (Ylera et al., 2013). The KDs for 3H3 binding to Aβ40 fibrils (2.00 × 10−10 m) and Aβ42 fibrils (1.78 × 10−10 m) were calculated based on kon and koff rates.
Immunohistochemical assessment of 3H3 binding to diverse amyloids in diseased tissues
We used ELISA to test whether 3H3 could bind Aβ amyloid from AD patient brains and LC amyloid from AL patients. 3H3 bound to AD patient fibrils from congophilic neuritic plaque cores as well as to different LC species isolated from two AL patients, one a κ1 LC and the other a λ3 LC (Fig. 1d). Calculated EC50 values were AD plaques: 14 ± 0.05 nm, κ1 LC fibrils: 14 ± 0.15 nm, and λ3 LC fibrils: 123 ± 10 nm, which are comparable to those observed with the synthetic amyloids.
We assessed binding of 3H3 to pathologic amyloids in brain tissues from patients with neurologic amyloid diseases. 3H3 bound plaques and blood vessels in the AD brain (Fig. 2b,e), but not in normal brain (Fig. 2a,d). The 30B control mAb did not bind any structures in the AD brain (Fig. 2c,f). 3H3 also bound to amyloid deposited in brain tissue of patients with multiple systems atrophy, which is characterized by α-synuclein-positive glial cytoplasmic inclusions, AD with CAA, and progressive supranuclear palsy, with neurofibrillary tangles containing tau, whereas the pooled human IgG control did not bind to these tissues (Fig. 2m–o). Naturally occurring antibodies in serum that react with α-synuclein or Aβ plaques have been described in healthy subjects (Britschgi et al., 2009; Besong-Agbo et al., 2013). 3H3 also bound to Aβ plaques deposited in the brains of TgCRND8 mice, which express a mutant (K670N/M671L and V717F) human βAPP695 transgene (Janus et al., 2000; Fig. 2).
Figure 2.



3H3 binding to AD brain and AL kidney tissues. Normal (a, d) and AD brains (b, c, e, f) were stained with 3H3 (a, b, d, e) or the human nonbinding control IgG mAb, 30B (c, f). Plaques (P) and blood vessels (V) are shown with arrows. (400×). 3H3 was also tested for binding to kidney samples from a patient with κ1 LC deposition (g), λ8 LC deposition (h), and a normal subject (i). Pooled human serum IgG did not bind these tissues (j–l). (200×). (m–t) 3H3 (bottom row) was compared with pooled human IgG (top row) by immunohistochemistry for binding to cortical tissue from patients and-transgenic mice. MSA (m, q) Multiple Systems Atrophy α-synuclein positive glial cytoplasmic inclusions; CAA (n, r) AD with CAA; PSS (o, s) Progressive Supranuclear Palsy, tau-containing neurofibrillary tangles, CRND8 (p, t) mouse, the TgCNRD8-transgenic mouse model with Aβ plaques. The brown color indicates binding. Scale bar, 500 μm, insets 50 μm.
We tested binding of 3H3 to non-neurological amyloids. The 3H3 mAb bound to kidney samples from PSA patients, one with a κ1 LC (Fig. 2g) and the other a λ8 LC (Fig. 2h), whereas it did not bind to normal kidney (Fig. 2i) and pooled human IgG did not bind to any of these tissues (Fig. 2j–l). 3H3 bound cardiac tissue affected by transthyretin deposition, as well as pathological λ1 amyloid in kidney and λ3 amyloid in spleen (data not shown). These data suggest that the 3H3 mAb recognizes an amyloid-specific conformational epitope that is present in amyloids of different primary sequences in diseased tissue.
Inhibition of fibril formation
Antibody binding to amyloid epitopes can correlate with the ability to interfere with de novo growth (O'Nuallain et al., 2006; Adekar et al., 2010). Therefore, we tested the ability of the 3H3 mAb to inhibit elongation of fibrils formed from Aβ40 or JTO. We first mixed Aβ40 monomers with ThT, a molecule that fluoresces quantitatively when bound to amyloid fibrils, in the presence of serially diluted 3H3 or the 30B control mAb (Fig. 3a,b). At the higher mAb concentrations tested, 0.25–1.0 μm, the 3H3 inhibited Aβ40 fibril elongation, whereas the 30B mAb did not. At the lower concentrations, 0.06 and 0.13 μm, the time course for fibril formation was delayed, although there was an increase in the total amount of ThT fluorescence relative to the control. Similar results were seen with JTO fibrils (Fig. 3c,d), with inhibition of fibril growth at 0.5 and 1.0 μm and elongation delay at 0.25 μm, which also gave a greater final level of ThT fluorescence than the control. 3H3 concentrations of 0.06 and 0.13 μm did not alter the rate of ThT uptake by JTO. These findings indicate that the 3H3 mAb can inhibit Aβ40 and JTO fibril growth.
