

Abstract
Axons in the adult CNS have poor ability to grow after injury, impeding functional recovery in patients of spinal cord injury. This has been attributed to both a developmental decline in neuron-intrinsic growth ability and the presence of extrinsic growth inhibitors. We previously showed that genetic deletion of Nogo, an extrinsic inhibitor, promoted axonal sprouting from uninjured corticospinal tract (CST) neurons but not regeneration from injured CST neurons, whereas genetic deletion of PTEN, an intrinsic inhibitor, promoted both CST sprouting and regeneration. Here we test the hypothesis that combining an elevation of neuron-intrinsic growth ability and a reduction of extrinsic growth inhibition by genetic codeletion of PTEN and Nogo may further improve injury-induced axonal growth. In an apparent paradox, additionally deleting Nogo further enhanced CST regeneration but not sprouting in PTEN-deleted mice. Enhanced CST regeneration and sprouting in PTEN and PTEN/Nogo-deleted mice were associated with no or only temporary improvement in functional recovery. Our data illustrate that neuron-intrinsic and -extrinsic factors regulate axon regeneration and sprouting in complex ways and provide proof-of-principle evidence that targeting both can further improve regeneration. Neuron-intrinsic growth ability is an important determinant of neuronal responsiveness to changes in extrinsic growth inhibition, such that an elevated intrinsic growth state is a prerequisite for reducing extrinsic inhibition to take effect on CST regeneration. Meanwhile, additional strategies are required to unleash the full potential for functional recovery with enhanced axon regeneration and/or sprouting.
Keywords: axon regeneration, axon sprouting, CNS repair, extrinsic growth inhibition, neuron-intrinsic growth ability, spinal cord injury
Introduction
Increasing axonal growth is important for promoting functional recovery after CNS injury, such as a spinal cord injury. Two different forms of axonal growth may contribute to functional recovery: regeneration, the growth from injured neurons; and sprouting, the compensatory growth from uninjured neurons (Tuszynski and Steward, 2012; Geoffroy and Zheng, 2014). Whereas regenerating axons often need to travel a long distance to be functionally beneficial, sprouting of spared axons may originate distal to the injury site, bypassing the need to grow past the lesion. A baseline level of sprouting occurs naturally after CNS injury and may underlie some of the spontaneous recovery after anatomically incomplete injuries (Bradbury and McMahon, 2006). In contrast, baseline regeneration is much more limited in the CNS.
Both neuron-extrinsic and -intrinsic factors may impede axonal growth. Earlier studies emphasized the role of extrinsic axon growth inhibitors (Silver and Miller, 2004; Schwab and Strittmatter, 2014). Nogo represents one prototypical extrinsic inhibitor associated with the CNS myelin. Although function-blocking antibodies and peptide antagonists against Nogo have been shown to promote axonal regeneration and functional recovery (Schwab and Strittmatter, 2014), our genetic studies indicated that deleting Nogo is insufficient to promote axon regeneration from the corticospinal tract (CST), a functionally important descending tract (Zheng et al., 2003; Lee et al., 2009, 2010; Lee and Zheng, 2012). In contrast, manipulating Nogo pharmacologically or genetically has consistently led to enhanced axon sprouting (Thallmair et al., 1998; Raineteau et al., 2001; Cafferty and Strittmatter, 2006; Cafferty et al., 2010; Lee et al., 2010). Thus, the main effect of targeting Nogo, at least alone, appears to be on promoting axon sprouting rather than regeneration.
Meanwhile, recent studies highlighted the increasing importance of neuron-intrinsic growth regulators (Lu et al., 2014). We previously showed that genetically deleting neuronal PTEN (phosphatase and tensin homolog), a tumor suppressor and negative regulator of the mTOR (mammalian target of rapamycin) signaling pathway, promotes CST regeneration after experimental spinal cord injury (Liu et al., 2010). mTOR globally regulates protein synthesis and cell growth (Ma and Blenis, 2009). Neuronal mTOR activity is downregulated as development proceeds and, intriguingly, is further downregulated after axonal injury in the adult (Park et al., 2008; Liu et al., 2010). This further diminished mTOR activity presumably makes injured neurons less capable of axonal growth than their uninjured counterparts and potentially renders them less responsive to a reduction in extrinsic growth inhibition.
Although both regeneration and sprouting may serve as important anatomical substrates for functional recovery, their regulation by neuron-intrinsic and -extrinsic factors may differ. Here we codeleted PTEN and Nogo as proof-of-principle experiments to target neuron-intrinsic and -extrinsic factors simultaneously. We hypothesized that elevating intrinsic growth ability by deleting PTEN may render injured adult CST neurons more responsive to a reduction in extrinsic growth inhibition. Our results illustrate that neuron-intrinsic and -extrinsic factors regulate axon regeneration and sprouting in complex ways and that intrinsic growth ability is an important determinant of neuronal responsiveness to changes in extrinsic growth inhibition.
Materials and Methods
Mutant mice
The Nogo null mutant (null for all the three major isoforms of Nogo, Nogo-A-C) has been described previously (Lee et al., 2009). The PTEN conditional allele (PTENflox, or PTENf) was obtained from The Jackson Laboratory (B6.129S4-Ptentm1Hwu/J; stock #006440) (Lesche et al., 2002). The single-mutant mice, after being backcrossed to C57BL/6 for at least 9 generations, were bred to each other to obtain compound heterozygous mutants. These mice were then intercrossed to obtain PTENf/f;Nogo−/− and PTENf/f;Nogo+/− mice, which were then crossed to each other to obtain experimental mice of the same genotypes. AAV-Cre or control virus AAV-GFP were injected into the sensorimotor cortex of P1 or young adult (4- to 6-week-old) mice to obtain the following four experimental groups: control mice (PTENf/f;Nogo+/−, AAV-GFP), Nogo single-mutant mice (PTENf/f;Nogo−/−, AAV-GFP), PTEN single-mutant mice (PTEN−/−;Nogo+/−, AAV-Cre), and PTEN/Nogo double-mutant mice (PTEN−/−;Nogo−/−, AAV-Cre).
Viral production and injection
AAV-Cre and control AAV-GFP (Liu et al., 2010) were produced at the Salk Institute Viral Vector Core. Viral concentrations were titered by qPCR. AAV-Cre reached a titer of 0.5 × 1012 TU/ml and AAV-GFP 0.7 × 1012 TU/ml. For neonatal AAV injections, postnatal day 1 (P1) PTENf/f;Nogo−/− and PTENf/f;Nogo+/− mice were cryo-anesthetized, placed on an ice-cold pad, and injected with 1 μl of either AAV-GFP or AAV-Cre into the right sensorimotor cortex. A modified 10 μl of Hamilton syringe attached to a fine glass pipette was mounted on a stereotaxic device for injection. After injection, pups were placed on a 37°C warming pad and covered with some home cage bedding to decrease the risk of rejection by the mother. Pups were returned to their mothers once they regained consciousness, normal color, and behavior.
For young adult AAV injections, 4- to 6-week-old PTENf/f;Nogo−/− and PTENf/f;Nogo+/− mice were anesthetized with freshly prepared 2.5% Avertin (Sigma). A total of 1.2 μl of AAV-GFP or AAV-Cre was injected into the sensorimotor cortex at 6 sites (0.2 μl/site). For the dorsal hemisection model, the right sensorimotor cortex targeting the left hindlimb was injected (0.1, 0.6, and 1.1 mm posterior to the bregma; 1.0 and 1.4 mm lateral, 0.7 mm deep). For the pyramidotomy model, the right sensorimotor cortex targeting the left forelimb was injected (0.5 and 0.1 mm anterior, and 0.3 mm posterior to the bregma; 1.2 and 2.2 mm lateral, 0.7 mm deep).
Surgical procedures for injury production and axon tracing
Dorsal hemisection spinal cord injury, pyramidotomy, and biotinylated dextran amine (BDA) tracer injection into the sensorimotor cortex were performed as described previously (Lee et al., 2010; Liu et al., 2010), with minor modifications as highlighted below. Adult female mice were used for spinal cord injury experiments; adult male mice were used for pyramidotomy. In total, 169 mice of various genotypes were used in surgical experiments. Surgical procedures for spinal cord injury or pyramidotomy were performed 6 weeks after AAV injections in P1 mice and 4 weeks after AAV injections in young adult mice. For consistency throughout the study, all pyramidotomy surgeries were performed by one surgeon; all dorsal hemisection surgeries were performed by another surgeon. Surgeons performing the surgeries were blinded to the genotypes.
Pyramidotomy.
Mice were anesthetized with 2.5% Avertin and an incision made on the left side of the trachea. Skull base was exposed, and craniotomy of the occipital bone was performed using laminectomy forceps to expose the underlying pyramidal tract of adult male mice (Starkey et al., 2005). A micro feather scalpel was used to puncture the dura and lesion the entire left pyramidal tract. Wound was closed with 6.0 sutures and Dermabond. mice were perfused 4 weeks after injury.
Dorsal hemisection spinal cord injury.
A laminectomy at T8 was performed to expose the spinal cord. The dura was punctured bilaterally with a 30G needle at appropriate locations. A pair of superfine iridectomy scissors was then used to cut the dorsal half of the spinal cord at a depth of 0.8 mm. Finally, a micro feather ophthalmic scalpel was used to retrace the lesion to ensure its completeness. The muscle layers were sutured with 6.0 sutures, and the skin was secured with wound clips and Dermabond. Mice were perfused 6 weeks after injury.
BDA tracing.
Two weeks before death (i.e., 2 weeks after pyramidotomy, or 4 weeks after dorsal hemisection), mice received stereotaxic injection of BDA (10%, Invitrogen) to label CST axons by anterograde tracing. The same injection volumes and coordinates were used for BDA as for young adult AAV injections (see above).
Behavioral assessment
All behavioral tests were performed and quantified by observers blinded to the genotypes. Three different behavioral tests were performed for each injury model.
1. Rearing test (for pyramidotomy only).
This test assesses the forepaw preference for touching and leaning against the wall when mice explore around in a new cage while standing on their hindlimbs (i.e., rear limbs) (Starkey et al., 2005; Lee et al., 2010). A 15 min videotape was taken. An observer blinded to the genotypes scored forepaw usage for weight support against the cage wall while rearing on its hindlimbs for the first 25 times. Mice were tested before injury (day −1) and on day 2, 7, 14, 21, and 28 after injury. The percentage of using left forepaw only, right forepaw only, or both forepaws for weight support while rearing on hindlimbs was measured. Incidences of using both forepaws were subdivided by analyzing the video in slow motion into right forepaw touching the wall first, left forepaw touching the wall first, and both forepaws touching the wall simultaneously (when it was indistinguishable which forepaw touched the wall first).
2. Tape removal test (for pyramidotomy only).
This test was performed as described previously (Starkey et al., 2005) with some modifications. Mice were habituated to the testing environment by being handled as they would during testing to decrease the risk of anxiety. This consisted of handling the mice and touching their forepaws as it would be done to attach a sticker for the actual test. Mice were then left for 2 min in a cylinder. In actual tests, precut round stickers (Office Depot, ¼” diameter) were applied in 2 trials for each forepaw, alternating between left and right. A video camera was used to videotape mice until the sticker was removed, or for a maximum length of 2 min. Mice were tested on day −1 (negative 1, as preinjury baseline), 2, 7, 14, 21, and 28 after injury, by the same experimenters, at about the same time of the day. An observer blinded to the mouse genotypes later analyzed the video recordings. The sensory score is the amount of time (in seconds) for a mouse to notice the sticker (as indicated by shaking the paw, bringing paw to the mouth, scrubbing the paw on the head); the motor score is the time (in seconds) for a mouse to remove the sticker after noticing it.
