

Abstract
Presynaptic terminal cAMP elevation plays a central role in plasticity at the mossy fiber-CA3 synapse of the hippocampus. Prior studies have identified protein kinase A as a downstream effector of cAMP that contributes to mossy fiber LTP (MF-LTP), but the potential contribution of Epac2, another cAMP effector expressed in the MF synapse, has not been considered. We investigated the role of Epac2 in MF-CA3 neurotransmission using Epac2−/− mice. The deletion of Epac2 did not cause gross alterations in hippocampal neuroanatomy or basal synaptic transmission. Synaptic facilitation during short trains was not affected by loss of Epac2 activity; however, both long-term plasticity and forskolin-mediated potentiation of MFs were impaired, demonstrating that Epac2 contributes to cAMP-dependent potentiation of transmitter release. Examination of synaptic transmission during long sustained trains of activity suggested that the readily releasable pool of vesicles is reduced in Epac2−/− mice. These data suggest that cAMP elevation uses an Epac2-dependent pathway to promote transmitter release, and that Epac2 is required to maintain the readily releasable pool at MF synapses in the hippocampus.
Keywords: cAMP, Epac2, mossy fiber, readily releasable pool
Introduction
cAMP plays a role in regulated exocytosis of both secretory granules and synaptic vesicles. Its major downstream target is commonly identified as protein kinase A (PKA), but it has recently emerged that alternate cAMP targets, the guanine nucleotide exchange factors (GEFs) Epac1 and 2 (exchange protein directly activated by cAMP) (Kawasaki et al., 1998; de Rooij et al., 1998), can also modulate vesicle exocytosis (Seino and Shibasaki, 2005). These ubiquitous proteins have been ascribed many roles as cAMP effectors in both the periphery and the CNS, but in both neuronal and non-neuronal cells there is evidence that a primary role of Epac activity is regulation of exocytosis (Gloerich and Bos, 2010). Initial evidence of this emerged from studies of insulin secretion (Kashima et al., 2001), and subsequently from studies at the neuromuscular junction (Zhong and Zucker, 2005; Cheung et al., 2006) and central synapses in the brainstem (Kaneko and Takahashi, 2004; Gekel and Neher, 2008). These studies have demonstrated a conserved role for Epac signaling in enhancing neurotransmitter release (Ozaki et al., 2000). Less is known about the role of Epac signaling in neurosecretory processes at cortical synapses. Regulation of transmitter release by cAMP is not ubiquitous, but there are several well characterized central synapses where cAMP elevation in the presynaptic terminal increases transmitter release. In the hippocampus, mossy fiber (MF) synapses formed between the granule cells in the dentate gyrus and the proximal dendrites of the CA3 pyramidal neurons are subject to regulation by cAMP (Weisskopf et al., 1994).
Pharmacological and molecular genetic approaches have established the importance of a cAMP-dependent PKA signaling pathway for long-term potentiation of mossy fiber synapses (MF-LTP; Weisskopf et al., 1994; Huang et al., 1995). However, investigation of potential presynaptic PKA substrates has not been able to fully account for cAMP-dependent potentiation (Castillo et al., 1997, 2002; Kaeser-Woo et al., 2013), suggesting that other downstream effectors may also play a role in cAMP-mediated potentiation of neurotransmitter release at MF terminals. A possible role for Epac at MF synapses has not been established, but it is possible that this effector has a central, and as yet unrecognized, function in neurotransmitter release. Epac is directly activated by cAMP and has been characterized as a GEF for the GTPases Rap1 and Rap2 (Kawasaki et al., 1998; de Rooij et al., 2000), although it is not clear how it may regulate exocytosis. The two Epac isoforms have distinct and differential expression patterns with some overlap. Epac1 is enriched in the kidney and gonadal tissue with limited expression in the CNS (Kawasaki et al., 1998). Epac2 is prominently expressed in the pancreas and brain, where it is abundant in the cortex and hippocampus and is particularly enriched within the stratum lucidum of the hippocampus in the terminal field of the MF synapse, suggesting that Epac2 is expressed at high levels in the presynaptic terminal boutons of MF axons (Kawasaki et al., 1998).
Here we tested whether Epac2 has a role in MF synaptic transmission by making use of mice in which the primary hippocampal isoform of Epac, Epac2, is ablated (Epac2−/−). We found that there were no major alterations in basal synaptic transmission or in short-term facilitation of MF synapses in Epac2−/− mice. However, there were clear deficits in the extent of synaptic potentiation induced by pharmacological elevation of cAMP, or after tetanus-induced MF-LTP. Sustained stimulation of MF inputs demonstrated a significantly greater depression during long trains of synaptic activation in Epac2−/− mice. Analysis of the steady-state depression during these long trains strongly suggested that the readily releasable pool of vesicles is reduced in Epac2−/− mice. These observations are the first evidence of a significant role for Epac2 in regulating MF synaptic transmission and plasticity, and describe a novel role for Epac2 in maintaining the vesicular pools required for sustained release of neurotransmitter.
Materials and Methods
Animals.
Epac2−/− mice were obtained (from S. Seino, Kobe University, Japan; Shibasaki et al., 2007) and backcrossed for at least nine generations onto a congenic C57BL/6 background. Breeders were maintained as heterozygous crosses and all experiments were performed blind to genotype in age-matched littermates of either sex. Tail biopsies were used to perform post hoc genotyping of all mice used in the study.
Slice preparation and electrophysiological recordings.
Horizontal slices from the ventral hippocampus (350 μm) were prepared from mice aged P14–P28 as previously described (Fernandes et al., 2009). Briefly, animals were anesthetized with isoflurane and decapitated. The brain was rapidly removed under ice-cold, oxygenated sucrose-slicing ACSF containing the following (in mm): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2, 4 MgCl2, 0.5 ascorbic acid, 0.01 dl-APV, and 0.1 kynurenic acid, equilibrated with 95% O2/5% CO2. Slices were heated to 28.5°C before being returned to room temperature while slowly exchanging sucrose-slicing ACSF for sodium-ACSF solution containing the following (in mm): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 1 CaCl2, 2 MgCl2, 0.01 dl-APV, and 0.1 kynurenic acid. After a 60 min recovery, individual slices were transferred to a recording chamber and continuously perfused with oxygenated sodium ACSF containing 2 mm CaCl2 and 1 mm MgCl2. All recordings were performed at 30°C. For whole-cell voltage-clamp recordings borosilicate glass electrodes (resistance 2.5–4.5 MΩ) were filled with an internal solution containing the following (in mm): 95 CsF, 25 CsCl, 10 Cs-HEPES, 10 Cs-EGTA, 2 NaCl, 2 Mg-ATP, 10 QX-314, 5 TEA-Cl, and 5 4-AP, pH adjusted to 7.3 with CsOH. Series resistance was continuously monitored using hyperpolarizing voltage steps generated by pClamp 9 software (Molecular Devices), and recordings were discarded if there was a >20% change during the experiment. MF synaptic currents were evoked using a monopolar glass electrode filled with oxygenated ACSF positioned in the stratum lucidum. Criteria for acceptable MF EPSCs were as previously reported (Contractor et al., 2001; Armstrong et al., 2006). EPSCs were isolated using the GABAA receptor antagonists bicuculline (10 μm) and picrotoxin (50 μm), and NMDAR antagonist d-APV (50 μm). For experiments monitoring progressive block of NMDAR EPSCs, MF EPSCs were initially recorded at −60 mV, and the AMPA component was fully blocked with GYKI 53655 (50 μm). Following a 15 min incubation of the slice in MK-801 (40 μm) without synaptic stimulation, NMDAR EPSCs were evoked every 30 s in the continued presence of MK-801, while stepping Vm to +40 mV.