Figure 3.

Dose-dependent inhibition of de novo Aβ and LC amyloid fibril formation by subequimolar 3H3. Aβ and LC monomers were incubated in the presence of dilution series of the 3H3 and 30B mAbs. The progress curves for Aβ (A, B) and LC (C, D) amyloid fibril formation show that 3H3 dose-dependently inhibited the formation of ThT positive amyloidogenic aggregates, whereas the control 30B did not. The aggregation reactions were performed using 15 and 5 μm Aβ monomers and JTO λ LC, respectively, in PBS containing 10 μm ThT, pH 7.2.
Abrogation of the disruption of hippocampal synaptic plasticity by soluble Aβ in AD brain extract in vivo
Intracerebral injection of water-soluble Aβ oligomers potently inhibits LTP of synaptic transmission in the hippocampus, a neurophysiological correlate of hippocampus-dependent memory (Walsh et al., 2002; Shankar et al., 2008). Here, we assessed the ability of 3H3 to acutely abrogate the inhibition of LTP by AD-patient-derived soluble Aβ oligomers at CA3-to-CA1 synapses in the hippocampus of urethane-anesthetized rats (Cullen et al., 1997; Klyubin et al., 2005). Electrically evoked field EPSPs (EPSPs) were recorded for 1 h before the application of conditioning HFS. Whereas LTP decayed gradually in animals that received an intracerebroventricular injection of soluble Aβ-containing AD brain extract 15 min before HFS, an identical HFS protocol induced a stable, persistent increase in magnitude of CA1 EPSPs in animals that received an injection of the same AD patient brain soluble extract that had been immunodepleted of Aβ (p < 0.01%, 107 ± 4.3%, and 127.6 ± 4.5% of pre-HFS baseline, respectively, at 3 h post-HFS, n = 7 per group; Fig. 4a,c). The Aβ-containing AD brain extract no longer inhibited LTP (119.3 ± 3.0%, n = 7, p < 0.05 compared with AD group) when coadministered with a dose of the 3H3 mAb that did not affect control LTP (p > 0.05 compared with the immunodepleted-alone group, 123.2 ± 1.3%, n = 6; Fig. 4b,c). Coadministration of the same amount of the control 30B mAb did not affect the ability of the soluble Aβ in AD brain extract to inhibit LTP (106.2 ± 4.4%, n = 5, data not shown). These results suggest that 3H3 mitigates the disruptive effect of AD-patient-derived, soluble Aβ oligomers on hippocampal LTP.
Figure 4.
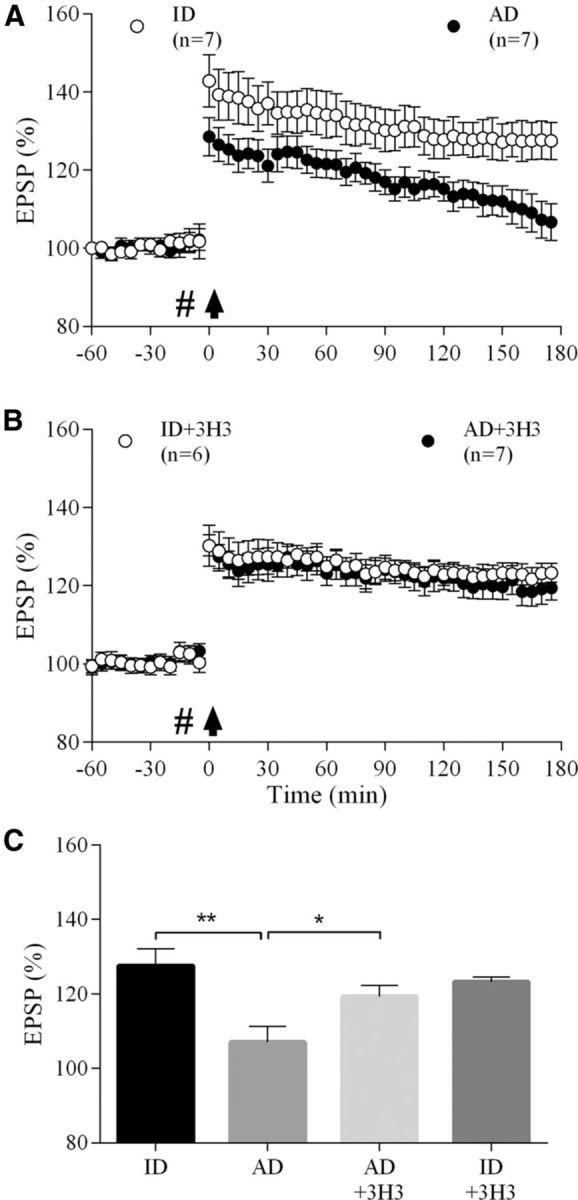
3H3 abrogates the disruption of hippocampal synaptic plasticity by soluble Aβ in AD brain extract. A, Synaptic field potentials (EPSPs) were recorded from the CA1 area of anesthetized rats. The acute intracerebroventricular injection (#) of AD brain TBS extract (AD, filled circles, n = 7) 15 min prior high-frequency stimulation (HFS) conditioning (arrow) strongly inhibited LTP compared with brain extract that had been effectively immunodepleted (ID) of all detectable Aβ, see (Hu et al., 2014) (ID, open circles, n = 7). B, Coinjection of 3H3 antibody (10 μg) prevented the inhibition of LTP by soluble Aβ-containing AD brain extract (AD+3H3, closed circles, n = 7). When coinjected with ID extract, 3H3 did not affect LTP (ID+3H3, open circles, n = 6). C, Summary LTP data at 3 h post-HFS for all experimental groups shown in (A, B). *p < 0.05 and **p < 0.01. Values are mean ± SEM.