3. Ladder rung test (for both pyramidotomy and dorsal hemisection).
This test was performed as described previously (Metz and Whishaw, 2009) with some modifications. Briefly, mice were habituated and trained to run on the apparatus for 5 consecutive days before injury. On the day before injury and on selected days after injury, mice were tested. An irregular rung pattern (with uneven spacing between rungs) is used each time to prevent the mice from compensating for their limb impairment by learning a particular pattern. The same set of irregular patterns was used for all the mice based on the time point relative to the injury (such that on day −1, all mice were tested using Pattern 1; on day 2 after injury, Pattern 2; day 7, Pattern 3, etc.). A video camera was positioned from the side of a horizontal ladder at a slight ventral angle, so the positions of all four limbs could be recorded simultaneously. Testing consisted of having the mice walk for a minimum of 6 passages in front of the video camera to obtain sufficient numbers of steps (a minimum of 30 steps) for analysis. For each injury model, all recordings were performed by the same experimenters, at approximately the same time of the day. The video recordings were analyzed by an observer blinded to the genotypes using frame-by-frame analysis at 30 f/s as described previously (Metz and Whishaw, 2009). A foot fault scoring system provided a score from 0 (total miss step) to 6 (perfect placement) for each consecutive step for individual paws, giving the percentage of missteps as well as an average score. For dorsal hemisection, the percentage of correct steps instead of missteps was plotted.
4. Basso Mouse Scale (BMS) (for dorsal hemisection only).
The BMS assesses the locomotion behaviors of mice in an open field (Basso et al., 2006). Mice were observed for 5 min by two observers. Many features were noted, including ankle movements, stepping pattern, coordination, paw placement, trunk instability, and tail position, with a minimum score of 0 (no movement) to a maximum score of 9 (normal locomotion). Mice were tested before injury (day −1) and at day 1, 7, 14, 21, 28, 35, and 42 after spinal cord injury. Mice with a BMS score of >3 (some stepping) at 1 d after injury were excluded from the analysis.
5. Rotarod (for dorsal hemisection only).
Mice were placed on a rod (Ugo Basile) rotating at increasing speeds from 5 to 50 rpm in 3 min with constant acceleration as described previously (Lee et al., 2010). The latency to fall (in seconds) was averaged between two trials per session. Mice were first acclimated to the test for two sessions for 5 d the week before injury and one additional session 1 d before injury (baseline). Mice were then tested at day 1, 7, 14, 21, 28, 35, and 42 after spinal cord injury.
Tissue processing
Tissue processing was performed as described with minor modifications (Lee et al., 2009, 2010). Mice were given lethal dose of Fatal plus, and perfused transcardially with 4% PFA. Brain and spinal cord were dissected out, and the tissues were postfixed overnight at 4°C in the same fixative solution. Tissues were incubated in 30% sucrose for 3 d for cryo-protection. The bulk of the brain, medulla, and different segments of the spinal cord were embedded in OCT compound, and snapped frozen on dry ice. For the pyramidotomy groups, C3-C7 spinal cord was embedded for transverse sections. For dorsal hemisection, an ∼8 mm segment from 4 mm rostral and 4 mm caudal to the injury was embedded for sagittal sections; the spinal cord tissues just rostral and caudal to this 8 mm segment were embedded for transverse sections to evaluate axon labeling efficiency (based on the rostral tissue sections) and lesion completeness (based on the caudal tissue sections). Tissues were sectioned with a cryostat at 20 μm thickness for further processing.
Transverse sections of the medullary pyramids were processed to obtain the total number of CST axons labeled to control for labeling efficiency (see below). For the dorsal hemisection groups, floating step serial (every other) sagittal sections centered at the injury site were stained for BDA and GFAP. Sections were incubated in Vectastain ABC solution (Vector Laboratories) overnight at 4°C, washed in PBS, and mounted on gelatin-coated slides. BDA was detected with TSA Plus Fluorescein System (10 min, room temperature, 1:200, PerkinElmer). Sections were costained with polyclonal rabbit anti-GFAP (overnight incubation, 1:500, Dako) after blocking in PBS with 0.4% Triton X-100 and 5% horse serum for 2 h at room temperature. The next day, sections were washed and incubated with secondary anti-Rabbit AlexaFluor-488 (Invitrogen) for 1 h at room temperature. After several washes with PBS, sections were coverslipped with Fluoromount-G (Southern Biotechnology). For immunohistochemistry, purified polyclonal rabbit anti-Nogo-A (1:500) as described previously (Zheng et al., 2003), rat anti-GFAP (1:500, Invitrogen), rat anti-myelin basic protein (MBP, 1:500, Millipore), mouse anti-CNPase (1:50, Sigma), mouse anti-NG2 (1:50, Millipore), mouse anti-NeuN (1:100, Millipore), chicken anti-HF-H (1:200, Millipore), and rat anti-CD68 (1:400, AbD Serotec) were used. For the pyramidotomy groups, medullas and transverse sections of the cervical C7 were processed similarly, but without GFAP costaining. Selected transverse sections of cervical spinal cord (C7) were immunostained for PKCγ (Santa Cruz Biotechnology, 1:100) to examine the completeness of the lesion (see Fig. 1G,H) for every single animal, as described previously (Lee et al., 2010). Mice with incomplete lesion were excluded from the study. A total of 5 of 103 mice stained for PKCγ were removed, including 2 control, 1 Nogo KO, and 2 PTEN KO mice. Coronal brain sections were stained for pS6 (Cell Signaling Technology) as described previously (Liu et al., 2010).
Figure 1.
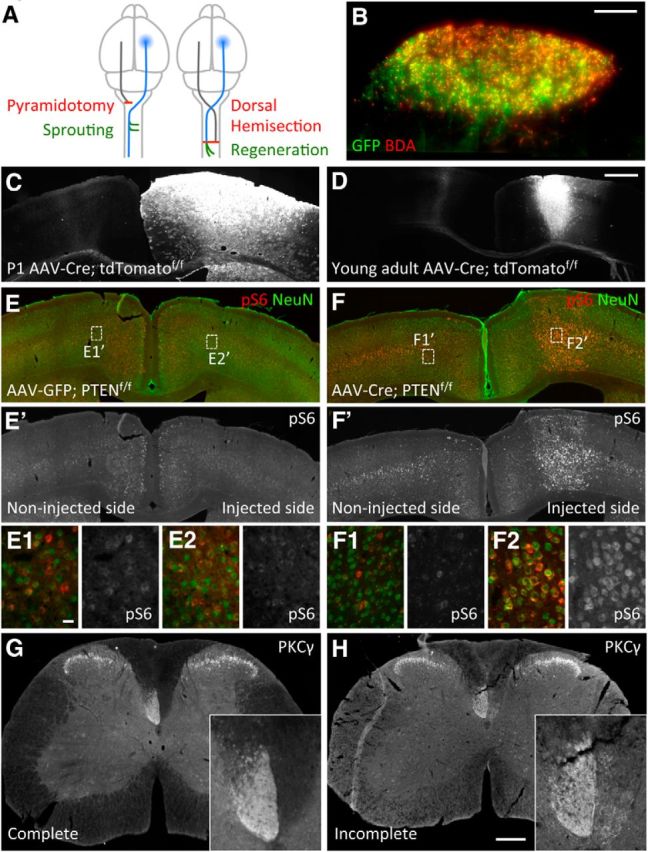
Schematic representation of the injury models to study CST axon sprouting and regeneration and pS6 upregulation in PTEN-deleted mice. A, Dorsal view of the brain and part of the spinal cord illustrating the unilateral pyramidotomy injury model (left) and the dorsal hemisection spinal cord injury (right). Unilateral pyramidotomy (red line) severs all CST axons originating from the left side of the brain (gray line, not traced with BDA) at the pyramidal level before the axons cross the midline, leading to denervation of the right side of the spinal cord. Axon tracer BDA is injected into the right sensorimotor cortex (blue shaded area) to label the uninjured CST axons (blue line), which may sprout (green lines) across the midline into the denervated side in the cervical cord. Dorsal hemisection at T8 (red line) severs the vast majority of CST axons in the cord bilaterally. Axon tracer BDA is injected into the right sensorimotor cortex (blue shaded area) to label the injured CST axons (blue line) on the left side of the spinal cord to detect any regeneration (green lines) below the injury site. The other side of the CST is also injured but not traced with BDA (gray line). In both models, AAV-Cre (or AAV-GFP control) is also injected into the right sensorimotor cortex (blue shaded area) in neonatal or young adult mice to initiate PTEN deletion, before injury. B, Example of a medulla section from a mouse injected with control AAV-GFP at a young adult age followed by BDA injection 4 weeks later, showing significant overlap between GFP and BDA labeling (in this animal, ∼42% of BDA-labeled CST axons in the medulla were GFP positive). C, D, Representative images of coronal brain sections 4 weeks after unilateral AAV-Cre injection into the right cortex of P1 (C) or 4-week-old (D) ROSA26-loxP-STOP-loxP-tdTomatof/f mice, illustrating a wider distribution of AAV-Cre mediated tdTomato reporter gene activation in P1-injected mice than in young adult-injected mice. E, E′, F, F′, Representative images of coronal brain sections 4 weeks after unilateral AAV injection into the right cortex of young adult PTENf/f;Nogo+/− mice, showing pS6 and NeuN costaining (E,F) or pS6 signals only (E′,F′). E1, E2, F1, F2, High-magnification images of the regions depicted in E and F. Note the higher level of pS6 and the larger soma size associated with pS6 immunoreactivity on the right injected side with AAV-Cre. G, H, PKCγ staining 4 weeks after unilateral pyramidotomy showing an example of a successful injury (G) and an example of an incomplete lesion that was removed from the study (H). Scale bars: B, 100 μm, C–F′, 1 mm; E1–F2, 20 μm; G, H, 200 μm.
Quantification of axons
Images were taken with a digital camera mounted on a Zeiss epifluorescence microscope using the 10×, 20×, or 100× (medulla only) objective lenses. Axon densities and/or numbers were quantified using ImageJ. Both the initial observation of the stained sections and later quantification of axon densities/numbers were conducted in a blinded fashion in which the observers did not have a priori knowledge of the genotypes of the sections being examined or quantified. The average total number of axons labeled in the medulla did not differ significantly among genotypes: (1) 3228 ± 295 (control), 3082 ± 322 (Nogo KO), 2793 ± 210 (PTEN KO), and 2724 ± 284 (double KO) with the dorsal hemisection model and neonatal AAV injection; (2) 3086 ± 258 (control), 3317 ± 263 (Nogo KO), 2917 ± 312 (PTEN KO), and 2989 ± 530 (double KO) with the pyramidotomy model and neonatal AAV injection; (3) 2831 ± 356 (control), 2778 ± 282 (Nogo KO), 2609 ± 138 (PTEN KO), and 2687 ± 204 (double KO) with the dorsal hemisection model and young adult AAV injection; (2) 2814 ± 450 (control), 2550 ± 352 (Nogo KO), 2574 ± 299 (PTEN KO), and 2740 ± 297 (double KO) with the pyramidotomy model and young adult AAV injection (all numbers are given as mean ± SEM). Three mice that received AAV-GFP injection as young adults followed by BDA injection 2 weeks later were assessed for GFP/BDA colabeling efficiency in the medulla as exemplified in Figure 1B. The axon numbers with GFP/BDA colabeling, BDA labeling, and GFP labeling are as follows: Mouse 1, 499, 2333, 2137; Mouse 2, 653, 1552, 1856; Mouse 3, 869, 2800, 3732. For statistical analyses, we used GraphPad Prism 5 software. A D'Agostino-Pearson normality test was initially performed before appropriate one-way or two-way ANOVA followed by Bonferroni's post hoc test.