To evoke asynchronous EPSCs (aEPSCs), sodium-ACSF was exchanged for strontium-ACSF containing the following (in mm): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 6 SrCl2, 0.5 CaCl2, and 2 MgCl2. Events were considered aEPSCs if rise time was <2 ms and integrated area was ≥10. aEPSCs were quantified within a 500 ms window, beginning 50 ms after the stimulus artifact.
Associational commissural synaptic responses (CA3-CA3) were evoked using a monopolar glass electrode filled with oxygenated ACSF and positioned within the stratum radiatum. Data collection and analysis were performed with pClamp 9, Microsoft Excel, and GraphPad Prism software. aEPSCs were analyzed using Mini Analysis (Synaptosoft). Two sample comparisons were made using unpaired two-tailed Student's t test and multiple comparisons were made using a one-way ANOVA. The n value represents number of individual recordings. Only one recording was made from each slice and recordings from at least three animals were used for each experiment.
CA1 field recordings.
Animals were anesthetized using ketamine/xylazine, and rapid cardiac perfusion was performed with ice-cold sucrose ACSF containing the following (in mm): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2, 4 MgCl2, 0.5 ascorbic acid, 0.1 kynurenic acid, and 0.01 dl-APV, equilibrated with 95% O2 and 5% CO2 before decapitation and removal of the brain. Horizontal slices from ventral hippocampus were prepared as described above, and transferred to a holding chamber maintained at 28.5°C until used for experiments. Individual slices were transferred to a recording chamber, perfused with regular ACSF [containing the following (in mm): 125 NaCl, 2.4 KCl, 1.2 Na2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2] and visualized under DIC optics. For extracellular field potential recordings, electrodes were fabricated from borosilicate glass at a resistance of 3–5 MΩ and filled with regular ACSF. EPSPs were evoked using a monopolar glass electrode filled with regular ACSF and placed in the stratum radiatum in the CA1 region to stimulate Schaffer collateral inputs. fEPSP responses were elicited with clearly discernable stimulation artifact, fiber volley, and EPSP components. After 10 min of stable (<15% variability) baseline responses were obtained, LTP was induced using theta-burst stimulation consisting of 10 bursts of five stimuli at 100 Hz and delivered with an interburst interval of 200 ms. LTP was quantified during the period 30–40 min following induction. Experiments were performed at 30°C. Data were collected at 20 kHz and filtered at 4 kHz using pClamp 10 software (Axon Instruments). Sampled data were analyzed off-line using Clampfit 10.2. The 10–50% rise slope of fEPSP responses was measured and normalized to the calculated average 10–50% rise slope obtained during the 10 min baseline period.
Immunohistochemistry.
Immunostaining for synaptoporin was performed on sections from paired age-matched 2- to 6-month-old Epac2+/+ and Epac2−/− mice. Mice were perfused with ice-cold PBS followed by 4% PFA, and brains were extracted and stored overnight in 4% PFA at 4°C. Coronal sections were cut between bregma −1.50 and −2.30 mm on a Leica vibratome at 50 μm, permeabilized in 0.1% Triton X-100 in PBS for 90 min at room temperature (RT), and incubated overnight in PBS at 4°C with rabbit anti-synaptoporin (Synaptic Systems; 1:1000). Sections were washed in PBS and incubated with Alexa Red 594-conjugated donkey anti-rabbit secondary IgG (1:2000) for 90 min at RT. Sections were washed in PBS, transferred onto glass slides to dry, and mounted in Vectashield medium containing DAPI. Measurements of the infrapyramidal blade (IPB) and suprapyramidal blade (SPB) lengths were performed on a minimum of three sections per animal from three animals.
Synaptosomal preparation.
Hippocampi were dissected from age-matched Epac2+/+ and Epac2−/− mice and homogenized in buffer (15 mm Na TES, 0.3 m sucrose, and 1 mm MgSO4, pH 7.4) using 0.25 mm clearance and 0.15 mm clearance dounces. Homogenates were filtered through 100 and 70 μm nylon filters (H1) and centrifuged at 900 × g for 10 min at 0–5°C. Pellets were washed in ice-cold buffer and the centrifugation and wash steps repeated. Pellets were suspended in 18% (w/v) Ficoll in cold buffer and centrifuged at 7500 × g for 40 min. The supernatant was collected and diluted with 2 volumes ice-cold buffer and centrifuged at 13,000 × g for 20 min. The resulting pellet (P3) was suspended in 500 μl Krebs bicarbonate medium (5 mm Na2CO3, 20 mm TES salt, 0.5 mm KH2P04, 122 mm NaCl, 3.1 mm KCl, and 11 mm d-glucose, pH 7.4) and centrifuged at 20,800 × g for 15 min at 4°C. Pellets were snap frozen and stored at −80°C until used for Western blot analysis.
Western blot.
Ten microgram protein aliquots were mixed with NuPAGE LDS Sample Buffer (4×) and 0.5 m DTT and boiled at 95°C before separating on 4%–12% NuPAGE Bis-Tris gels in 1× MOPS running buffer (Invitrogen). Proteins were transferred to Millipore Immobilon-P polyvinylidene difluoride membranes (Millipore), stained with Ponceau S, and imaged on a scanner. Membranes were then blocked in 5% nonfat dry milk in Tris-buffered saline (TBS) and 0.1% Tween 20 (TBST; Sigma) for 1 h at RT, then incubated in primary antibody (Epac1,Santa Cruz Biotechnology, 1:1000; Rap1, Millipore, 1:500; Rab3a, Santa Cruz Biotechnology, 1:1000; synaptoporin, Synaptic Systems, 1:1000; PSD-95, NeuroMab, 1:1000; CASK, NeuroMab, 1:1000; RIM1αβ, Synaptic Systems, 1:1000; munc-13, Synaptic Systems, 1:500). Blots were washed in TBST and incubated for 1 h in HRP-conjugated (Jackson ImmunoResearch Laboratories) secondary antibody (1:10,000) in 5% milk in TBST. Immunosignals were detected using enhanced chemiluminescence (Luminata Forte; EMD Millipore) and quantified using a Kodak Image Station 400R imager. Densitometric analyses of immunoblots and images of Ponceau S-stained blots were performed using Kodak Molecular Imaging Software SE. Immunosignals were normalized to the measured intensity of whole-lane Ponceau S (total protein) staining.
