

Abstract
Growth-associated protein 43 (GAP43) is known to regulate axon growth, but whether it also plays a role in synaptogenesis remains unclear. Here, we found that GAP43 regulates the aggregation of gephyrin, a pivotal protein for clustering postsynaptic GABAA receptors (GABAARs), in developing cortical neurons. Pharmacological blockade of either protein kinase C (PKC) or neuronal activity increased both GAP43-gephyrin association and gephyrin misfolding-induced aggregation, suggesting the importance of PKC-dependent regulation of GABAergic synapses. Furthermore, we found that PKC phosphorylation-resistant GAP43S41A, but not PKC phosphorylation-mimicking GAP43S41D, interacted with cytosolic gephyrin to trigger gephyrin misfolding and its sequestration into aggresomes. In contrast, GAP43S41D, but not GAP43S41A, inhibited the physiological aggregation/clustering of gephyrin, reduced surface GABAARs under physiological conditions, and attenuated gephyrin misfolding under transient oxygen-glucose deprivation (tOGD) that mimics pathological neonatal hypoxia. Calcineurin-mediated GAP43 dephosphorylation that accompanied tOGD also led to GAP43-gephyrin association and gephyrin misfolding. Thus, PKC-dependent phosphorylation of GAP43 plays a critical role in regulating postsynaptic gephyrin aggregation in developing GABAergic synapses.
INTRODUCTION
Proper development of inhibitory GABAergic synapses is critical for establishing an excitatory/inhibitory balance in the neural network (1, 2). The impairment of postsynaptic GABAA receptor (GABAAR) activity is a major cause of neuronal hyperactivity, affecting cognitive development and psychosocial behaviors (3, 4). Postsynaptic surface insertion and clustering of GABAARs determine the efficacy of GABAergic synapses (4, 5). Gephyrin, a microtubule-associated protein, is a key scaffolding protein that requires the GABAAR γ2 subunit for clustering GABAARs at the postsynaptic membrane (6, 7). The lack of neuronal gephyrin reduces postsynaptic GABAAR clustering, thereby impairing inhibitory synaptic transmission (8, 9).
In central neurons, gephyrin monomers oligomerize to form a hexagonal lattice, also called gephyrin clusters, underneath the cell surface membrane to anchor postsynaptic GABAARs (10). However, numerous studies have shown that gephyrin is an aggregation-prone protein that forms large clumps when expressed in nonneural cells or cell-free systems (11, 12). Instead, gephyrin in neurons forms small aggregates/clusters in both the cytosol and submembrane domain for receptor clustering, suggesting a neuronal machinery that regulates gephyrin clustering. To date, a postsynaptic protein, collybistin, a GDP-GTP exchanging factor, is the only gephyrin-interacting protein that can effectively disperse gephyrin clumps into oligomeric clusters in HEK293T cells (13). Gephyrin scaffolding in neurons depends on the dynamic rearrangement of microtubules and actin microfilaments at postsynaptic sites (14, 15). Whether cytoskeleton-associated proteins are involved in regulating gephyrin aggregation between clumps and clusters remains unclear.
Growth-associated protein 43 (GAP43) is an activity-dependent phosphoprotein that mediates neurite outgrowth in developing neurons by stabilizing actin filaments (16–18). The actin-stabilizing activity of GAP43 depends on its phosphorylation at serine 41 (S41), a protein kinase C (PKC)- and calcineurin-specific site (19–21). GAP43 also regulates synaptic plasticity for memory storage in adult brains (22). The reduction of GAP43 expression in mice causes abnormal barrel cortex organization (23) and an increase in neuronal excitation related to hyperactivity and autism-like behaviors (24, 25). Excessive GAP43 in the brain is also a pathological indication of plasticity-associated aberrant sprouting and is positively correlated with the severity of memory deficit in Alzheimer's disease patients (26). While F-actin also is present at postsynaptic sites and PKC is pivotal for postsynaptic plasticity (27), it remained unclear whether and how PKC-phosphorylated GAP43 enriched in growing neurites (20, 28) contributes to the postsynaptic plasticity involving the organization of postsynaptic receptor scaffolding during synapse development.
In this study, we found that GAP43 is associated with gephyrin in early-developing cortical neurons. This association could be enhanced by pharmacological blockade of PKC and neuronal activities, as well as pathological insults that activate calcineurin. We found that these effects could be attributed to the phosphorylation status of GAP43-S41, which modulates GAP43-gephyrin association. Further studies revealed a novel regulatory action of GAP43 on gephyrin aggregation between misfolded aggregates and physiological clusters. This PKC-dependent phosphorylation of GAP43 may contribute to the proper development of GABAergic synapses.
MATERIALS AND METHODS
Animals.
Pregnant female Sprague-Dawley (SD) rats, obtained from BioLASCO Taiwan Co., were used to harvest embryonic rats for the primary culture of cortical neurons. Postnatal day 2 (P2) SD rat pups were used in neonatal hypoxia experiments. Animal experimentation procedures were reviewed and approved by the Experimental Animal Review Committee at National Yang-Ming University and were according to the Guide for the Care and Use of Laboratory Animals (29) and the U.S. National Institutes of Health guidelines for the care and use of animals for experimental procedures.
Drug treatment.
Pharmacological compounds, including PKC inhibitor Ro318220 (Ro), PKA inhibitor Rp-8-Br–cyclic AMP (cAMP), mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitor PD-98059, calcineurin inhibitor FK506, and calcium ionophore ionomycin were obtained from Sigma-Aldrich. GABAA receptor agonist muscimol and tetrodotoxin (TTX) were obtained from Tocris Bioscience. Fura-2 acetoxymethyl ester (AM) was obtained from Molecular Probes.
Primary culture of rat cortical neurons and HEK293T cell culture.
Primary cultured cortical neurons were prepared from embryonic day 17 (E17) fetal rats as previously described (30). Cortical neurons were cultured in neurobasal medium (Invitrogen) in 5% CO2 at 37°C for 4 days in vitro (DIV) or 11 DIV, which represent early- and late-developing neurons, respectively. HEK293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) with 10% fetal bovine serum in 5% CO2 at 37°C.
LC-MS/MS analysis for identifying GAP43-interacting proteins.
Total cell lysate of primary cultured rat cortical neurons at 4 DIV were harvested and incubated with anti-GAP43 antibody. Immune complex was purified and separated by SDS-PAGE, followed by in-gel trypsin digestion. Peptide was analyzed by using nano-liquid chromatography-nano electrospray ionization tandem mass spectrometry (nano-LC-nano-ESI-MS/MS) analysis on a nanoAcquity system (Waters, Milford, MA) connected to an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a nanospray interface (Proxeon, Odense, Denmark). The mass spectrometer was operated in the data-dependent mode. Raw files were transformed to msm files using RAW2MSM v.1.10 software (Matthias Mann) using default parameters and without any filtering, charge state deconvolution, or deisotoping. The msm files were searched against the Swiss-Prot Rattus database (7,917 sequences) using a Mascot Daemon 2.4.0 server. In order to estimate the false discovery rate (FDR), the decoy search option was allowed. The significance threshold was adjusted to keep an FDR of <1% for the orbitrap data in this study.
Co-IP, Western blotting, and biotinylation assay.
The coimmunoprecipitation (co-IP) and Western blotting was performed as previously described (31). For coimmunoprecipitation, 600 to 1,200 μg total proteins of cortical neuronal lysate were incubated with 2 μg anti-GAP43 (Millipore), antigephyrin (Santa Cruz), or anti-Flag (Sigma) antibodies and protein G-Sepharose beads, and the immune complex was washed and eluted for Western blotting. Primary antibodies used for Western blotting were mouse anti-GAP43 (Millipore), rabbit anti-S41-phosphorylated GAP43 (pGAP43-S41; Millipore), rabbit or mouse antigephyrin (Santa Cruz), mouse anti-Na+/K+-ATPase α3 (Santa Cruz), rabbit anti-GABAARγ2 (Alpha Diagnostic), mouse anti-Flag M2 (Sigma), mouse anti-PSD95 (NeuroMab), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Biogenesis) antibodies. The immune complex was further probed with horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson) and then visualized using HRP-reactive ECL reagents (GE Healthcare). The protein bands were detected by NightOWL LB 981, and the intensity was analyzed and quantified by Berthold WinLight32 (Berthold Technologies). Surface GABAAR expression was detected by using a biotinylation assay using a Pierce cell surface protein isolation kit to obtain surface protein (Thermo) as described previously (30). The biotinylated surface proteins were Western blotted using rabbit anti-GABAARγ2 or mouse anti-Na+/K+-ATPase α3 antibody.
Immunofluorescence and confocal microscopic imaging.
