

Abstract
Molecular chaperones monitor the proper folding of misfolded proteins and function as the first line of defense against mutant protein aggregation in neurodegenerative diseases. The eukaryotic chaperonin TRiC is a potent suppressor of mutant protein aggregation and toxicity in early stages of disease progression. Elucidation of TRiC functional regulation will enable us to better understand the pathological mechanisms of neurodegeneration. We have previously shown that vaccinia-related kinase 2 (VRK2) downregulates TRiC protein levels through the ubiquitin-proteasome system by recruiting the E3 ligase COP1. However, although VRK2 activity was necessary in TRiC downregulation, the phosphorylated substrate was not determined. Here, we report that USP25 is a novel TRiC interacting protein that is also phosphorylated by VRK2. USP25 catalyzed deubiquitination of the TRiC protein and stabilized the chaperonin, thereby reducing accumulation of misfolded polyglutamine protein aggregates. Notably, USP25 deubiquitinating activity was suppressed when VRK2 phosphorylated the Thr680, Thr727, and Ser745 residues. Impaired USP25 deubiquitinating activity after VRK2-mediated phosphorylation may be a critical pathway in TRiC protein destabilization.
INTRODUCTION
Protein misfolding and aggregation causes several neurodegenerative diseases. For example, aggregation of huntingtin (Htt) protein leads to Huntington's disease (HD). An expanded mutation in a polyglutamine (polyQ) stretch (>35 residues) at the N-terminal of Htt produces mutant Htt (mHtt), which is easily misfolded and amyloidogenic (1, 2). The number of glutamines in the polyQ stretch positively correlates with symptom severity and inversely correlates with the age of symptom onset (2). mHtt aggregates sequester many transcriptional factors via aberrant protein interactions, and these sequestrations cause transcriptional inhibition of genes such as brain-derived neurotrophic factor (BDNF) and its cognate receptors, which are responsible for neuronal survival and function (3–6). Moreover, mHtt aggregates impair axonal transport and mitochondrial function, resulting in striatal neuron cell death in the basal ganglia and cortex (7, 8). As a result, HD patients exhibit obvious symptoms, including chorea, psychiatric impairments, and cognitive deficits (1).
To cope with aggregate-mediated cytotoxicity, cells possess defense machinery called molecular chaperones. Within cells, molecular chaperones regulate proper folding of misfolded proteins into their correct conformations (9). Studies have revealed that molecular chaperones alleviate neurodegeneration by modulating aberrant protein interactions in the early stages of aggregation (10, 11). Therefore, it is necessary to understand the upstream pathways controlling molecular chaperone protein levels in order to develop novel therapeutics. Because expression of chaperones such as heat shock proteins (HSPs) is affected by heat shock factor protein 1 (HSF1), various pharmacological agents have been developed to enhance HSF1 transcriptional activity by inhibiting HSP90, which negatively regulates HSF1 activation (12, 13). However, expression of eukaryotic chaperonin TCP-1 ring complex (TRiC), a potent suppressor of polyQ aggregation and toxicity, is not regulated by HSF1 transcriptional activity. TRiC protein levels are elevated by inhibition of the degradation pathway. Although the novel vaccinia-related kinase 2 (VRK2) is involved in TRiC turnover, the molecular mechanisms of TRiC protein degradation remain unclear (14). It is, therefore, necessary to examine VRK2 function in TRiC protein degradation to provide better therapeutic insights into TRiC-assisted alleviation of polyQ toxicity.
VRK2 is highly expressed in proliferating cells such as cancer cells and negatively regulates mitogen-activated protein kinase signaling by interacting with the scaffold proteins JNK-interacting protein (JIP1) and kinase suppressor of ras 1 (KSR1) (15, 16). VRK2 also promotes cancer cell invasion by elevating the transcriptional activity of nuclear factor of activated T cells 1 (NFAT1) by VRK2-mediated phosphorylation (17). In addition, VRK2 prevents apoptosis by interacting with Bcl-xL, an antiapoptotic Bcl-2 homology (BH) domain protein, which is involved in BAX gene expression and regulates mitochondrial function (18). Recently, VRK2 function in the brain has been noted in psychiatric disorders, including schizophrenia and epilepsy. Using genome-wide associated analyses, researchers observed a single-nucleotide polymorphism, rs2312147, located upstream of VRK2 among patients with schizophrenia that conferred an increased risk of schizophrenia (19, 20).
