

Abstract
Alzheimer's disease (AD) is characterized by a robust inflammatory response elicited by the accumulation and subsequent deposition of amyloid (Aβ) within the brain. The brain's immune cells migrate to and invest their processes within Aβ plaques but are unable to efficiently phagocytose and clear plaques from the brain. Previous studies have shown that treatment of myeloid cells with nuclear receptor agonists increases expression of phagocytosis-related genes. In this study, we elucidate a novel mechanism by which nuclear receptors act to enhance phagocytosis in the AD brain. Treatment of murine models of AD with agonists of the nuclear receptors PPARγ, PPARδ, LXR, and RXR stimulated microglial phagocytosis in vitro and rapidly induced the expression of the phagocytic receptors Axl and MerTK. In murine models of AD, we found that plaque-associated macrophages expressed Axl and MerTK and treatment of the cells with an RXR agonist further induced their expression, coincident with the rapid reduction in plaque burden. Further characterization of MerTK+/Axl+ macrophages revealed that they also expressed the phagocytic receptor TREM2 and high levels of CD45, consistent with a peripheral origin of these cells. Importantly, in an ex vivo slice assay, nuclear receptor agonist treatment reversed the AD-related suppression of phagocytosis through a MerTK-dependent mechanism. Thus, nuclear receptor agonists increase MerTK and Axl expression on plaque-associated immune cells, consequently licensing their phagocytic activity and promoting plaque clearance.
Keywords: Alzheimer's disease, microglia, nuclear receptors, phagocytosis
Introduction
Inflammation plays critical roles in the initiation and progression of Alzheimer's disease (AD) (Wyss-Coray, 2006). This has been highlighted by genetic studies that identify numerous immune mediators linked to AD risk (Jones et al., 2010; Hollingworth et al., 2011; Naj et al., 2011). Notably, variants of the microglial cell surface receptor TREM2 confer dramatically elevated risk for AD and other neurodegenerative diseases (Cuyvers et al., 2014). The AD brain is typified by the presence of abundant “activated” inflammatory macrophages that are found principally associated with extracellular deposits of amyloid (Akiyama et al., 2000). An enigmatic feature of the disease is that these “activated” macrophages are unable to mount a phagocytic response directed toward amyloid deposits, despite expression of phagocytic receptors, including TREM2, Axl, and MerTK. This results in the progressive accumulation of plaques within the brain.
The phenotypes of microglia and other myeloid cells are governed by the actions of heterodimeric Type II nuclear receptors, most prominently PPARγ:RXR, PPARδ:RXR, and LXR:RXR (Glass and Saijo, 2010). These ligand-activated transcription factors act to “license” complex cellular responses through their ability to coordinately regulate arrays of functionally allied genes (Hucke et al., 2012, 2013). In myeloid cells, nuclear receptors drive the acquisition of “alternative activation” states that are responsible for the resolution of inflammation (Glass et al., 2010), and the promotion of tissue repair and phagocytosis (Nagy et al., 2012). In murine models of AD, nuclear receptor agonist treatment suppresses inflammation, reduces amyloid accumulation, and improves cognition (Skerrett et al., 2014). The reduction in plaque burden is due to nuclear receptor stimulation of phagocytosis, and the present study implicates the phagocytic receptors Axl and MerTK in this response. Importantly, the expression of MerTK, Axl (Mukundan et al., 2009; A-Gonzalez et al., 2009), and TREM2 (Daniel et al., 2014) is directly regulated by nuclear receptor agonists.
The phagocytic receptors TREM2, MerTK, and Axl are expressed on myeloid cells, including microglia (Gautier et al., 2012; Butovsky et al., 2014). Axl and MerTK are TAM-family receptor tyrosine kinases that are required for the phagocytosis of apoptotic cells (Lemke, 2013). These receptors recognize exposed phosphatidylserine on the surface of apoptotic cells through the adaptor molecules Protein S or Gas6 (Hafizi and Dahlbäck, 2006). In tissue, Gas6 is often constitutively bound to Axl but does not signal in the absence of phosphatidyl serine (Zagórska et al., 2014). Gas6 binds to and can activate all TAM receptors, whereas Protein S cannot activate Axl efficiently, and the relevant ligand domains have recently been elucidated (Lew et al., 2014). TREM2 has been postulated to be a phagocytic receptor whose expression is also associated with suppression of inflammatory gene expression (Turnbull et al., 2006; Hsieh et al., 2009; N′Diaye et al., 2009).
Although MerTK, Axl, and TREM2 are expressed on plaque-associated macrophages in murine models of AD, these cells are phagocytically inactive. MerTK- and TREM2-expressing macrophages are also CD45hi, a canonical marker of peripherally derived “inflammatory” monocytes (Sedgwick et al., 1991). In allied work (Jay et al., 2015), the presence of plaque-associated macrophages in the AD brain was reliant upon the expression of TREM2, which was found to be exclusively expressed by CD45hi macrophages. Importantly, nuclear receptor activation enables phagocytic machinery within brain macrophages, licensing phagocytosis, resulting in the reduction in plaque burden in vivo.
Materials and Methods
Animals.
B6C3-Tg (APP/PS1Δe9) 85Dbo/J mice (Jankowsky et al., 2001) as well as B6SJL-Tg(5XFAD) mice (Oakley et al., 2006) were obtained from The Jackson Laboratory. At 12 months of age, APP/PS1Δe9 animals were treated with 100 mg/kg bexarotene (Targretin) or water by oral gavage for 5 d before slice phagocytosis, or 7 d (Cramer et al., 2012) before death. Targretin (75 mg) was dispersed in H2O to a concentration of 30 mg/ml, and animals received 100 μl of solution by oral gavage, resulting in a treatment of 100 mg/kg/d. At 4 months of age, 5XFAD mice were treated with 100 mg/kg/d bexarotene, 30 mg/kg/d GW0742 (GlaxoSmithKline), or water for 14 d. At 8 months of age, 5XFAD mice were treated with 100 mg/kg/d bexarotene for 7 d. An equal number of male and female animals were used for each experiment. Animals were housed and cared for according to Case Western Reserve University's Institutional Animal Care and Use Committee committee.
Nuclear receptor agonists.
The following nuclear receptor agonists were used: LXR agonist GW3965, PPARγ agonist pioglitazone, PPARδ agonist GW0742, and RXR agonist bexarotene. Drugs were dispersed in water for oral gavage (GW0742, bexarotene) or dissolved in DMSO for tissue culture.
Tissue collection.
Four hours after their last gavage, animals were killed and transcardially perfused with saline. The left hemispheres were fixed overnight in 4% PFA in phosphate buffer, while the cortices and hippocampi were dissected from the right hemispheres and processed for protein and Aβ extraction. The tissue was homogenized in tissue homogenization buffer (20 mm Tris, pH 7.4, 250 mm sucrose, 0.5 mm EDTA, 0.5 mm EGTA) using a tissue homogenizer. A sequential extraction of Aβ was performed and Aβ40 and 42 levels were analyzed by ELISA as previously described (Jiang et al., 2008). A second cohort of 12-month-old animals were processed for adult microglial isolation.
Microglial/macrophage isolation, RNA extraction, and qRT-PCR.
Adult microglia/macrophages were isolated as described previously (Cardona et al., 2006). Briefly, cortices and hippocampi were removed and dissociated using a sterile razorblade and enzymatically digested in RPMI media containing collagenase Type 3 (0.2%, Worthington) and dispase (2 U/ml, Invitrogen) for 45 min at 37°C. Digestion was stopped by adding 15 ml of PBS containing 2.5 mm EDTA and 1% FBS. Homogenates were passed through a 35 μm filter and centrifuged. Pellets were resuspended in a 30% Percoll solution and centrifuged to separate microglia and macrophages from other cell types. The purity of CD11b+/CD45+ macrophages was ∼70% as assessed by flow cytometry. After several PBS washes, cells were pelleted and frozen immediately at −80°C for mRNA extraction. mRNA was extracted using RNEasy spin columns (QIAGEN), genomic DNA was eliminated, and mRNA was reverse transcribed using a quantitect reverse transcription kit (QIAGEN). The resulting cDNA was preamplified using TaqMan preamp master mix (Applied Biosystems) and detected using a Step One plus and TaqMan assays (Applied Biosystems). Data presented represent relative quotient with 95% confidence intervals, and statistical analyses were performed on ΔCt values (Yuan et al., 2006).