CST sprouting after pyramidotomy.
Sprouting Axon Density Index and Sprouting Axon Number Index were quantified. Images of transverse cervical (C7) spinal cord sections were taken. Axon density in each side of the gray matter (denervated or not) was measured after subtraction of the background using ImageJ, as described previously (Grider et al., 2006). Five randomly selected sections between C6/C7 per animal were quantified and averaged. The ratio of contralateral (denervated) and ipsilateral counts was taken as the measure for Sprouting Axon Density Index.
To quantify the Sprouting Axon Number Index, lines were drawn through the central canal and across the dorsoventral axis, at 50 μm from the midline, then every 100 μm laterally in the denervated side of the gray matter. Numbers of axons crossing theses lines were averaged from five sections and normalized against total axon count in medulla (Lee et al., 2010) to obtain the Sprouting Axon Number Index, which was plotted as a function of the distance from the midline.
CST regeneration after dorsal hemisection spinal cord injury.
We assessed the severity of the injuries by measuring the maximal depth of the lesions. We did not observe any significant differences in lesion depths among the different genotypes: 711 ± 21 (control), 702 ± 22 (Nogo KO), 673 ± 35 (PTEN KO), and 700 ± 19 (double KO) (mean ± SEM). The density of axons rostral to the injury site was quantified by measuring the BDA staining intensity as described previously (Liu et al., 2010). A series of rectangles of 100 μm wide covering the entire dorsoventral axis of the spinal cord were superimposed onto sagittal sections, starting from the injury site (delimited by GFAP staining) all the way to 1.5 mm rostral to the injury. After subtraction of the background, intensity was measured in every rectangle using ImageJ; and the pixel value of each segment was normalized against the intensity at 1.5 mm rostral to injury. The ratio was taken as the Rostral Axon Density Index and was plotted as function of the distance to the injury. Three adjacent sagittal sections containing the main CST were quantified and averaged for each animal.
The number of axons present in all regions containing BDA-labeled axons caudal to injury was quantified to assess regeneration. Vertical lines 100 μm apart were drawn caudal to the injury site covering the dorsoventral axis of the spinal cord, and the total number of axons between two adjacent lines was quantified. The axon numbers on every other sagittal section throughout the mediolateral axis were summed up, normalized against the total number of axons labeled in the medulla (Lee et al., 2010), and expressed as a function of the distance from the injury site to obtain the Caudal Axon Number Index. BDA-labeled axons exiting the GFAP sparse lesion core and entering the GFAP dense astroglial reactive area immediately caudal to the injury were considered regenerated. In addition, the total number of axons present within the lesion core was quantified and normalized to the number of axons at the medulla level.
Results
The primary goal of this study was to assess whether manipulating both neuron-intrinsic (PTEN) and extrinsic (Nogo) factors could further increase the sprouting and regeneration of CST axons following injury beyond what is already observed with manipulating either factor alone. While definitions of axon regeneration versus sprouting vary in the literature (Cafferty et al., 2008), here we used the injury status of the neurons as the primary criterion in distinguishing regeneration from sprouting: regeneration is axonal growth from injured neurons and sprouting is axonal growth from uninjured neurons (Tuszynski and Steward, 2012; Geoffroy and Zheng, 2014). We used two different injury models, a unilateral pyramidotomy model and a dorsal hemisection spinal cord injury (Fig. 1A), to assess CST sprouting and regeneration, respectively (Starkey et al., 2005; Zheng et al., 2006). Age-, sex-, and genetic background-matched littermates (backcrossed ≥9 generations into C57BL/6) were used to facilitate a direct comparison among different genotypes. In addition, PTEN deletion was initiated in either neonatal or young adult mice in separate experiments to examine the effect of the timing of PTEN deletion. We followed functional recovery with several behavioral assays, including the ladder rung, rearing, and tape removal tests after pyramidotomy and the open field locomotion (BMS score), rotarod, and ladder rung tests after dorsal hemisection spinal cord injury. In addition, our study represented the first time where PTEN deletion and Nogo deletion could be compared side by side in their effect on axon regeneration and sprouting after CNS injury.
We applied unilateral AAV-Cre (adeno-associated virus carrying a Cre expression construct) injection into the sensorimotor cortex of PTENf/f (homozygous floxed, PTEN conditional mutant) mice (Lesche et al., 2002) to induce PTEN deletion as described previously (Liu et al., 2010). We evaluated the effectiveness of AAV infection in two ways. First, we estimated the percentage of GFP-positive axons among axon tracer BDA-labeled CST axons in the medulla at ∼30% in mice that received control AAV-GFP injection as young adults (4–6 weeks old) followed by BDA injection later (21%, 31%, and 42% for the three mice assessed, with the last example shown in Fig. 1B). Second, we injected AAV-Cre into the conditional ROSA26-loxP-STOP-loxP-tdTomatof/f reporter mice (Madisen et al., 2010) at either the neonatal (postnatal day 1 [P1]) or young adult stage. This led to strong tdTomato expression, with a wider diffusion of the virus in the P1 infected brains than in the young adult infected brains (Fig. 1C,D). Subsequent BDA injection in these mice allowed us to quantify the percentage of BDA-labeled axons at the medulla that coexpressed tdTomato. This percentage was higher in P1 infected mice (∼70%) than in young adult infected mice (∼45%). We anticipate that these numbers are more indicative of the efficiency of AAV-Cre mediated PTEN deletion among BDA-labeled axons because Cre mediated gene deletion/activation is expected to be more efficient than axon labeling with the virally encoded GFP. As described previously (Liu et al., 2010), unilateral injection of AAV-Cre into the (right) sensorimotor cortex of PTENf/f mice led to the upregulation of phospho-S6 (pS6), a downstream indicator of mTOR activity, and an accompanied increase in neuronal soma size, as exemplified with mice that received AAV-Cre injection at the young adult stage and a pyramidotomy injury later (Fig. 1E–F2).
Nogo deletion did not further increase CST sprouting that is already enhanced by neonatal PTEN deletion
Initiating PTEN deletion at P1 has previously been shown to promote robust CST sprouting across the midline after unilateral pyramidotomy in adult mice (Liu et al., 2010). We therefore first examined the effects of Nogo and PTEN codeletion on CST sprouting after pyramidotomy in mice where PTEN deletion had been initiated by AAV-Cre injection at P1. Six weeks later, a pyramidotomy was performed on the left side of the medullary pyramids, which severs CST axons on this side just before they cross the midline (Fig. 1A). Lesion completeness was verified by PKCγ immunostaining on transverse sections at the cervical level 7 (C7) (Fig. 1G,H), as described previously (Lee et al., 2010). Two weeks later, we injected BDA into the right sensorimotor cortex to label the uninjured CST axons to visualize their sprouting across the midline into the denervated side of the spinal cord (Fig. 1A). Mice were perfused 2 additional weeks later. No significant differences in the total number of BDA-labeled CST axons at the medulla level were noted among different genotypes (for details, see Materials and Methods).
For simplicity, the following nomenclatures are used to describe the four genotypes: (1) control mice (i.e., control) are PTENf/f;Nogo+/− mice injected with AAV-GFP control virus; (2) PTEN-deleted mice (i.e., PTEN knock-out [KO]) are PTENf/f;Nogo+/− mice injected with AAV-Cre; (3) Nogo-deleted mice (i.e., Nogo KO) are PTENf/f;Nogo−/− mice injected with AAV-GFP; and (4) PTEN/Nogo doubly deleted mice (i.e., double KO) are PTENf/f;Nogo−/− mice injected with AAV-Cre. The Nogo mutant allele carries an ∼40 kb genomic deletion within the Nogo locus that is a null for all known Nogo isoforms, as described previously (Lee et al., 2009).
To assess axon sprouting across the midline, we applied two different quantitative measures on C7 transverse sections: the Sprouting Axon Density Index as the ratio of total BDA labeling intensity in the gray matter of the denervated side over that of the intact side and the Sprouting Axon Number Indices as the total numbers of axons crossing a series of vertical lines within the denervated gray matter at defined distances from the midline normalized to the total CST axon number labeled in the medulla.
Nogo-deleted mice exhibited a trend for increased CST sprouting compared with the control mice (Fig. 2A,B) based on both the Sprouting Axon Density Index (Fig. 2E; control, 0.066 ± 0.006; Nogo KO, 0.088 ± 0.013, representing a ∼33% increase. p = 0.13) and the Sprouting Axon Density Indices at 50 and 150 μm from the midline (Fig. 2F; e.g., control, 0.009 ± 0.001; Nogo KO, 0.012 ± 0.001 at 50 μm, p = 0.059) that approached statistical significance. This is in line with our previous study where a modest increase of CST sprouting (<50%) was observed in Nogo-deleted mice (Lee et al., 2010). Initiating PTEN deletion at P1 led to significantly more CST sprouting than control mice by both measures (Fig. 2C,E,F), again consistent with our previous PTEN study (Liu et al., 2010). The Sprouting Axon Density Index in PTEN-deleted mice (0.142 ± 0.014) was ∼2× greater than control mice and ∼1.5× greater than Nogo-deleted mice (Fig. 2E). The Sprouting Axon Number Indices on the denervated side were significantly higher in PTEN-deleted mice than control or Nogo-deleted mice, from 50 to 450 μm off midline (Fig. 2F; e.g., 0.017 ± 0.002 at 50 μm). However, additional Nogo deletion did not noticeably further increase the level of sprouting already enhanced by PTEN deletion (Fig. 2D), as quantified by either the Sprouting Axon Density Index (Fig. 2E; double KO, 0.152 ± 0.02) or the Sprouting Axon Number Indices (Fig. 2F; e.g., 0.020 ± 0.003 for double KO at 50 μm).
Figure 2.

CST axon sprouting after unilateral pyramidotomy with neonatal PTEN deletion. A–D, Representative images of C7 spinal cord transverse sections from mice of different genotypes after a left pyramidotomy. PTEN deletion was initiated on P1 by injecting AAV-Cre (or AAV-GFP as control) into the sensorimotor cortex of PTENf/f mice. *Location of central canal. Scale bars: A–D, 300 μm; A′–D′, 100 μm (Higher-magnification images of boxed areas in A–D). E, Sprouting Axon Density Index, measuring the ratio of axon tracer BDA staining intensity in the denervated over nondenervated gray matter. F, Sprouting Axon Number Index, measuring the numbers of axons crossing specific distances in the denervated gray matter of the spinal cord, normalized against the total number of CST axons labeled in the medulla. N = 15 (control), N = 13 (Nogo KO), N = 16 (PTEN KO), N = 9 (double KO). For Sprouting Axon Density Index and Sprouting Axon Number Index, five random sections collected from C6 to C7 were quantified. *p < 0.05. ***p < 0.001. One-way (E) or two-way (F–H) ANOVA followed by Bonferroni's post hoc test. ns, Not significant.
To our knowledge, this was the first time Nogo-deleted mice and PTEN-deleted mice were compared side by side for CST sprouting in the same study, especially as age-, sex-, and genetic background-matched littermates. Together, these data indicate that modulating the neuron-intrinsic PTEN signaling pathway may lead to a more robust axon sprouting response from uninjured CST neurons than modulating Nogo-mediated extrinsic inhibition. In addition, Nogo deletion does not lead to a detectable level of further enhancement in CST sprouting in mice where PTEN deletion has been initiated at P1.
Axonal sprouting has been associated with behavioral improvement in the literature (Bradbury and McMahon, 2006). To assess the functional consequence of enhanced CST sprouting in Nogo and/or PTEN-deleted mice, we examined forelimb function recovery by uneven spacing ladder rung, rearing, and tape removal tests. We used these tests because the same or similar tests have been shown to reveal a persistent deficit after pyramidotomy in mice (Starkey et al., 2005).