Immunofluorescent staining of GAP43 and gephyrin in cultured cortical neurons was performed as previously described (30). Fluorescence-conjugated secondary antibodies used for labeling primary antibody-bound GAP43 or gephyrin were fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG, Texas Red-conjugated goat anti-rabbit IgG, Texas Red-conjugated goat anti-mouse IgG (Jackson), and Alexa Fluor 633–goat anti-mouse IgG (Invitrogen). 4′,6-Diamidino-2-phenylindole (DAPI) (Abcam) was used for nuclear staining. For determination of misfolded protein aggregates and aggresomes, fixed cells were stained using the aggresome detection reagent (ADR) Proteostat (Enzo), a molecular rotor dye that becomes fluorescent when binding to the β-sheet structure of misfolded proteins (32), according to the manufacture's protocol (Enzo). The fluorescent images and colocalization signals of the merged images (based on colocalized pixel areas) were processed and analyzed by confocal microscopy (FV1000; Olympus) and MetaMorph 7.7.0.0 software (Molecular Device). For the measurement of misfolded gephyrin aggregates and physiological gephyrin clusters in neurons, the signal of gephyrin immunostaining was separated with the color separate tool of MetaMorph Display software from the selected cell area. Thresholded images of gephyrin staining then were used to show all of the analyzed clusters/aggregates. The Integrated Morphometry Analysis tool of MetaMorph measurement software was used to count the total number of gephyrin clusters/aggregates with sizes larger than 0.04 μm2. The total area of gephyrin clusters/aggregates per neuron also was measured. Misfolded gephyrin aggregate signals then were obtained by masking the total gephyrin signal with the ADR-stained signal to obtain the number of misfolded gephyrin aggregates per neuron. The properly folded gephyrin cluster number and total area per neuron were obtained by subtracting the data for misfolded gephyrin clusters/aggregates from the data for total gephyrin clusters/aggregates. The size distribution of gephyrin clusters under each condition also was analyzed and expressed as cumulative frequency.
Site-directed mutagenesis of rat GAP43.
A Ser41-to-Ala GAP43 mutant (GAP43S41A) and a Ser41-to-Asp GAP43 mutant (GAP43S41D) were generated as described previously, with minor modifications (33, 34). The S41 residue of GAP43 was replaced with an alanine or aspartic acid residue by priming with oligonucleotide 5′-GCAACCAAAATTCAGGCTGCCTTCCGTGGACACATA ACA-3′ or 5′-GCAACCAAAATTCAGGCTGACTTCCGTGGACACATAACA-3′, respectively. The Flag-tagged GAP43 plasmids, including pCMV-Tag2B-GAP43S41A, pCMV-Tag2B-GAP43S41D, and pCMV-Tag2B-GAP43-WT, and the Myc-tagged gephyrin plasmid pCMV-Tag3B-gephyrin were constructed by inserting the DNA fragment encoding rat GAP43, GAP43S41A, GAP43S41D, or gephyrin between the EcoRI and XhoI sites of pCMV-Tag2B and pCMV-Tag3B (Stratagene), respectively. The plasmid pEGFPN1-GAP43-WT (encoding enhanced green fluorescent protein [EGFP]-tagged wild-type GAP43), pEGFPN1-GAP43-S41A, and pEGFPN1-GAP43-S41D were constructed by inserting the DNA fragments encoding the rat wild-type GAP43, GAP43S41A, or GAP43S41D between the XhoI and EcoRI sites of pEGFPN1 (Clontech). For DNA transfection, Lipofectamine 2000-mediated transfection was performed in Opti-MEM medium (Invitrogen) and neurobasal medium (Invitrogen) for transfection into HEK293T and 4-DIV cortical neurons, respectively. Five hours after the transfection, the medium of transfected HEK293T was changed to DMEM containing 10% fetal bovine serum, and medium of transfected cortical neurons was changed to neurobasal medium. The cultures were subjected to further treatments 48 h after the transfection. In immunostaining with GFP-tagged GAP43 cotransfected with myc-tagged gephyrin, cells without GFP expression in the transfection were termed mock cells, which served as an internal control to compare cells with and without overexpression of transfected GAP43 in the same transfection.
RNA knockdown.
Small interfering RNA (siRNA) was used to knock down the expression of rat gephyrin (Gphn; accession no. NM_022865) as described previously (35). In brief, the cultured neurons (4 DIV) were transfected with siRNAs (s134499, 5′-GAUAGUUCCUCAUCACAUATT-3′ and 5′-UAUGUGAUGAGGAACUAUCTT-3′; s134500, 5′-CGAUCAACACUUCUAGCAATT-3′ and 5′-UUGCUAGAAGUGUUGAUCGAT-3′; Ambion) or scrambled RNA produced by Silencer-predesigned siRNA (Ambion) using Lipofectamine 2000 reagent for 72 h. The knockdown efficiency was examined by Western blotting.
Measurement of intracellular Ca2+ concentrations.
Intracellular calcium imaging of cultured neurons expressing GFP was modified from previous reports (30, 36). In brief, cultured neurons transfected with empty vector or GFP-tagged GAP43 DNA constructs, or pretreated with PKC inhibitor Ro318220, were incubated with 2.5 μM Fura-2-AM for 30 min at 37°C and then treated with 10 μM muscimol, followed by real-time intracellular calcium concentration ([Ca2+]i) imaging of GFP-expressing neurons with an inverted microscope (Axiovert 200; Zeiss) using a charge-coupled device (CCD) camera (CoolSNAP HQ; Photometrics). The digital image processing and analysis were performed using MetaFluor (Universal Imaging). The time-lapse changes of F340/F380 [Δ(F340/F380)], which indicate the changes of [Ca2+]i, were calculated and plotted by subtracting the ratio obtained at each time point from the average ratio of the baseline.
Transient hypoxia-ischemic brain injury in neonatal rats.
Transient hypoxic ischemia (tHI), which was applied 45 min after HI, followed by reperfusion in neonatal rat brains was performed as previously described, with minor modifications (37, 38). Since we found that GAP43-gephyrin interaction was induced during the reoxygenation/reperfusion period in the in vitro tHI (see Fig. 7B), transient common carotid artery (CCA) ligation, instead of permanent CCA ligation, which is used to induce severe brain damage in neonatal HI studies (39), was used to examine the tHI-induced GAP43-gephyrin interaction in vivo. P2 SD rat pups with the brain development stage close to the E17 embryo-derived 4- to 6-DIV cultured neurons were used. The rat pups were separated into sham-operated and tHI groups with 5 pups in each group. The tHI rats were anesthetized by 1% isoflurane during the surgical procedure of right CCA (rCCA) isolation and ligation with double live knots using a 5-0 chromic gut (Syneture). The entire procedure was completed within 5 min to keep the consistency of the isoflurane exposure time in each pup. Sham-operated pups were subjected to the same procedures of anesthesia, surgical incision, and right CCA isolation. Rat pups then were placed in a water bath in a chamber kept at a 37°C, and the chamber air was switched to 7.5% oxygen and 92.5% nitrogen for 45 min for hypoxic insult. At the end of hypoxia, the pup was recovered in room air with the knots on the right CCA untied for reperfusion, and the incision was closed with fast glue. The pups then were allowed to recover in a cage with the air temperature maintained at 30°C for 12 h and then killed with an overdose of isoflurane to harvest the left and right cerebral cortex separately for Western blotting and co-IP.
FIG 7.

Calcineurin inhibitor and S41-phosphorylated GAP43 attenuated tOGD-induced GAP43-gephyrin colocalization and gephyrin misfolding. (A) Biotinylation assay of surface GABAARγ2 expression in tOGD-treated 4-DIV neurons with or without FK506 cotreatment. Na+/K+ ATPase-α3 served as a surface protein control. (B) Time-dependent increase of GAP43-gephyrin (Geph) colocalization in 4-DIV cortical neurons after tOGD. Cortical neurons were under normal conditions (Ctrl) or were subjected to 1 h of tOGD, followed by 5, 11, and 23 h of recovery (6, 12, and 24 h in total, as indicated) under normal oxygen/glucose conditions. Immunofluorescent confocal images (top) and a quantitative graph for the cocalization signals (bottom) are shown. ###, P < 0.001 versus the tOGD 6-h group; n = 6. (C) Immunofluorescent images of GAP43 (green), Geph (red), and misfolded protein aggregates (ADR; blue) in cortical neurons with or without FK506 treatment under tOGD. (D) Cortical neurons were transfected with GFP-GAP43WT (WT), GFP-GAP43S41A (S41A), or GFP-GAP43S41D (S41D), and tOGD insult was applied 24 h after the transfection. The neurons were immunolabeled with GFP (green), Geph (red), misfolded protein aggregates (ADR; blue), and cell nuclei (DAPI; gray). *, GFP-positive neurons; m, mock cell. Arrowheads in panels C and D indicate GAP43-Geph or GFP-Geph colocalization signals (yellow); dashed arrows, Geph-ADR signals (purple); solid arrows, Geph cluster signals (red clusters in Geph/ADR). (E and H) The percentage of GAP43-Geph or GFP-Geph colocalization signals; n = 5. (F and I) The population of misfolded Geph-containing neurons; n = 7. (G and J) The number of Geph clusters per neuron. Total numbers of cells counted were 164 (Ctrl), 199 (tOGD), 129 (tOGD+FK506), 6 (tOGD-mock), 6 (tOGD-WT), 8 (tOGD-S41A), and 11 (tOGD-S41D). For panels E, F, and G, P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) versus Ctrl or WT; P < 0.05 (#), P < 0.01 (##), P < 0.001 (###) versus tOGD or mock cells or between S41A and S41D. For panels H, I, and J, P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) by unpaired t test.
Transient oxygen-glucose deprivation in cultured neurons.
Transient oxygen-glucose deprivation treatment (tOGD), an in vitro tHI model, was performed as previously described, with minor modifications (40). Briefly, cortical neuron cultures were incubated in glucose-free Earle's balanced salt solution in a hypoxia chamber (BioSpherix) with the chamber air at 1% O2 and 37°C for 1 h, and then the cultures were recovered in normal oxygen/glucose-containing neurobasal medium at 37°C for 24 h. FK506 (1 μM) treatments were applied during the recovery period.
Statistics.