We have addressed here whether VRK2 regulates TRiC protein degradation, which affects polyQ aggregation. In our previous study, VRK2 increased TRiC protein degradation by promoting ubiquitination, and TRiC ubiquitination was dependent on VRK2 enzymatic activity (14). However, the molecular mechanism of VRK2 and its putative substrates have remained largely unexplored. Here, we identified ubiquitin-specific protease 25 (USP25) as a VRK2 substrate that acts on TRiC deubiquitination. These findings suggest that VRK2 is important for the eukaryotic chaperonin TRiC protein degradation pathway and is involved in polyQ protein aggregation.
MATERIALS AND METHODS
Plasmids.
VRK2 (accession number NM_027260) and TRiC expression constructs were generated by PCR amplification of mouse full-length VRK2 and TRiC subunits (CCT1 to CCT8) from a day 16 mouse embryo cDNA library (Clontech, Mountain View, CA). The USP25 construct was purchased from DNASU plasmid repository (clone ID HSCD00442918). For mammalian expression constructs, VRK2, USP25, and TRiC subunits were subcloned into pFlag-CMV2 (Sigma, St. Louis, MO), pDsRed1-N1, and pDsRed1-C1 (BD Biosciences, San Jose, CA) vectors, as well as vector pcDNA3.1 containing hemagglutinin (HA) sequences. For expression in Escherichia coli, VRK2 and USP25 were subcloned into pProEX, pGEX-4T-1, and pGEX-4T-3 (Amersham, Piscataway, NJ) vectors. USP25 mutants were generated by site-directed mutagenesis. Htt-exon1-GFP-pcDNA3.1 (length of polyQ tract = 25 or 103) was a gift from Judith Frydman (Stanford University, Stanford, CA). The HA-ubiquitin construct was a gift from Joo-Yeon Yoo (Postech, Pohang, South Korea).
Biochemical methods.
Protein extraction and preparation, glutathione S-transferase (GST) pulldown, and immunoblotting were performed as previously described (14).
In vitro kinase assays.
Kinase assays utilized 1 μg of recombinant GST (or His)-VRK2 and 1 μg of recombinant GST-USP25 (full length, fragments, or mutants) as the substrates. The reaction was performed at 30°C in 20-μl reaction mixtures containing kinase buffer (20 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 0.5 mM dithiothreitol, 150 mM KCl, and [γ-32P]ATP). After 30 min, the reactions were resolved by SDS-PAGE and visualized with autoradiography.
In vitro deubiquitination assays.
Ubiquitinated CCTs were immunoprecipitated from cells transfected with HA-ubiquitin and treated with MG132 using the anti-CCT antibody. Ubiquitinated CCTs and 1 μg of GST-USP25 or USP25 mutants (C178S or phosphomimic) were added in a buffer containing 20 mM Tris (pH 7.5) and 0.5 mM dithiothreitol. Incubation was performed at 37°C for 2 h, followed by 16°C overnight.
Fluorescence microscopy.
Mammalian cells were grown on coverslips until they reached 50 to 60% confluence. Transfected cells were maintained for 24 h, fixed with 4% paraformaldehyde for 15 min, and if necessary, immunostained with appropriate primary and fluorescence-conjugated secondary antibodies. Coverslips were mounted onto slides using Fluoromount (Sigma). An Axioplan2 fluorescence imaging microscope (Carl Zeiss, Jena, Germany) equipped with an ApoTome (Carl Zeiss) was used to obtain fluorescence or differential interference contrast images.
Cell culture, transfection, and immunoprecipitation.