Flow cytometry.
Adult microglia and macrophages were isolated and resuspended in FACS buffer (1% FBS in PBS) containing FC block for 15 min before incubation with appropriate antibodies. Cells were incubated with tagged antibodies for 30 min before washing, fixing, and analysis using a FACSAria. Analysis was done using FlowJo software. Cells were gated on CD11b+ populations and further separated into CD45lo and CD45hi cells. The following antibodies were used: anti-mouse TREM2 (R&D Systems, clone 237920), anti-mouse MerTK (R&D Systems, clone 108928), anti-mouse CD45 (eBioscience, clone 30-F11), and anti-mouse CD11b (eBioscience, clone M1/70).
Immunohistochemistry.
Tissue sections (10 μm) were rehydrated in PBS, followed by antigen retrieval in 10 mm sodium citrate or Reveal Decloaker (Biocare Medical) at 80°C for 20 min. Sodium citrate was used for all images where fluorescence intensity was measured, whereas Reveal Decloaker was used for TREM2 and confocal images. Sections were incubated in blocking solution (2% donkey serum and 0.1% Triton X-100 in PBS) for 1 h followed by incubation with primary antibody in blocking solution overnight at 4°C. Following several PBS rinses, sections were incubated with appropriate secondary antibodies (Invitrogen) and stained with DAPI. TREM2 fluorescence was visualized using tyramide amplification according to the manufacturer's instructions (PerkinElmer). Images were analyzed using ImageJ software. Mean fluorescence intensity of plaque-associated cells was measured and normalized to the vehicle-treated animals. The experimenter was blinded to genotype and treatment groups. The following antibodies were used: anti-mouse TREM2 (LSBio), anti-mouse MerTK (R&D Systems), anti-mouse Axl (R&D Systems), anti-mouse Gas6 (R&D Systems), anti-mouse Iba1 (Wako), and anti-mouse CD45 (BD Biosciences).
Cell culture.
The N9 mouse microglial cell line was grown in RPMI containing 5% heat-inactivated FBS and 1% penicillin/streptomycin. Cells were plated at a density of 500,000 cells/well in a 6-well plate for in vitro phagocytosis assays. Primary microglia were cultured from P0–3 C57BL/6J pups as previously described (McDonald et al., 1997). Briefly, neonatal pups were killed, and their brains removed, mechanically dissociated, and digested in 0.5% Trypsin-EDTA for 20 min at 37°C. Digestion was stopped by addition of 10% FBS in DMEM/F12 media containing 1% penicillin/streptomycin, and the homogenate was plated on 150 mm plates and allowed to grow for 14–21 d at 37°C and 5% CO2. Microglia were separated using a mild trypsin protocol (Saura et al., 2003) and switched to serum-free media 24 h before drug treatments.
Western blotting.
Protein levels of cell lysates or brain extracts were measured by BCA, and equal amounts of protein from each sample were resolved on 4%–12% Bis/Tris gels (Invitrogen). The following primary antibodies were used: goat anti-MerTK (R&D Systems), goat anti-Mouse Gas6 (R&D Systems), and goat anti-β-actin (Santa Cruz Biotechnology).
In vitro phagocytosis assay.
Amyloid was fibrillized by dissolving 1 mg Aβ1–42 in 220 μl endotoxin-free water and incubating for 5 d at 37°C. N9 or primary microglia were incubated with nuclear receptor agonists in serum-free media for 18 h. Following drug pretreatment, cells were treated ±2.5 μm fAβ or 40 nm Gas6 for 15 min before incubation with 1 μm Nile red-labeled polystyrene beads (Invitrogen) for 15 min. Immune IgG was used as a positive control to drive phagocytosis (Koenigsknecht-Talboo and Landreth, 2005). Cells were washed thoroughly with PBS, fixed in 2% PFA in PBS, and phagocytic cells were measured via flow cytometry using a BD FACSAria. Data were analyzed using FlowJo software and normalized to nontreated control cells.
Ex vivo phagocytosis assay.
Phagocytosis assays were was performed in brain slices as described previously (Krabbe et al., 2013). Briefly, mice were treated with vehicle or 100 mg/kg/d bexarotene for 5 d before death. Mice were lightly anesthetized with isoflurane, killed by cervical dislocation, and brains were removed and 150 μm slices prepared using a Leica vibratome. After resting in RPMI for 30–60 min, slices were incubated with 2 μm fluorescent beads alone or in the presence of recombinant Gas6 or MerTK function blocking antibodies. Following incubation, slices were washed for 1 h, fixed, and stained using anti-Iba1 to identify myeloid cells. Slices were imaged on a Leica confocal microscope and analyzed using ImageJ. The number of beads per cell were counted, and the phagocytic index was calculated by determining the percentage of cells containing 0, 1–4, 5–7, 8–10, or >10 beads. The percentage of cells in each group was then multiplied by the grade of phagocytosis (1–4:1, 6–7:2, 8–10:3, >10:4), and the sum of the products in each group was then normalized to values obtained from the nontransgenic animal. The experimenter was blinded to mouse genotype and treatment.
Statistical analysis.
All values reported as mean ± SEM. Statistics were evaluated using Graphpad Prism software and Student's t test or one-way ANOVA with Tukey's post hoc where noted.
Results
We use the term “microglia” to refer to resident myeloid cells of the CNS parenchyma that are derived from yolk sac progenitors that invade the neuroepithelium early (E8.5) in development (Ginhoux et al., 2010; Schulz et al., 2012), whereas “macrophages” refer to activated parenchymal myeloid cells, which can be derived either from peripherally derived CD45hi monocytes or resident microglial cells (Ransohoff and Cardona, 2010; Mildner et al., 2011; Prinz et al., 2011; Prinz and Priller, 2014).
Nuclear receptor agonist treatment of microglia increases phagocytosis
We and others have reported that nuclear receptors act broadly to ameliorate AD-related deficits in murine models of the disease (Skerrett et al., 2014). The agonists lower amyloid burden, and the reduction in amyloid levels was correlated with increased numbers of Aβ-containing myeloid cells in mice treated with nuclear receptor agonists. This led us to test whether Type II nuclear receptor agonists could directly stimulate phagocytosis. Phagocytosis is the process by which microglia and other macrophages take up large (defined as >1 μm) particles (Aderem and Underhill, 1999). We treated the immortalized N9 microglial cell line with vehicle, the RXR agonist bexarotene (bex), the PPARγ agonist pioglitazone (pio), the LXR agonist GW3965, or the PPARδ agonist GW0742 for 18 h before testing the cells' phagocytic capacity with polystyrene beads (Fig. 1B). The nuclear receptor agonists increased basal levels of phagocytosis (Fig. 1B). As has been previously reported, incubation with fibrillar amyloid (Paresce et al., 1996; Bamberger et al., 2003; Reed-Geaghan et al., 2009; Stewart et al., 2010) also stimulated N9 cell phagocytosis (Fig. 1A,C). Furthermore, pretreatment of N9 microglia with nuclear receptor agonists increased their phagocytic response to fibrillar amyloid (Fig. 1A,C) and the TAM receptor ligand Gas6, an adaptor molecule that recognizes apoptotic cells (Fig. 1D). These data demonstrate that nuclear receptor agonists directly enhance the phagocytic activity of N9 cells, as does activation of the TAM receptors.