Statistical analysis was performed using Prism 5 software (GraphPad Software). Data were expressed as means ± standard errors of the means (SEM), and the number of batches for each experimental condition was indicated as n. Data were analyzed by one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple-comparison post hoc test, and statistical significance was set at P < 0.05, P < 0.01, or P < 0.001 versus the respective control (Ctrl) group or the designated pair of groups unless stated otherwise.
RESULTS
LC-MS/MS analysis identified gephyrin in GAP43-IP complex.
Previous works have characterized several GAP43-associated proteins, including actin and calmodulin, in neurons under physiological conditions with the roles limited to the regulation of actin cytoskeletons in axon growth upon PKC-dependent phosphorylation (17, 41). In order to search for novel GAP43-interacting proteins under pathological conditions, we exposed cultured cortical neurons (at 4 DIV) to the PKC inhibitor Ro318220 and subjected the cell lysate to anti-GAP43 immunoprecipitation and LC-MS/MS analysis. As shown in Table 1, we identified within the GAP43-IP complex abundant actin, alpha-actinin 4, spectrin, and ELAV-like protein 2, RNA-binding proteins that stabilize GAP43 mRNA (42). In addition, we also found proteins associated with the ubiquitin-proteosome pathway, chaperon proteins that prevent protein misfolding, and microtubule-related proteins, including kinesin heavy chain isoform 5A (KIF5A), microtubule-associated protein 1B (MAP1B), and gephyrin, the GABAA receptor scaffolding protein. We were particularly intrigued by the presence of gephyrin, a postsynaptic protein that was not expected to interact with GAP43, which generally is considered to be axonal. This led to the following study on the role of GAP43 in regulating GABAergic synapses.
TABLE 1.
Proteins identified with LC-MS/MS from anti-GAP43 antibody- immunoprecipitated protein complex of PKC inhibitor-treated primary cultured rat cortical neurons at 4 DIV
| Protein name | Swiss-Prot identifier | Protein mass (Da) | Score | No. of matching peptides |
|---|---|---|---|---|
| Heat shock protein HSP 90-alpha (Hsp90aa1) | HS90A_RAT | 84,762 | 1,114 | 48 |
| Ubiquitin-like modifier-activating enzyme 1 (Uba1) | UBA1_RAT | 117,713 | 510 | 21 |
| Actin, cytoplasmic 1 (Actb) | ACTB_RAT | 41,710 | 622 | 19 |
| Heat shock cognate 71-kDa protein (Hspa8) | HSP7C_RAT | 70,827 | 351 | 12 |
| 26S proteasome non-ATPase regulatory subunit 2 (Psmd2) | PSMD2_RAT | 100,124 | 259 | 8 |
| ELAV-like protein 2 (Elavl2) | ELAV2_RAT | 39,481 | 189 | 7 |
| Spectrin alpha chain, non-erythrocytic 1 (Sptan1) | SPTN1_RAT | 284,462 | 166 | 7 |
| Heat shock 70-kDa protein 4 (Hspa4) | HSP74_RAT | 93,997 | 152 | 7 |
| Gephyrin (Gphn) | GEPH_RAT | 83,213 | 135 | 7 |
| Neuromodulin (Gap43) | NEUM_RAT | 23,589 | 151 | 5 |
| Kinesin heavy-chain isoform 5A (Kif5a) | KIF5A_RAT | 116,843 | 228 | 5 |
| Alpha-actinin-4 (Actn4) | ACTN4_RAT | 104,849 | 133 | 5 |
| Microtubule-associated protein 1B (Map1b) | MAP1B_RAT | 269,334 | 88 | 3 |
PKC blockade induced GAP43-gephyrin association in developing cortical neurons.
We further confirmed the proteomic finding described above by using coimmunoprecipitation (co-IP) of GAP43. We found that GAP43-gephyrin interaction was markedly increased up to 4.8-fold in cultured neurons treated with the PKC inhibitor Ro318220 for 24 h (P = 0.002) (Fig. 1A and D). The effect of other signaling kinases known to be involved in the phosphorylation of GAP43 and gephyrin, such as PKA and MAPK, was also examined. As shown in Fig. 1B, treatment with PKA inhibitor Rp-8-Br (for 24 h) had a much smaller effect than Ro318220 in elevating the GAP43-gephyrin interaction. MAPK inhibitor PD-98059 appeared to reduce GAP43-gephyrin interaction, although the level of GAP43 expression was increased (Fig. 1B). Further confirmation with antigephyrin-based co-IP indicated that Ro318220 treatment increased gephyrin-GAP43 interaction by 2.7-fold (P = 0.002) but decreased gephyrin-GABAARγ2 interaction to 63.8% (P = 0.006) (Fig. 1C and D). Notably, total cell lysate Western blotting (WB) indicated that 24 h of Ro318220 treatment increased the total level of GAP43 by 1.8-fold (P < 0.05) (Fig. 1A), which could be due to compensation for the decrease in GAP43 activity but was less than the fold increase in the GAP43-gephyrin association. The total levels of gephyrin and GABAARγ2 were not affected by the Ro318220 treatment.
FIG 1.

Inhibition of PKC activity induced GAP43 interaction with gephyrin in developing cortical neurons. (A) Primary rat cultured cortical neurons at 4 DIV were treated with vehicle control (Ctrl; 0.01% dimethylsulfoxide [DMSO]) or PKC inhibitor Ro318220 (Ro; 100 nM) for 2 or 24 h and then subjected to immunoprecipitation (IP) with mouse anti-GAP43 antibody (α-GAP43), followed by Western blotting (WB) with rabbit anti-GAP43 (α-GAP43) or antigephyrin (anti-Geph) antibody. Total cell lysate WB for the total level of GAP43 and gephyrin and the 24-h Ro group was quantified. (B) Neurons were treated with PKA inhibitor Rp-8-Br (Rp; 100 μM), MAPK/ERK inhibitor PD-98059 (PD; 20 μM), or Ro (100 nM) for 24 h, followed by total cell lysate WB with GAP43 and gephyrin or IP with anti-GAP43 and then WB with anti-Geph or anti-GAP43. (C and E) Immunoprecipitation with mouse anti-Geph or normal mouse IgG, followed by WB with anti-GAP43, anti-Geph, or anti-GABAARγ2 antibody in neurons at 4 (C) or 11 (E) DIV after the Ro318220 treatments. IgG served as the loading control. The total levels of GAP43, gephyrin, GABAARγ2, and GAPDH under each condition were shown in the cell lysate WB. (D) Quantification of the ratio of the two coimmunoprecipitated protein band intensities in panels A and C. (F) Differential expressions of GAP43, phosphorylated GAP43-S41 (pGAP43), gephyrin, and PSD95 in 4- and 11-DIV neurons. (G) Quantification of the relative levels of pGAP43, GAP43, and pGAP43-41/GAP43 in 4- and 11-DIV neurons. *, P < 0.05; **, P < 0.01; ***, P < 0.001 for Ctrl versus Ro in panels A and D (n = 4) and for 4 versus 11 DIV in panel G (n = 3) by unpaired t test.
The observations described above were obtained from cultured cortical neurons at 4 DIV, which is considered to be a stage prior to synaptogenesis (35). We then examined whether the PKC inhibitor-induced GAP43-gephyrin association would occur at 11 DIV, when synapses are formed. Immunoblotting results showed that GAP43-gephyrin association in 11-DIV neurons was barely detectable in both Ro318220- and vehicle-treated groups (Fig. 1E). This difference may result from differential expression levels of GAP43 or gephyrin at these two developmental stages. Indeed, we found that the levels of both GAP43 and its S41-phosphorylated form (pGAP43) were higher at 4 than 11 DIV (4 versus 11 DIV, P = 0.012 for GAP43, P < 0.0001 for pGAP43, and P = 0.005 for pGAP43/GAP43.) (Fig. 1F and G), whereas gephyrin and PSD95 (an excitatory postsynaptic receptor scaffolding protein) were more abundant at 11 DIV (Fig. 1F). Thus, the higher levels of GAP43 and pGAP43 may account for the robust effect of the PKC inhibitor on GAP43-gephyrin interaction.
Blocking PKC/neuronal activities promoted GAP43-gephyrin colocalization and gephyrin misfolding.
Based on the GAP43/gephyrin co-IP results, we further investigated the subcellular localization of GAP43 and gephyrin in cultured cortical neurons. Using confocal microscopy, we found that GAP43 and gephyrin immunoreactivities distributed in relatively segregated small clusters in the submembranous region of neuronal soma in 4-DIV neurons, and the GAP43 immunoreactivities became much weaker in 11-DIV neurons, where prominent gephyrin clusters were observed along the somatodendritic surface (Fig. 2A). Treatment with Ro318220 (24 h) enhanced GAP43-gephyrin colocalization to a much greater degree in 4- than 11-DIV neurons (P < 0.001 between 4- and 11-DIV Ro groups) (Fig. 2A and C). Notably, the gephyrin immunoreactivities in the Ro318220-treated neurons appear to form cytosolic aggregates in irregular shapes, while the neurons appeared to be morphologically intact. In order to further define the nature of gephyrin aggregates, we used an aggresome detection reagent (ADR), which labels misfolded proteins, together with immunostaining of gephyrin. The gephyrin aggregates with and without ADR costaining represent misfolded aggregates and properly folded clusters, respectively. As shown in Fig. 2A (Geph/ADR images), Ro318220 treatment markedly increased the percentage of neurons exhibiting misfolded gephyrin aggregates at both 4 and 11 DIV (in the 4-DIV control, 4.8%; Ro, 42.7%; P < 0.001; in the 11-DIV control, 9.2%; Ro, 53.9%; P < 0.001.) (Fig. 2D). Notably, GAP43 was found in the ADR-labeled gephyrin aggregates at both stages (Fig. 2A and E). Correspondingly, properly folded gephyrin clusters, which were not labeled by ADR, were greatly reduced by the Ro318220 treatment (P < 0.05 for Ctrl versus Ro at both 4 and 11 DIV) (Fig. 2F).