In the ladder rung test, unilateral lesion of the CST significantly increased the percentage of missteps on the injured side 2 d after injury (Fig. 3B). For all four genotypes, this difference persisted for the 4 week testing period, and no differences among genotypes were observed at any of the time points. There was a slight deficit in the percentage of missteps on the uninjured side that persisted for the duration of the experiment in all genotypes (Fig. 3A). We also used a forepaw digit scoring system (Metz and Whishaw, 2009) to assess more subtle differences in functional deficits and recovery (Fig. 3C,D). There was a slight dip in the average score on the uninjured side at day 2 that restored to preinjury level at later time points (Fig. 3C). In contrast, the injured side showed a significant and persistent deficit from 2 to 28 d after injury (Fig. 3D). Again, no differences among genotypes were observed at any time point examined. In the rearing test, we assessed forepaw preferences for touching the wall during vertical exploration while mice were standing on their hindlimbs. No differences were observed among genotypes for any of the quantifications performed (data not shown).
Figure 3.
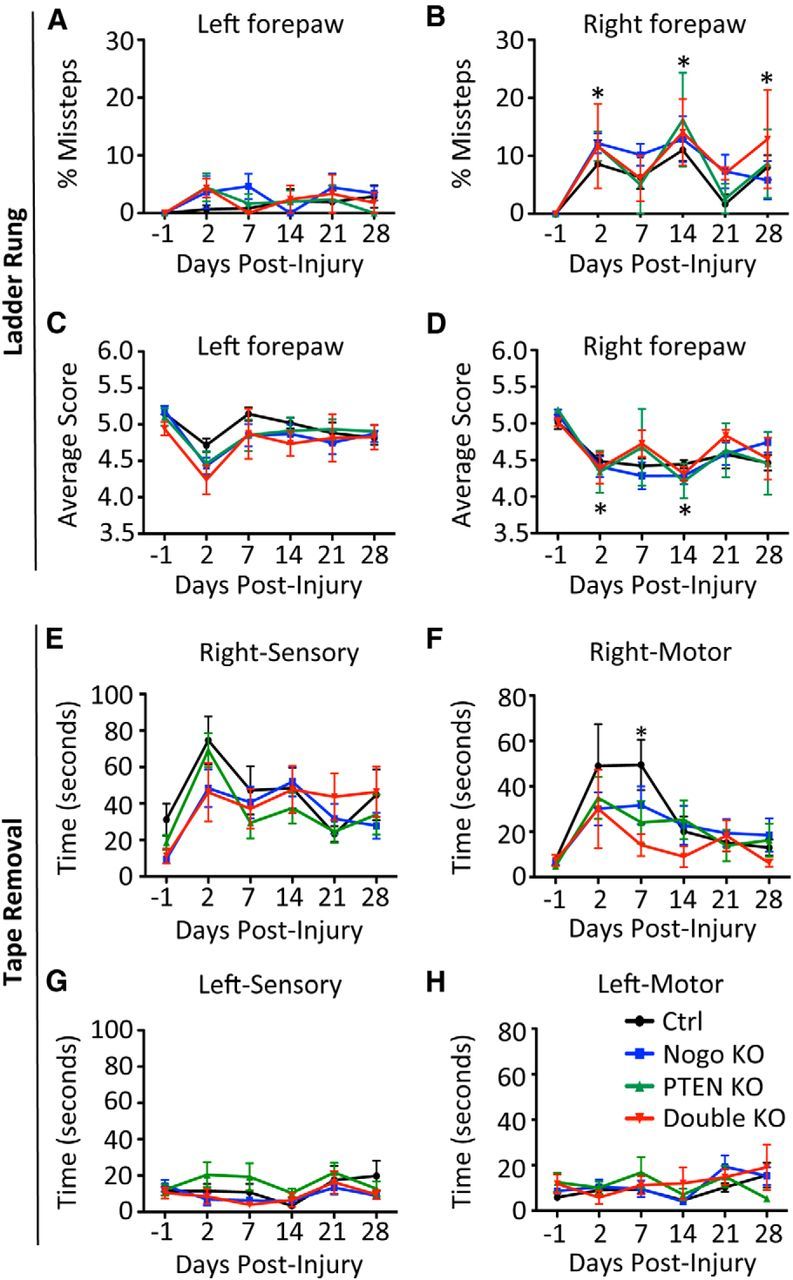
Assessing recovery of forelimb function after neonatal PTEN deletion and unilateral pyramidotomy. A–D, Ladder Rung test. There was an increase in the number of missteps for the injured side (B, right forepaw) compared with the uninjured side (A, left forepaw). No improvement in the percentage of missteps (B) or the average walking score (D) was observed during the period of recovery for all genotypes. E–H, Tape removal test. Sensory and motor scores for the right forepaw (injured side, E, F) and the left (uninjured side, G, H) for the tape removal assay. Sensory score is the time taken by a mouse to notice a sticky tape placed under the forepaw; motor score is the time taken to remove the tape after the mouse notices it. All mice improved motor function over time for the injured side, but PTEN KO and double KO mice exhibited an accelerated improvement 7 d after injury compared with control mice. Unlike the right, injured side, there was little sensory or motor deficit on the left, uninjured side after unilateral pyramidotomy, which served as a control. N = 15 (control), N = 13 (Nogo KO), N = 16 (PTEN KO), N = 9 (double KO). B, D, *Time point when all genotypes are significantly different (p < 0.05) from preinjury levels (day −1). Two-way ANOVA followed by Bonferroni's post hoc test.
The tape removal test has both a sensory and a motor component for forelimb function. Unilateral lesion of the CST impaired the ability of mice to sense a sticker attached to their injured forepaw, as reflected by the worse sensory score (i.e., a longer time for mice to notice the sticker) after injury in all four genotypes (Fig. 3E). Despite some level of spontaneous recovery, this impairment persisted over the 4 week testing period for all genotypes, with no significant differences among genotypes for any of the time points tested.
Likewise, the motor score, given by the latency to remove the tape after sensation, revealed a significant impairment specifically for the injured (right) side, which recovered over time in all genotypes (Fig. 3F). Interestingly, at day 7 after injury, PTEN and PTEN/Nogo-deleted mice displayed a significantly improved motor score (i.e., faster removal once the mouse senses the tape), whereas Nogo-deleted mice exhibited a nonsignificant trend for faster tape removal, compared with control mice (Fig. 3F; control, 50 ± 19 s; Nogo KO, 32 ± 8 s; PTEN KO, 24 ± 10 s; double KO, 14 ± 5 s). At later time points, mice of all genotypes recovered to a similar degree and approached or equaled preinjury levels. Thus, it appears that PTEN and PTEN/Nogo-deleted mice exhibited an accelerated recovery that allowed them to reach a plateau sooner than control mice, which also recovered over time.
Nogo deletion did not further enhance CST sprouting that is already enhanced by young adult PTEN deletion
As neonatal PTEN deletion may result in confounding gene compensation and developmental alterations, we also initiated PTEN deletion in young adult mice (4–6 weeks old). Four weeks later, pyramidotomy was performed and sprouting of BDA-labeled CST axons was examined four additional weeks later as described above. Qualitatively similar results were obtained as those from the neonatal PTEN deletion study above (Fig. 4A–D′). There was a modest and nonsignificant increase in the Sprouting Axon Density Index in Nogo-deleted mice compared with the control mice (Fig. 4E; control, 0.043 ± 0.003; Nogo KO, 0.063 ± 0.006, representing a ∼47% increase; p = 0.13). Both PTEN and PTEN/Nogo-deleted mice presented a significant increase in the Sprouting Axon Density Index compared with either control or Nogo-deleted mice (Fig. 4E). The Density Index of either PTEN or PTEN/Nogo-deleted mice was ∼3× that of the control mice and ∼2× that of the Nogo-deleted mice (PTEN, 0.124 ± 0.008; double KO, 0.136 ± 0.037). Thus, deleting PTEN in young adult mice also promoted CST axon sprouting after injury. However, codeleting Nogo and PTEN did not further increase CST sprouting compared with PTEN deletion alone (Fig. 4E).
Figure 4.
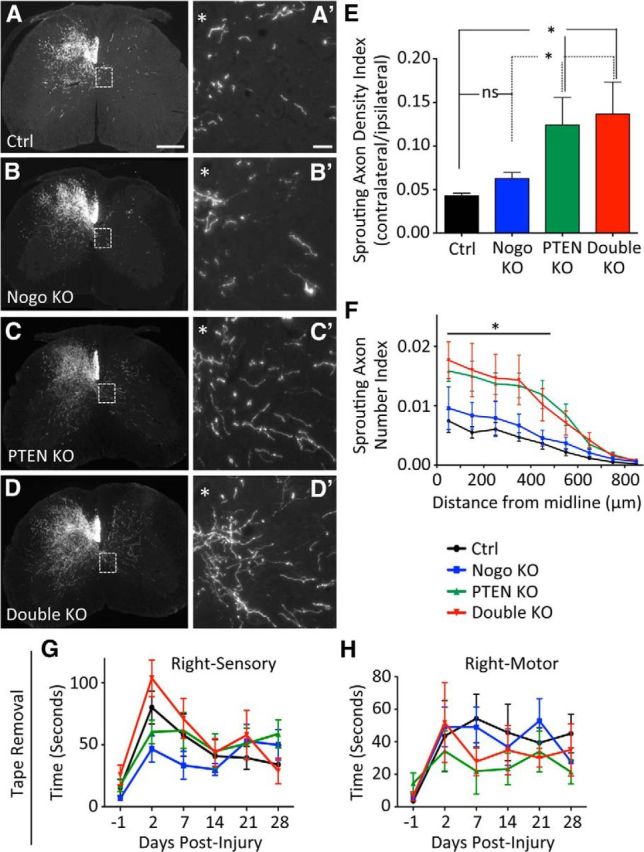
CST axon sprouting after unilateral pyramidotomy with young adult PTEN deletion. A–D, Representative images of C7 spinal cord transverse sections from mice of different genotypes after a left pyramidotomy. PTEN deletion was initiated in young adult mice by injecting AAV-Cre (or AAV-GFP control) into the sensorimotor cortex of 4- to 6-week-old PTENf/f mice. *Location of central canal. Scale bars: A–D, 300 μm; A′–D′, 100 μm. E, F, Quantification of Sprouting Axon Density Index (E) and Sprouting Axon Number Index (F). G, H, Tape removal assay showing the sensory score (G) and the motor score (H) for the right forepaw (injured side). N = 8 (control), N = 10 (Nogo KO), N = 8 (PTEN KO), N = 8 (double KO). For Sprouting Axon Density Index and Sprouting Axon Number Index, five random sections collected from C6 to C7 were quantified. *p < 0.05. One-way (E) or two-way (F) ANOVA followed by Bonferroni's post hoc test. ns, Not significant.
Quantification using the Sprouting Axon Number Indices confirmed these results: Nogo-deleted mice exhibited a modest, statistically nonsignificant increase in CST sprouting compared with control mice across the different distances (Fig. 4F; e.g., control, 0.007 ± 0.002; Nogo KO, 0.010 ± 0.002 at 50 μm), whereas both PTEN and PTEN/Nogo-deleted mice showed significantly enhanced CST sprouting (Fig. 4F; e.g., PTEN KO, 0.016 ± 0.002 at 50 μm; double KO, 0.018 ± 0.003 at 50 μm). Both PTEN and PTEN/Nogo-deleted mice had slightly >2× the amount of CST sprouting as in control mice and slightly <2× the amount of sprouting as in Nogo-deleted mice from the midline all the way to 450 μm on the denervated side. Again, there were no noticeable differences between PTEN and PTEN/Nogo-deleted mice, indicating that codeleting Nogo and PTEN did not further enhance CST sprouting compared with deleting PTEN alone.