HEK293A, HEK293T, and SH-SY5Y cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 100 U/ml each of penicillin G and streptomycin. Transient transfections were performed using Metafectene reagent (Biontex, Munich, Germany) or Microporator (Invitrogen) as described in the manufacturer's protocols. For immunoprecipitation, cell extracts were incubated with rotation with the indicated antibody at 4°C for 1 h and precipitated with protein G-agarose beads (Roche, Mannheim, Germany) overnight. Immunoprecipitates were washed five times with lysis buffer and subjected to immunoblotting.
siRNA sequences.
The small interfering RNA (siRNA) duplex pool targeting human USP25 was obtained from Dharmacon (Lafayette, CO). The siRNA sequence for the nonspecific control oligonucleotide was CCUACGCCACCAAUUUCGUdTdT.
Antibodies.
Commercially available antibodies were as follows: anti-Flag epitope (M2) from Sigma; anti-HA epitope from Roche (Mannheim, Germany); anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) from Calbiochem (San Diego, CA); anti-CCT1, anti-CCT2, anti-CCT4, and anti-USP25 from Abcam (Cambridge, United Kingdom); anti-VRK2, anti-green fluorescent protein (anti-GFP), antiubiquitin, anti-His epitope, and anti-GST from Santa Cruz Biotechnology (Santa Cruz, CA); anti-Hsp70 from Enzo Life Sciences (Farmingdale, NY); and antiactin from MP Biomedicals (Santa Ana, CA). All fluorescence-conjugated secondary antibodies were purchased from Invitrogen.
Quantitative real-time reverse transcription-PCR.
Total RNA was isolated with TRI Reagent (Molecular Research Center). Isolated RNA was reverse transcribed by using an ImProm-II reverse transcription system (Promega) according to the manufacturer's instructions. For detection and quantification, the StepOnePlus real-time PCR system (Applied Biosystems) was used. The sequences of the forward and reverse primers were as follows: human hsp70 (5′-AGCCAAGAAGGCAAAAGTGA-3′ and 5′-CCACTGCGTTCTTAGCATCA-3′) and human gapdh (5′-GCCATCAATGACCCCTTCATT-3′ and 5′-GCTCCTGGAAGATGGTGATGG-3′).
Statistical analyses.
All quantitative data are presented as means ± the standard deviations (SD) or standard errors of the mean (SEM). Statistical analyses were performed using one-way analyses of variance and post hoc Tukey's multiple-comparison tests and GraphPad software. Differences were considered significant when the P value was <0.05.
RESULTS
USP25 interacts with the chaperonin TRiC.
Because increased CCT4 ubiquitination is dependent on VRK2 enzymatic activity, we sought to identify substrate proteins that function in the ubiquitination of chaperonin (14). To delineate the molecular mechanisms of TRiC protein degradation, we attempted to identify a novel binding partner of TRiC. After lysates of TRiC subunit-expressing cells (CCT2, CCT4, and CCT8) were incubated with the anti-HA antibody, proteins associating with HA-CCT2, HA-CCT4, and HA-CCT8 were immunoprecipitated. Using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) analyses, cytoplasmic USP25 was determined to be a novel interacting protein of the chaperonin subunits (Fig. 1A). To further confirm the binding between CCT4 and USP25, we performed immunoprecipitation assays with cells transfected with Flag-USP25 and HA-CCT4. The coprecipitated CCT4 was hardly detectable without proteasomal inhibitor MG132, but it was evident in the presence of MG132, which results in the accumulation of ubiquitinated proteins (Fig. 1B and C). In order to determine which subunit of TRiC binds to USP25, immunoprecipitation experiments were performed with TRiC subunits (CCT1 to CCT8) in HEK293A cells. When HA-TRiC subunits CCT1, CCT3, CCT4, CCT5, and CCT7 were coexpressed with Flag-USP25, they were efficiently coimmunoprecipitated using anti-HA antibody (Fig. 1D and E). Notably, the interaction between USP25 and TRiC was detected in the presence of MG132 (Fig. 1C), suggesting that USP25 recognizes ubiquitinated TRiC rather than its native form. USP25 has one ubiquitin-associated domain (UBA) and two ubiquitin-interacting motifs (UIMs) (21), which are important for recognition of ubiquitinated substrates. To map the USP25 binding domains for TRiC, we performed pulldown assays to observe the interactions between CCT4 and various USP25 fragments. We found that the second UIM deletion fragment (147 to 1055) lost binding capacity to CCT4, whereas the UBA domain deletion fragment (91 to 1055) or both UBA domain and the first UIM deletion fragment (121 to 1055) still had interaction with CCT4 (Fig. 1F; also see Fig. 4F). These data suggest that USP25 interacts with TRiC subunits and that the second UIM is critical for recognition of ubiquitinated CCT4.