FIG 2.

Blockade of PKC or neuronal activity induced GAP43-gephyrin colocalization and gephyrin misfolding in developing cortical neurons. Immunofluorescent confocal microscopic images of 4-DIV (A) or 11-DIV (B) cortical neurons treated with vehicle (Ctrl), Ro, or TTX (1 μM) for 24 h. The merged images were from the respective dashed-line-framed area in the single-color images. Green, GAP43; red, gephyrin (Geph); blue, ADR-labeled misfolded protein. Arrowheads, GAP43-Geph colocalization (yellow signals in the GAP43/Geph panel); dashed arrows, misfolded Geph (purple signals in the Geph/ADR panel); solid arrows, red clusters in the Geph-ADR panel indicating physiological gephyrin cluster signals. (C) Quantitative results of the GAP43-Geph colocalization (n = 5 for 4 DIV; n = 15 to 35 for 11 DIV). (D) The percentage of misfolded Geph aggregate-containing neurons in each group (n = 6 for 4 DIV; n = 3 to 5 for 11 DIV). (E) The populations of misfolded Geph-containing neurons that showed GAP43+ or GAP43− immunoreactivities in the misfolded Geph aggregates under each condition. Numbers of cells counted under each condition were 91 (4 DIV-Ctrl), 119 (4 DIV-Ro), 98 (4 DIV-TTX), 77 (11 DIV-Ctrl), 46 (11 DIV-Ro), and 48 (11 DIV-TTX). n.s., not significant. (F) The number of Geph clusters per neuron in each treatment, determined by counting the red cluster signals with a diameter of >0.04 μm in the Geph-ADR merged images using MetaMorph Imaging (n = 6 for 4 DIV; n = 3 in 11 DIV). P < 0.05 (&), P < 0.01 (&&), and P < 0.001 (&&&) between Ro and TTX groups of the same DIV. P < 0.05 (#) and P < 0.001 (###) between 4 and 11 DIV of the same treatment. Fisher's exact test was used for panel E.
Since neuronal activity is the major physiological signal regulating PKC activity in developing neurons, we further examined whether blocking neuronal activity with the Na+ channel blocker tetrodotoxin (TTX; 1 μM, 24 h) can induce GAP43-gephyrin colocalization and gephyrin misfolding. Interestingly, we found that TTX-treated 4-DIV neurons exhibited gephyrin-containing aggresome-like structures at the perinuclear region and along neurites, and GAP43 immunoreactivities were absent from these aggresomes (Fig. 2B and D). GAP43 and gephyrin immunoreactivities were more segregated in 4- but not 11-DIV neurons that survived well under the treatment. As a result, the TTX-induced increase of GAP43/gephyrin colocalization at 4 DIV was less than that found in the Ro318220 treatment (Fig. 2B and C) (for TTX versus Ro, P < 0.001 at 4 DIV), whereas their effects were similar at 11 DIV. TTX also reduced the properly folded gephyrin clusters at both stages (P < 0.05 for TTX versus the control in both 4- and 11-DIV neurons) (Fig. 2F).
Together with the information shown in Fig. 1, these immunostaining data further suggest that blocking PKC/neuronal activities increased GAP43-gephyrin colocalization/interaction mainly in the soma of GAP43-enriched developing neurons. Moreover, both treatments reduced gephyrin clusters, possibly via inducing gephyrin misfolding into aggregates, albeit with differential subcellular localizations and interaction with GAP43.
GAP43 with S41A and S41D mutations differentially associated with gephyrin and affected gephyrin aggregation.
Gephyrin clustering is known to be regulated by actin polymerization in the postsynaptic microfilaments (15), but whether the phosphorylation status of GAP43 at S41, which regulates actin polymerization, is involved in the gephyrin clustering by interacting with gephyrin was unknown. We found that both Ro318220 and TTX treatments reduced the GAP43-S41 phosphorylation to 0.68-fold (P = 0.0026) and 0.64-fold (P < 0.001), respectively, compared to that of the control group in 4-DIV cortical neurons (Fig. 3A). Thus, we generated Flag- and GFP-tagged expression constructs of wild-type GAP43 (GAP43WT) and two GAP43 mutants, with the replacement of S41 with alanine (GAP43S41A) or with glutamate (GAP43S41D) mimicking S41-unphosphorylated and -phosphorylated GAP43, respectively, and transfected them into 4-DIV neurons to further investigate their association with the endogenous gephyrin. The co-IP data showed that Flag-tagged GAP43S41A could bond more strongly with gephyrin than GAP43S41D or GAP43WT (GAP43S41D, 0.91-fold; GAP43S41A, 1.48-fold; P < 0.01 versus GAP43WT) (Fig. 3B). Notably, none of these GAP43 transfections affected the total level of gephyrin as examined by cell lysate WB (Fig. 3B). Immunostaining of GFP-tagged GAP43 and endogenous gephyrin in combination with ADR staining further indicated that the GFP-gephyrin colocalization signals were stronger in GAP43S41A- than in GAP43WT- or GAP43S41D-expressing neurons (GAP43WT, 26.84%; GAP43S41A, 52.80%; P < 0.001 versus GAP43WT) (Fig. 3C and D). In addition, only GAP43S41A overexpression, but not GAP43S41D or GAP43WT overexpression, induced gephyrin misfolding (Fig. 3C and E). Notably, GAP43S41A induced gephyrin-containing perinuclear aggresome formation, and GAP43S41A/gephyrin colocalization was found mainly along the outer surface but not within the gephyrin aggresome.
FIG 3.

GAP43-S41 mutants differentially interacted with gephyrin and affected gephyrin aggregation in cortical neurons. (A) Western blotting of pGAP43-S41, total GAP43, and GAPDH in 4-DIV neurons treated with Ro (0.1 μM) or TTX (1 μM). The pGAP43-S41/GAP43 ratio (normalized by GAPDH) was used to indicate phosphorylated GAP43 at S41. **, P < 0.01; ***, P < 0.001 by unpaired t test; n = 3. (B) Coimmunoprecipitation of Flag-tagged GAP43WT (WT) or the GAP43S41A (S41A) or GAP43S41D (S41D) mutant with gephyrin (Geph) in Flag-GAP43-transfected neurons. The IgG signal served as the loading control. **, P < 0.01 versus the WT; n = 3. Total cell lysate WB of Geph showed no difference among groups. (C) Immunofluorescent confocal images of neurons transfected with GFP-tagged WT, S41D, or S41A. Green, GFP; red, Geph; blue, ADR-labeled misfolded protein; gray, DAPI. Arrowheads, GFP-Geph colocalization (yellow signals in GFP/Geph panel); arrows, physiological Geph clusters (red puncta in the Geph/ADR panel); dashed arrows, misfolded Geph (purple signals in the Geph/ADR panel); m, mock cells. (D) The percentage of GFP-Geph colocalization signals in panel C. Cell numbers counted were the following: WT, 20; S41D, 15; S41A, 18. ***, P < 0.001 versus WT results. (E) The percentage of misfolded Geph aggregate-containing neurons. W-mock, D-mock, and A-mock indicate mock cells in WT, S41D, and S41A transfection, respectively. **, P < 0.01 versus the respective mock group; P < 0.01 (##) and P < 0.001 (###) versus S41A; n = 5 to 8. (F) The number of Geph clusters per neuron. Cell numbers counted were the following: WT, 12; W-mock, 10; S41D, 5; D-mock, 5; S41A, 7; A-mock, 10. (G) Cumulative frequency of the Geph cluster area per neuron in each transfection condition. Data for the mock group were obtained from mock cells in all transfection conditions. A Kolmogorov-Smirnov test was used.
We further examined the effect of mutated GAP43 on the physiological gephyrin clusters. Surprisingly, the image analysis result indicated that the numbers of gephyrin clusters per neuron were preserved in the GAP43S41A-expressing neurons but were reduced in GAP43WT and mostly eliminated by GAP43S41D compared with their levels in the respective mock cells (Fig. 3C and F). Significant difference was observed between the GAP43S41A and GAP43S41D groups (P = 0.007) but not between GAP43WT and either of these two mutants. Notably, GAP43S41D induced the reduction of gephyrin immunoreactivities but did not do so in the total cell lysate WB (Fig. 3B), which could be due to the low immunoreactivities of unaggregated gephyrin or the additional signals from nontransfected cells in Western blotting. We further analyzed the size distribution of gephyrin clusters in all GFP-positive neurons of each transfection group. Our data showed that GAP43WT, but not GAP43S41A or GAP43S41D, significantly reduced gephyrin cluster size population-wise compared with those of mock cells, as indicated by a leftward shift of the cumulative frequency curve (P < 0.05 between GAP43WT and mock cells) (Fig. 3G). The size distribution of the gephyrin cluster in the GAP43S41D group also was smaller than that of the GAP43S41A group (P < 0.01), but none of them was significantly different from that of the GAP43WT group.