Figure 1.

Bexarotene increases microglial phagocytosis in vitro. N9 microglia were incubated with 1 μm polystyrene beads, and their uptake was measured with flow cytometry (A). N9 cells were treated with or without nuclear receptor agonists (50 nm Bex, RXR agonist; 100 nm pio, PPARγ agonist; 100 nm GW3965, LXR agonist; GW0742, PPARδ agonist) for 18 h before incubation with 1 μm polystyrene beads, and the fold change in phagocytic index was measured (B). fAβ (2.5 μm) was added 30 min before bead uptake (C). Gas6 (40 nm) was added 30 min before bead uptake (D). Data represent three separate experiments; 10,000 cells were analyzed via flow cytometry. *p < 0.05. **p < 0.01. ***p < 0.001.
Nuclear receptor agonist treatment increases expression of MerTK and Axl in vitro
The receptor tyrosine kinases MerTK and Axl are direct genetic targets of nuclear receptor activation in macrophages (A-Gonzalez et al., 2009; Röszer et al., 2011; Daniel et al., 2014). Given that we observed an elevated phagocytic response to the TAM ligand Gas6, we next sought to determine whether this effect was directly mediated though their induction by nuclear receptor agonists.
We treated cultured N9 microglia with the RXR agonist bexarotene for 18 h and found that treated microglia expressed higher levels of MerTK and Axl mRNA (Fig. 2A,B) than naive microglia. Treating microglia with the proinflammatory polarizing agent LPS reduced MerTK mRNA levels, and pretreatment with bexarotene was able to prevent this reduction (Fig. 2A). Interestingly, LPS treatment did not affect Axl mRNA levels, although LPS treatment reversed nuclear receptor mediated increases in Axl mRNA expression (Fig. 2B). We next examined MerTK protein levels in microglia treated with the RXR agonist bexarotene or the LXR agonist GW3965. Microglia treated with either bexarotene or GW3965 showed increased levels of MerTK protein expression (Fig. 2C,D). Furthermore, N9 microglia treated with bexarotene, GW3965, the PPARγ agonist pioglitazone, or the PPARδ agonist GW0742 also showed increased levels of MerTK protein (Fig. 2E,F). These data provide direct evidence that microglial MerTK expression is regulated by nuclear receptors and this response is sensitive to the inflammatory status of the cells and their environment.
Figure 2.

Nuclear receptor agonist treatment increases MerTK and Axl receptor expression in microglia. At 24 h after addition of bexarotene, N9 microglia were incubated in the presence or absence of 10 ng/ml LPS for an additional 18 h before analysis of MerTK mRNA levels (A) and Axl mRNA levels (B). Protein levels of MerTK in primary microglia were evaluated by Western analysis 18 h after treatment with 50 nm bexarotene or 500 nm GW3965 (C, quantified in D). Protein levels of MerTK in N9 microglia were probed via Western blot 18 h after treatment with 50 nm bexarotene, 100 nm pioglitazone, 100 nm GW3965, or 100 or 500 nm GW0742 (E, quantified in F). Data represent three separate experiments. *p < 0.05. **p < 0.01. ***p < 0.001.
Bexarotene treatment increases MerTK and Axl expression in two different models of AD
We next investigated the ability of bexarotene to regulate MerTK levels in vivo. Interestingly, mRNA levels of MerTK are significantly lower in microglia isolated from 12-month-old APP/PS1Δe9 mice than control mice (Fig. 3A). To determine whether bexarotene could regulate MerTK levels in vivo, we treated 12-month-old APP/PS1Δe9 mice for 7 d with 100 mg/kg/d bexarotene or vehicle by oral gavage. Indeed, MerTK mRNA levels were returned to nontransgenic levels after 7 d of bexarotene treatment (Fig. 3A). Interestingly, Axl expression was higher in microglia isolated from APP/PS1Δe9 mice than nontransgenic mice and was further increased upon 7 d of bexarotene treatment (Fig. 3A). Furthermore, the levels of MerTK in combined cortical and hippocampal homogenates were measured by Western analysis. Animals treated with bexarotene had significantly higher levels of MerTK than vehicle-treated control animals (Fig. 3B,C).
Figure 3.

Bexarotene treatment increases MerTK but not Gas6 expression in APP/PS1Δe9 mice. APP/PS1Δe9 mice (12mo) were treated with 100 mg/kg/d bexarotene or vehicle for 7 d by oral gavage before tissue collection. Microglia were isolated using a Percoll gradient, and MerTK and Axl mRNA levels were investigated using qRT-PCR (A). Brain homogenate was probed for MerTK protein levels by Western blot (7 or 8 animals/group) (B, quantified in C). Levels of Gas6 and Protein S mRNA in microglia isolated from 12-month-old APP/PS1de9 animals treated with bexarotene or vehicle for 7 d before tissue collection were investigated using qRT-PCR (D). Brain homogenate was probed for Gas6 protein levels by Western blot (7 or 8 animals per group) (E, quantified in F). *Difference from nontransgenic animal. #Difference from nontreated APP/PS1Δe9. *p < 0.05. **p < 0.01. ***p < 0.001. #p < 0.05. ##p < 0.01. ###p < 0.001.
Because levels of MerTK and Axl were increased by bexarotene treatment of 12-month-old APP/PS1Δe9 mice, we next investigated the effect of genotype and bexarotene treatment on the expression of the two principal TAM receptor ligands, Gas6 and Protein S. Microglia isolated from 12-month-old APP/PS1Δe9 contain similar amounts of Protein S and Gas6 mRNA, regardless of bexarotene treatment (Fig. 3D). However, microglia are not the sole source of TAM ligands within the CNS. We investigated the levels of Gas6 in combined cortical and hippocampal homogenates by Western blot (Fig. 3E, quantified in Fig. 3F) and found no significant differences based on genotype or drug treatment.
In nontransgenic mice, MerTK expression was below the level of detection by immunohistochemistry. However, in both APP/PS1Δe9 and 5XFAD animals, MerTK colocalized with plaque-associated myeloid cells (Fig. 4A,B,E–G). Additionally, MerTK and Gas6 were coexpressed on plaque-associated macrophages in 12-month-old APP/PS1Δe9 animals (Fig. 4E). Significantly, MerTK expression increased in Iba1+ cells after 7 d of bexarotene treatment (Fig. 4A). Similarly, in plaque-associated cells, MerTK expression was also higher in 4-month-old 5XFAD mice treated with bexarotene or the PPARδ agonist GW0742 for 14 d compared with vehicle treated animals (Fig. 4F). These results demonstrate that nuclear receptors increased MerTK expression surrounding plaques in AD mouse models.
Figure 4.