FIG 1.

USP25 interacts with TRiC via the second UIM domain. (A) Lysates from HEK293T cells transfected with HA-CCT2, HA-CCT4, and HA-CCT8 were divided into cytoplasmic and nuclear fractions and then incubated with anti-HA antibody. Precipitated proteins were visualized by silver staining and analyzed with MALDI-TOF. Cytoplasmic USP25 was determined to be a novel interacting protein of TRiC by immunoprecipitation with CCT2, CCT4, and CCT8. (B and C) Immunoprecipitation assays using HEK293T cells cotransfected with Flag-USP25 and HA-CCT4 vectors in the absence (B) or presence (C) of MG132 (10 μM). HA-CCT4 was coimmunoprecipitated with anti-Flag antibody. (D) TRiC subunits 1, 3, 4, 5, and 7 were efficiently coimmunoprecipitated by USP25. (E) Calculated amounts of bound USP25 with TRiC subunits. Error bars represent the means ± the SEM (n = 3). (F) Serial deletions of domains that are responsible for substrate binding which indicated the second UIM of USP25 is important for TRiC binding.
FIG 4.

VRK2 binds to and phosphorylates USP25. (A) Immunoprecipitation assays using HEK293T cells cotransfected with Flag-VRK2 and HA-USP25 vectors. (B) Colocalization of VRK2 and USP25. HEK293A cells were cotransfected with EGFP-VRK2 and Flag-USP25 and stained for Flag (red) and nuclei (Hoechst; blue). Scale bar, 20 μm. (C) In vitro kinase assays with His-VRK2 and GST-USP25 derived from E. coli. (D) Site-directed mutagenesis of candidate phosphorylation sites in USP25 fragment 3-1, followed by in vitro kinase assays with His-VRK2. (E) In vitro kinase assays with His-VRK2 and the triple site mutant (Thr680, Thr727, and Ser745 to Ala). (F) Diagram of USP25 and fragments. UBA, ubiquitin-associated domain; SIM, SUMO-interacting motif; UIM, ubiquitin-interacting motif.
USP25 deubiquitinates CCT4.
USP25 is a deubiquitinating enzyme (DUB) that catalyzes removal of ubiquitin from its substrates. Ubiquitination of target proteins is important for substrate recognition of DUBs (22). To this end, we investigated whether USP25 could deubiquitinate CCT4. Ubiquitinated CCT4 was incubated with USP25 or enzymatically inactive USP25 (C178S) (21) proteins that were purified from Escherichia coli. Although the purified proteins did not change polyubiquitination of CCT2, which had no interaction with USP25, CCT4 polyubiquitination was decreased when incubated with USP25 but not with enzymatically inactive USP25 (C178S) (Fig. 2A; see Fig. S1 in the supplemental material). Conversely, USP25 knockdown enhanced CCT4 polyubiquitination and reduced CCT4 protein levels (Fig. 2B). These data suggest that catalytically active USP25 deubiquitinates CCT4 and stabilizes it. To further confirm the role of the deubiquitinating enzyme USP25 against other TRiC subunits, we examined polyubiquitination of CCT1, CCT2, and CCT4 in cells transfected with Flag-USP25. Based on the interaction of USP25 with TRiC subunits (Fig. 1D and E), we found that USP25 catalyzed the deubiquitination of CCT1 and CCT4 but not CCT2 (Fig. 2C). Taken together, these results show that USP25 functions as a DUB for ubiquitinated TRiC, which is dependent on ubiquitinated substrate recognition.
FIG 2.