Together, these results suggest that dephosphorylated GAP43 at S41 strongly interacts with gephyrin and triggers gephyrin aggresome formation, leading to the protection of physiological gephyrin clusters. In contrast, an increase of phosphorylated GAP43-S41 may lead to the negative regulation of gephyrin clustering in developing neurons.
S41-phosphorylated GAP43 inhibited gephyrin aggregation in HEK293T cells.
Although gephyrin forms submembranous clusters in neurons, it is also known to form large aggregates/clumps in nonneuronal cells (13, 43). We examined how GAP43 and its two S41 mutants could differentially affect gephyrin folding and aggregation in HEK293T cells. Immunostaining results indicated that cells monotransfected with Flag-tagged GAP43S41A or GAP43S41D showed diffused cytoplasmic GAP43 immunoreactivities (Fig. 4A). Cells monotransfected with myc-tagged gephyrin (myc-Geph) exhibited gephyrin immunoreactivities in both diffused form and spherical aggregates/clumps in the cytoplasm (Fig. 4B), similar to results of other reports (11, 13). Notably, neither form of gephyrin was colabeled with ADR, as shown by the lack of purple signals in the merged Geph-ADR image, suggesting that gephyrin clumps were derived from polymerization of properly folded, rather than misfolded, gephyrin. No immunoreactivity of gephyrin or GAP43 was detected in the GAP43 mutant- or gephyrin-monotransfected cells, confirming that they are not endogenously expressed in HEK293T cells. In the gephyrin-GAP43 cotransfection, we found distinct effects of the two GAP43-S41 mutations on gephyrin aggregation. First, the GFP-gephyrin colocalization signals were higher in the cells expressing GAP43S41A (49.95%) than in cells expressing GAP43WT (33.92%) or GAP43S41D (29.96%), whereas no significant difference was observed between the GAP43S41D and GAP43WT groups (P < 0.01 for GAP43S41A versus GAP43WT, P > 0.05 for GAP43S41D versus GAP43WT, and P < 0.001 for GAP43S41A versus GAP43S41D) (Fig. 4B, GFP-Geph, and C). Notably, the GFP-GAP43S41A colocalization was found mainly in the diffusively distributed but not aggregated gephyrin in the cytoplasm. The co-IP result of Flag-GAP43 and gephyrin further indicated that GAP43S41A was more abundant than GAP43S41D in the antigephyrin-IP complex (Fig. 4F) when the total input levels of the two GAP43 mutants were similar (Fig. 4G). Second, gephyrin aggregates were found mostly diminished in the gephyrin/GAP43WT- and gephyrin/GAP43S41D-cotransfected cells while being observed in the gephyrin/GAP43S41A-cotransfected cells (Fig. 4B), similar to their inhibitory effect on gephyrin clustering in neurons. Quantitative analysis further indicated that the percentage of gephyrin aggregate-containing cells was much lower in the GAP43S41D-gephyrin cotransfection than in the gephyrin monotransfection or gephyrin-GAP43S41A cotransfection (Fig. 4C). Lastly, unlike in the gephyrin-monotransfected cells, we found that the spherical/perinuclear aggregates and the diffusively distributed forms of gephyrin both were colabeled with ADR in GAP43S41A (Fig. 4B, Geph-ADR). Quantitative analysis indicated that 61% of gephyrin clumps in the GAP43S41A transfection were misfolded, while only 35% and 18.75% in the GAP43WT- and GAP43S41D-expressing cells were misfolded, respectively (P < 0.001 in GAP43S41A versus GAP43WT, P < 0.05 in GAP43S41D versus GAP43WT and GAP43S41A, and P < 0.001 in GAP43S41D versus GAP43S41A) (Fig. 4E).
FIG 4.

Distinct effects of GAP43 S41 mutants on gephyrin aggregation in HEK293T cell line. Immunofluorescent confocal microscopy of HEK293T cells transfected with (A) Flag-GAP43WT (Flag-WT), Flag-GAP43S41A (Flag-S41A), or Flag-GAP43S41D (Flag-S41D) or (B) Myc-Geph, Myc-Geph and Flag-WT, Myc-Geph and Flag-S41A, or Myc-Geph and Flag-S41D. Transfected cells were immunostained for Geph and GAP43. Gephyrin clumps were noted in both Myc-Geph- and Myc-Geph/Flag-S41A-transfected cells but not in the Myc-Geph/Flag-S41D-transfected cells. Dashed arrows, Geph aggregates; solid arrows, GFP-Geph colocalization. (C) GFP-Geph colocalization signals from panel B. **, P < 0.01; ###, P < 0.001; n = 15 (Geph/WT), n = 90 (Geph/S41A), and n = 88 (Geph/S41D). (D) Cell population that expressed only diffused Geph without (w/o) clumps in the cytosol or with (w/) additional Geph clumps under each condition. The number of cells counted in each group is indicated. ***, P < 0.001; n.s., not significant. (E) Population of Geph clumps colabeled with ADR or not colabeled under each condition. The numbers of Geph clumps counted in each group were the following: n = 25 (Geph), n = 20 (Geph/WT), n = 18 (Geph/S41A), and n = 16 (Geph/S41D). *, P < 0.05. (F) Co-IP of Flag-tagged GAP43 WT or S41 mutants with Geph in transfected HEK293T cells. IgG served as the loading control. (G) Western blotting of Geph, Flag, and GAPDH in the cell lysate used for panel F. Fisher's exact test was used for panels D and E.
Thus, these results provide direct support to the notion that S41-phosphorylated GAP43 can reduce gephyrin aggregation as its inhibitory effect on neuronal gephyrin clustering. Similar to the effect in neurons, S41-dephosphorylated GAP43 can strongly interact with gephyrin to induce gephyrin misfolding and subsequent gephyrin aggresome formation to exclude GAP43 in HEK293T cells.
S41-phosphorylated GAP43 reduced surface expression of GABAA receptor.
Gephyrin clustering is important for the surface expression and clustering of the postsynaptic GABAARs. Thus, we investigated the relationship between gephyrin and surface GABAAR expression in developing neurons by using siRNA knockdown of gephyrin expression, followed by biotinylation assay to examine surface GABAARγ2, a critical subunit for gephyrin clustering of GABAARs (1, 6). The result showed that the transfection of gephyrin siRNA that reduced the total gephyrin expression by 20% (P = 0.008) decreased the biotinylated surface GABAARγ2 by 22% compared with the level for the scrambled RNA-transfected group (Fig. 5A) (P = 0.012), indicating that surface GABAAR expression is indeed gephyrin dependent. We further examined whether overexpressing GAP43 S41 mutants would affect the surface GABAARs. Our data indicated that the surface GABAARγ2 was increased by the GAP43S41A transfection up to 1.26-fold (P < 0.01), whereas it was decreased by GAP43S41D to 84.7% (P < 0.05) compared to that of the vector-transfected control (Fig. 5B). Fura-2 calcium imaging was performed to further examine the responses of neurons expressing each GFP-GAP43 mutant to GABAAR agonist treatment. GABAAR is excitatory in early-developing neurons (44), and its agonist muscimol could increase intracellular calcium concentration [Ca2+]i in 4-DIV cortical neurons (Fig. 5C, control group). Both the representative traces and compiled data indicated that the muscimol-induced [Ca2+]i elevation was higher in GAP43S41A and lower in GAP43S41D than in the GAP43WT transfection (P < 0.05 for GAP43S41A versus GAP43WT and GAP43S41D versus GAP43WT, P < 0.01 for GAP43S41D versus GAP43S41A) (Fig. 5C and D). GAP43WT overexpression did not significantly affect this response compared to the response for the vector-transfected (Vec) or no-transfection (Ctrl) groups. These results correlated well with the biotinylation assay result.
FIG 5.

Gephyrin knockdown and GAP43S41D overexpression reduced surface GABAAR expression. (A) Biotinylation assay of surface GABAARγ2 in gephyrin siRNA (siGeph)- and scrambled RNA (Scr)-transfected cortical neurons at 4 DIV. Na+/K+ ATPase-α3 served as the surface protein control. Total levels of GABAARγ2 and Geph were determined by cell lysate WB. P < 0.05 (*) and P < 0.01 (**) versus the Scr group by unpaired t test; n = 3. (B) Biotinylation assay of surface GABAARγ2 in cortical neurons transfected with GAP43S41A (S41A), GAP43S41D (S41D), or the vector (Vec). P < 0.05 (*) and P < 0.01 (**) versus Vec; ###, P < 0.001 between S41A and S41D groups; n = 4. (C) Representative traces and (D) compiled data of GABAAR agonist muscimol-induced [Ca2+]i increase in GFP-tagged GAP43-transfected neurons using Fura-2 calcium imaging. The calcium ionophore ionomycin (10 μM) applied at the end of each measurement confirmed the responsiveness of the observed cells. Data represent the increased ratio of Ca2+-bound to Ca2+-free Fura-2 [Δ(F340/F380)]. *, P < 0.05 versus Ctrl or Vec; P < 0.05 (#) and P < 0.01 (##) versus S41A; n = 3.
Thus, these data suggested that the surface expression of GABAARs in developing cortical neurons is gephyrin dependent and can be negatively regulated by the phosphorylated GAP43-S41, which inhibited gephyrin clustering. Dephosphorylated GAP43-S41, which induced gephyrin aggresome formation without disrupting gephyrin clusters, seemed to facilitate surface GABAAR expression.
tHI induced calcineurin-dependent dephosphorylation of GAP43 and GAP43-gephyrin association.