Transmission electron microscopy.
Age-matched mice (P25) of each genotype (n = 3 per genotype) were anesthetized by intraperitoneal injection of a ketamine (0.5 mg/ml)/xylazine (5 mg/ml) 0.9% saline solution (0.1 ml/g body weight) and transcardially perfused with 30 ml ice-cold PBS (0.1 m), followed by 50 ml ice-cold fixative [4% PFA and 1% glutaraldehyde (GA) in PBS]. Brains were removed and stored at 4°C overnight in fixative. Horizontal hippocampal slices (350 μm) were prepared the next day in the same orientation as used for electrophysiological recordings and rectangular sections of hippocampal tissue containing the CA3 region were dissected. Tissue sections were postfixed in a 0.1 m Na-cacodylate solution (2.5% GA, 1% osmium tetroxide). Samples were dehydrated through a graded ethanol series and embedded in an epoxy resin. Before embedding, tissue sections were cut and stained with cresyl violet to confirm proper orientation for visualization of mossy fiber synapses. After embedding, ultrathin (90 nm) sections were cut using a Leica ultramicrotome, stained with 2% uranyl acetate and lead solution, and mounted on grids. Sections were visualized using an FEI Tecnai Spirit G2 120 kV TEM. TEM images were captured at magnifications from 13,000× to 30,000× and digitally stored for analysis. MF synapses were visually identified and contained characteristic morphological features including numerous synaptic vesicles, mitochondria, irregular shaped terminals, and complex postsynaptic spines with multiple postsynaptic densities. Analysis of TEM images was performed blind. FIJI software was used to measure active zone (AZ) length and synaptic vesicle diameter, calculated by averaging two measurements per vesicle taken at perpendicular axes.
Results
Epac2 is the primary Epac isoform expressed in the brain and mRNA for Epac2 is particularly enriched in the DG and CA3 regions of the hippocampus (Kawasaki et al., 1998). To determine whether Epac2 protein is localized to MF synapses we isolated the P3 fraction from MF synaptosome preparations (Armstrong et al., 2006), which have been demonstrated to be enriched in proteins from the large MF boutons (Terrian et al., 1988). Consistent with the mRNA distribution we found that Epac2 protein was present in the MF bouton-enriched P3 fraction in Epac2+/+ mice, but was absent in the synaptosomes from Epac2−/− mice (Fig. 1A). cAMP signaling can promote neurite outgrowth (Song et al., 1997), therefore, it is possible that ablation of Epac2 might affect MF axonal development and targeting. The MF axons project to two distinct regions of the CA3; the SPB terminates in stratum lucidum and the IFB traverses the stratum oriens. Immunocytochemical staining with the presynaptic marker synaptoporin, which is abundant in MFs, enabled visualization and calculation of the SPB/IFB ratio, which might be expected to be disrupted in the case of axon guidance or pruning deficits (Bagri et al., 2003; Fig. 1B). Measurement of IFB and SPB lengths in knock-out and control mice did not demonstrate any difference in the length of either tract, or in the ratio of SPB/IPB length (Fig. 1C), indicating that loss of Epac2 activity does not disrupt appropriate targeting or pruning of MF inputs to the CA3 region.
Figure 1.

MF axon targeting is not affected by loss of Epac2. A, Representative Western blots of Epac2 expression from mossy fiber synaptosomal fractions from Epac2+/+ and Epac2−/− mice. Epac2 expression was not detected in samples obtained from Epac2−/− mice (n = 3 paired preparations). B, Immunofluorescence staining of mossy fiber marker synaptoporin (red) and principal cell nuclei (DAPI) in hippocampal sections from Epac2+/+ and Epac2−/− mice. Measurements of the SPB and IPB are indicated by dotted lines. C, Ratios of SPB to IPB bundle length were not significantly different between Epac2+/+ and Epac2−/− mice (n = 4 paired experiments, p > 0.05). H, homogenate; P2, small synaptosomes; P3, large synaptosomes/mossy fiber giant boutons.
Elevation of cAMP by the application of the diterpene forskolin (FSK) produces a long-lasting increase in glutamate release from MF synapses (Weisskopf et al., 1994). To determine whether Epac2 contributes to cAMP-induced potentiation of MF neurotransmitter release, we made whole-cell voltage-clamp recordings from CA3 pyramidal neurons and evoked MF EPSCs (Fernandes et al., 2009). Application of FSK (50 μm) to stimulate cAMP production produced a long-lasting elevation in the EPSC amplitude in Epac2+/+. The peak potentiation during FSK application at 20 min after onset of administration was 562.8 ± 43.6% (n = 5; Fig. 2A). In recordings from Epac2−/− mice, FSK also enhanced release; however, the peak potentiation of the EPSC 20 min after onset of application was lower (350.7 ± 28.5%, n = 7; p = 0.0017, unpaired t test). Similarly the long-lasting potentiation 20–30 min after washout of FSK was significantly lower than observed in Epac2+/+ mice (Epac2+/+: 445.5 ± 66.4%, n = 5; Epac2−/−: 276.4 ± 27.8%, n = 7, p = 0.0252, unpaired t test; Fig. 2A–C). This demonstrates that a component of cAMP-mediated potentiation of MF synapses requires Epac2. The paired-pulse ratio (PPR), which can be used as a proxy for the relative release probability, was not different between the two genotypes during baseline (PP40 Epac2+/+: 3.06 ± 0.58, n = 5; PP40 Epac2−/−: 2.72 ± 0.33, n = 7; p = 0.594 by unpaired t test; Fig. 2D). The PPRs decreased significantly from their baseline values in both genotypes, but were not different compared with each other at 20–30 min following washout of FSK (Epac2+/+: 1.76 ± 0.45, n = 5; Epac2−/−: 1.50 ± 0.20, n = 7, p = 0.577, unpaired t test; Fig. 2D), indicating that in both genotypes the primary action of FSK was presynaptic in elevating release probability. To determine whether the residual potentiation seen in the Epac2−/− mice was mediated by PKA, we measured FSK potentiation of MF synapses in the presence of the selective PKA inhibitor KT-5720 (1 μm, pre-incubated for ≥1 h). Surprisingly we found that KT-5720 had very little effect on FSK potentiation in slices from Epac2+/+ mice (Epac2+/+: 406.1 ± 112%, n = 8). However, in slices from Epac2−/− mice FSK potentiation was almost completely blocked by KT-5720 (Epac2−/−: 128.2 ± 29.7%, n = 4, p = 0.044, unpaired t test; Fig. 2E,F), further supporting a role for both Epac2 and PKA in cAMP-dependent facilitation of MF transmission.
Figure 2.