To determine whether there was any functional improvement associated with enhanced CST sprouting, we performed the same battery of behavioral tests as in the neonatal PTEN deletion study above. We found that the behavioral deficit in rearing and ladder rung tests persisted in mice of all genotypes, and there was no difference in recovery among the genotypes (data not shown). As in the neonatal PTEN deletion study, the tape removal assay did not show improvement of the sensory score for PTEN or PTEN/Nogo-deleted mice compared with control or Nogo-deleted mice for any of the time points examined (Fig. 4G). However, there was a trend for an improved motor score in PTEN and PTEN/Nogo-deleted mice at day 7 after injury (control, 54 ± 15 s; Nogo KO, 49 ± 12 s; PTEN KO, 22 ± 14 s; double KO, 28 ± 9 s. p = 0.14) but not at later time points (Fig. 4H). Together, these data are consistent with the neonatal PTEN deletion study and point to an accelerated motor recovery associated with enhanced CST sprouting in PTEN and PTEN/Nogo-deleted mice.
Nogo deletion further enhanced CST regeneration elicited by neonatal PTEN deletion
Although both neonatal and young adult PTEN deletion led to enhanced CST sprouting across the midline, this was not further enhanced by Nogo deletion. However, it remains possible that PTEN and Nogo deletions may cooperate in enhancing axon regeneration from injured CST neurons, as elevating neuron-intrinsic growth ability may render injured neurons more sensitive to a reduction in growth inhibition, as we hypothesized. As described for the pyramidotomy model, we first initiated PTEN deletion in neonatal mice, because this paradigm had previously been shown to produce the most robust effect on CST regeneration (Liu et al., 2010). At 6 weeks of age, mice underwent a dorsal hemisection spinal cord injury at thoracic level 8 (T8) as described previously (Zheng et al., 2006; Lee et al., 2010; Liu et al., 2010). Four weeks later, CST axons were traced with unilateral BDA injection in the sensorimotor cortex (on the side where AAV-Cre or control AAV-GFP had been injected), followed by two additional weeks of survival time (Fig. 1A). We first looked at transverse sections 4 mm caudal to the injury site to ensure completeness of the lesion. There were no mice with spared CST axons in the dorsal main CST (Zheng et al., 2006). A total of three mice with labeled dorsolateral CST axons below the level of injury, possibly spared, were excluded from the analyses below.
In mice, after a penetrating spinal cord injury such as a dorsal hemisection, a GFAP sparse lesion core forms at the injury site that is surrounded by a GFAP dense area, which reflects reactive astrogliosis (Herrmann et al., 2010). Injured CST axons typically die back for hundreds of micrometers at the injury site. Examination of the tissue rostral to injury on sagittal sections by observers blinded to the genotypes immediately revealed clear distinctions between groups of mice that turned out to be PTEN dependent. Whereas the typical axon die-back was observed in control and Nogo-deleted mice, both PTEN and PTEN/Nogo-deleted mice exhibited extensive CST labeling deep into the GFAP dense area immediately rostral to the lesion core (Fig. 5A–D′). We quantified this observation by measuring the BDA labeling intensity at different distances rostral to the injury site to obtain the axon Density Indices. Consistent with the blind observation mentioned above, PTEN and PTEN/Nogo-deleted mice presented significantly higher axon Density Indices than control and Nogo-deleted mice from the rostral end of the lesion core all the way to 0.5 mm rostral to injury (Fig. 5E). We have previously shown that this increased rostral axon density in PTEN-deleted mice is due to enhanced CST regeneration but not a reduction in the initial die-back following injury (Liu et al., 2010). However, no difference in axon Density Index was observed between PTEN-deleted and PTEN/Nogo-deleted mice, indicating that the neuron-intrinsic growth ability following PTEN deletion is the main driver for axon growth rostral to injury. Furthermore, as reported previously (Liu et al., 2010), large bulbous endings of BDA-labeled axons characteristic of the retraction bulbs were much less noticeable in PTEN-deleted groups than non–PTEN-deleted groups (data not shown).
Figure 5.
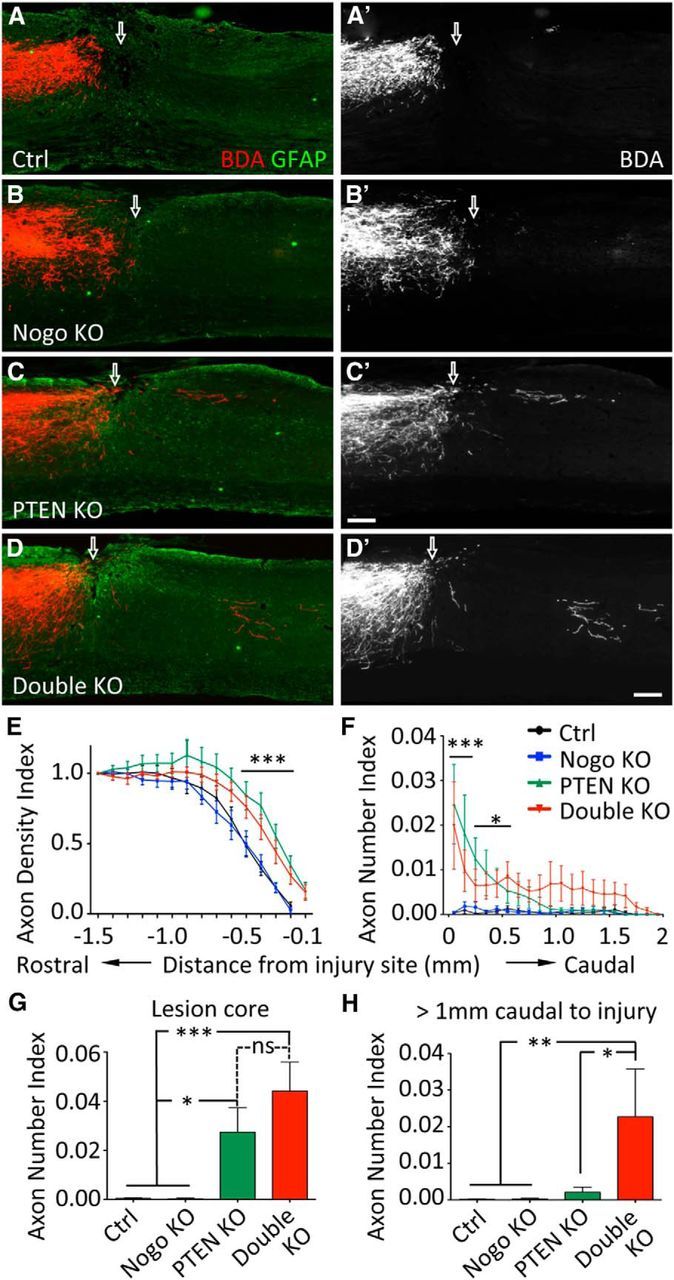
CST axon regeneration after dorsal hemisection spinal cord injury with neonatal PTEN deletion. A–D, Representative images of thoracic spinal cord sagittal sections from mice of different genotypes after a T8 dorsal hemisection. PTEN deletion was initiated in neonatal mice. Scale bar, 200 μm. Arrow indicates injury site. Note the larger gap between BDA-labeled CST axon endings and the injury site in A and B compared with that in C and D. E, Axon Density Index rostral to the injury site, quantified as the staining intensity of axon tracer BDA in function to the distance from the injury site, averaged for three adjacent sagittal sections containing the main CST and normalized against the level at 1.5 mm rostral to injury for each mouse. F, Axon Number Index caudal to the injury site, quantified as the number of axons crossing specific distances, summed from every other sagittal section throughout the mediolateral axis and then normalized against the total number of CST axons labeled in the medulla for each mouse. Axons need to exit the injury site and extend into the GFAP dense area caudal to the injury site to be considered regenerated. G, Axon Number Index within the GFAP sparse lesion core. H, Cumulative Axon Number Index past 1 mm caudal to the injury. N = 14 (control), N = 11 (Nogo KO), N = 11 (PTEN KO), N = 9 (double KO). *p < 0.05. **p < 0.01. ***p < 0.001. Two-way (E, F) or one-way (G, H) ANOVA followed by Bonferroni's post hoc test.
At, around, and beyond the injury site, Nogo-deleted mice did not exhibit any significant CST regeneration compared with control mice (Fig. 5A–B′), consistent with our previous studies (Lee et al., 2009, 2010). Occasionally, few axons were observed to pass beyond the injury site via the ventral white matter (data not shown), but this occurred in both Nogo-deleted and control mice and presumably reflected a low baseline level of spontaneous regeneration (Steward et al., 2008). In contrast, BDA-labeled axons could readily be seen across and beyond the injury site, and extending into the GFAP dense area caudal to the injury site in PTEN and PTEN/Nogo-deleted mice (Fig. 5C–D′). These axons often took a route in the dorsal part of the spinal cord after they passed the injury site (Fig. 5C–D′), supporting the notion that they had been regenerated rather than spared. To assess axon regeneration, we quantified the axon Number Index as the number of axons crossing specific distances in relevance to the injury site, summed from every other sagittal section throughout the mediolateral axis and then normalized against the total number of CST axons labeled in the medulla for each mouse.
Within the GFAP sparse lesion core, while there were barely any axons detected within the lesion core in control or Nogo-deleted mice, both PTEN and PTEN/Nogo-deleted mice exhibited a high number of CST axons in the lesion core (Fig. 5G). There was a trend for a higher number of CST axons within the lesion core in PTEN/Nogo-deleted mice compared with PTEN-deleted mice that approached significance (PTEN KO, 0.027 ± 0.010; double KO, 0.044 ± 0.011. p = 0.087).
Caudal to the injury site, there were minimal numbers of axons in control or Nogo-deleted mice (Fig. 5F). In contrast, both PTEN and PTEN/Nogo-deleted mice exhibited significantly higher numbers of axons beyond the injury site (Fig. 5F). Interestingly, whereas most regenerating axons in PTEN-deleted mice dropped off by ∼1 mm beyond injury at 6 weeks after injury, some CST axons clearly extended beyond 1.5 mm in PTEN/Nogo doubly deleted mice. Thus, PTEN/Nogo-deleted mice appeared to extend their axons for longer distances than PTEN-deleted mice (Fig. 5F). Indeed, beyond 1 mm caudal to injury (approximately half of the maximal regeneration length), the cumulative axon Number Index was significantly higher in PTEN/Nogo-deleted mice than PTEN-deleted mice (0.022 ± 0.013 and 0.002 ± 0.001, respectively, p < 0.05; Fig. 5H). At 1250 μm caudal to the injury, ∼55% of PTEN/Nogo-deleted mice had regenerating axons, whereas only 27% of PTEN-deleted mice did, reinforcing the notion that additional Nogo deletion increased regeneration in PTEN-deleted mice.
Because of the significant increase of axon regeneration in the PTEN and PTEN/Nogo-deleted mice, we tested whether this regeneration was linked with improved functional recovery. Several behavioral tests were performed, including the open field locomotion test (the BMS score and subscore) (Basso et al., 2006), the rotarod test (Lee et al., 2010), and the uneven spacing ladder rung test (Metz and Whishaw, 2009). None of these tests revealed an improved functional recovery for PTEN or PTEN/Nogo-deleted mice (Fig. 6), despite their significantly enhanced CST regeneration.
Figure 6.