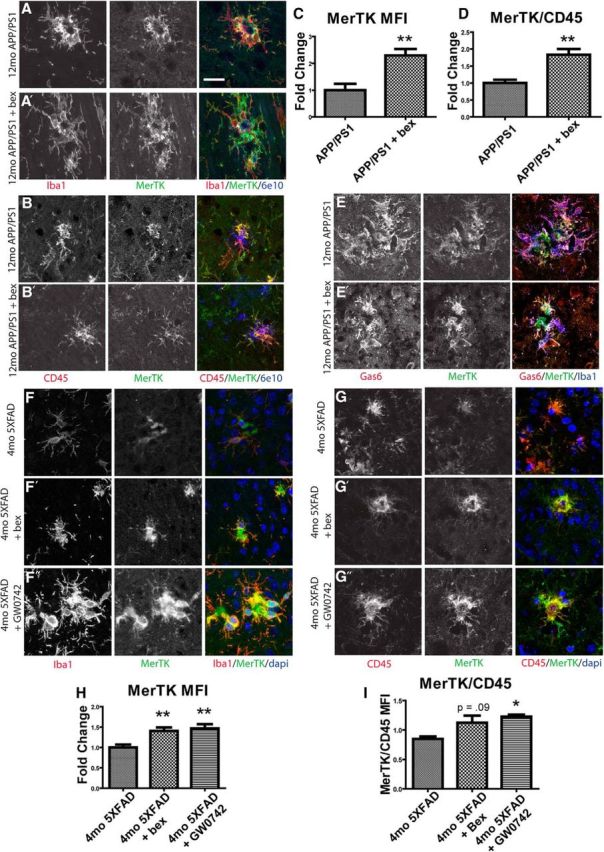
MerTK is expressed by microglia and macrophages in AD model mice and is increased by NR agonist treatment. APP/PS1Δe9 mice (12mo) were treated with vehicle (A, B, E) or the RXR agonist bexarotene (A′, B′, E′) for 7 d by oral gavage. Sections were stained for MerTK (green), Iba1 (red), and amyloid (6e10, blue) (A). MerTK mean fluorescence intensity was quantified (C). MerTK (green) colocalizes with CD45 (red), and the ratio of MerTK/CD45 mean fluorescence intensity was quantified (B, D). MerTK (green) colocalizes with Gas6 (red) and Iba1 (blue) (E). 5XFAD mice (4mo) were treated with vehicle (F, G), bexarotene (F′, G′), or the PPARδ agonist GW0742 (F″, G″) for 14 d. Sections were stained for MerTK (green) and Iba1 (red), and MerTK mean fluorescence intensity was quantified (F, H). MerTK (green) colocalizes with CD45 (red), and the ratio of MerTK/CD45 mean fluorescence intensity was quantified (G, I). Scale bar, 20 μm. Four to six animals were analyzed per group. *p < 0.05. **p < 0.01.
To further determine the cell-type specific expression of MerTK, we assessed levels of the marker CD45 on MerTK+, plaque-associated cells. CD45 is a canonical marker of myeloid cells (Sedgwick et al., 1991). MerTK colocalized with CD45-expressing cells surrounding amyloid deposits in both 5XFAD and APP/PS1Δe9 animals (Fig. 4B,G). Moreover, those MerTK+/CD45+ cells surrounding plaques were more abundant in 12-month-old APP/PS1Δe9 animals treated with bexarotene (Fig. 4B, quantified in Fig. 4D). Similarly, 4-month-old 5XFAD animals also showed increased ratios of MerTK/CD45 mean fluorescence intensity in response to 14 d of treatment with bexarotene or GW0742 (Fig. 4G, quantified in Fig. 4I). These results further characterize the MerTK+ plaque-associated cell population that is increased by nuclear receptor agonist treatment.
Because Axl has been shown to have complementary roles to MerTK in mediating phagocytosis (Lemke, 2013), we next investigated whether nuclear receptor treatment would affect Axl expression similarly to MerTK expression in vivo. Axl expression was not detected in the nontransgenic brain. However, Axl was abundantly expressed in cells surrounding plaques in both 12-month-old APP/PS1Δe9 and 4-month-old 5XFAD animals (Fig. 5A,C). MerTK and Axl are expressed by the same Iba1+ cells in APP/PS1Δe9 animals (Fig. 5A). Axl expression was increased in 4-month-old 5XFAD mice treated with bexarotene or PPARδ agonist GW0742 for 14 d compared with vehicle-treated animals (Fig. 5D). Similar to what we observed with the expression pattern of MerTK, Axl also colocalized with CD45 expressing cells in both 5XFAD and APP/PS1Δe9 animals (Fig. 5B,E). Upon quantification, 4-month-old 5XFAD animals demonstrate higher Axl/CD45 mean fluorescence intensity in response to 14 d of treatment with either bexarotene or PPARδ agonist GW0742 (Fig. 5F). Together, these data support the hypothesis that MerTK and Axl are similarly regulated by nuclear receptors, and that they're expressed on a common population of Iba1+, CD45+ cells surrounding Aβ deposits.
Figure 5.
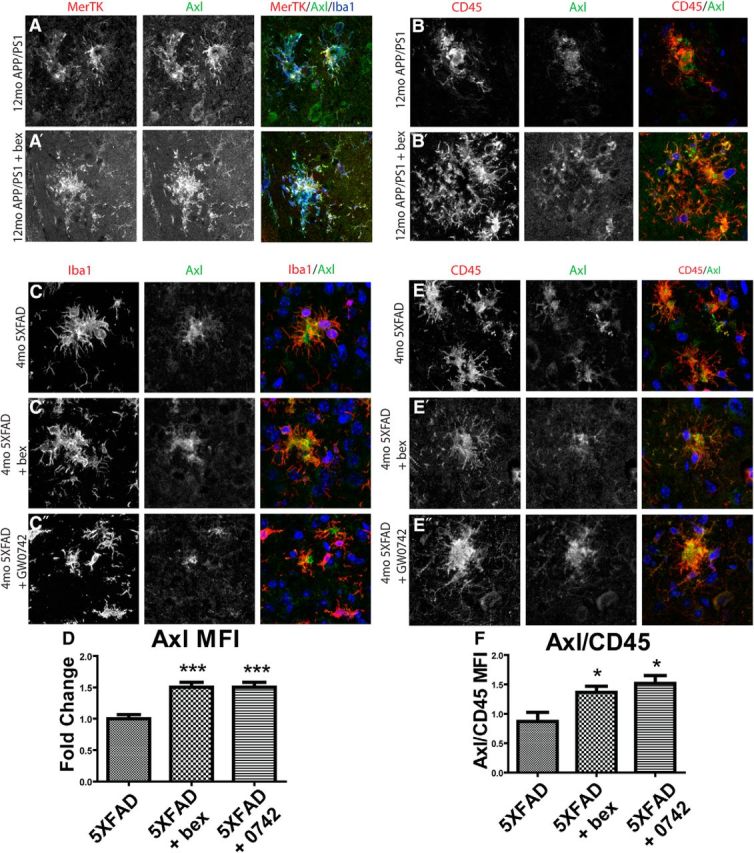
Axl is coexpressed with MerTK by microglia and macrophages in AD model mice and is increased with nuclear receptor agonist treatment. APP/PS1Δe9 mice (12mo) were treated with vehicle (A, B) or the RXR agonist bexarotene (A′, B′) for 7 d by oral gavage. Sections were stained for Axl (green), MerTK (red), and Iba1 (blue) (A). 5XFAD mice (4mo) were treated with vehicle (C, E), bexarotene (C′, E′), or PPARδ agonist GW0742 (C, D″, E″) for 14 d before sectioning and staining. Sections were stained for Axl (green) and Iba1 (red), and Axl mean fluorescence intensity was quantified (D). Axl (green) colocalizes with CD45 (red) and the ratio of Axl/CD45 mean fluorescence intensity was quantified (F). Scale bar, 20 μm. Four to six animals were analyzed per group. *p < 0.05. ***p < 0.001.
TREM2 and MerTK are coexpressed in plaque associated macrophages
TREM2 is expressed exclusively by microglia in the brain, but also by peripheral monocytes and macrophages (Colonna, 2003). We found that the same population of cells in the brain, which are MerTK+, CD45+ also expressed TREM2. Indeed, MerTK and TREM2 colocalize in cells surrounding plaques in both APP/PS1Δe9 (Fig. 6A) and 5XFAD (Fig. 6B) mice.
Figure 6.