Studies of in vivo expression of 3H3 scFv in the TgCRND8 and ADan mice
We converted the 3H3 into an scFv for in vivo expression using the AAV system. Neonatal injection of AAVs into the brains of mice evades the host immune response and enables persistent transgene expression. We cloned the variable regions of the 3H3 scFv into the mammalian expression vector pSecTag and the plasmid, which drives expression from the CBA promoter and provides C-terminal c-Myc and 6xHis tags. After transfection of HEK cells with pSecTag-3H3 and pAAV-3H3, analysis of scFv secreted in cell culture media was performed by ELISA in triplicate. Secreted scFv preferentially bound to Aβ fibrils relative to monomers in an ELISA: pSecTag-3H3 (OD450 fibrils: 0.354 ± 0.024, OD450 monomer: 0.056 ± 0.012), pAAV-3H3 (OD450 fibrils: 1.089 ± 0.067, OD450 monomer: 0.187 ± 0.037), pan-Ab scFv9 (OD450 fibrils: 0.542 ± 0.034, OD450 monomer: 0.983 ± 0.005; Levites et al., 2006a). This confirms that the scFv replicated the binding specificity of the intact IgG and suggests that the recognition of Aβ aggregates by 3H3 results from canonical Ig binding activity.
Four microliters of a pAAV-3H3 scFv solution (1013 genomes/ml) was delivered into the cerebral ventricles of newborn TgCRND8 pups. At 5 months, an age by which substantial amyloid pathology typically develops in these mice, we extracted brains and processed them for immunohistochemical and biochemical analysis. We evaluated scFv expression in the brains of the injection group using the anti-c-Myc antibody (Fig. 5a–c). All mice in this group had widespread brain expression of the scFv, particularly in the hippocampus and cortex. We assessed the effects of 3H3 scFv expression on Aβ plaque deposition. We stained brain sections from PBS-injected (Fig. 5d,f) or pAAV-3H3 scFv-injected mice (Fig. 5e,g) with either a pan-Aβ mAb (Fig. 5d,e) or an Aβ40-specific mAb (Fig. 5f,g). We did not observe clear differences in the number of plaques present. However, the plaque morphology in the pAAV-3H3 scFv-injected mice was more diffuse (Fig. 5f,g). In the injected mouse brains, the plaques were decorated by the 3H3 scFv, as visualized by anti-c-Myc staining around the plaques (Fig. 5h).
Figure 5.