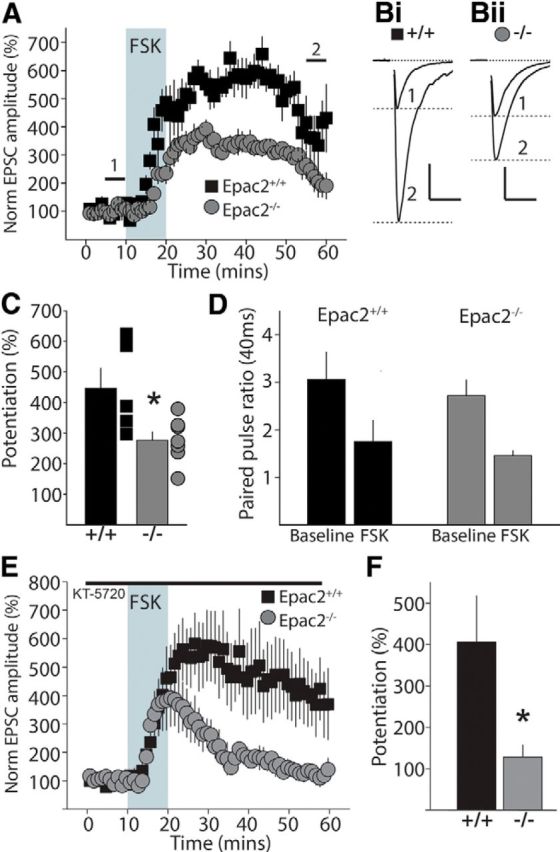
cAMP-mediated potentiation of transmitter release is impaired in Epac2−/− mice. A, Grouped data of time course of MF facilitation by diterpene FSK in hippocampal slices from Epac2+/+ and Epac2−/− mice. Shaded area represents application of 50 μm FSK. B, Representative EPSC traces from Epac2+/+ (Bi) and Epac2−/− (Bii). Traces are shown for baseline period (1) and after potentiation (2). Calibration: Bi, 200 pA, 20 ms; Bii, 400 pA, 20 ms. C, Summary of all data of FSK potentiation calculated as the potentiation of the EPSC amplitude between 50 and 60 min compared with the baseline amplitude (0–10 min), *p < 0.05 by unpaired t test. D, Paired-pulse ratio of EPSCs (PP40 ms) for each recording during baseline and after FSK potentiation for Epac2+/+ (black) and Epac2−/− (gray) mice. E, Time course for FSK potentiation of MF transmission in the presence of the PKA inhibitor KT-5720 (1 μm, pre-incubated for at least 1 h). F, Summary of all data of FSK potentiation calculated as the potentiation of normalized EPSC amplitude between 50 and 60 min (*p < 0.05 by unpaired t test).
MF-LTP is dependent on presynaptic elevations of cAMP and is occluded by prior application of FSK (Weisskopf et al., 1994). PKA activity has been directly implicated in MF-LTP (Huang et al., 1995), but it is also suggested that noncanonical cAMP targets are involved (Mellor et al., 2002; Huang and Hsu, 2003; but also see Chevaleyre and Castillo, 2002). To determine whether Epac2 deletion has an impact on MF-LTP we made voltage-clamp recordings of MF EPSCs and induced LTP using tetanic stimulation (three 1 s trains at 100 Hz, given at 0.05 Hz) in the presence of the NMDAR antagonist d-APV (50 μm). In recordings from Epac2+/+ mice the EPSC measured 20–30 min after LTP induction was potentiated to 183.1 ± 22.5% (n = 9). In interleaved recordings from Epac2−/− mice the amplitude of LTP was significantly lower (130.2 ± 10.6%, n = 11, p = 0.0366, unpaired t test; Fig. 3A–C). The PPR of EPSCs was significantly decreased in recordings from both genotypes (Epac2+/+: baseline PP40 2.45 ± 0.32, LTP PP40 1.57 ± 0.24, n = 8 cells; Epac2−/−: baseline PP40 2.82 ± 0.34, LTP PP40 1.57 ± 0.20, n = 11; Fig. 3D). In some recordings the potentiation immediately following LTP induction (post-tetanic potentiation, PTP) looked lower in Epac2−/−, although this was not significantly different from recording in Epac2+/+ mice (PTP Epac2+/+: 760.2 ± 221%, n = 9 cells; PTP Epac2−/−: 401.9 ± 76.5%, n = 11, p = 0.116, unpaired t test; Fig. 3A,B). To determine whether LTP deficits in these mice were more widespread, we also examined LTP in the Schaffer collateral CA1 synapse where LTP uses different signaling pathways and is expressed postsynaptically (Bliss and Collingridge, 1993). We observed no genotypic differences in the magnitude of CA1 LTP (CA1 LTP Epac2+/+: 135.9 ± 8.4%, n = 5; CA1 LTP Epac2−/−: 128.0 ± 2.2%, n = 4, p > 0.511) consistent with a prior study in an independently engineered Epac2 knock-out mouse (Yang et al., 2012). These results indicate Epac2 is a cAMP target in the mossy fiber synapse and contributes to cAMP-dependent presynaptic potentiation of synaptic transmission.
Figure 3.

MF-LTP is impaired in Epac2−/− mice. A, Time course of MF-LTP in Epac2+/+ mice. Black bar represents application of group II mGluR agonist DCG-IV at the end of each experiment. Inset shows representative traces from one recording during baseline (1) and after 20–30 min after LTP induction (2). B, Average time course of LTP for all recordings from Epac2−/− mice. The magnitude of LTP measured as the potentiation 20–30 min after induction compared with the baseline amplitude (0–10 min) is significantly reduced in the knock-out recordings. C, Cumulative probability distribution of MF-LTP of all recordings. D, Paired-pulse ratios measured at 40 ms interpulse intervals during baseline and after LTP induction. Calibration for EPSC traces: 200 pA, 20 ms.
Epac2 may play a role at several distinct points of the synaptic vesicle cycle that can affect other release parameters including synaptic vesicle release probability (Pr) or vesicle trafficking from distinct pools. To further define how Epac2 might regulate release at this synapse we monitored short-term plasticity (STP), which is largely defined by the basal Pr and is dependent upon elevations in presynaptic calcium (Regehr et al., 1994; Salin et al., 1996). Pairing synaptic stimulation at several interstimulus intervals allowed us to calculate the PPR. We found no difference in the average PPR between interleaved recordings in Epac2+/+ and Epac2−/− littermates (Fig. 4A), indicating that lack of Epac2 activity does not affect basal Pr. Similarly, when we delivered a transient increase in stimulation frequency from 0.05 to 1 Hz to facilitate release (Regehr et al., 1994), we found no significant difference in short-term frequency facilitation (FF) between the two genotypes (FF Epac2+/+: 293 ± 37%, n = 16; FF Epac2−/−: 253 ± 22%, n = 16, p = 0.290 unpaired t test; Fig. 4B). In addition, short trains at higher stimulation frequencies (five stimuli at 10 Hz or 20 Hz) also facilitated synaptic transmission equivalently in recordings from both genotypes (10 Hz; Epac2+/+: 3.09 ± 0.4, n = 7; Epac2−/−: 2.58 ± 0.3, n = 9, p = 0.0967, unpaired t test; 20 Hz Epac2+/+: 2.68 ± 0.3, n = 7; Epac2−/−: 2.72 ± 0.5, n = 9, p = 0.568, unpaired t test; Fig. 4C). Thus the ability of MF synapses to transiently facilitate across a range of stimulus frequencies is not affected by the loss of Epac2 activity, suggesting MF synapses in Epac2−/− mice have normal release probability. As a second way to assess any potential effect that Epac2 knock-out has on MF release probability, we measured the rate of NMDAR block by the use-dependent antagonist MK-801 (Rosenmund et al., 1993; Weisskopf et al., 1994). In the presence of MK-801, synaptic NMDARs are progressively blocked during repetitive stimulation at a rate proportional to the probability of transmitter release. Therefore, a faster rate of NMDAR block would indicate a higher probability of release. The rate of MK-801 block of MF NMDARs was no different between genotypes (number of events to reach 50% inhibition, Epac2+/+: 20.6 ± 4.0, n = 8 cells; Epac2−/−: 18.1 ± 3.0, n = 5 cells; p = 0.458, unpaired t test), suggesting that Epac2 knock-out does not alter vesicular Pr (Fig. 4D).