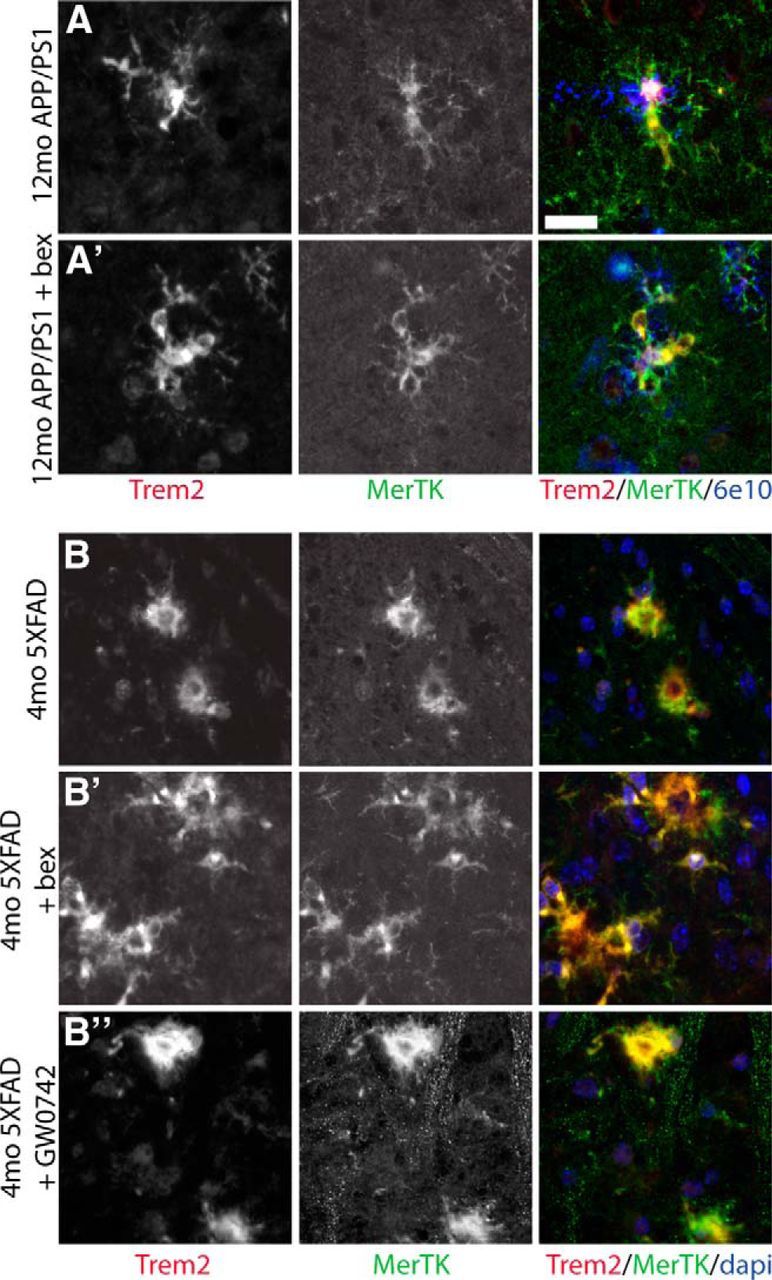
MerTK and TREM2 are expressed by plaque-associated macrophages. MerTK (green) colocalizes with TREM2 (red) and surround 6e10+ plaques (blue) in 12-month-old APP/PS1Δe9 mice treated with vehicle (A) or bexarotene (A′) for 7 d. MerTK (green) also colocalizes with TREM2 (red) in 4-month-old 5XFAD mice treated with vehicle (B), bexarotene (B′), or GW0742 (B″). Scale bar, 20 μm.
Jay et al. (2015) have reported that plaque-associated macrophages are derived from blood-borne monocytes that infiltrate the brain and are identified by their high levels of CD45 expression. CD45 is expressed by myeloid cells (Sedgwick et al., 1991), and its expression levels have traditionally been used to discriminate between microglia (CD45lo) and peripherally derived myeloid cells (CD45hi) (El Khoury et al., 2007; Hickman et al., 2013; Butovsky et al., 2014). Because MerTK and TREM2 colocalized with plaque-associated CD45-expressing cells, we were interested in determining whether MerTK was selectively expressed in either microglia or macrophages derived from infiltrating monocytes. FACS analysis revealed that MerTK expression was largely restricted to the CD45hi cells in 5XFAD animals (Fig. 7C). We treated 8-month-old 5XFAD mice with bexarotene or vehicle for 7 d before isolating myeloid cells from the brain. We gated the CD11b+ population into CD45lo and CD45hi populations (Fig. 7A). Indeed, 35%–40% of CD11b+/CD45hi cells in 8-month-old 5XFAD mice expressed TREM2, whereas <1% of CD11b+/CD45hi cells in nontransgenic littermates were TREM2+ (Fig. 7B), demonstrating that TREM2 expression is restricted to peripherally derived cells infiltrating the CNS. Less than 5% of CD11b+/CD45lo cells of any genotype or treatment were TREM2+ (data not shown), indicating that the resident micro2+ glia did not express TREM2 at detectable levels regardless of disease status. There was no effect of bexarotene treatment on the percentage of CD45hi cells expressing TREM2 (Fig. 7B). However, MerTK was expressed at a higher level in CD11b+/CD45hi cells in 5XFAD mice, but not in nontransgenic littermates (Fig. 7C), coincident with MerTK/CD45 coexpression reported on plaque associated cells (Fig. 4B,G). Unsurprisingly, MerTK was expressed at a higher level in TREM2+ cells as well (Fig. 7D,E), indicating that peripherally derived CD45hi, TREM2+ macrophages also express detectable levels of MerTK. Bexarotene treatment increased both the total MerTK levels, as well as the MerTK levels in TREM2+ cells (Fig. 7E), indicating RXR agonist treatment increased MerTK levels of both CD45lo and CD45hi cells in the brains of 5XFAD mice.
Figure 7.
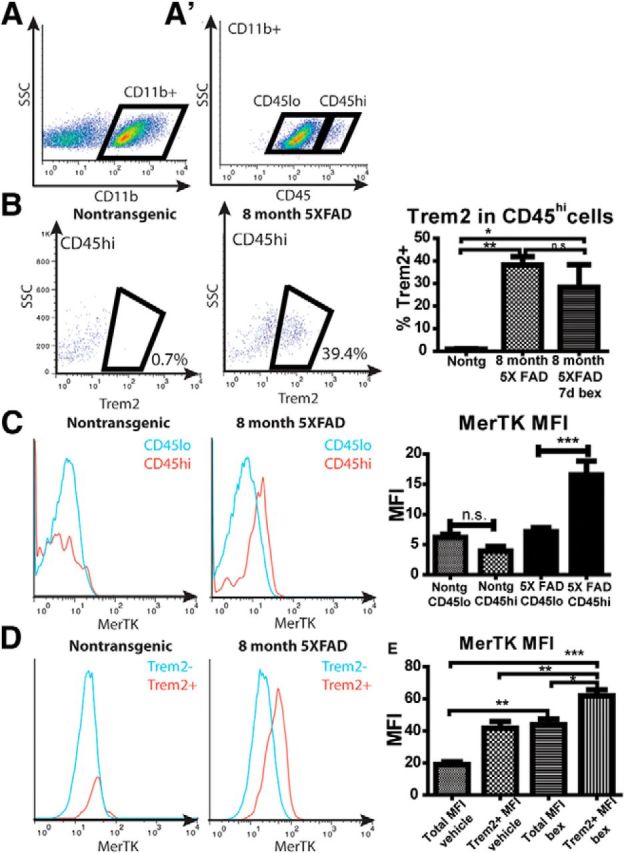
MerTK and TREM2 are expressed by peripherally derived myeloid cells. Microglia were isolated from 8-month-old 5XFAD mice (4–6 per group) and gated on CD11b+/CD45lo and CD11b+/CD45hi cell populations (A, A′). B, TREM2 expression in the CD11b+/CD45hi cells was quantitated. C, Mean fluorescence intensity of MerTK was measured in CD11b+/CD45lo and CD11b+/CD45hi populations in both transgenic and nontransgenic mice. Mean fluorescence intensity of MerTK was measured in the TREM2 expressing population (D, quantified in E). *p < 0.05. **p < 0.01. ***p < 0.001. n.s., Not significant.