USP25 catalyzes deubiquitination of TRiC. (A) In vitro deubiquitination assays with purified GST-USP25 or mutant USP25 (CS, C178S) and ubiquitinated CCT4 from cells treated with MG132. Polyubiquitinated CCT4 was immunoblotted with anti-HA antibody to detect HA-ubiquitin. (B) Immunoprecipitation assays with anti-CCT4 and SH-SY5Y cells transfected with HA-ubiquitin and siRNA against USP25. Cells were treated with MG132 (10 μM) before harvest, followed by incubation of the cell lysates with the anti-CCT4 antibody. Immunoprecipitated CCT4 was subjected to Western blot analyses with the anti-HA antibody (upper panel). Endogenous CCT4 protein in cells transfected with siUSP25 was investigated by Western blotting analyses with the anti-CCT4 antibody (bottom panel). (C) Polyubiquitination levels of CCT1, CCT2, and CCT4 in cells expressing USP25 were analyzed by Western blotting with anti-CCT1, -CCT2, and -CCT4 antibodies.
Physiological role of USP25 in polyQ aggregation.
To further examine the physiological role of USP25 on TRiC protein stability, we evaluated polyQ aggregation using the polyQ-expanded Htt fragment fused to GFP when USP25 protein levels were modulated. Growing evidences reveal that functional TRiC inhibits polyQ aggregation at early stages and alleviates soluble or insoluble toxic species-mediated harmful effects (10, 14). Notably, overexpression of either a specific TRiC subunit or recombinant CCT1 significantly suppresses polyQ aggregation and neuronal cell death (23, 24). We coexpressed Flag-USP25 or enzymatically inactive USP25 (C178S) with pathogenic HttQ103-GFP in HEK293A cells. Because USP25 stabilizes TRiC protein levels by detaching polyubiquitin chains from the TRiC proteins, the formation of HttQ103-GFP aggregates was decreased in Flag-USP25-overexpressing cells (Fig. 3A and B). In addition, the number of double-labeled cells containing large foci of aggregated HttQ103-GFP in wild-type USP25-expressing cells reduced ca. 40% compared to control (Fig. 3C and D). Moreover, mutant USP25 (C178S) did not significantly affect polyQ aggregation and CCT4 protein levels, suggesting that the deubiquitinating enzyme activity of USP25 is important for polyQ aggregation. Conversely, HttQ103-GFP aggregate formation was increased in SH-SY5Y cells transfected with siRNA against USP25, which reduced CCT4 protein levels (Fig. 3E and F). Consistent with the immunoblot observations, USP25 knockdown increased the number of cells containing large foci of aggregated HttQ103-GFP, as detected by fluorescence imaging (Fig. 3G and H).
FIG 3.

USP25 enhances the chaperone activity of TRiC in reducing polyQ-expanded Htt aggregation in mammalian cells. (A) HttQ103-GFP with Flag-USP25 or the inactive mutant USP25 (C178S) were coexpressed in HEK293A cells, followed by Western blotting to detect polyQ aggregates. (B) Calculated ratio of insoluble to soluble mHtt levels. Error bars represent the means ± the SD (n = 4). *, P < 0.05; ***, P < 0.001. (C) Fluorescence microscopic images of HttQ103-GFP and Flag-USP25 or USP25 (C178S) expressed in HEK293A cells. (D) Percentages of double-labeled cells containing large foci with USP25. Medians, confidence intervals, and the SD are represented (n = 3). *, P < 0.05. Total counted cell numbers are shown at the bottom. (E) HttQ103-GFP, with or without siRNA against USP25, was cotransfected into SH-SY5Y neuroblastoma cells, and HttQ103 aggregates were investigated by immunoblotting. (F) Calculated ratio of insoluble to soluble mHtt levels. Error bars represent the means ± the SD (n = 3). **, P < 0.01. (G) Fluorescence microscopy of HttQ103-GFP-expressing SH-SY5Y cells treated with siUSP25 or siCont. USP25 knockdown increased number of cells containing HttQ103-GFP aggregates. (H) The percentage of aggregate-positive cells from panel G was calculated. Median, confidence intervals, and the SD are represented (n = 4). **, P < 0.01. Total counted cell numbers are shown at the bottom. Scale bars represent 60 μm in panels C and G.