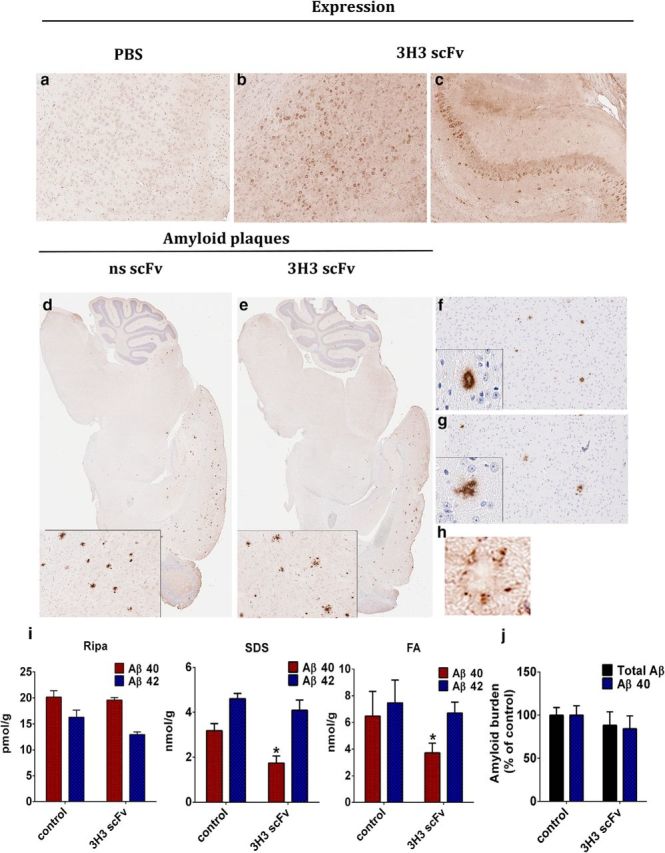
3H3 scFvs attenuates Aβ deposition in 5 month-old CRND8 mice. Newborn CRND8 mice were injected ICV with AAV 3H3 scFv (4 × 1010 genomes/mouse). Control mice received PBS. Five months later, the mice were killed and analyzed. One hemibrain was used for immunohistochemistry and the other for biochemical analysis. scFv expression in the brain was detected by staining for the c-Myc tag. The photomicrographs are representative sections of groups of 7–10 mice: (a) PBS (b, c) 3H3 scFv. Magnification 10×. Representative brain sections from PBS (d, f) or 3H3 scFv (e, g) injected mice were stained with pan-Aβ mAb (33.1.1) (d, e) or the Aβ40 specific mAb (13.1.1) (f, g). Magnification 2× and 5× (insets 5× and 20×). (h) 3H3 scFv expression surrounding amyloid plaques was visualized by staining for c-Myc tag. Magnification 20× (i) Aβ40 and Aβ42 levels in brain extracts solubilized with RIPA buffer, SDS, or formic acid (FA), analyzed by sandwich ELISA n = 5. *p < 0.05 vs control. (j) Aβ plaque burden analysis was performed on three nonconsecutive sections from each mouse in the group. n = 7–10.
We assessed the total amyloid burden by measuring amyloid Aβ levels in RIPA-soluble, SDS-soluble, and FA-soluble brain fractions by ELISA (Fig. 5i). Aβ40 levels were significantly lower in the SDS and FA extracts from the AAV-3H3 scFv-injected mice, compared with the control mice, but a statistically significant change was not observed in Aβ42 levels in the SDS and FA extracts or in the Aβ40 or Aβ42 levels in the RIPA-extracted fractions. Quantification of the total Aβ40 plaque burden on brain sections of control and scFv-expressing mice also did not show a significant difference (Fig. 5j).
We extended these results by delivering the pAAV-3H3 scFv in ADan mice, in which the Danish amyloid subunit comprising the last 34 aa of human BRI2 is expressed under the control of the mouse prion protein promoter. ADan mice show significant intracellular and extracellular deposition of oligomeric forms of ADan, including vascular amyloid deposition, parenchymal ADan deposition, and amyloid associated gliosis (Vidal et al., 2009). We injected newborn ADan mice with 4 μl of pAAV-3H3 scFv (1013 genomes/ml). Control mice were injected with PBS. Expression of scFv was confirmed by anti-c-Myc brain staining 3 weeks after injection in 3 mice and remaining mice were aged to 12 months. At that time, brains were harvested and effects of scFv on development of pathology were evaluated. Control mice developed significant Thio-S-positive CAA in the meninges, thalamus, and cerebellum, whereas 3H3 scFv-expressing mice had less CAA pathology in these tissues (Fig. 6a). Quantification of reduction in Thio-S-positive total positive signal as well as the number of Thio-S-labeled blood vessels revealed statistically significant reductions in the 3H3 scFv-expressing mouse brains (Fig. 6b).
Figure 6.

3H3 scFv expression prevents development of pathology in ADan-transgenic mice. (a). Newborn ADan-transgenic mice were injected into cerebral ventricles with pAAV - 3H3 scFv (4 × 1010 genomes/mouse) and killed at 12 months of age. CAA pathology in blood vessels was visualized with Thio-S in the hippocampus (top panels), thalamus (middle panels), and cerebellum (bottom) of mice injected with PBS or pAAV-3H3 scFv. (b) Quantification of fluorescence positive signal (left) and number of Thio-S positive blood vessels (right), showing decreases in the 3H3-scFv group compared with PBS-injected mice (n = 5–7/group). (*p < 0.05, t test). Data are shown as the mean ± SEM (c) An anti-ADan polyclonal antibody 1700 was used to detect ADan peptide accumulation in the hippocampus and cerebellar blood vessels of PBS treated and 3H3-scFv-expressing mice. (d). GFAP was analyzed by immunohistochemistry in brains of 3H3-scFv expressing mice, compared with age-matched controls, in hippocampus (top), thalamus (middle) and cerebellum (bottom).