Figure 4.

MF STP and AMPA/NMDA ratios are unaffected in Epac2−/− mice. A, Paired-pulse facilitation over a range of interpulse intervals at MF-CA3 synapses in Epac2+/+ and Epac2−/− mice. A representative example of a recording from an Epac2−/− mouse is shown at the top. Calibration: 50 ms. B, Frequency facilitation at MF-CA3 synapses. Bi, Time course of FF from a single representative experiment in Epac2−/− mouse. Bii, Representative MF EPSC traces recorded at 0.05 and 1 Hz. Calibration: 200 pA, 10 ms. Biii, Summary of all frequency facilitation recordings. No significant difference was observed in 1 Hz FF between the two genotypes (n = 16 for both Epac2+/+ and Epac2−/−). C, Facilitation of EPSCs to short stimulus trains at 10 and 20 Hz. The facilitation ratio was calculated as the amplitude of the fifth EPSC in the train to the first EPSC in the train. No significant difference was observed in short-term facilitation between genotypes at 10 Hz or 20 Hz. D, Progressive block of synaptic NMDAR currents by 40 μm MK-801 at MF synapses in slices from Epac2+/+ and Epac2−/− mice. Inset, Representative traces from a single experiment of the initial NMDAR current (sweep 1) and after block (sweep 60). Calibration: 100 pA, 10 ms. Ei, Representative MF EPSC traces recorded at −70 mV and +40 mV in Epac2+/+ (left) and Epac2−/− slices (right). The amplitude of the AMPA component was measured as the peak at −70 mV and the NMDA component was measured in a 2.5 ms window, 60 ms after the onset of the outward current at +40 mV. Calibration: Epac2+/+, 100 pA; Epac2−/− 200 pA, 20 ms. Eii, Grouped data for all AMPA/NMDA recordings. There was no significant difference between the two genotypes (p > 0.05, t test).
A recent study reported that knock-out of both Epac1 and Epac2 isoforms reduced the strength of excitatory synapses in the CA1 region of the hippocampus, although Epac2 knock-out alone had no effect (Yang et al., 2012). To determine whether there were changes in postsynaptic strength at the MF synapse, we recorded MF EPSCs with cells voltage clamped at −70 mV to measure the peak AMPA component and at +40 mV when the outward NMDA current can be measured 60 ms after the onset of the outward current (Marie et al., 2005). A comparison of the AMPA to NMDA EPSC ratio in the two genotypes did not uncover any differences (Fig. 4E), indicating that there are no major alterations in postsynaptic glutamate receptors at MF synapses. Measuring the decay of the responses, we found no differences in the deactivation kinetics of either EPSC component (tau decay Epac2+/+: AMPA 8.4 ± 0.6, NMDA 40.0 ± 2.1 ms; n = 10 cells; Epac2−/−: AMPA 8.9 ± 0.5, NMDA 34.0 ± 2.5 ms; n = 7 cells; p = 0.093, unpaired t test).
In pancreatic β-cells Epac2 interacts with two regulators of vesicle priming, RIM and Munc13, to potentiate insulin secretion (Ozaki et al., 2000). In the CNS disrupting the function of either RIM1αβ or Munc13-1 impairs MF-LTP without disrupting STP (Castillo et al., 2002; Yang and Calakos, 2011), findings that are similar to what we have observed in Epac2−/− mice. It is possible that Epac2, RIM1αβ, and Munc13-1 function in concert in MF terminals and ablation of Epac2 might result in compensatory changes in the expression of other presynaptic proteins. Therefore, we performed Western blot analysis on hippocampal MF synaptosomes (in the P3 fraction) to determine protein expression of several presynaptic proteins in Epac2+/+ and Epac2−/− mice. Interestingly, there were no significant differences detected in expression of RIM1αβ and Munc13-1 or in the low-abundant isoform cAMP-GEF Epac1 (Fig. 5A,B). We also did not observe a difference in the expression of the small GTPase Rap1, which is one downstream effector of Epac signaling, or the postsynaptic protein PSD-95, which also is found in the P3 synaptosomal fraction (Armstrong et al., 2006). However, we did observe a small, but significant, reduction in the expression of three of the presynaptic proteins that we probed, including the scaffolding protein CASK (p = 0.0369, paired t test, n = 3), the synaptic vesicle membrane protein synaptoporin (p = 0.002, paired t test, n = 3), and most notably the vesicle-associated protein Rab3A (p = 0.0315, n = 3), an RIM1 interacting protein proposed as a direct target for Epac (Branham et al., 2009; Fig. 5A,B).
Figure 5.

Expression levels of Rab3A, CASK, and synaptoporin (Synprn) are significantly reduced in MF synaptosomes from Epac2−/− mice. A, Representative Western blots of P3 synaptosomal fractions from three independent synaptosomal preparations, run in parallel, from Epac2+/+ and Epac2−/− mice. B, Quantification of grouped Western blot data from MF synaptosomal preparations. Expression level of proteins of interest normalized to total protein. *p < 0.05, paired t test.
To assess whether there were morphological changes in synaptic structure in Epac2−/− mice that might affect synaptic properties, we used transmission electron microscopy to visualize MF terminals, presynaptic AZs, and synaptic vesicles. Mossy fiber boutons were clearly identifiable from their unique architecture and the presence of a large number of synaptic vesicles and AZs were visible as electron-dense regions on the presynaptic membrane (Fig. 6A,B). Measuring the cross-sectional area of the AZ in ultrathin sections from three animals per genotype (Epac2+/+: 35 MF boutons analyzed, 60 AZ total; Epac2−/−: 20 MF boutons analyzed, 46 AZ total) we found a small but significant reduction in the measured AZ lengths in Epac2−/− animals (Epac2+/+: 244.0 ± 12 nm; Epac2−/−: 206 ± 10 nm; p = 0.022, unpaired t test; Fig. 6C). There was no difference in the average diameter of individual vesicles measured in each genotype (Epac2+/+: 38 ± 1 nm, 3203 vesicles; Epac2−/−: 37 ± 1 nm, 1729 vesicles; p = 0.645, unpaired t test; Fig. 6D).