USP25 is phosphorylated by VRK2.
Since VRK2 reduced CCT4 protein levels by promoting ubiquitination in a kinase-dependent manner and triggered the accumulation of polyQ aggregates (14), we tested whether VRK2 could regulate USP25 deubiquitinating activity toward ubiquitinated CCT4. In order to confirm the interaction between VRK2 and USP25, we performed pulldown assays with cells cotransfected with Flag-VRK2 and HA-USP25. VRK2 weakly interacted with USP25 (Fig. 4A). Fluorescence imaging analyses previously showed that VRK2 is localized in the endoplasmic reticulum membrane due to its C-terminal transmembrane domain (25). Unlike VRK2, USP25 diffuses throughout the cytoplasm (Fig. 4B). Although the interaction is weak, VRK2 could phosphorylate USP25 (Fig. 4C). To clarify the effect of VRK2-mediated USP25 phosphorylation, we constructed three fragments of USP25 to find the specific phosphorylation sites (Fig. 4F). After purifying His-VRK2 protein and three fragments of USP25, we performed in vitro kinase assays. VRK2 phosphorylated fragment 3 (F3, amino acids [aa] 655 to 1055) that containing the regulatory domain, whereas fragment 1 (F1, aa 1 to 146), which includes one UBA and two UIM domains, and fragment 2 (F2, aa 147 to 654), which contains a ubiquitin-specific peptidase domain, were not phosphorylated by VRK2 (see Fig. S2A in the supplemental material). We further divided USP25 F3 into three fragments, F3-1 (aa 655 to 780), F3-2 (aa 781 to 910), and F3-3 (aa 911 to 1055), and purified them from transformed E. coli cultures. By in vitro kinase assays, we found that only USP25 F3-1 is phosphorylated by VRK2 (see Fig. S2B in the supplemental material). To determine the VRK2-specific phosphorylation sites of USP25, we generated USP25 mutants by replacing the putative Ser and Thr phosphorylation sites to Ala. Substitution mutants of Thr680, Thr727, and Ser745 to Ala showed reduced VRK2-mediated USP25 phosphorylation (Fig. 4D). When we performed in vitro kinase assays with VRK2 and the triple site mutant (Thr680, Thr727, and Ser745), phospho-USP25 was completely abrogated (Fig. 4E).
USP25 has decreased deubiquitinating activity when it is phosphorylated by VRK2.
In a previous report regarding posttranslational regulation of USP25 activity, sumoylation of USP25 within its two UIMs impaired its capability to bind ubiquitinated substrates (26). To further verify the effect of VRK2-mediated USP25 phosphorylation, we examined its ability to catalyze ubiquitinated CCT4 in cells treated with MG132 after USP25 was preincubated with or without VRK2 in the kinase reaction. Interestingly, we found that phosphorylated USP25 showed markedly impaired deubiquitination of CCT4 (Fig. 5A). Furthermore, the phosphomimic mutant (Thr680, Thr727, and Ser745 to Glu) was also unable to catalyze deubiquitination of CCT4 (Fig. 5B and C). However, there was no difference in substrate binding, protein stability and subcellular localization between phosphomimic mutant USP25 and wild-type USP25 (see Fig. S3A to C in the supplemental material). Because the USP25 phosphomimic mutant has defect on deubiquitinating activity, it did not affect polyQ aggregation and CCT4 protein levels (see Fig. S3C to E in the supplemental material). Collectively, our results suggest that VRK2 suppresses the deubiquitinating activity of USP25 via posttranslational modification and, as a result, destabilizes TRiC.
FIG 5.

USP25 is impaired by VRK2-mediated phosphorylation. (A) In vitro deubiquitination assays for purified GST-USP25 with or without kinase reaction of VRK2 were performed with ubiquitinated CCT4 from MG132-treated cells. Before incubation with ubiquitinated CCT4, USP25 was subjected to VRK2-mediated phosphorylation. Polyubiquitinated CCT4 was immunoblotted with anti-HA antibody to detect HA-ubiquitin. (B) Polyubiquitination levels of CCT4 in cells expressing USP25 and the phosphomimic mutant USP25 (PM, triple mutation to Glu) were analyzed by Western blotting with anti-HA antibody. (C) In vitro deubiquitination assays with purified GST-USP25 or PM and ubiquitinated CCT4 from cells treated with MG132. Polyubiquitinated CCT4 was investigated by immunoblotting.