RXR activation reduces plaque burden in 12 month APP/PS1Δe9 mice and restores phagocytosis in an ex vivo phagocytosis assay
We have demonstrated that nuclear receptor treatment both in vitro and in vivo increased levels of MerTK and Axl and enhanced phagocytosis. We wished to determine whether induction of TAM receptors was necessary for nuclear receptor-mediated increases in phagocytosis. To investigate this potential mechanism, we treated mice with bexarotene and assessed whether modulating signaling through MerTK would alter the ability of bexarotene treatment to enhance phagocytosis in an ex vivo phagocytosis assay.
First, we validated that bexarotene treatment induced plaque clearance and enhanced phagocytosis. RXR agonist treatment has been shown to improve behavior and decrease both soluble and fibrillar forms of Aβ in several mouse models of AD (Jiang et al., 2008; Cramer et al., 2012), although others have failed to observe these effects (Tesseur and De Strooper, 2013). We treated 12-month-old APP/PS1Δe9 animals with bexarotene by oral gavage for 7 d. We found acute bexarotene treatment reduced the number of plaques in both cortex and hippocampus by 20% and 30%, respectively, as measured by the number of thioflavin S-positive plaques (Fig. 8A–D). Additionally, insoluble levels of Aβ in combined cortical and hippocampal homogenates were reduced as shown by ELISA (Fig. 8E,F). These data demonstrate that RXR activation reduced plaque burden in aged APP/PS1Δe9 mice over a period of 1 week. The diminished number of plaques was correlated with the appearance of Iba1+ microglia containing intracellular amyloid in bexarotene-, but not vehicle-treated, mice (Fig. 8G).
Figure 8.
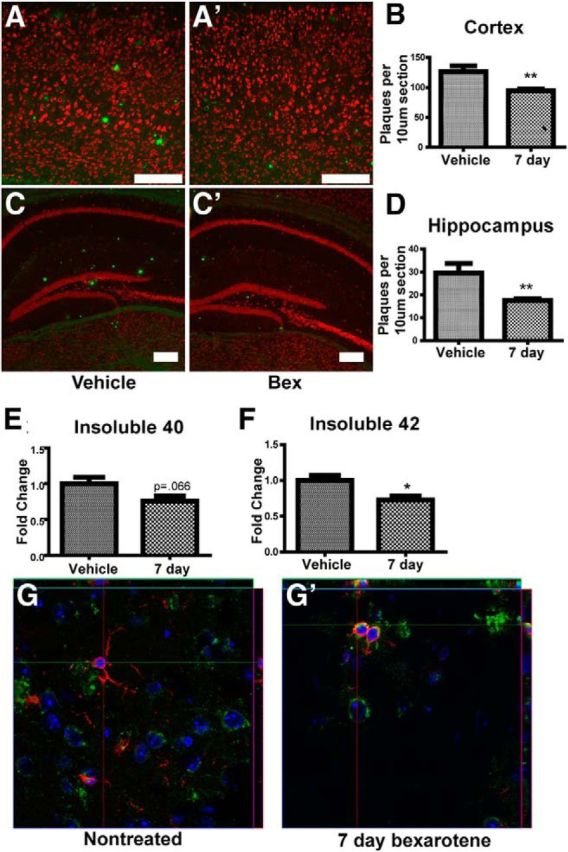
Bexarotene treatment decreases plaque burden in APP/PS1Δe9 animals. APP/PS1Δe9 mice (12 months) were treated with vehicle (A, C) or 100 mg/kg/d bexarotene (A′, C′) for 7 d by oral gavage before staining with Thioflavin S. Plaques in the cortex (B) and hippocampus (D) were counted from 5 animals. Scale bar, 200 μm. ELISAs of Aβ40 and Aβ42 were run on formic acid extractions of cortical homogenates from 7 or 8 animals per group to determine insoluble Aβ40 and Aβ42 levels (E, F). Confocal images show Aβ (6e10, green) inside microglia (Iba1, red) (G). *p < 0.05. **p < 0.01.
Phagocytosis is suppressed by the sustained presence of elevated amyloid levels in vivo (Krabbe et al., 2013). Because bexarotene treatment decreased plaque levels and increased macrophage levels of phagocytic receptors MerTK and Axl in a mouse model of AD, we were interested in investigating bexarotene's ability to increase the phagocytic competence of myeloid cells. Cultured microglia are competent phagocytes and readily take up fAβ introduced into the media (Koenigsknecht and Landreth, 2004). However, these cells are unable to efficiently take up and degrade amyloid in the AD brain, as evidenced by the progressive appearance of insoluble fAβ plaques throughout the course of the disease. For these reasons, we were interested in studying microglial phagocytosis in a system as close to in situ as possible.
We used an ex vivo slice phagocytosis assay (Gyoneva and Traynelis, 2013; Krabbe et al., 2013) to determine whether nuclear receptor activation could restore phagocytic competence to microglia residing in the AD brain. We treated 12-month-old APP/PS1Δe9 animals with bexarotene or vehicle for 5 d before preparing brain slices and performing a phagocytosis assay. Indeed, 12-month-old APP/PS1Δe9 animals show reduced phagocytosis compared with nontransgenic littermates (Fig. 9A,B), as previously reported in APPPS1 and APP23 mice (Krabbe et al., 2013). Remarkably, 5 d of oral bexarotene treatment restored phagocytic competence to nontransgenic levels (Fig. 9C). Furthermore, incubation with MerTK and Axl ligand Gas6 restored APP/PS1Δe9 microglial phagocytosis to nontransgenic levels (Fig. 9C). Most strikingly, incubation with a function-blocking anti-MerTK antibody reduced phagocytosis in both nontransgenic slices as well as those from APP/PS1Δe9 animals, which had been treated with bexarotene for 5 d before the assay (Fig. 9C). These data demonstrate that the increase in microglial phagocytosis seen upon bexarotene treatment is reliant on MerTK expression and function.
Figure 9.
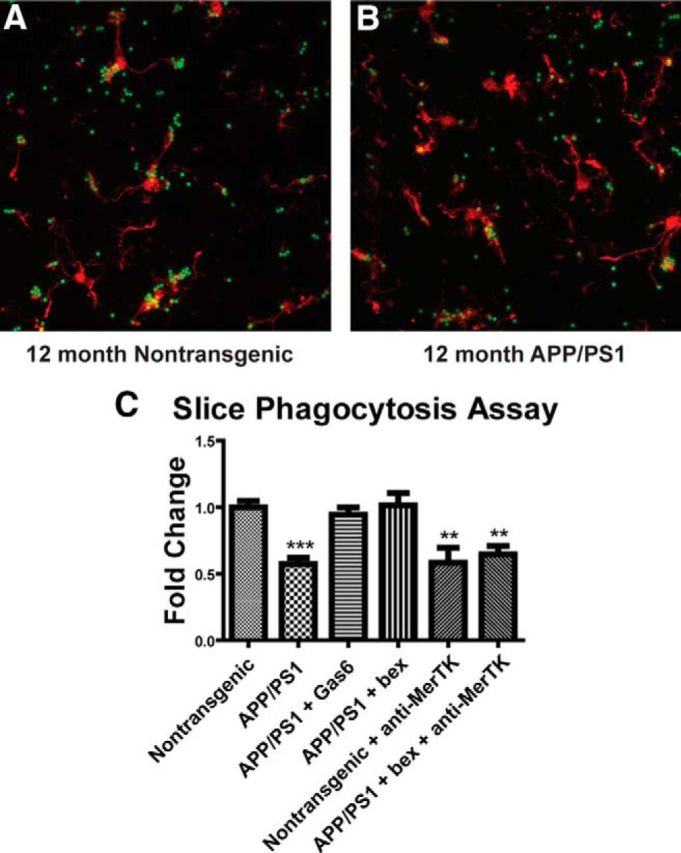
Bexarotene treatment increases phagocytosis in 12-month-old APP/PS1Δe9 mice in a MerTK-dependent manner. The 12-month-old APP/PS1 mice were treated with 100 mg/kg/d bexarotene or vehicle for 5 d by oral gavage before ex vivo slice culture phagocytosis assay. Slices were taken from nontransgenic (A) or APP/PS1Δe9 (B) animals treated with 100 mg/kg/d bexarotene or vehicle and incubated with Gas6 or function blocking anti-MerTK antibody and 2 μm polystyrene beads to determine their phagocytic index (C). Data represent three separate experiments. **p < 0.01. ***p < 0.001.