It has been shown that pathological insults, such as hypoxia ischemia (HI), cause dephosphorylation of GAP43 in developing brains via the activation of calcineurin (45). Therefore, it is possible that HI also induces GAP43-gephyrin interaction in a calcineurin-dependent manner. We examined this possibility by using a transient neonatal HI (tHI) animal model, in which neonatal rat pups were subjected to 7.5% hypoxia and right common carotid artery (rCCA) ligation for 45 min, followed by removal of rCCA ligation and recovery under normoxic conditions for 12 h. Western blotting and co-IP of tissue homogenates showed that the level of phosphorylated GAP43-S41 was lower and the GAP43-gephyrin association was more abundant on the right (ipsilateral) than the left (contralateral) hemisphere of cerebral cortex after tHI (P = 0.015 for pGAP43/GAP43 and P = 0.002 for GAP43/gephyrin) (Fig. 6A and B). We further used an in vitro neonatal tHI model, i.e., transient oxygen-glucose deprivation (tOGD), for 1 h, followed by recovery in normal oxygen/glucose medium for 23 h in 4-DIV neurons to examine the changes of GAP43-gephyrin interaction. The results indicated that tOGD decreased the S41-phosphorylated GAP43 (pGAP43/GAP43) by 20% (P < 0.05), and this effect can be blocked by cotreatment with a calcineurin inhibitor, FK506 (Fig. 6C). GAP43-gephyrin association was also enhanced by tOGD by 2.2-fold (P < 0.01 compared to the control), and this effect was partially inhibited by the FK506 treatment (P < 0.05 compared to tOGD) (Fig. 6D). Therefore, calcineurin-mediated GAP43 dephosphorylation may contribute in part to the GAP43-gephyrin association as induced by the pathological tHI/tOGD insult in developing neurons.
FIG 6.

Transient hypoxia ischemia (tHI)-induced calcineurin-dependent GAP43-S41 dephosphorylation and GAP43-gephyrin association in developing neurons. Neonatal P2 rat pups were subjected to unilateral tHI or sham operation, and both contralateral (Cont) and ipsilateral (Ipsi) cerebral cortex were subjected to Western blotting of pGAP43/GAP43 in total cell lysate (A) and co-IP for GAP43-Geph association (B). (Top) representative blot; (bottom) quantification graph. The graph in panel B illustrates the data from each individual (left) and the compiled data (right). An unpaired t test was used for panel A and the compiled data in panel B (*, P < 0.05; **, P < 0.01 versus sham treatment), and paired t test was used for the individual data graph in panel B (*, P < 0.05; ns, not significant); n = 5. (C and D) Transient oxygen-glucose deprivation (tOGD), an in vitro tHI model, was applied to 4-DIV cortical neurons for 1 h, followed by 23 h of recovery under normal oxygen/glucose conditions with or without the calcineurin inhibitor FK506 (1 μM). Cell lysate WB for pGAP43-S41 and total GAP43 (C) and co-IP of GAP43 and Geph (D) were performed. ##, P < 0.01 versus tOGD; n = 4.
Calcineurin inhibitor and GAP43S41D diminished tOGD-induced gephyrin misfolding and preserved gephyrin clusters.
Dephosphorylated GAP43 that associated with gephyrin appears to induce gephyrin aggresome formation while preserving gephyrin clusters under physiological conditions (Fig. 3). However, we found that tOGD that induced GAP43 dephosphorylation reduced the surface GABAAR level in an FK506-reversible manner (Fig. 7A) and increased GAP43-gephyrin colocalization during the recovery period 5 and 11 h after 1 h of OGD (Fig. 7B). Therefore, we further examined how inhibition of GAP43 dephosphorylation would affect tOGD-induced GAP43-gephyrin colocalization and gephyrin aggregation. Immunofluorescent triple labeling of GAP43, gephyrin, and misfolded proteins of tOGD-treated neurons with or without FK506 treatment showed that, indeed, tOGD not only induced GAP43-gephyrin colocalization from 9.5% to 38.8% (Fig. 7C and E) but also increased ADR-labeled misfolded gephyrin in the cytoplasm, without forming perinuclear gephyrin aggresome, from 14.51% to 62.20% (Fig. 7C and F). Cotreatment with calcineurin inhibitor FK506 markedly decreased tOGD-induced GAP43-gephyrin colocalization to 15.43% (P < 0.001) and misfolded gephyrin signals to 32.24% (P < 0.001) (Fig. 7C, E, and F). Notably, FK506 treatment did not reduce the properly folded gephyrin clusters in either the sham- or tOGD-treated neurons (Fig. 7C, red cluster signals in the Geph/ADR images, and G). These results suggested that GAP43-gephyrin colocalization and gephyrin misfolding were inducible by tOGD in a calcineurin-dependent manner, and the misfolded gephyrin was not recruited to aggresomes.
The rescue effect of FK506 on the tOGD-induced gephyrin misfolding may be attributed to its inhibition of GAP43 dephosphorylation. Thus, overexpression of GAP43S41D to specifically increase the phosphorylated GAP43 in tOGD-treated neurons might attenuate gephyrin misfolding. We examined the effects of GAP43S41A and GAP43S41D overexpression on the gephyrin misfolding/aggregation and clustering in neurons after tOGD. GFP-tagged GAP43 mutants were transfected into 4-DIV cortical neurons 48 h prior to tOGD, and the neurons were immunostained for GFP-gephyrin and ADR at 23 h after tOGD. Immunofluorescent images and quantitative results indicated that the GAP43S41D-transfected tOGD neurons (Fig. 7D), similar to the FK506-treated tOGD neurons, showed much less GFP-gephyrin colocalization than the GAP43S41A- and GAP43WT-transfected tOGD neurons (P < 0.05 for GAP43S41A versus GAP43WT and GAP43S41D versus GAP43WT, P < 0.01 for GAP43S41A versus GAP43S41D) (Fig. 7D and E). Notably, the GAP43WT colocalization with gephyrin was 31.69%, which is lower than that for GAP43S41A (55.83%) and higher than that for GAP43S41D (16.11%), possibly due to the partial phosphorylation of GAP43WT in transfected neurons. The percentage of misfolded gephyrin-containing neurons under tOGD also was lower in GAP43WT- or GAP43S41D-overexpressing cultures than in the mock or GAP43S41A-overexpressing cultures (P < 0.001 for GAP43WT and GAP43S41D versus mock and GAP43S41A and for GAP43S41A versus GAP43WT) (Fig. 7D and F). Furthermore, we found that GAP43S41A but not GAP43S41D overexpression reduced the physiological gephyrin clusters compared to either mock or GAP43WT neurons under tOGD (P < 0.05 for GAP43S41A versus mock or versus GAP43WT, P < 0.05 for GAP43S41A versus GAP43S41D) (Fig. 7D and G). Notably, GAP43S41D seemed to preserve gephyrin clusters in tOGD-treated neurons (Fig. 7G), which is different from its effect on gephyrin clustering in unchallenged neurons (Fig. 3C). Further comparison of GAP43 and its mutants with or without tOGD revealed that tOGD increased gephyrin misfolding in all three transfection conditions but did not further affect their colocalization with gephyrin (Fig. 7H and I). Interestingly, neurons under tOGD showed more gephyrin clusters than those without tOGD when GAP43WT (P < 0.01) and GAP43S41D (P < 0.001) were overexpressed, whereas GAP43S41A had no such effect (Fig. 7J). Thus, phosphorylated GAP43 seems to facilitate gephyrin clustering in developing neurons under tOGD insult.
Altogether, these data suggest that pathological tOGD insult that reduces surface GABAAR can induce GAP43-gephyrin colocalization and gephyrin misfolding in a calcineurin-dependent manner. Misfolded gephyrin aggregates in tOGD-treated neurons failed to be sequestered into aggresomes, which may lead to neurotoxicity. Different from the effect in unchallenged neurons, an increase of S41-phosphorylated GAP43 in tOGD-treated neurons is beneficial for gephyrin clusters by preventing gephyrin misfolding to facilitate gephyrin clustering in developing GABAergic synapses under pathological insult.
DISCUSSION
The present study revealed for the first time that the PKC-dependent phosphorylation and calcineurin-dependent dephosphorylation of GAP43 at S41 is causally related to gephyrin folding and clustering, which regulates surface expression of GABAAR during synapse development. We found that gephyrin associates with dephosphorylated GAP43-S41, which occurred mainly in developing neurons prior to synaptogenesis. This GAP43-gephyrin interaction is inducible under hypoxia/ischemia insult and may lead to gephyrin misfolding. Intriguingly, S41-phosphorylated GAP43, the active form of GAP43 that promotes axon growth, was effective in preventing gephyrin misfolding under pathological conditions, yet it negatively regulates gephyrin clustering under physiological conditions. Thus, we proposed that both dephosphorylated and phosphorylated GAP43 actively participate in the folding and aggregation processes of gephyrin, which may contribute to their distinct effects on surface GABAAR expression during the development of inhibitory synapses (Fig. 8).
FIG 8.