Mutant ADan amyloid deposits in ADan mice can be visualized by binding with antibody 1700 (rabbit polyclonal; Vidal et al., 2009). In the control mice, typical staining was observed in the hippocampus as well as in the blood vessels in the cerebellum, whereas mice expressing 3H3 scFv had virtually no amyloid-like deposits in the hippocampus and less ADan-positive CAA in the cerebellum (Fig. 6c). Importantly, 3H3 scFv expression correlated with reduced astrocytosis in the brains of the mice, as demonstrated by decreased GFAP staining in the hippocampus, midbrain, and cerebellum (Fig. 6d).
Discussion
The 3H3 mAb is a human monoclonal antibody, cloned from a healthy subject, that binds multiple amyloids, regardless of the primary sequence of the protein subunits. The 3H3 demonstrated anti-amyloid activity in vitro and in three in vivo model systems. Therefore, the 3H3 recapitulates essential features of amyloid-binding IgGs previously identified in polyclonal IVIG (O'Nuallain et al., 2006; Britschgi et al., 2009; Dodel et al., 2011). Because 3H3 was identified by its recognition of JTO Ig LC and Aβ42 amyloids, it represents just one of the many types of anti-amyloid antibodies that may exist in IVIG. Nonetheless, the 3H3 demonstrates that potentially therapeutic features of IVIG can be embodied in a human mAb that may be adaptable for therapeutic use.
The 3H3 originated from a CD27+ peripheral blood B cell and has somatically mutated variable domains, indicating that it had undergone T-cell-dependent antibody affinity maturation and is therefore the product of the adaptive immune response. The amyloid-specific binding of the 3H3 scFv indicates that recognition of the conformational amyloid epitope occurs through the canonical CDR-binding domains, not Fc-mediated interactions, as has been reported for at least one amyloid protein (Huang et al., 1993). The observation that IgGs with similar features can be found in children, in addition to IVIG, suggests that IgG-binding conformational amyloid epitopes are the products of the normal immune response to amyloids (Baril et al., 2004).
In the live rat model of hippocampal LTP, 3H3 abrogated the synaptotoxicity of soluble Aβ oligomers contained in an AD-brain extract. Soluble Aβ species extracted from human brains are pathophysiologically relevant material for modeling key features of AD (Shankar et al., 2008). The ability of the 3H3 mAb to inhibit their toxicity on a living brain demonstrates one mechanism whereby naturally occurring, conformation-specific antibodies in IVIG may exert a protective function in AD. Murine conformation-specific mAbs have also shown activity in the rat model of Aβ synaptotoxicity (Klyubin et al., 2008; O'Nuallain et al., 2011).
In the TgCRND8-transgenic mouse model of CNS Aβ deposition, expression of the 3H3 scFv significantly reduced the levels of SDS- and FA-extractable Aβ40, but had no statistically significant change in Aβ42 levels. This selective effect on Aβ40 is consistent with selective targeting of Aβ amyloid because Aβ40 is more selectively deposited in cored amyloid plaques, whereas Aβ42 deposits in both cored amyloid plaques and more diffuse immunoreactive deposits that are not congophilic. Indeed, a change in plaque morphology was noted in the mice expressing the 3H3 scFV because the plaques appeared less compact and more diffuse. The 3H3 scFV was observed to surround Aβ plaques in the brain, suggesting that it may inhibit amyloid formation by plaque binding. In brains of AD patients, colocalization of IgGs with Aβ neuritic plaques has been observed and patients with high IgG plaque labeling had significantly reduced Aβ plaque burdens (Kellner et al., 2009).
In the FDD model of CAA deposition in ADan mice, 3H3 scFv expression reduced amyloid deposition and CAA while reducing inflammation-associated expression of GFAP. Others have shown that AAV delivery of an anti-amyloid scFv preferentially targeting Aβ42 oligomers can reduce Aβ42 in the hippocampus in the TgAβPPswe/PS1dE9 mouse model of AD, although it exacerbates CAA (Kou et al., 2011). These different results may be attributable to particular binding properties of the scFvs and the use of different models. It is unlikely that the 3H3 scFv could induce glial clearance of Aβ because it lacks effector functions. Additional efforts to add effector functions, increase affinity, or increase the concentrations of the scFv in vivo may enhance its anti-amyloid activity. In any case, our data with the 3H3 scFv indicates that pan-amyloid scFv antibodies can attenuate deposition of different amyloids in vivo.