Discussion
Understanding of the complex biology of microglia has been hampered by the lack of a conceptual framework through which to interpret how myeloid cell phenotype is regulated in the normal and diseased brain. The triggers of inflammation, such as elevated soluble and deposited amyloid levels in the AD brain, are well studied (Akiyama et al., 2000). However, the chronic adaptations of myeloid cells to the diseased brain environment are not well understood. A central role for inflammation in the pathogenesis of Alzheimer's disease and other neurodegenerative diseases has been buttressed by a succession of studies identifying genetically linked risk factors that regulate or affect the activity of immune cells in the brain (Hollingworth et al., 2011; Naj et al., 2011). Indeed, the cell biological evidence, embodied within a large literature (Akiyama et al., 2000), and in directed studies focused on newly identified risk genes, such as TREM2, CD33, CD300a, and others, is largely consistent with the actions of immune responses in the brain that confer elevated disease risk (Jones et al., 2010; Guerreiro et al., 2013; Jonsson et al., 2013).
One of the unexplained aspects of AD pathogenesis is why microglia fail to effectively phagocytose fibrillar amyloid deposits, reflecting the disengagement of the phagocytic machinery directed toward this target. These cells express an array of receptors that act in an ensemble to recognize fibrillar forms of amyloid, including Scara1, CD47, α6β1 integrin, TLRs 2/4/6, and their coreceptors CD36 and CD14 (Paresce et al., 1996; Bamberger et al., 2003; Reed-Geaghan et al., 2009; Stewart et al., 2010). Importantly, microglia in the AD brain are phagocytically competent, as opsonization of amyloid with antibodies (Bard et al., 2000; Demattos et al., 2012) or complement (Fu et al., 2012) can effectively stimulate phagocytosis and plaque clearance. The presence of amyloid acts to suppress macrophage phagocytic function (Krabbe et al., 2013), whereas the interaction of macrophages with plaques elicits the secretion of an array of inflammatory cytokines, which directly suppress phagocytosis (Koenigsknecht-Talboo and Landreth, 2005).
The present study describes a mechanism whereby macrophages in the AD brain are induced to phagocytose and clear amyloid deposits through the action of nuclear receptors, which regulate the expression of arrays of functionally allied genes to license this response. Phagocytes use a diverse cohort of receptors that recognize and engulf cellular fragments and apoptotic cells whose membrane lipids serve as direct ligands of the nuclear receptors. However, until now, it has not been clear exactly how various nuclear receptors mediate enhanced phagocytosis. Here, we show that the enhanced phagocytic capacity is subserved by the induction of the phagocytic receptors Axl and MerTK, as well as a cohort of other genes supporting this function (Daniel et al., 2014).
There is an extensive literature on the salutary actions of nuclear receptors in murine models of AD and other CNS disorders, where they act to promote the conversion of microglia from proinflammatory to anti-inflammatory phenotypes (Skerrett et al., 2014). Studies using PPARγ, PPARδ, LXR, and RXR agonists have consistently reported amelioration of AD-related phenotyes and behavioral impairments. PPARγ and RXR have been shown to increase phagocytosis of amyloid in a CD36-dependent mechanism (Yamanaka et al., 2012), as well as control C1 and FcγR transcript levels on peripheral macrophages (Röszer et al., 2011). Plaque reduction was observed in the majority of these studies but was the most variable outcome and is influenced by age and aggressiveness of the disease models. The nuclear receptor agonists in these studies were typically delivered chronically through the diet over a period of months. We demonstrated that acute (3–9 d) treatment with PPARγ (Mandrekar-Colucci et al., 2012) or RXR agonists (Cramer et al., 2012) was sufficient to induce plaque loss, but this effect was highly sensitive to drug formulation and pharmacodynamics (Landreth et al., 2013; Tesseur and De Strooper, 2013; Chen et al., 2014). The present work identifies a more complex biology associated with the phagocytic clearance of amyloid deposits from the brain because of the identification of the peripheral origins of the plaque-associated macrophages and their responses to nuclear receptor agonists within the brain.
We report here and in related work (Jay et al., 2015) that plaque-associated macrophages express markers consistent with myeloid cells derived from peripheral monocytes. Blood-borne ‘inflammatory’ monocytes are CD45hi, whereas microglia are CD45lo. CD45 expression has historically been used to discriminate between these cells, which arise from entirely distinct lineages and more recently has allowed the delineation of transcriptional differences between microglia and other myeloid populations (Gautier et al., 2012; Hickman et al., 2013; Butovsky et al., 2014; Yamasaki et al., 2014). The suggestion that monocytes infiltrate the AD brain and have salutary effects on AD pathogenesis is not new (Simard et al., 2006; El Khoury and Luster, 2008), but these studies were confounded by technical considerations (Morganti et al., 2014). However, El Khoury et al. (2007) and El Khoury and Luster (2008) reported that peripheral myeloid cells infiltrated the AD brain in a CCL2-dependent manner, and loss of CCR2 was associated with fewer parenchymal macrophages, increased plaque burden, and increased levels of Aβ peptides. Our observation that MerTK+/Axl+ plaque-associated macrophages are also CD45hi and TREM2+ argues strongly that these cells are derived from peripheral monocytes. Importantly, our data show that nuclear receptor agonists increase phagocytic receptor expression on both TREM2− and TREM2+ populations and enhance phagocytosis in these different populations.
The question of whether MerTK+/Axl+ plaque-associated macrophages are derived from resident microglia or peripheral monocytes will be important to explore further as they may have distinct roles in disease and be regulated differently by nuclear receptor agonist treatment. The roles of resident microglia and infiltrating monocytes have only recently begun to be delineated (Yamasaki et al., 2014). New work suggests that the action of nuclear receptors in circulating myeloid subpopulations may license a different array of genes that subserve the emigration of these cells into tissues to sites of inflammation and their subsequent differentiation (Daniel et al., 2014). Analysis of RXR-regulated gene expression in peripheral monocytes has revealed the coordinate induction of genes associated with extravasation through the endothelium (CD300a, Ninj1, CCL2), differentiation into macrophages (MafB, IRF8), and regulation of phagocytosis (TREM2, MerTK, Axl, CD300a, C1q, CD36) (Daniel et al., 2014). Nuclear receptor actions in tissue macrophages have been observed on a cohort of genes that are specifically associated with “alternative activation” and phagocytosis (Odegaard et al., 2007; A-Gonzalez et al., 2009; Chinetti-Gbaguidi et al., 2011; Röszer et al., 2013).