Figure 6.

MF synaptic ultrastructure in Epac2−/−. A, B, Representative electron micrographs of MF synapses from an Epac2−/− mouse brain, taken at 23,000× (Ai) and 13,000× (Bi); Aii and Bii are magnified areas from Ai and Bi, respectively, to illustrate AZ length measurements (between hashed red lines). Scale bars: Ai, Aii, 200 nm; Bi, 500 nm; Bii, 200 nm. Arrowheads indicate sites where AZs are apposed to PSDs. C, Analysis of AZ length from all experiments, *p < 0.05, unpaired t test. D, Measurement of vesicle diameter in MF bouton from Epac2+/+ and Epac2−/− mice. Sp, spine; MFB, mossy fiber bouton.
Given that we had observed deficits in long-term enhancement of MFs but not STP, one possibility was that Epac2 ablation might impact the size of the vesicular pool required to maintain synaptic strength during sustained periods of elevated release. In addition, prior work has demonstrated that elevation of cAMP increases the readily releasable pool (RRP) of vesicles in MF terminals (Lonart et al., 1998). It is therefore possible that the impact of Epac2 ablation on reducing forskolin potentiation and MF-LTP might result from alteration in the size of, or trafficking between, the RRP or reserve pool (RP; Zucker and Regehr, 2002). To estimate the total number of releasable vesicles (the RRP plus the RP), we used an electrophysiological approach analyzing the cumulative EPSC during a long train of stimuli. This analysis assumes that depression of the EPSC during the train occurs as the RRP is depleted, and the product of N (total number of releasable vesicles) and q (the quantal size) can be estimated from the zero time intercept of the linear regression fit to the cumulative EPSC amplitude plot (Schneggenburger et al., 1999; Kaneko and Takahashi, 2004). Although this method does not give an absolute value for N, assuming that q is not altered, a relative comparison can be made between genotypes to determine whether there are likely differences in the RRP. To determine Nq we applied long trains (300 stimuli at 10 Hz) to stimulate MF EPSCs and analyzed the cumulative synaptic current. During the train MF EPSCs were facilitated, but the extent of facilitation gradually reduced during the train (Fig. 7A). Comparing recordings from Epac2−/− and Epac2+/+ mice there was a clear reduction in both the peak facilitation early in the train and in the response during the later part of the train in the Epac2−/− mice (Fig. 7A). We plotted the normalized cumulative EPSC and used a linear regression fit of the last 50 events in the train to obtain a functional estimate of Nq. There was a significant difference in the y-intersect of the extrapolated fit in recordings from Epac2−/− mice or littermate controls (Epac2+/+: 423.8 ± 90.3, n = 9; Epac2−/−: 196.7 ± 32.5, n = 10; p = 0.0246, unpaired t test; Fig. 7B,Ciii). To experimentally determine the value of quantal size (q), we could not simply measure spontaneous mEPSC amplitude because CA3 pyramidal neurons receive multiple inputs from other synapses. Therefore, to measure the quantal size of MF synapses, we selectively activated the MF pathway and evoked asynchronous release by replacing extracellular Ca2+ with a Sr2+-containing ACSF (Bekkers and Clements, 1999; Lawrence et al., 2004). aEPSCs were detected up to several hundred milliseconds following stimulation of the MF inputs (Fig. 7Ci). The mean amplitude of the aEPSCs was not significantly different in Epac2+/+ and Epac2−/− (Epac2+/+: 14.9 ± 3.5 pA, n = 3 cells, 1611 events; Epac2−/−: 16.6 ± 1.2 pA, n = 5 cells, 3276 events; p = 0.603, unpaired t test; Fig. 7Cii). Therefore q is not altered in MF synapses in Epac2−/− mice. As we did not find a difference in q, these data indicate that N, the number of readily releasable vesicles, is significantly reduced in Epac2−/− mice. To investigate whether this reduction in the RRP was a general synaptic dysfunction linked to the loss of Epac2, we also analyzed the cumulative EPSC during long trains (300 stimuli at 10 Hz) at associational-commissural (A/C) synapses between CA3 pyramidal neurons (CA3-CA3). We found the functional estimate of N*q at this synapse was not significantly different between Epac2+/+ and Epac2−/− (Epac2+/+: 51.3 ± 7.7, n = 8 cells; Epac2−/−: 66.9 ± 21.4, n = 6 cells; p = 0.569 by unpaired t test). This study proposes a novel mechanism for enhancement of transmitter release at MF synapses involving a noncanonical cAMP target and uncovers a novel role for Epac2 as a synapse-specific regulator of the RRP of vesicles.
Figure 7.

Functional estimates of the total number of synaptic vesicles available for release in Epac2+/+ and Epac2−/− MF boutons. A, Time course of EPSC facilitation during trains in Epac2+/+ and Epac2−/− mice. B, Analysis of cumulative MF EPSCs in Epac2+/+ (black) and Epac2−/− (gray). Red lines indicate linear regression fit to the last 50 events and extrapolated to time = 0 to provide estimates of N*q. Ci, Cii, Measurement of quantal content (q) by evoking aEPSCs. Ci, Representative examples of evoked synchronous release events in normal extracellular Ca2+ and asynchronous release events (inset, indicated by gray arrows) in Sr2+ from Epac2−/− mice. Red arrows indicate time of stimulus. Calibration: 100 pA, 50 ms. Cii, Group data, all experiments for measurement of amplitude of quantal MF events (p > 0.05, unpaired t test). Ciii, Group data of measured N*q for Epac2+/+ and Epac2−/− (*p < 0.05, unpaired t test). D, Analysis of cumulative EPSCs during trains at A/C synapses (CA3-CA3) in Epac2+/+ (black) and Epac2−/− (gray). Inset, Summary of N*q product from A/C recordings (p > 0.05, unpaired t test).
Discussion
The MF-CA3 pyramidal cell synapse in the hippocampus is notable not only for its dynamic STP (Salin et al., 1996) but also for a form of LTP dependent on elevations of cAMP in the presynaptic terminal (Weisskopf et al., 1994). Prior studies using pharmacological approaches have demonstrated that PKA is a critical downstream target of cAMP (Huang et al., 1994; Weisskopf et al., 1994), and it has been proposed that enhancement of PKA activity is the principal pathway for the facilitation of neurotransmitter release from MF terminals following LTP induction (Huang et al., 1994; Weisskopf et al., 1994; Lonart et al., 1998). In this study we investigated the potential contribution of an alternate cAMP substrate, Epac2, to MF synaptic transmission and plasticity. We found that there were deficits in LTP of MF synapses in Epac2−/− mice, which might be explained by an alteration in the available pool of releasable vesicles. These results propose a novel role for Epac2 as a major regulator of synaptic plasticity in the CA3 region of the hippocampus.