The finding that 3H3 bound to plate-immobilized monomeric Aβ in a direct ELISA may seem paradoxical given the strong evidence for its binding to a pan-amyloid conformational epitope; for example, binding to diverse amyloid oligomers and preferential binding to oligomeric Aβ in competition binding studies. One explanation is that, when Aβ binds the 3H3 antibody, the bound Aβ has an amyloid-like conformation that was induced by plate immobilization (O'Nuallain et al., 2007; Szabo et al., 2010). The differential effects of 3H3 at different concentrations on Aβ and JTO amyloid formations provide indirect support for this assertion. At low concentrations, 3H3 delayed fibril formation but increased the total amount of amyloid formed, whereas at higher concentrations, amyloid formation was inhibited. It is possible that such effects could be explained by 3H3 converting monomeric Aβ and JTO into more uniform amyloid seeds, which could increase the lag time for aggregation but, once seeded, enable more uniform and efficient assembly. These 3H3-induced seeds could be structurally different from the uninhibited amyloid rections, resulting in slower reactions and different ThT reaction product yields. At higher 3H3 concentrations, sequestration of JTO and Aβ would lower concentrations of free monomer and inhibit overall amyloid formation.
The potent in vitro and in vivo anti-amyloid activities of 3H3 demonstrate that amyloids can be therapeutically targeted through epitopes that are independent of the primary sequence of the amyloid polypeptide. This offers the potential to treat amyloid diseases with mAbs that specifically bind toxic oligomeric forms and not to nontoxic, nonaggregated forms. A mAb with specificity for multimeric Aβ, similar to the 3H3 in competition assays, reduced amyloid deposition and Aβ protofibril levels in a transgenic mouse model expressing both the Arctic and Swedish Aβ mutations (tg-ArcSwe) (Englund et al., 2007; Lord et al., 2009).
In this study, 3H3 demonstrated activity in animal models intended to reproduce essential features of AD and FDD. Binding to patient-derived Ig LC amyloids in vitro and in pathological tissue sections further suggests that 3H3 may be helpful for treatment of other amyloid diseases such as primary amyloidosis, in which the LC amyloids are deposited in extracellular tissues that are accessible to the blood circulation. Proof-of-concept of the ability of a mAb specific for an amyloid epitope to clear LC amyloids in vivo has been established using a mouse model in which patient-derived amyloids were introduced by subcutaneous injection (Hrncic et al., 2000). mAb opsonization and macrophage-dependent clearance of tissue-deposited amyloids has been demonstrated in a mouse model of amyloidosis induced by overexpression of serum amyloid P component (Bodin et al., 2010).
In conclusion, we have isolated an affinity matured human mAb that binds a pan-amyloid epitope and has binding specificity for aggregated amyloid forms. The mAb has anti-amyloid activities in vitro and in vivo, suggesting that naturally occurring anti-amyloid antibodies such as 3H3 may be protective and have potential as anti-amyloid therapeutics.
Footnotes
This work was supported by Sharpe-Strumia Research Foundation (S.K.D.), the Edward N. and Della Thome Memorial Foundation (T.G. and S.K.D.), NIH Grant AG18454 (T.G.), and Science Foundation Ireland (M.J.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Gordon Freeman (Dana Farber/Partners Cancer Care) for the tCD40L cell line, Brenda D. Moore for preparing and characterizing various generic amyloids and fibrillar Aβ, Chandana Devi Kattala for laboratory assistance, and Gwen Gilliard with help with the brain IHC. Human tissues used in Figure 2, m–t, were obtained from the University of Florida Neuromedicine Human Brain Tissue Bank.
S.K.D. has stock ownership in a company (Immunome, Inc.) that has licensed the human monoclonal antibody cloning method and the 3H3 antibody described here. He and S.P.A. are also inventors on patents related to the hybridoma method. The remaining authors declare no competing financial interests.