Assessing recovery of hindlimb function after neonatal PTEN deletion and T8 dorsal hemisection spinal cord injury. A, B, Open field locomotion test, giving the BMS score (A) and the BMS subscore (B). C, Rotarod test. Latency to fall is plotted as a percentage of the preinjury level. D, Ladder rung test, plotting the percentage of correct steps. N = 14 (control), N = 11 (Nogo KO), N = 11 (PTEN KO), N = 9 (double KO). For all the tests performed, there were no significant difference among genotypes at any time point. Two-way ANOVA followed by Bonferroni's post hoc test.
Nogo-A is upregulated at and around the injury site
The data above suggest that Nogo may act at and/or around the injury site specifically to suppress axonal growth. To examine this possibility, we analyzed Nogo-A expression pattern at and around the injury site over time after T8 dorsal hemisection injury, as there were conflicting data on whether Nogo-A is upregulated after injury (Huber et al., 2002; Wang et al., 2002). In wild-type mice, in addition to the diffuse pattern of normal Nogo-A expression in the gray and white matter, Nogo-A was also upregulated at and around the dorsal hemisection injury site (Fig. 7A–C). As a control, Nogo-A immunoreactivity was not detected beyond background levels anywhere in the cord before or at any of the time points examined after injury in Nogo KO mice (e.g., compare Fig. 7D with 7A; and 7E with 7B).
Figure 7.
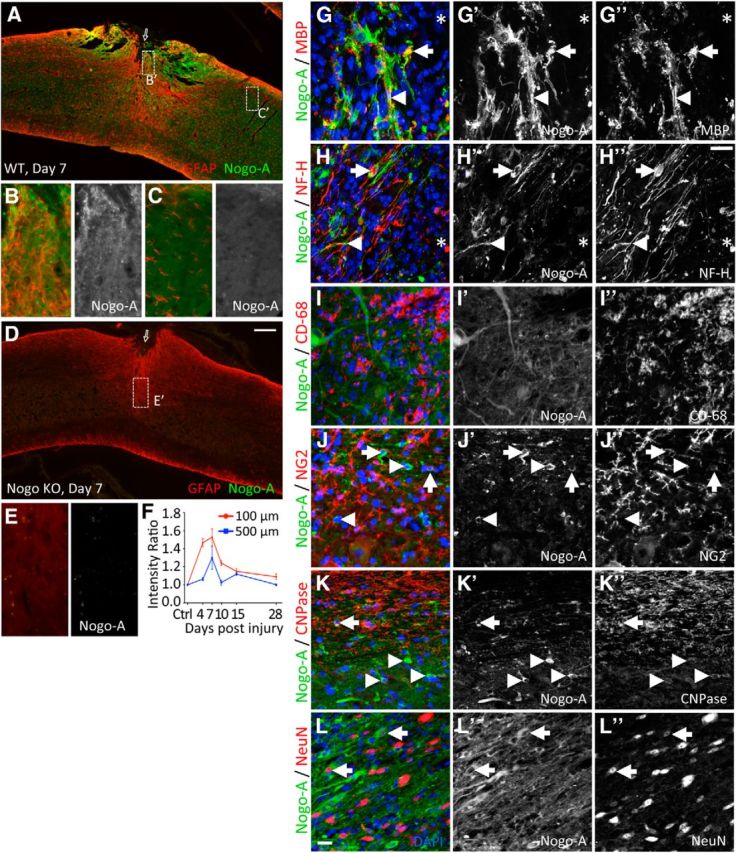
Characterization of Nogo-A expression after T8 dorsal hemisection spinal cord injury. A–C, Representative images of spinal cord sagittal sections from a wild-type (WT) mouse costained for GFAP and Nogo-A 7 d after T8 dorsal hemisection. B, C, Larger-magnification images of boxes (B′,C′, ∼100 μm and 1 mm, respectively, from the injury site, marked by an arrow) in A. D, E, Nogo KO as negative control for Nogo-A immunoreactivity. F, Quantification of Nogo-A immunoreactivity (arbitrary unit) at 100 and 500 μm caudal to the injury site, normalized to Nogo-A immunoreactivity at 1 mm caudal to injury. Three mice were analyzed per time point, with three sections examined per mouse. G–L, Confocal images sagittal section of wild-type spinal cords examining coexpression of Nogo-A with various cellular markers at (G, H) or close to (I–L) the injury site 7 d after injury. G–H″, *Injury site. G, MBP, myelin marker. Arrow indicates coexpression. Arrowhead indicates closely juxtaposed signals. H, NF-H, marker for neurons, including neuronal processes. Arrow indicates coexpression in neuronal soma. Arrowhead indicates colocalization in axon. I, CD-68, macrophage/microglia marker. No colocalization with Nogo-A. J, NG2, marker for NG2 or oligodendrocyte precursor cells. Arrow indicates coexpressing cells. Arrowhead indicates Nogo-A-positive but NG2-negative cells. K, CNPase, oligodendrocyte marker. Arrow indicates coexpressing cells in the white matter. Arrowhead indicates coexpressing cells in the gray matter. L, NeuN, Neuronal nuclear antigen. Arrow indicates coexpressing cells in the gray matter. Scale bars: A, D, 200 μm; B, C, E, G–H″, I–L″, 20 μm.
To quantify this upregulation, we measured Nogo-A immunoreactivity intensity at 100 and 500 μm away from the GFAP sparse lesion core over multiple time points after dorsal hemisection injury. Nogo-A expression around the injury site rapidly increased at 4 d and peaked at 7 d after injury (Fig. 7F). The level of Nogo-A immunoreactivity dropped significantly by day 28, but this region remained slightly more immunoreactive for Nogo-A relative to the distant areas or preinjury levels even at 42 d after injury (data not shown). Together, this temporal and spatial expression pattern of Nogo-A indicates that a local upregulation of Nogo-A at and around the injury site may limit the number of axons growing in this region, and that its removal might allow CST axons from PTEN deleted neurons to regenerate further.
We next characterized the cell type origin of Nogo-A upregulation, focusing on day 7 after injury when Nogo-A expression appeared to be at its maximum. Nogo-A and GFAP did not colocalize. Rather, Nogo-A positive processes and GFAP-positive processes interdigitated at and around the injury site (e.g., Fig. 7B), indicating distinct cell type origins for the two proteins. Likewise, the increase of Nogo-A expression at the injury site was neither associated with macrophages/microglia as indicated by CD68 immunoreactivity (Fig. 7I–I″) nor with meningeal fibroblasts as indicated by fibronectin immunoreactivity (data not shown).
Nogo-A has been previously associated with oligodendrocyte markers (Wang et al., 2002; Simonen et al., 2003). Indeed, we found CNPase-positive cells coexpressing Nogo-A in both the white and gray matter, as illustrated in high-magnification confocal images (Fig. 7K–K″). As previously shown (Huber et al., 2002), MBP and Nogo-A were mostly nonoverlapping, but a small amount of overlap could be seen along with prevalent juxtaposed signals (Fig. 7G–G″). NG2-positive cells occasionally coexpressed Nogo-A (Fig. 7J–J″).
Consistent with previous reports on neuronal and axonal expression of Nogo-A (Wang et al., 2002; Vajda et al., 2015), we found some NF-H (neurofilament heavy) and Nogo-A coexpression at the vicinity of the injury site in both cell bodies and axon-like processes (Fig. 7H–H″). In the gray matter close to the injury site, strong Nogo-A expression was also clearly associated with a subset of NeuN-positive cells where Nogo-A was predominantly cytoplasmic and NeuN was predominantly nuclear (Fig. 7L–L″). Together, these data indicate that Nogo-A upregulation around the injury site was of oligodendroglial and neuronal origins but not of astrocytic, macrophagial/microglial, or fibroblastic origins.
Nogo deletion further enhanced CST regeneration elicited by young adult PTEN deletion
Neonatal PTEN deletion elicited a robust regenerative response in CST neurons, which may have produced a ceiling effect on the regeneration observed, blunting the effect of Nogo deletion. In addition, assessing the effect of adult PTEN deletion is more important, as neonatal PTEN deletion may lead to gene compensation and developmental alterations that could confound the interpretation of regeneration data. We therefore also tested the effect of deleting PTEN in young adult mice in combination with Nogo deletion.
Similar to what is described above after neonatal PTEN deletion, we observed a clear distinction in the density of BDA-labeled CST axons just rostral to the injury site that proved to be PTEN dependent (Fig. 8A–D′). The usual die-back pattern for hundreds of micrometers rostral to the injury site in control and Nogo-deleted mice were supplanted by robust growth at high density into the GFAP dense area of reactive astrogliosis, abutting the GFAP sparse lesion core (Fig. 8A–D′). Quantitative analyses confirmed that PTEN and PTEN/Nogo-deleted mice exhibited significantly higher axon Density Indices than control and Nogo-deleted mice, starting from the rostral end of the lesion core all the way to 0.6 mm rostral to injury. However, further deleting Nogo did not increase CST axon growth rostral to the injury site (Fig. 8E), as we had observed with neonatal PTEN deletion (see above). It is noteworthy that the values of axon Density Indices are comparable with those from the neonatal PTEN-deleted groups, suggesting a similar effect on axon growth rostral to injury between neonatal and young adult PTEN deletion.
Figure 8.
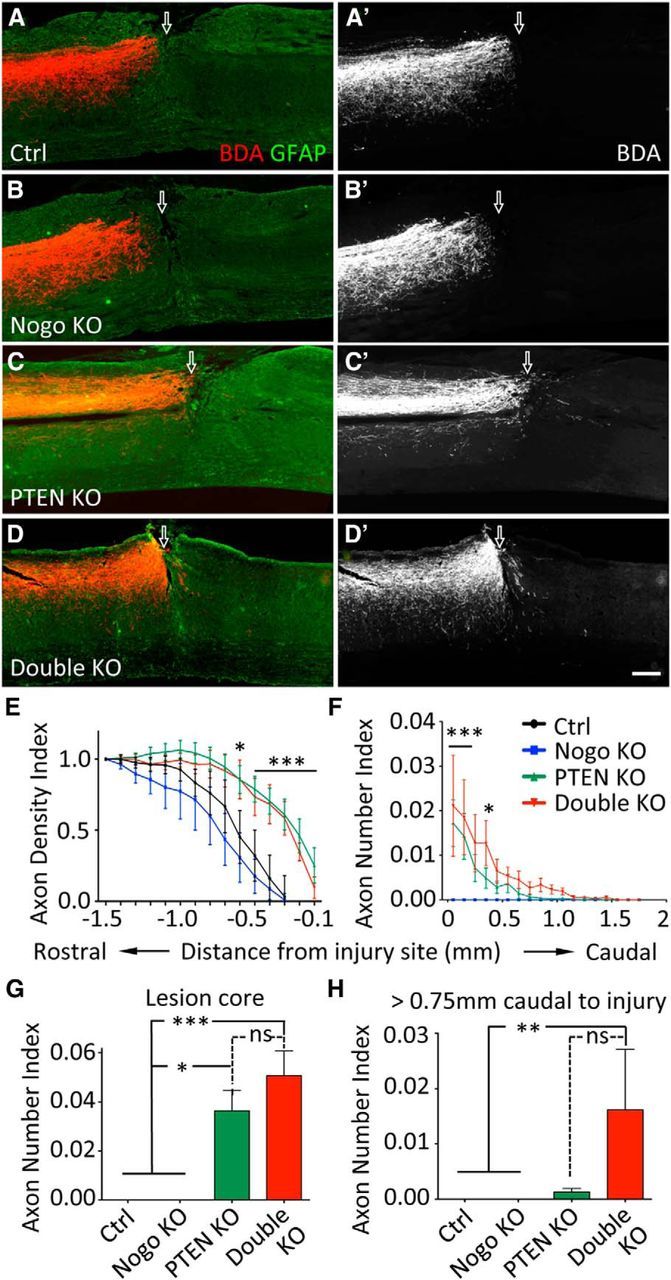
CST axon regeneration after dorsal hemisection spinal cord injury with young adult PTEN deletion. A–D, Representative images of thoracic spinal cord sagittal sections from mice of different genotypes after a T8 dorsal hemisection. PTEN deletion was initiated in 4- to 6-week-old mice. Scale bar, 200 μm. Arrow indicates injury site. Note the larger gap between BDA-labeled CST axon endings and the injury site in A and B compared with that in C and D. E, Axon Density Index rostral to the injury site, quantified as the staining intensity of axon tracer BDA in function to the distance from the injury site, averaged for three adjacent sagittal sections containing the main CST and normalized against the level at 1.5 mm rostral to injury for each mouse. F, Axon Number Index caudal to the injury site, quantified as the number of axons crossing specific distances, summed from every other sagittal section throughout the mediolateral axis and then normalized against the total number of CST axons labeled in the medulla for each mouse. Axons need to exit the injury site and extend into the GFAP dense area caudal to the injury site to be considered regenerated. G, Axon Number Index within the lesion core. H, Cumulative Axon Number Index past 0.75 mm caudal to the injury. N = 8 (control), N = 6 (Nogo KO), N = 14 (PTEN KO), N = 9 (double KO). *p < 0.05. **p < 0.01. ***p < 0.001. Two-way (E, F) or one-way (G,H) ANOVA followed by Bonferroni's post hoc test. ns, Not significant.