A number of recent studies concerning modulation of neurotransmission by Epac proteins have provided some indication of the physiological functions that these molecules fulfill as mediators of cAMP-dependent signaling. Initial investigations used pharmacological approaches in in vitro systems to dissect the role of Epac in enhancing neurotransmission. In pituitary melanotrophs Epac and PKA function cooperatively to enhance secretion, acting on distinct phases (Sedej et al., 2005). Epac mediates serotonergic enhancement of synaptic transmission at the crayfish neuromuscular junction in a cAMP-dependent fashion, but the mechanisms of action remain unknown (Zhong and Zucker, 2005). At the calyx of Held, application of FSK, cAMP, or Epac-selective agonists increased both release probability and the total number of releasable synaptic vesicles, independent of any effect on presynaptic Ca2+ or K+ currents, suggesting that Epac is a major effector of cAMP-induced synaptic facilitation (Kaneko and Takahashi, 2004). Similar findings were reported in autaptic murine hippocampal cultures, where application of the Epac-selective cAMP analog 8-pCPT was found to mimic FSK by acting presynaptically to increase both evoked EPSC amplitudes and mEPSC frequency (Gekel and Neher, 2008). These lines of evidence suggest a broad role for Epac as an effector for cAMP in promoting synaptic vesicle exocytosis.
Both the size and refill rate of the RRP at the MF synapse is critical to maintaining high-fidelity neurotransmission (Suyama et al., 2007); therefore, any factor that affects the size of the RRP could potentially impact expression of MF-LTP. We have found that Epac2 plays a role in maintaining the size of the RRP in MF synapses similar to already described roles for Epac2 in mediating a cAMP-dependent increase in the RRP of β-islet cells (Seino and Shibasaki, 2005; Shibasaki et al., 2007) and at the calyx of Held (Kaneko and Takahashi, 2004). This role of Epac is synapse specific as we did not observe a change in RRP size at A/C synapses in Epac2−/− mice; however, it would be interesting in future work to test for a similar role at synapses that are known to be regulated by presynaptic cAMP.
We examined the effect of Epac2 ablation on STP at the MF-CA3 synapse because it was possible that changes in basal neurotransmission in the knock-out mice affect synaptic release probability. We did not observe any significant deficits in several measures of stereotypical STP of MF-CA3 synapses in Epac2−/− mice. It is not surprising that Epac2 ablation does not directly affect STP since, unlike LTP, transient facilitation of transmitter release is not dependent on elevations of cAMP in the presynaptic terminal, but rather on acute elevations of Ca2+ within the terminal bouton (Regehr et al., 1994; Salin et al., 1996). Additionally we found that genetic deletion of Epac2 did not produce a compensatory change in Epac1 expression or in two critical presynaptic proteins that interact with Epac2, Rim1, and Munc13-1 (Kwan et al., 2007). This is particularly important because knock-out or knockdown of these two proteins results in impaired MF-LTP (Castillo et al., 2002; Yang and Calakos, 2011). We did see small but significant change in expression of Rab3A, Cask, and synaptoporin in MF synaptosome fractions, and thus far it is unclear whether these presynaptic proteins interact in any way with Epac2. The role of Rab3A is of particular interest as MF-LTP is abolished in the Rab3A−/− mouse, although FSK-mediated synaptic potentiation remains fully intact (Castillo et al., 1997). It is difficult to speculate whether this ∼20% reduction in expression of Rab3A directly impacts MF synaptic transmission, and there have been no published analyses of heterozygous Rab3A+/− mice (in which, presumably, there is a ∼50% decrease in Rab3A expression). Therefore, further detailed study is required to determine how Rab3A and Epac2 interact with each other and control transmitter release at MF synapses.
In agreement with a role for both PKA and Epac2 as effectors for cAMP-mediated synaptic potentiation, we found that inhibition of PKA, using the selective inhibitor KT-5720 in slices from Epac2−/− mice, almost completely eliminated FSK-induced MF potentiation. Surprisingly, we observed no significant inhibition of FSK enhancement in slices from Epac2+/+ mice. Two prior studies had reported that KT-5720 partially inhibits MF-LTP (Weisskopf et al., 1994; Huang et al., 1995); however, it was not known whether KT-5720 can block FSK-induced potentiation. Our results suggest that there is likely a complex dual role for PKA and Epac2 in FSK potentiation that intriguingly may differ from the mechanisms engaged when MF-LTP is engaged by synaptic stimulation.
One possible explanation is that both cAMP effectors act in parallel, and that Epac2 compensates fully if PKA is inhibited during FSK potentiation but not after LTP induction. Another possibility is that cAMP signaling in MF terminals is normally compartmentalized to specific subdomains allowing for spatial segregation of distinct second messenger pathways, as is found in cardiac myocytes (Vandecasteele et al., 2006) and β-islet cells (Kashima et al., 2001). Thus FSK potentiation and synaptic stimulation may activate different but overlapping pathways at MF synapses.
Previous studies have demonstrated that the contribution of Epac proteins to neurotransmission is not restricted to presynaptic terminals, or to promoting excitatory neurotransmission. Application of an Epac agonist, 8-pCPT, to hippocampal slices produced Schaffer collateral-CA1 LTD, which involved a postsynaptic mechanism requiring internalization of synaptic AMPARs (Ster et al., 2009). In cortical cultures activation of Epac produces shrinkage of spines and internalization of AMPARs (Woolfrey et al., 2009). We did not find any obvious changes in postsynaptic parameters at MF synapses in Epac2−/− mice. Both the AMPA/NMDA ratio of MF EPSCs and the amplitude of quantal MF EPSCs were not changed in the knock-out mice, suggesting no major changes in the MF post synapse. It is possible that both Epac isoforms, which would be activated by 8-pCPT, are required to effect changes in postsynaptic glutamate receptors, and this is supported by evidence that at Schaffer collateral synapses deletion of both Epac1 and Epac2 was required to produce an observable postsynaptic phenotype (Yang et al., 2012). Together with our findings, these studies suggest that the Epac proteins can have multiple effects on hippocampal excitatory synaptic transmission, which are dependent on the synapse and on the presence of different Epac isoforms.