References
- Adekar SP, Jones RM, Elias MD, Al-Saleem FH, Root MJ, Simpson LL, Dessain SK. A human monoclonal antibody that binds serotype A botulinum neurotoxin. Hybridoma. 2008a;27:11–17. doi: 10.1089/hyb.2007.0536. [DOI] [PubMed] [Google Scholar]
- Adekar SP, Jones RM, Elias MD, Al-Saleem FH, Root MJ, Simpson LL, Dessain SK. Hybridoma populations enriched for affinity-matured human IgGs yield high-affinity antibodies specific for botulinum neurotoxins. J Immunol Methods. 2008b;333:156–166. doi: 10.1016/j.jim.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Adekar SP, Klyubin I, Macy S, Rowan MJ, Solomon A, Dessain SK, O'Nuallain B. Inherent anti-amyloidogenic activity of human immunoglobulin gamma heavy chains. J Biol Chem. 2010;285:1066–1074. doi: 10.1074/jbc.M109.044321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem. 2003;278:16462–16465. doi: 10.1074/jbc.C300049200. [DOI] [PubMed] [Google Scholar]
- Baril L, Nicolas L, Croisile B, Crozier P, Hessler C, Sassolas A, McCormick JB, Trannoy E. Immune response to Abeta-peptides in peripheral blood from patients with Alzheimer's disease and control subjects. Neurosci Lett. 2004;355:226–230. doi: 10.1016/j.neulet.2003.10.071. [DOI] [PubMed] [Google Scholar]
- Besong-Agbo D, Wolf E, Jessen F, Oechsner M, Hametner E, Poewe W, Reindl M, Oertel WH, Noelker C, Bacher M, Dodel R. Naturally occurring alpha-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology. 2013;80:169–175. doi: 10.1212/WNL.0b013e31827b90d1. [DOI] [PubMed] [Google Scholar]
- Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ, Minogue S, Dhillon AP, Taylor GW, Bradwell AR, Petrie A, Gillmore JD, Bellotti V, Botto M, Hawkins PN, Pepys MB. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–97. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi M, Olin CE, Johns HT, Takeda-Uchimura Y, LeMieux MC, Rufibach K, Rajadas J, Zhang H, Tomooka B, Robinson WH, Clark CM, Fagan AM, Galasko DR, Holtzman DM, Jutel M, Kaye JA, Lemere CA, Leszek J, Li G, Peskind ER, et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:12145–12150. doi: 10.1073/pnas.0904866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Zelenetz AD, Levy S, Levy R. Use of family specific leader region primers for PCR amplification of the human heavy chain variable region gene repertoire. Mol Immunol. 1992;29:193–203. doi: 10.1016/0161-5890(92)90100-C. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C, Crosby K, Jansen K, Borchelt DR, Kim JY, Jankowsky JL, Golde TE, Levites Y. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Coronella JA, Telleman P, Truong TD, Ylera F, Junghans RP. Amplification of IgG VH and VL (Fab) from single human plasma cells and B cells. Nucleic Acids Res. 2000;28:E85. doi: 10.1093/nar/28.20.e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Diamandis EP. Immunoassays with time-resolved fluorescence spectroscopy: principles and applications. Clin Biochem. 1988;21:139–150. doi: 10.1016/0009-9120(88)90001-x. [DOI] [PubMed] [Google Scholar]
- Dodel R, Balakrishnan K, Keyvani K, Deuster O, Neff F, Andrei-Selmer LC, Röskam S, Stüer C, Al-Abed Y, Noelker C, Balzer-Geldsetzer M, Oertel W, Du Y, Bacher M. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer's disease. J Neurosci. 2011;31:5847–5854. doi: 10.1523/JNEUROSCI.4401-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B, Bales K, Gao F, Moeller HJ, Oertel W, Farlow M, Paul S. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57:801–805. doi: 10.1212/WNL.57.5.801. [DOI] [PubMed] [Google Scholar]
- Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, Lannfelt L, Pettersson FE. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103:334–345. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- Gu H, Zhong Z, Jiang W, Du E, Dodel R, Liu J, Farlow MR, Zheng W, Du Y. The role of choroid plexus in IVIG-induced beta-amyloid clearance. Neuroscience. 2014;270:168–176. doi: 10.1016/j.neuroscience.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrncic R, Wall J, Wolfenbarger DA, Murphy CL, Schell M, Weiss DT, Solomon A. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol. 2000;157:1239–1246. doi: 10.1016/S0002-9440(10)64639-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Martin M, Hu D, Roses AD, Goldgaber D, Strittmatter WJ. Binding of IgG to amyloid beta A4 peptide via the heavy-chain hinge region with preservation of antigen binding. J Neuroimmunol. 1993;48:199–203. doi: 10.1016/0165-5728(93)90192-2. [DOI] [PubMed] [Google Scholar]
- Hu NW, Nicoll AJ, Zhang D, Mably AJ, O'Malley T, Purro SA, Terry C, Collinge J, Walsh DM, Rowan MJ. mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat Commun. 2014;5:3374. doi: 10.1038/ncomms4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, Glatzel M. Autoantibodies against beta-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol. 2009;65:24–31. doi: 10.1002/ana.21475. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679–1689. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, Rowan MJ. Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. Eur J Neurosci. 2004;19:2839–2846. doi: 10.1111/j.1460-9568.2004.03389.x. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou J, Kim H, Pattanayak A, Song M, Lim JE, Taguchi H, Paul S, Cirrito JR, Ponnazhagan S, Fukuchi K. Anti-amyloid-beta single-chain antibody brain delivery via AAV reduces amyloid load but may increase cerebral hemorrhages in an Alzheimer's disease mouse model. J Alzheimers Dis. 2011;27:23–38. doi: 10.3233/JAD-2011-110230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Giudicelli V, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clément O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005;33:D593–597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine H., 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/S0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, Golde TE. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006a;26:11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006b;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, Kim J, McGowan E, Reed DK, Rosenberry TL, Das P, Golde TE. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer's disease mouse models. FASEB J. 2006c;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Lord A, Gumucio A, Englund H, Sehlin D, Sundquist VS, Söderberg L, Möller C, Gellerfors P, Lannfelt L, Pettersson FE, Nilsson LN. An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;36:425–434. doi: 10.1016/j.nbd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T, Härtig W, Grosche J, Goldsteins G, Tanila H, Koistinaho J, Koistinaho M. Human intravenous immunoglobulin provides protection against Abeta toxicity by multiple mechanisms in a mouse model of Alzheimer's disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel D, Röskam S, Neff F, Balakrishnan K, Deuster O, Gold M, Oertel WH, Bacher M, Bach JP, Dodel R. Naturally occurring autoantibodies interfere with beta-amyloid metabolism and improve cognition in a transgenic mouse model of Alzheimer's disease 24 h after single treatment. Translational Psychiatry. 2013;3:e236. doi: 10.1038/tp.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A. 2002;99:1485–1490. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B, Hrncic R, Wall JS, Weiss DT, Solomon A. Diagnostic and therapeutic potential of amyloid-reactive IgG antibodies contained in human sera. J Immunol. 2006;176:7071–7078. doi: 10.4049/jimmunol.176.11.7071. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Allen A, Kennel SJ, Weiss DT, Solomon A, Wall JS. Localization of a conformational epitope common to non-native and fibrillar immunoglobulin light chains. Biochemistry. 2007;46:1240–1247. doi: 10.1021/bi0616605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B, Acero L, Williams AD, Koeppen HP, Weber A, Schwarz HP, Wall JS, Weiss DT, Solomon A. Human plasma contains cross-reactive Abeta conformer-specific IgG antibodies. Biochemistry. 2008;47:12254–12256. doi: 10.1021/bi801767k. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B, Klyubin I, Mc Donald JM, Foster JS, Welzel A, Barry A, Dykoski RK, Cleary JP, Gebbink MF, Rowan MJ, Walsh DM. A monoclonal antibody against synthetic Abeta dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J Neurochem. 2011;119:189–201. doi: 10.1111/j.1471-4159.2011.07389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin N. Clinical trials of intravenous immunoglobulin for Alzheimer's disease. J Clin Immunol. 2014;34:S74–S79. doi: 10.1007/s10875-014-0041-4. [DOI] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- St-Amour I, Paré I, Tremblay C, Coulombe K, Bazin R, Calon F. IVIg protects the 3xTg-AD mouse model of Alzheimer's disease from memory deficit and Abeta pathology. J Neuroinflammation. 2014;11:54. doi: 10.1186/1742-2094-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Greenstein A, Wilcock DM. Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Abeta in APP/PS1 mice along a different time course than anti-Abeta antibodies. J Neurosci. 2013;33:9684–9692. doi: 10.1523/JNEUROSCI.1220-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P, Mujalli DM, Rotondi ML, Sharma R, Weber A, Schwarz HP, Weksler ME, Relkin N. Measurement of anti-beta amyloid antibodies in human blood. J Neuroimmunol. 2010;227:167–174. doi: 10.1016/j.jneuroim.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Urashima M, Chauhan D, Uchiyama H, Freeman GJ, Anderson KC. CD40 ligand triggered interleukin-6 secretion in multiple myeloma. Blood. 1995;85:1903–1912. [PubMed] [Google Scholar]
- Vidal R, Barbeito AG, Miravalle L, Ghetti B. Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol. 2009;19:58–68. doi: 10.1111/j.1750-3639.2008.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry. 1999;38:14101–14108. doi: 10.1021/bi991131j. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Ylera F, Harth S, Waldherr D, Frisch C, Knappik A. Off-rate screening for selection of high-affinity anti-drug antibodies. Anal Biochem. 2013;441:208–213. doi: 10.1016/j.ab.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Zhang X, Sun XX, Xue D, Liu DG, Hu XY, Zhao M, Yang SG, Yang Y, Xia YJ, Wang Y, Liu RT. Conformation-dependent scFv antibodies specifically recognize the oligomers assembled from various amyloids and show colocalization of amyloid fibrils with oligomers in patients with amyloidoses. Biochim Biophys Acta. 2011;1814:1703–1712. doi: 10.1016/j.bbapap.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Zhao M, Wang SW, Wang YJ, Zhang R, Li YN, Su YJ, Zhou WW, Yu XL, Liu RT. Pan-amyloid oligomer specific scFv antibody attenuates memory deficits and brain amyloid burden in mice with Alzheimer's disease. Curr Alzheimer Res. 2014;11:69–78. doi: 10.2174/15672050113106660176. [DOI] [PubMed] [Google Scholar]