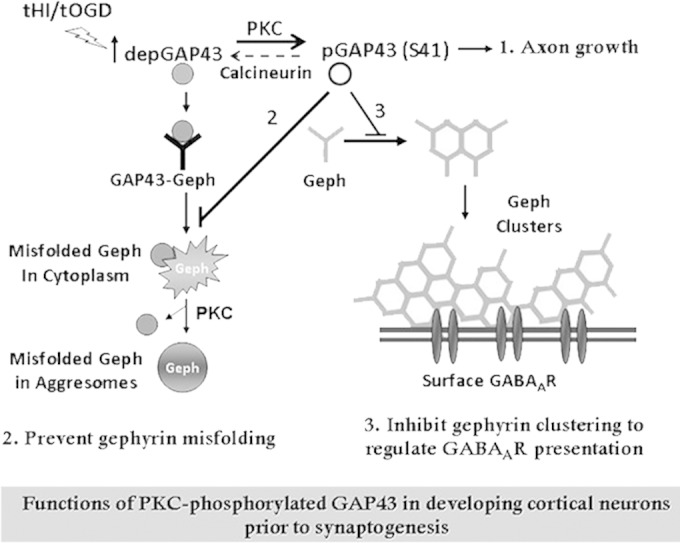
Proposed mode of action of GAP43 in regulating gephyrin aggregation during GABAergic synapse development. Developing cortical neurons at the presynaptogenesis stage are enriched with PKC/activity-dependent S41-phosphorylated GAP43 [pGAP43(S41)], which is known for axon growth (1). When GAP43 is dephosphorylated under physiological or pathological stimuli that inhibit PKC or activate calcineurin, dephosphorylated GAP43 (depGAP43) interacts with gephyrin (Geph) and triggers gephyrin misfolding to form cytosolic aggregates, which can be sequestered into aggresomes and then dissociated from GAP43 in a PKC-dependent manner. In contrast, pGAP43(S41) negatively regulates gephyrin clustering and surface GABAAR expression under normal development (2), yet it can effectively prevent gephyrin misfolding and facilitate gephyrin clustering under tHI insult (3). As a result, the PKC-dependent phosphorylation of GAP43 functions to regulate gephyrin-dependent surface GABAAR expression and prevent gephyrin misfolding during GABAergic synapse development.
Gephyrin misfolding can be triggered by dephosphorylated GAP43 but can be prevented by phosphorylated GAP43, suggesting that GAP43 is involved in the folding processes of gephyrin. GAP43S41A in HEK293T cells also can induce gephyrin misfolding in the cytoplasm and proceed to aggresome formation, as in neurons. One possibility for this mode of action of GAP43 is that it interferes with the gephyrin E-domain, the key region for the intermolecular dimerization of gephyrin; thus, it prevents cytosolic gephyrin aggregation (46). Although we found that HSC70, a gephyrin-interacting protein known to decrease synaptic localization of gephyrin (47), also was associated with GAP43 (Table 1), it interacts with the gephyrin G-domain but not the E-domain and did not inhibit gephyrin aggregation in nonneuronal cells. The treatment of PKC inhibitor and TTX led to gephyrin misfolding at similar degrees in both 4- and 11-DIV neurons, unlike their age-dependent effect on GAP43-gephyrin interaction. It is likely that gephyrin misfolding in mature neurons can be induced independently of the GAP43-gephyrin interaction. It is noted that the limitation of using ADR to label misfolded protein is that one cannot distinguish whether the gephyrin-ADR colocalization is due to the misfolding of gephyrin by itself or its associated proteins. Additional observation of ADR-colabeled gephyrin in aggresomes with GAP43 excluded in TTX-treated and GAP43S41A-overexpressing neurons further strengthened the conclusion that gephyrin underwent misfolding. Further investigation is warranted to unravel the molecular mechanism of how gephyrin folding can be regulated by GAP43 and other GAP43-independent factors in both developing and mature neurons.
Another important finding of the present study is that the misfolded gephyrin aggregates in the PKC inhibitor- or tOGD-treated neurons were GAP43 associated and exhibit irregular shapes, whereas those in TTX-treated or GAP43S41A-expressing neurons were in aggresome-like structures and did not trap GAP43 (Fig. 3). This finding is important, because aggresome formation is an important process for removing misfolded protein aggregates from the cytosol to prevent cytotoxicity (48, 49). Inhibition of PKC and tOGD insult may block this protective process while inducing gephyrin misfolding by dephosphorylated GAP43. This idea is supported by our data showing that GAP43S41A overexpression reduced gephyrin clusters in neurons under tOGD insult (Fig. 7) but not in neurons without insult (Fig. 3). Therefore, the present study revealed a novel feature of the phosphorylation status of GAP43-S41 beyond its role in axon growth, serving as a regulator of protein quality control for gephyrin in developing GABAergic synapses.
The inhibitory effect of phosphorylated GAP43 on the physiological clustering/aggregation of gephyrin correlated well with other studies demonstrated that postsynaptic microfilament dynamics are critical for the scaffolding process of gephyrin (14, 15). It remains unresolved how GAP43 S41A mutation can increase surface levels of GABAAR γ2 while both PKC and calcineurin can directly affect GABAAR phosphorylation (50, 51). Indeed, we found that GAP43 was enriched in the submembrane region of neuronal soma in 4-DIV developing neurons (Fig. 2A). Lipid anchoring of gephyrin on the plasma membrane is important for its receptor-clustering property (10), and GAP43 phosphorylation also can be induced by lipid-derived fatty acids (52). In addition, our mass spectrum analysis showed that KIF5A, an adaptor protein on the microtubules for anterograde transport of gephyrin-GABAAR (53), was associated with GAP43 (Table 1). It is likely that the GAP43-KIF5A association is involved in the GAP43-regulated gephyrin clustering. We found that the spontaneously formed gephyrin aggregates/clumps in HEK293T were not misfolded, and GAP43S41D can inhibit this process, similar to its inhibitory effect on gephyrin clustering in neurons, suggesting that S41-phosphorylated GAP43 directly regulates gephyrin polymerization. This inhibitory effect of pGAP43 may function to negatively regulate excitatory GABAAR presentation at the postsynaptic membrane to for the adjustment of the excitatory input prior to synaptogenesis while promoting axon growth for target reaching. This brake would be released when GAP43 declines during synaptogenesis, thereby promoting gephyrin clustering for GABAergic synapse formation. Further investigation using electrophysiological approaches is needed to determine the function of GAP43 in the regulation of GABAergic synapse development.
Increased GAP43 phosphorylation prevents gephyrin misfolding induced by tHI insult, suggesting a novel activity-dependent neuroprotective mechanism for injured neonatal brain. Note that FK506, which inhibits GAP43-S41 dephosphorylation and gephyrin misfolding, cannot completely reverse tOGD-induced GAP43-gephyrin association, suggesting that other posttranslational modifications on GAP43 and/or gephyrin are involved in the tOGD/tHI-induced GAP43-gephyrin association. Mild neonatal hypoxia, although not causing significant neuronal loss, impairs the development of GABAergic synapses and leads to neurological deficits, such as neonatal seizures (54). Furthermore, we also found that most of the tOGD-treated neurons that contain misfolded gephyrin aggregates were viable at the time of the observation (Fig. 7C). This finding is encouraging, as the affected neurons might be rescuable by proper interventions that increase phosphorylated GAP43-S41. Cell type-specific inhibition of calcineurin activity or activation of neuronal activity also may be applied to the therapy of neurodevelopmental disorders that involve perinatal brain injury-induced inhibitory synapse abnormalities, such as childhood epilepsy, attention deficit hyperactivity disorders, and autism spectrum disorders.
ACKNOWLEDGMENTS
We thank Mu-Ming Poo for his critical comments and suggestions on this study and Chien-Chang Chen for suggestions and providing materials for the surface GABAAR experiment. The LC-MS/MS data analysis by LTQ-Orbitrap XL was performed by the Academia Sinica Common Mass Spectrometry Facilities at the Institute of Biological Chemistry.
This study was financially supported by grants NSC 99-2321-B-010-015, 96-2320-B-010-039-MY3, 100-2320-B-010-033-MY2, and MOST 103-2325-B-039-006 from the National Science Council, Aim for the Top University Plan from the Ministry of Education, and Department of Health Clinical Trial and Research Center of Excellence (DOH 101-TD-B-111-004, 104-TDU-B-212-113002, and BM104010092), Taiwan.
We have no conflicts of interest to declare.