The primary finding in our study is that potentiation of MF synapses both by tetanic stimulation and FSK application was impaired in the Epac2−/− mice. No prior studies have addressed a potential role for Epac in regulating MF synapses, but there was reason to believe that cAMP substrates other than PKA might be important. Several studies have demonstrated that FSK continues to transiently or persistently potentiate MF-CA3 synaptic transmission in the absence of several potential PKA targets that are critical to the expression of MF-LTP (Castillo et al., 1997, 2002; Kaeser-Woo et al., 2013), indicating the presence of a PKA-independent pathway in MFs for the enhancement of neurotransmitter release. Moreover, our experiments suggest that Epac2 has a role in regulating the RRP of vesicles at MF synapses. A similar mechanism for the potentiation of vesicle release exists in β-islet cells of the pancreas, where a component of the enhancement of insulin secretion in response to elevations in blood glucose has been shown to depend on Epac2 activity regulating the releasable pool of vesicles (Ozaki et al., 2000; Kashima et al., 2001; Kwan et al., 2007). Insulin secretion may provide a model for investigating the dual cAMP-dependent pathways (Epac2 and PKA) for enhancement of neurotransmitter release from MF boutons, as there are distinct parallel components of the potentiation of insulin granule exocytosis dependent on each pathway, including the involvement of Munc13-1 and Rim proteins (Kwan et al., 2007), which are both required for long-term enhancement of MF-LTP (Castillo et al., 2002; Yang and Calakos, 2011). One possibility is that the PKA and Epac2 pathways converge at the interaction between Rab3A, Rim1, and Munc13-1 at presynaptic AZs. Knock-out of Rab3A and Rim1 in knock-out mice, and knockdown of Munc13-1, results in normal STP but produces deficits in MF-LTP (Castillo et al., 1997, 2002; Yang and Calakos, 2011). We found very similar alterations in synaptic function in Epac2−/− mice with no observable changes in STP, but prominent deficits in LTP and FSK induced potentiation. Further studies are required to determine exactly how these presynaptic proteins partner to effect changes in transmitter release.
In summary, we have demonstrated a central role for Epac2 in regulating transmitter release through an effect on the readily releasable pool of synaptic vesicles at MF synapses. This role of Epac2 does not impact basal synaptic parameters but has a large effect on transmitter release during sustained transmission or after induction of MF-LTP. These studies add to the growing evidence for an important role for Epac proteins in regulating both presynaptic and postsynaptic neurotransmission at central synapses.
Footnotes
This work was funded by grants from National Institutes of Health–National Institute of Mental Health (R01MH099114) and the McKnight Foundation (to A.C.). H.B.F. was funded by a fellowship from the Canadian Institutes of Health Research. C.L.R. was funded by an institutional T32 award (T32 MH067564). We thank Professor Susumu Seino for Epac2−/− mice and Anna Toth for help with initial CA1 LTP experiments. Imaging was performed in the Northwestern University Cell Imaging Facility, which is supported by National Institutes of Health–National Cancer Institute (P30 CA060553) to the Robert H Lurie Comprehensive Cancer Center.
The authors declare no competing financial interests.
References
- Armstrong JN, Saganich MJ, Xu NJ, Henkemeyer M, Heinemann SF, Contractor A. B-ephrin reverse signaling is required for NMDA-independent long-term potentiation of mossy fibers in the hippocampus. J Neurosci. 2006;26:3474–3481. doi: 10.1523/JNEUROSCI.4338-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagri A, Cheng HJ, Yaron A, Pleasure SJ, Tessier-Lavigne M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 2003;113:285–299. doi: 10.1016/S0092-8674(03)00267-8. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol. 1999;516:227–248. doi: 10.1111/j.1469-7793.1999.227aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Branham MT, Bustos MA, De Blas GA, Rehmann H, Zarelli VE, Treviño CL, Darszon A, Mayorga LS, Tomes CN. Epac activates the small G proteins Rap1 and Rab3A to achieve exocytosis. J Biol Chem. 2009;284:24825–24839. doi: 10.1074/jbc.M109.015362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Südhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Cheung U, Atwood HL, Zucker RS. Presynaptic effectors contributing to cAMP-induced synaptic potentiation in Drosophila. J Neurobiol. 2006;66:273–280. doi: 10.1002/neu.20218. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci U S A. 2002;99:9538–9543. doi: 10.1073/pnas.142213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/S0896-6273(01)00191-X. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Catches JS, Petralia RS, Copits BA, Xu J, Russell TA, Swanson GT, Contractor A. High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron. 2009;63:818–829. doi: 10.1016/j.neuron.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekel I, Neher E. Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci. 2008;28:7991–8002. doi: 10.1523/JNEUROSCI.0268-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Reexamination of the role of hyperpolarization-activated cation channels in short- and long-term plasticity at hippocampal mossy fiber synapses. Neuropharmacology. 2003;44:968–981. doi: 10.1016/S0028-3908(03)00098-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Younts TJ, Yang X, Zhou P, Wu D, Castillo PE, Südhof TC. Synaptotagmin-12 phosphorylation by cAMP-dependent protein kinase is essential for hippocampal mossy fiber LTP. J Neurosci. 2013;33:9769–9780. doi: 10.1523/JNEUROSCI.5814-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Takahashi T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci. 2004;24:5202–5208. doi: 10.1523/JNEUROSCI.0999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII–Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kwan EP, Xie L, Sheu L, Ohtsuka T, Gaisano HY. Interaction between Munc13-1 and RIM is critical for glucagon-like peptide-1 mediated rescue of exocytotic defects in Munc13-1 deficient pancreatic beta-cells. Diabetes. 2007;56:2579–2588. doi: 10.2337/db06-1207. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Janz R, Johnson KM, Südhof TC. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 1998;21:1141–1150. doi: 10.1016/S0896-6273(00)80631-5. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/S0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Sedej S, Rose T, Rupnik M. cAMP increases Ca2+-dependent exocytosis through both PKA and Epac2 in mouse melanotrophs from pituitary tissue slices. J Physiol. 2005;567:799–813. doi: 10.1113/jphysiol.2005.090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Ster J, de Bock F, Bertaso F, Abitbol K, Daniel H, Bockaert J, Fagni L. Epac mediates PACAP-dependent long-term depression in the hippocampus. J Physiol. 2009;587:101–113. doi: 10.1113/jphysiol.2008.157461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama S, Hikima T, Sakagami H, Ishizuka T, Yawo H. Synaptic vesicle dynamics in the mossy fiber-CA3 presynaptic terminals of mouse hippocampus. Neurosci Res. 2007;59:481–490. doi: 10.1016/j.neures.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Terrian DM, Johnston D, Claiborne BJ, Ansah-Yiadom R, Strittmatter WJ, Rea MA. Glutamate and dynorphin release from a subcellular fraction enriched in hippocampal mossy fiber synaptosomes. Brain Res Bull. 1988;21:343–351. doi: 10.1016/0361-9230(88)90146-3. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Rochais F, Abi-Gerges A, Fischmeister R. Functional localization of cAMP signalling in cardiac myocytes. Biochem Soc Trans. 2006;34:484–488. doi: 10.1042/BST0340484. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci. 2009;12:1275–1284. doi: 10.1038/nn.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Munc13-1 is required for presynaptic long-term potentiation. J Neurosci. 2011;31:12053–12057. doi: 10.1523/JNEUROSCI.2276-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, Xu X, Tian Q, Zhang J, Qian K, Wang YX, Petralia RS, Tu W, Zhu LQ, Wang JZ, Lu Y. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron. 2012;73:774–788. doi: 10.1016/j.neuron.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Zucker RS. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci. 2005;25:208–214. doi: 10.1523/JNEUROSCI.3703-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]