MerTK and Axl are necessary for the phagocytosis of apoptotic cells but also act broadly to regulate the innate immune response in tissue macrophages (Lemke, 2013). These receptors exhibit important anti-inflammatory actions, and their expression serves as a marker of “alternative activation” (Zizzo et al., 2012). Axl/MerTK have recently been shown to be expressed in a mutually exclusive manner on bone marrow-derived macrophages. MerTK acts in a homeostatic role and is induced in immunosuppressive environments, whereas Axl levels are increased by proinflammatory factors (Zagórska et al., 2014). Both receptors act as negative feedback regulators to reduce inflammation (Rothlin et al., 2007); however, neither has been studied in the context of AD. We found that plaque-associated macrophages express high levels of both MerTK and Axl in the AD brain, which are further increased through the activation of nuclear receptors. Importantly, our data suggest that MerTK activity is necessary for amyloid-stimulated phagocytosis, and it will be important to understand the mechanistic basis of this phenomenon.
TREM2 has classically been considered a phagocytic receptor, based largely on in vitro studies demonstrating its requirement for the phagocytosis of apoptotic neurons and bacteria. However, we demonstrate that TREM2+ cells accumulate on plaques where they exhibit an inflammatory phenotype and are phagocytically ineffective, as evidenced by the progressive increase in plaque burden throughout the course of the disease. Similarly, it is widely held that TREM2 acts to suppress inflammatory gene expression, based largely on data demonstrating that knockdown or genetic knock-out of TREM2 resulted in higher levels of proinflammatory cytokines (Takahashi, 2005; Turnbull et al., 2006; Sharif et al., 2014). This view is inconsistent with the observation of inflammatory macrophages associated with plaques, virtually all of which express high levels of TREM2. TREM2 has recently been reported to have analogous proinflammatory actions in murine models of stroke (Sieber et al., 2013), colitis (Correale et al., 2013), and lung inflammation (Sharif et al., 2014). Importantly, Jay et al. (2015) have shown a dramatic decrease in monocyte infiltration in TREM2−/− AD model mice and an absence of plaque-associated cells, suggesting that TREM2 has previously unappreciated roles in peripheral monocytes and their subsequent actions within the brain.
This study provides direct evidence that nuclear receptors act to promote the clearance of amyloid deposits from the brain, in part, through their ability to induce the expression of the phagocytic receptors Axl and MerTK on plaque-associated macrophages. The recognition that these latter cells are likely derived from circulating monocytes and express TREM2 force a substantial reevaluation of both the actions of TREM2 and the roles of myeloid cells in AD pathogenesis.
Footnotes
This work was supported by an Alzheimer's Association Multi-Center Program Grant to B.T.L., G.E.L., and R.M.R., the Jane and Lee Seidman Fund, a generous donation from Chet and Jane Scholtz, National Institute on Aging Grant AG023012 to B.T.L., National Institute of Neurological Disorders and Stroke Grants NS047804 and NS087298 to B.T.L. and R.M.R., Department of Defense Grant W81XWH12-1-0629 to B.T.L., Alzheimer's Association Zenith Award, gifts from the Roby and Taft funds to G.E.L., National Institute on Aging Grant AG043522 to G.E.L., and a National Research Service Award T32 NS067431 to T.J. We thank the members of the B.T.L. and G.T.L. laboratories for technical assistance; Maryanne Pendergast of the Neurosciences Imaging Center for technical assistance; John Collins for GW0742; and Colleen Karlo for copyediting the manuscript.
G.E.L. is the founding scientist of ReXceptor, Inc, a biotechnology company developing RXR agonists for the treatment of neurodegenerative diseases. The remaining authors declare no competing financial interests.
References
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat Protoc. 2006;1:1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang Y, Zhang J, Hao L, Guo H, Lou H, Zhang D. Bexarotene nanocrystal: oral and parenteral formulation development, characterization and pharmacokinetic evaluation. Eur J Pharm Biopharm. 2014;87:160–169. doi: 10.1016/j.ejpb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, Haulon S, Zawadzki C, Jude B, Staels B. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3:445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- Correale C, Genua M, Vetrano S, Mazzini E, Martinoli C, Spinelli A, Arena V, Peyrin-Biroulet L, Caprioli F, Passini N, Panina-Bordignon P, Repici A, Malesci A, Rutella S, Rescigno M, Danese S. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology. 2013;144:346–356.e3. doi: 10.1053/j.gastro.2012.10.040. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, Engelborghs S, Vandenbulcke M, Van Dongen J, Geerts N, Maes G, Mattheijssens M, Peeters K, Cras P, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Cruts M, Sleegers K. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014;35:726.e11–726.e19. doi: 10.1016/j.neurobiolaging.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Daniel B, Nagy G, Hah N, Horvath A, Czimmerer Z, Póliska S, Gyuris T, Keirsse J, Gysemans C, van Ginderachter JA, Balint BL, Evans RM, Barta E, Nagy L. The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 2014;28:1562–1577. doi: 10.1101/gad.242685.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76:908–920. doi: 10.1016/j.neuron.2012.10.029. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma'ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288:15291–15302. doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafizi S, Dahlbäck B. Gas6 and protein S: vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006;273:5231–5244. doi: 10.1111/j.1742-4658.2006.05529.x. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucke S, Floßdorf J, Grützke B, Dunay IR, Frenzel K, Jungverdorben J, Linnartz B, Mack M, Peitz M, Brüstle O, Kurts C, Klockgether T, Neumann H, Prinz M, Wiendl H, Knolle P, Klotz L. Licensing of myeloid cells promotes central nervous system autoimmunity and is controlled by peroxisome proliferator-activated receptor γ. Brain. 2012;135:1586–1605. doi: 10.1093/brain/aws058. [DOI] [PubMed] [Google Scholar]
- Hucke S, Wiendl H, Knolle P, Klotz L. Nuclear receptor control of myeloid cell responses: implications for CNS autoimmunity. Rheumatol Curr Res. 2013:3. [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. 2015;212:287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, Gerrish A, Pahwa JS, Jones N, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One. 2010;5:e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth GE, Cramer PE, Lakner MM, Cirrito JR, Wesson DW, Brunden KR, Wilson DA. Response to comments on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models.”. Science. 2013;340:924. doi: 10.1126/science.1234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5:a009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew ED, Oh J, Burrola PG, Lax I, Zagórska A, Través PG, Schlessinger J, Lemke G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife. 2014:3. doi: 10.7554/eLife.03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Jopson TD, Liu S, Gupta N, Rosi S. Cranial irradiation alters the brain's microenvironment and permits CCR2+ macrophage infiltration. PLoS One. 2014;9:e93650. doi: 10.1371/journal.pone.0093650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YPS, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, Chawla A. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–1272. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Szanto A, Szatmari I, Széles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–789. doi: 10.1152/physrev.00004.2011. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N′Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, Seaman WE. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184:215–223. doi: 10.1083/jcb.200808080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/S0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. nrn3722. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–631. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röszer T, Menéndez-Gutiérrez MP, Cedenilla M, Ricote M. Retinoid X receptors in macrophage biology. Trends Endocrinol Metab. 2013;24:460–468. doi: 10.1016/j.tem.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif O, Gawish R, Warszawska JM, Martins R, Lakovits K, Hladik A, Doninger B, Brunner J, Korosec A, Schwarzenbacher RE, Berg T, Kralovics R, Colinge J, Mesteri I, Gilfillan S, Salmaggi A, Verschoor A, Colonna M, Knapp S. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumonia. PLoS Pathog. 2014;10:e1004167. doi: 10.1371/journal.ppat.1004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8:e52982. doi: 10.1371/journal.pone.0052982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Skerrett R, Malm T, Landreth G. Nuclear receptors in neurodegenerative diseases. Neurobiol Dis. 2014;72:104–116. doi: 10.1016/j.nbd.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, De Strooper B. When the dust settles: what did we learn from the bexarotene discussion? Alzheimers Res Ther. 2013;5:54. doi: 10.1186/alzrt218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JS, Reed A, Chen F, Stewart CN., Jr Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagórska A, Través PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15:920–928. doi: 10.1038/ni.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzo G, Hilliard BA, Monestier M, Cohel PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189:3508–3520. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]