REFERENCES
- 1.Alldred MJ, Mulder-Rosi J, Lingenfelter SE, Chen G, Luscher B. 2005. Distinct gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J Neurosci 25:594–603. doi: 10.1523/JNEUROSCI.4011-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Ari Y. 2002. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 3.Mohler H. 2007. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem 102:1–12. doi: 10.1111/j.1471-4159.2007.04454.x. [DOI] [PubMed] [Google Scholar]
- 4.Fritschy JM, Brunig I. 2003. Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther 98:299–323. doi: 10.1016/S0163-7258(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 5.Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG, Moss SJ. 2001. GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat Neurosci 4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- 6.Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. 1998. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci 1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- 7.Fritschy JM, Harvey RJ, Schwarz G. 2008. Gephyrin: where do we stand, where do we go? Trends Neurosci 31:257–264. doi: 10.1016/j.tins.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Kneussel M, Brandstatter JH, Laube B, Stahl S, Muller U, Betz H. 1999. Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J Neurosci 19:9289–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, Moss SJ. 2005. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci 25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kneussel M, Betz H. 2000. Clustering of inhibitory neurotransmitter receptors at developing postsynaptic sites: the membrane activation model. Trends Neurosci 23:429–435. doi: 10.1016/S0166-2236(00)01627-1. [DOI] [PubMed] [Google Scholar]
- 11.Saiyed T, Paarmann I, Schmitt B, Haeger S, Sola M, Schmalzing G, Weissenhorn W, Betz H. 2007. Molecular basis of gephyrin clustering at inhibitory synapses: role of G- and E-domain interactions. J Biol Chem 282:5625–5632. doi: 10.1074/jbc.M610290200. [DOI] [PubMed] [Google Scholar]
- 12.Zita MM, Marchionni I, Bottos E, Righi M, Del Sal G, Cherubini E, Zacchi P. 2007. Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J 26:1761–1771. doi: 10.1038/sj.emboj.7601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter SE, Pearce BR, Lundgren J, Owen MJ, Smart TG, Luscher B, Rees MI, Harvey RJ. 2004. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci 24:5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bausen M, Fuhrmann JC, Betz H, O'Sullivan GA. 2006. The state of the actin cytoskeleton determines its association with gephyrin: role of ena/VASP family members. Mol Cell Neurosci 31:376–386. doi: 10.1016/j.mcn.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Giesemann T, Schwarz G, Nawrotzki R, Berhorster K, Rothkegel M, Schluter K, Schrader N, Schindelin H, Mendel RR, Kirsch J, Jockusch BM. 2003. Complex formation between the postsynaptic scaffolding protein gephyrin, profilin, and Mena: a possible link to the microfilament system. J Neurosci 23:8330–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strittmatter SM, Fankhauser C, Huang PL, Mashimo H, Fishman MC. 1995. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell 80:445–452. doi: 10.1016/0092-8674(95)90495-6. [DOI] [PubMed] [Google Scholar]
- 17.He Q, Dent EW, Meiri KF. 1997. Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J Neurosci 17:3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benowitz LI, Routtenberg A. 1997. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20:84–91. doi: 10.1016/S0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 19.Liu YC, Storm DR. 1989. Dephosphorylation of neuromodulin by calcineurin. J Biol Chem 264:12800–12804. [PubMed] [Google Scholar]
- 20.Skene JH. 1989. Axonal growth-associated proteins. Annu Rev Neurosci 12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- 21.Chan SY, Murakami K, Routtenberg A. 1986. Phosphoprotein F1: purification and characterization of a brain kinase C substrate related to plasticity. J Neurosci 6:3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holahan M, Routtenberg A. 2008. The protein kinase C phosphorylation site on GAP-43 differentially regulates information storage. Hippocampus 18:1099–1102. doi: 10.1002/hipo.20486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIlvain VA, Robertson DR, Maimone MM, McCasland JS. 2003. Abnormal thalamocortical pathfinding and terminal arbors lead to enlarged barrels in neonatal GAP-43 heterozygous mice. J Comp Neurol 462:252–264. doi: 10.1002/cne.10725. [DOI] [PubMed] [Google Scholar]
- 24.Dubroff JG, Stevens RT, Hitt J, Hodge CJ Jr, McCasland JS. 2006. Anomalous functional organization of barrel cortex in GAP-43 deficient mice. Neuroimage 29:1040–1048. doi: 10.1016/j.neuroimage.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 25.Zaccaria KJ, Lagace DC, Eisch AJ, McCasland JS. 2010. Resistance to change and vulnerability to stress: autistic-like features of GAP43-deficient mice. Genes Brain Behav 9:985–996. doi: 10.1111/j.1601-183X.2010.00638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rekart JL, Quinn B, Mesulam MM, Routtenberg A. 2004. Subfield-specific increase in brain growth protein in postmortem hippocampus of Alzheimer's patients. Neuroscience 126:579–584. doi: 10.1016/j.neuroscience.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 27.Linden DJ, Routtenberg A. 1989. The role of protein kinase C in long-term potentiation: a testable model. Brain Res Brain Res Rev 14:279–296. doi: 10.1016/0165-0173(89)90004-0. [DOI] [PubMed] [Google Scholar]
- 28.Benowitz LI, Apostolides PJ, Perrone-Bizzozero N, Finklestein SP, Zwiers H. 1988. Anatomical distribution of the growth-associated protein GAP-43/B-50 in the adult rat brain. J Neurosci 8:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 30.Lin CH, Chen CC, Chou CM, Wang CY, Hung CC, Chen JY, Chang HW, Chen YC, Yeh GC, Lee YH. 2009. Knockdown of the aryl hydrocarbon receptor attenuates excitotoxicity and enhances NMDA-induced BDNF expression in cortical neurons. J Neurochem 111:777–789. doi: 10.1111/j.1471-4159.2009.06364.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang CY, Liang YJ, Lin YS, Shih HM, Jou YS, Yu WC. 2004. YY1AP, a novel co-activator of YY1. J Biol Chem 279:17750–17755. doi: 10.1074/jbc.M310532200. [DOI] [PubMed] [Google Scholar]
- 32.Shen D, Coleman J, Chan E, Nicholson TP, Dai L, Sheppard PW, Patton WF. 2011. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem Biophys 60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meiri KF, Hammang JP, Dent EW, Baetge EE. 1996. Mutagenesis of ser41 to ala inhibits the association of GAP-43 with the membrane skeleton of GAP-43-deficient PC12B cells: effects on cell adhesion and the composition of neurite cytoskeleton and membrane. J Neurobiol 29:213–232. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura F, Strittmatter P, Strittmatter SM. 1998. GAP-43 augmentation of G protein-mediated signal transduction is regulated by both phosphorylation and palmitoylation. J Neurochem 70:983–992. [DOI] [PubMed] [Google Scholar]
- 35.Lin CH, Juan SH, Wang CY, Sun YY, Chou CM, Chang SF, Hu SY, Lee WS, Lee YH. 2008. Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J Neurochem 104:1415–1429. doi: 10.1111/j.1471-4159.2007.05098.x. [DOI] [PubMed] [Google Scholar]
- 36.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. 1996. Evidence for p53-mediated modulation of neuronal viability. J Neurosci 16:6753–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang D, Sun YY, Nemkul N, Baumann JM, Shereen A, Dunn RS, Wills-Karp M, Lawrence DA, Lindquist DM, Kuan CY. 2013. Plasminogen activator inhibitor-1 mitigates brain injury in a rat model of infection-sensitized neonatal hypoxia-ischemia. Cereb Cortex 23:1218–1229. doi: 10.1093/cercor/bhs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun YY, Morozov YM, Yang D, Li Y, Dunn RS, Rakic P, Chan PH, Abe K, Lindquist DM, Kuan CY. 2014. Synergy of combined tPA-edaravone therapy in experimental thrombotic stroke. PLoS One 9:e98807. doi: 10.1371/journal.pone.0098807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice JE III, Vannucci RC, Brierley JB. 1981. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 40.Yin KJ, Chen SD, Lee JM, Xu J, Hsu CY. 2002. ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke 33:2471–2477. doi: 10.1161/01.STR.0000030316.79601.03. [DOI] [PubMed] [Google Scholar]
- 41.Chao S, Benowitz LI, Krainc D, Irwin N. 1996. Use of a two-hybrid system to investigate molecular interactions of GAP-43. Brain Res Mol Brain Res 40:195–202. doi: 10.1016/0169-328X(96)00049-6. [DOI] [PubMed] [Google Scholar]
- 42.Pascale A, Amadio M, Scapagnini G, Lanni C, Racchi M, Provenzani A, Govoni S, Alkon DL, Quattrone A. 2005. Neuronal ELAV proteins enhance mRNA stability by a PKCalpha-dependent pathway. Proc Natl Acad Sci U S A 102:12065–12070. doi: 10.1073/pnas.0504702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayer S, Kumar R, Jaiswal M, Soykan T, Ahmadian MR, Brose N, Betz H, Rhee JS, Papadopoulos T. 2013. Collybistin activation by GTP-TC10 enhances postsynaptic gephyrin clustering and hippocampal GABAergic neurotransmission. Proc Natl Acad Sci U S A 110:20795–20800. doi: 10.1073/pnas.1309078110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. 2007. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- 45.Lyons WE, George EB, Dawson TM, Steiner JP, Snyder SH. 1994. Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proc Natl Acad Sci U S A 91:3191–3195. doi: 10.1073/pnas.91.8.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lardi-Studler B, Smolinsky B, Petitjean CM, Koenig F, Sidler C, Meier JC, Fritschy JM, Schwarz G. 2007. Vertebrate-specific sequences in the gephyrin E-domain regulate cytosolic aggregation and postsynaptic clustering. J Cell Sci 120:1371–1382. doi: 10.1242/jcs.003905. [DOI] [PubMed] [Google Scholar]
- 47.Machado P, Rostaing P, Guigonis J-M, Renner M, Dumoulin A, Samson M, Vannier C, Triller A. 2011. Heat shock cognate protein 70 regulates gephyrin clustering. J Neurosci 31:3–14. doi: 10.1523/JNEUROSCI.2533-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopito RR. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530. doi: 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 49.Johnston JA, Ward CL, Kopito RR. 1998. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song M, Messing RO. 2005. Protein kinase C regulation of GABAA receptors. Cell Mol Life Sci 62:119–127. doi: 10.1007/s00018-004-4339-x. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy I, Salter MM, Lu YM. 2003. Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci 23:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong KL, Murakami K, Routtenberg A. 1989. Dietary cis-fatty acids that increase protein F1 phosphorylation enhance spatial memory. Brain Res 505:302–305. doi: 10.1016/0006-8993(89)91456-X. [DOI] [PubMed] [Google Scholar]
- 53.Nakajima K, Yin X, Takei Y, Seog DH, Homma N, Hirokawa N. 2012. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 76:945–961. doi: 10.1016/j.neuron.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 54.Robinson S. 2005. Systemic prenatal insults disrupt telencephalon development: implications for potential interventions. Epilepsy Behav 7:345–363. doi: 10.1016/j.yebeh.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]