Within the injury site, PTEN-deleted mice had substantially more CST axons than control or Nogo-deleted mice. PTEN/Nogo doubly deleted mice had a slightly higher number of axons than PTEN-deleted mice, but this difference did not reach statistical significance (Fig. 8G; PTEN KO, 0.036 ± 0.08; double KO, 0.050 ± 0.010. p = 0.16). Caudal to the injury, PTEN and PTEN/Nogo-deleted mice exhibited significantly more axons than control or Nogo-deleted mice, and the number of regenerating axons appeared to drop more quickly in PTEN-deleted mice than PTEN/Nogo-deleted mice (Fig. 8F). When only axons growing beyond 0.75 mm past the injury site (approximately half of the maximal regeneration length observed) were quantified, there was a noticeable difference between PTEN and PTEN/Nogo-deleted mice that approached significance (PTEN KO, 0.0013 ± 0.006; double KO, 0.016 ± 0.011. p = 0.065) (Fig. 8H). At 0.75 mm caudal to the injury, 55% of PTEN/Nogo-deleted mice still had regenerating axons, whereas only 21% of PTEN-deleted mice did. Even though this was not a side-by-side comparison, the level of regeneration as quantified by distance and axon number was apparently lower in young adult PTEN-deleted mice than neonatal PTEN-deleted mice (compare Figs. 5F and 8F). Regardless of whether PTEN deletion was initiated at the neonatal or young adult stage, additionally deleting Nogo further enhanced CST regeneration that was elicited by PTEN deletion.
Finally, we determined whether this increase of CST regeneration was paralleled with improved functional recovery. As described for the neonatal PTEN deletion study, we performed the open field locomotion (BMS), rotarod, and uneven spacing ladder rung tests (Fig. 9). None of these tests detected improved functional recovery in PTEN or PTEN/Nogo-deleted mice despite their significantly enhanced CST regeneration.
Figure 9.

Assessing recovery of hindlimb function after young adult PTEN deletion and T8 dorsal hemisection spinal cord injury. A, B, Open field locomotion test, giving the BMS score (A) and the BMS subscore (B). C, Rotarod test. Latency to fall is plotted as a percentage of the preinjury level. D, Ladder rung test, plotting the percentage of correct steps. N = 8 (control), N = 6 (Nogo KO), N = 14 (PTEN KO), N = 9 (double KO). For all the tests performed, there were no significant difference among genotypes at any time point. Two-way ANOVA followed by Bonferroni's post hoc test.
Discussion
Here we tested the hypothesis that manipulating both neuron-intrinsic and -extrinsic factors may further enhance axon sprouting and regeneration after CNS injury by genetically codeleting PTEN and Nogo. In an apparent paradox with our previous studies of Nogo and PTEN single deletion mice (Lee et al., 2010; Liu et al., 2010), additionally deleting Nogo in PTEN-deleted mice further enhanced CST regeneration but not sprouting. These data indicate that neuron-intrinsic and -extrinsic factors regulate axon regeneration and sprouting in complex ways and provide proof-of-principle evidence that manipulating both can further improve axon regeneration. Despite clear enhancement in axonal growth, no or only limited improvement in behavioral recovery was detected, highlighting the need for additional strategies to fully use the enhanced axonal growth for functional gains.
A comparison of PTEN versus Nogo deletion on CST regeneration and sprouting
Our study represents the first side-by-side comparison between PTEN deletion and Nogo deletion on CST regeneration and sprouting, using age-, sex-, and background-matched littermates. Nogo deletion had a modest effect on CST sprouting (<50% enhancement), in general agreement with our previous study (Lee et al., 2010). Such a modest effect may not always be detected with statistical significance due to the inherent variability in CST labeling and baseline sprouting. Nevertheless, the trend for enhanced CST sprouting in Nogo-deleted mice is consistently observed across independent studies (Cafferty and Strittmatter, 2006; Lee et al., 2010). Other extrinsic inhibitors, such as chondroitin sulfate proteoglycans, may functionally compensate for the loss of Nogo (Starkey et al., 2012). A modest level of enhancement in sprouting, if widespread, may still have significant functional consequences. Deleting Nogo had no detectable effect on CST regeneration, consistent with our previous findings (Zheng et al., 2003; Lee et al., 2009, 2010). Although there are conflicting reports in the literature (Kim et al., 2003; Simonen et al., 2003; Zheng et al., 2003; Cafferty et al., 2007; Steward et al., 2007; for review, see Lee and Zheng, 2012), our results echo genetic studies of other components in the myelin inhibitory pathways, including MAG, OMgp, NgR1, p75NTR, and PirB (Bartsch et al., 1995; Kim et al., 2004; Song et al., 2004; Zheng et al., 2005; Ji et al., 2008; Nakamura et al., 2011).
In contrast, PTEN deletion had a strong effect on both CST sprouting and regeneration. Deleting PTEN in young adult mice enhances CST sprouting, reinforcing development-independent function of PTEN in regulating injury-induced axonal growth. Either neonatal or young adult PTEN deletion increased CST sprouting by twofold to threefold. Although PTEN deletion clearly and consistently promotes CST regeneration, the degree of regeneration in the current study may not appear as visually striking as in our previous study (Liu et al., 2010). A number of variables influence the observed CST regeneration after PTEN deletion, including gene deletion efficiency, injury type and depth, survival time after injury, axon tracing efficiency, and staining intensity. Even in the same study, the degree of regeneration observed may vary in different injury experiments due to such variables (Liu et al., 2010).
Initiating PTEN deletion in young adult mice led to less CST regeneration than initiating PTEN deletion in neonatal mice. AAV infection is more efficient and covers a larger cortical area in neonatal mice. Our unpublished data using the ROSA26-lsl-tdTomato reporter line indicate that AAV infection rate is significantly higher among the BDA-labeled axons in P1 infected mice than in young adult infected mice (∼70% vs ∼45%). In addition, the regeneration-inducing effect of PTEN deletion may decline with increasing age. This could be due to changes in extrinsic inhibitors, in neuronal responses to extrinsic inhibitors, and/or in other neuron-intrinsic pathways that regulate axon regeneration in an age-dependent manner (Byrne et al., 2014).
Concentration-dependent inhibitory effect of Nogo
Additionally deleting Nogo did not further enhance CST sprouting but further increased CST regeneration in PTEN-deleted mice. The effect of Nogo deletion was most noticeable at and around the injury site where regenerating CST axons traveled further in Nogo/PTEN doubly deleted mice. Intriguingly, Nogo-A expression was also upregulated in neurons and oligodendroglia at and around the injury site after dorsal hemisection. Together, our data are consistent with the scenario that PTEN-deleted CST neurons are not sensitive to the widespread, basal level of Nogo-A throughout the cord but remain sensitive to the highly concentrated Nogo-A at and around a spinal cord injury site. Consequently, the absence of this Nogo-A upregulation (by Nogo deletion) may allow regenerating axons in PTEN-deleted mice to grow further past the injury site. In contrast, sprouting CST axons in PTEN-deleted mice may have a sufficiently high level of growth ability that they could simply ignore the widespread, basal level of Nogo-A. Thus, it appears that a higher concentration of Nogo-A is required to inhibit axon growth from CST neurons upon PTEN deletion.
Neuron-intrinsic growth state determines neuronal responsiveness to changes in extrinsic inhibition
In the presence of PTEN, deleting Nogo has a detectable effect on the sprouting of uninjured CST neurons but not the regeneration of injured CST neurons. In the absence of PTEN where CST regeneration and sprouting are already enhanced, additionally deleting Nogo further increases CST regeneration but not sprouting. This complex pattern of regulation of CST regeneration and sprouting by axotomy, PTEN, and Nogo prompted us to propose a working model where the level of neuron-intrinsic growth ability determines neuronal sensitivity to extrinsic growth inhibition and consequently neuronal responsiveness to changes in extrinsic inhibition (Fig. 10).
Figure 10.

Neurons of different intrinsic growth abilities are differentially sensitive to extrinsic inhibition: a working hypothesis. Circle represents neuronal cell body. Line indicates axon. Axon growth from neurons of different intrinsic growth abilities gets inhibited at different concentrations of extrinsic inhibitors, as indicated by the places at which axon growth stops along a hypothetic upward gradient of extrinsic inhibitors from left to right. Neurons with a reduced (red), basal (yellow), and elevated (green) level of intrinsic growth ability are inhibited by a reduced, basal, and elevated level of extrinsic inhibitors, respectively. For the red neuron (e.g., injured adult CST neurons), a reduced level of extrinsic inhibition (Nogo deletion) may still be too challenging for axonal growth (regeneration); this neuron will appear unresponsive to a reduction in extrinsic inhibition. For the yellow neuron (e.g., uninjured adult CST neurons), reducing extrinsic inhibition from a basal level (Nogo deletion) would help axonal growth (sprouting). For the green neuron (e.g., PTEN-deleted CST neurons), reducing extrinsic growth inhibition from “elevated” to “basal” (Nogo deletion) would help axonal growth (regeneration); reducing extrinsic growth inhibition from “basal” to “reduced” (Nogo deletion) may not impact on axonal growth (sprouting). This complex interplay between neuron-intrinsic and -extrinsic control may also explain the apparently more robust effect of PTEN/Nogo codeletion with neonatal PTEN deletion compared with young adult PTEN deletion.
Intact adult CST neurons, with a basal level of intrinsic axon growth ability (Fig. 10, yellow neuron), are sensitive to the basal level of extrinsic growth inhibitors. Reducing this inhibition (e.g., by Nogo deletion) can enhance axon sprouting from these neurons. Axonal injury downregulates neuron-intrinsic growth ability of CST neurons (Fig. 10, red neuron). These neurons then become sensitive even to a reduced level of extrinsic inhibition such that they are unable to regrow axons even in the absence of Nogo. Following PTEN deletion, neurons acquire an elevated level of intrinsic growth ability (Fig. 10, green neuron), and consequently are only sensitive to an elevated level of growth inhibition, such as that provided by upregulated Nogo-A at and around an injury site. A reduction of extrinsic inhibition by Nogo deletion may thus further enhance CST regeneration. Meanwhile, these neurons are no longer sensitive to the basal level of extrinsic inhibitors encountered by sprouting axons such that Nogo deletion does not further enhance sprouting. Therefore, both axonal injury and molecular manipulations of signaling pathways (e.g., PTEN/mTOR) can alter neuron-intrinsic growth state, which in turn regulates neuronal responsiveness to changes in extrinsic inhibitors. This seemingly complex interplay between neuron-intrinsic and -extrinsic control of injury-induced axonal growth may unify many divergent observations in the field of CNS axon repair. The recognition that a certain level of neuron-intrinsic growth state is required before altering extrinsic influence can become effective may prove to be a guiding principle in developing therapeutic strategies to promote axonal repair after spinal cord injury.
Limited functional recovery associated with enhanced CST sprouting but not regeneration
CST sprouting and regeneration were significantly enhanced in PTEN and PTEN/Nogo-deleted mice. Associated with the enhanced sprouting was a modest, temporary improvement in one of three behavioral assays, whereas no improvement over control mice was observed in any of the three behavioral assays with the regeneration model. Thus, clear enhancement of CST regeneration and sprouting is not necessarily accompanied by robust and persistent improvement in behavioral recovery.
There are several considerations when interpreting these results. First, there is significant baseline sprouting from CST and other tracts (Raineteau et al., 2001, 2002; Morris et al., 2011; Hurd et al., 2013) that are associated with spontaneous functional recovery after pyramidotomy. Second, our tests might not be sensitive and/or specific enough to detect more subtle but persistent functional improvement in PTEN and PTEN/Nogo-deleted mice. Third, the number of regenerating CST axons in PTEN and PTEN/Nogo-deleted mice is still low (no more than a few percent compared with pre injury), and the distance of regeneration limited (<2 mm in 6 weeks). Fourth, deleting PTEN in both sides of the cortex may be required to promote functional recovery after spinal cord injury.
Regardless, the lack of robust behavioral improvement in mice where CST sprouting and regeneration are clearly enhanced after injury indicates the need for additional strategies to promote functional recovery. Recent studies implicated the importance of task-relevant rehabilitation (García-Alías et al., 2009), of the relative timing between growth promoting treatment and rehabilitative training (Maier et al., 2009; Marsh et al., 2011; Zhao et al., 2013; Wahl et al., 2014), and of an enhanced level of spinal cord excitability in combination with rehabilitative training (van den Brand et al., 2012; Angeli et al., 2014). These mechanisms remain to be explored to take full advantage of the anatomical substrates provided by enhanced CST regeneration and sprouting following manipulations of both neuron-intrinsic and -extrinsic factors as described here.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant R01NS054734, California Institute for Regenerative Medicine RB3-02143, Wings for Life Spinal Cord Research Foundation, and Roman Reed Spinal Cord Injury Research Fund of California to B.Z., C.G.G. was a recipient of a Craig H. Neilsen Foundation Postdoctoral Fellowship. A.O.L. was a recipient of NRSA Predoctoral Fellowship F31NS074867. Confocal imaging was performed at the University of California San Diego, Neuroscience Microscopy Imaging Core (P30NS047101). We thank Oliver Tress, Meifan Chen, and Jessica Meves for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Angeli CA, Edgerton VR, Gerasimenko YP, Harkema SJ. Altering spinal cord excitability enables voluntary movements after chronic complete paralysis in humans. Brain. 2014;137:1394–1409. doi: 10.1093/brain/awu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch U, Bandtlow CE, Schnell L, Bartsch S, Spillmann AA, Rubin BP, Hillenbrand R, Montag D, Schwab ME, Schachner M. Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron. 1995;15:1375–1381. doi: 10.1016/0896-6273(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, McMahon SB. Spinal cord repair strategies: why do they work? Nat Rev Neurosci. 2006;7:644–653. doi: 10.1038/nrn1964. [DOI] [PubMed] [Google Scholar]
- Byrne AB, Walradt T, Gardner KE, Hubbert A, Reinke V, Hammarlund M. Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron. 2014;81:561–573. doi: 10.1016/j.neuron.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Strittmatter SM. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci. 2006;26:12242–12250. doi: 10.1523/JNEUROSCI.3827-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Kim JE, Lee JK, Strittmatter SM. Response to correspondence: Kim et al., “axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38:187–199.”. Neuron. 2007;54:195–199. doi: 10.1016/j.neuron.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Duffy P, Huebner E, Strittmatter SM. MAG and OMgp synergize with Nogo-A to restrict axonal growth and neurological recovery after spinal cord trauma. J Neurosci. 2010;30:6825–6837. doi: 10.1523/JNEUROSCI.6239-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alías G, Barkhuysen S, Buckle M, Fawcett JW. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci. 2009;12:1145–1151. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- Geoffroy CG, Zheng B. Myelin-associated inhibitors in axonal growth after CNS injury. Curr Opin Neurobiol. 2014;27C:31–38. doi: 10.1016/j.conb.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grider MH, Chen Q, Shine HD. Semi-automated quantification of axonal densities in labeled CNS tissue. J Neurosci Methods. 2006;155:172–179. doi: 10.1016/j.jneumeth.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Shah RR, Chan AF, Zheng B. EphA4 deficient mice maintain astroglial-fibrotic scar formation after spinal cord injury. Exp Neurol. 2010;223:582–598. doi: 10.1016/j.expneurol.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brösamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd C, Weishaupt N, Fouad K. Anatomical correlates of recovery in single pellet reaching in spinal cord injured rats. Exp Neurol. 2013;247:605–614. doi: 10.1016/j.expneurol.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Ji B, Case LC, Liu K, Shao Z, Lee X, Yang Z, Wang J, Tian T, Shulga-Morskaya S, Scott M, He Z, Relton JK, Mi S. Assessment of functional recovery and axonal sprouting in oligodendrocyte-myelin glycoprotein (OMgp) null mice after spinal cord injury. Mol Cell Neurosci. 2008;39:258–267. doi: 10.1016/j.mcn.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPré T, Qiu D, Strittmatter SM. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron. 2003;38:187–199. doi: 10.1016/S0896-6273(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Lee JK, Zheng B. Role of myelin-associated inhibitors in axonal repair after spinal cord injury. Exp Neurol. 2012;235:33–42. doi: 10.1016/j.expneurol.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Chan AF, Luu SM, Zhu Y, Ho C, Tessier-Lavigne M, Zheng B. Reassessment of corticospinal tract regeneration in Nogo-deficient mice. J Neurosci. 2009;29:8649–8654. doi: 10.1523/JNEUROSCI.1864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Geoffroy CG, Chan AF, Tolentino KE, Crawford MJ, Leal MA, Kang B, Zheng B. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 2010;66:663–670. doi: 10.1016/j.neuron.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Belin S, He Z. Signaling regulations of neuronal regenerative ability. Curr Opin Neurobiol. 2014;27C:135–142. doi: 10.1016/j.conb.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier IC, Ichiyama RM, Courtine G, Schnell L, Lavrov I, Edgerton VR, Schwab ME. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain. 2009;132:1426–1440. doi: 10.1093/brain/awp085. [DOI] [PubMed] [Google Scholar]
- Marsh BC, Astill SL, Utley A, Ichiyama RM. Movement rehabilitation after spinal cord injuries: emerging concepts and future directions. Brain Res Bull. 2011;84:327–336. doi: 10.1016/j.brainresbull.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Metz GA, Whishaw IQ. The ladder rung walking task: a scoring system and its practical application. J Vis Exp. 2009;28:pii1204. doi: 10.3791/1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R, Tosolini AP, Goldstein JD, Whishaw IQ. Impaired arpeggio movement in skilled reaching by rubrospinal tract lesions in the rat: a behavioral/anatomical fractionation. J Neurotrauma. 2011;28:2439–2451. doi: 10.1089/neu.2010.1708. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Fujita Y, Ueno M, Takai T, Yamashita T. Paired immunoglobulin-like receptor B knockout does not enhance axonal regeneration or locomotor recovery after spinal cord injury. J Biol Chem. 2011;286:1876–1883. doi: 10.1074/jbc.M110.163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O, Fouad K, Noth P, Thallmair M, Schwab ME. Functional switch between motor tracts in the presence of the mAb IN-1 in the adult rat. Proc Natl Acad Sci U S A. 2001;98:6929–6934. doi: 10.1073/pnas.111165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O, Fouad K, Bareyre FM, Schwab ME. Reorganization of descending motor tracts in the rat spinal cord. Eur J Neurosci. 2002;16:1761–1771. doi: 10.1046/j.1460-9568.2002.02243.x. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Strittmatter SM. Nogo limits neural plasticity and recovery from injury. Curr Opin Neurobiol. 2014;27:53–60. doi: 10.1016/j.conb.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron. 2003;38:201–211. doi: 10.1016/S0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- Song XY, Zhong JH, Wang X, Zhou XF. Suppression of p75NTR does not promote regeneration of injured spinal cord in mice. J Neurosci. 2004;24:542–546. doi: 10.1523/JNEUROSCI.4281-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkey ML, Barritt AW, Yip PK, Davies M, Hamers FP, McMahon SB, Bradbury EJ. Assessing behavioural function following a pyramidotomy lesion of the corticospinal tract in adult mice. Exp Neurol. 2005;195:524–539. doi: 10.1016/j.expneurol.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Starkey ML, Bartus K, Barritt AW, Bradbury EJ. Chondroitinase ABC promotes compensatory sprouting of the intact corticospinal tract and recovery of forelimb function following unilateral pyramidotomy in adult mice. Eur J Neurosci. 2012;36:3665–3678. doi: 10.1111/ejn.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Zheng B, Banos K, Yee KM. Response to: Kim et al., “axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38, 187–199.”. Neuron. 2007;54:191–195. doi: 10.1016/j.neuron.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M, Hofstadter M, Sharp K, Yee KM. Regenerative growth of corticospinal tract axons via the ventral column after spinal cord injury in mice. J Neurosci. 2008;28:6836–6847. doi: 10.1523/JNEUROSCI.5372-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci. 1998;1:124–131. doi: 10.1038/373. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Steward O. Concepts and methods for the study of axonal regeneration in the CNS. Neuron. 2012;74:777–791. doi: 10.1016/j.neuron.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajda F, Jordi N, Dalkara D, Joly S, Christ F, Tews B, Schwab ME, Pernet V. Cell type-specific Nogo-A gene ablation promotes axonal regeneration in the injured adult optic nerve. Cell Death Diff. 2015;22:323–335. doi: 10.1038/cdd.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brand R, Heutschi J, Barraud Q, DiGiovanna J, Bartholdi K, Huerlimann M, Friedli L, Vollenweider I, Moraud EM, Duis S, Dominici N, Micera S, Musienko P, Courtine G. Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science. 2012;336:1182–1185. doi: 10.1126/science.1217416. [DOI] [PubMed] [Google Scholar]
- Wahl AS, Omlor W, Rubio JC, Chen JL, Zheng H, Schröter A, Gullo M, Weinmann O, Kobayashi K, Helmchen F, Ommer B, Schwab ME. Neuronal repair: asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 2014;344:1250–1255. doi: 10.1126/science.1253050. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao RR, Andrews MR, Wang D, Warren P, Gullo M, Schnell L, Schwab ME, Fawcett JW. Combination treatment with anti-Nogo-A and chondroitinase ABC is more effective than single treatments at enhancing functional recovery after spinal cord injury. Eur J Neurosci. 2013;38:2946–2961. doi: 10.1111/ejn.12276. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M. Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron. 2003;38:213–224. doi: 10.1016/S0896-6273(03)00225-3. [DOI] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M. Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Lee JK, Xie F. Genetic mouse models for studying inhibitors of spinal axon regeneration. Trends Neurosci. 2006;29:640–646. doi: 10.1016/j.tins.2006.09.005. [DOI] [PubMed] [Google Scholar]