DISCUSSION
HD is a late-onset and devastating neurodegenerative disorder. Although there have been many attempts to find a cure for the disease, no therapeutic medications have been developed. Although there are many pathological features, HD pathogenesis is ascribed to aggregation of mHtt, which leads to striatal neuron cell death in the basal ganglia. Therefore, targeted therapies for HD could inhibit mHtt aggregation or prevent cellular damage resulting from mHtt cytotoxicity. Approaches to decrease mHtt protein levels include allele-specific silencing expression of the mutant HTT gene (27), as well as the activation of autophagy (28) and the ubiquitin-proteasome system (29). In addition, inhibition of mHtt aggregation by elevating molecular chaperones, as well as antiaggregation drugs, is beneficial for the early stages of HD pathogenesis (13, 30). Trehalose was reported to reduce the symptoms of HD (31), and we previously found that guanidine-conjugated trehalose derivatives were delivered to the brain across the blood-brain barrier and exhibited improved therapeutic effects in HD model mice (30). Furthermore, there are approaches to alleviate the harmful effects of mHtt, including transcriptional dysregulation, mitochondrial impairment and excitotoxicity, or apoptotic cell death (32).
Although mHtt aggregate formation in the cortex and striatum is considered the hallmark of HD, a number of studies have revealed that the soluble oligomer is more toxic than insoluble aggregates (33, 34). Because molecular chaperones function in the proper folding of misfolded mutant proteins before formation of aberrant protein interactions, elevating the molecular chaperone protein levels would be a rational approach to reduce soluble mHtt oligomers. Although other molecular chaperones, such as heat shock proteins, could be easily induced by activating HSF1, expression of the eukaryotic chaperonin TRiC hetero-oligomeric complex is not regulated by HSF1 transcriptional activity. However, a recent study has revealed that HSF1 is regulated by TRiC (35). Because TRiC directly interacts with HSF1 and represses its transcriptional activity, disturbance of the interaction results HSF1 target gene expression, such as hsp70. Therefore, we examined whether VRK2-mediated USP25 phosphorylation, which results in TRiC destabilization, has effect on HSF1 activity. Interestingly, we found that hsp70 mRNA level was not changed by phosphomimic mutant USP25 expressing cells, but the protein level showed difference between wild-type USP25 and phosphomimic mutant USP25 (see Fig. S4A to C in the supplemental material). These data suggest that TRiC destabilization by VRK2-mediated USP25 phosphorylation does not affect HSF1 transcriptional activity but decreases the Hsp70 protein level. Since Hsp70 has been known to be polyubiquitinated and degraded by proteasome (36), its stability may be regulated by USP25 directly. Moreover, β-actin, a TRiC-client protein, was slightly decreased when phosphomimic mutant USP25 was overexpressed compared to wild-type USP25 (see Fig. S4D in the supplemental material). To summarize, elevating active chaperonin TRiC levels by inhibiting VRK2-mediated USP25 phosphorylation has beneficial effects on proper protein folding and stability. Elucidation of how TRiC protein is degraded will allow us to understand pathological disease mechanisms as well as suggest drug targets for the treatment of neurodegenerative diseases. We previously demonstrated that VRK2 plays an important role in TRiC protein turnover by promoting ubiquitin-proteasomal degradation (14). However, the exact function of VRK2 in TRiC protein degradation remained unclear.
Here, we extensively investigated whether VRK2 controls TRiC ubiquitination by inhibiting USP25 catalytic activity via posttranslational modification (Fig. 6). Notably, we demonstrated that USP25 is a novel deubiquitinating enzyme for TRiC. USP25 catalyzes the deubiquitination of CCT1 and CCT4, which are prominent subunits that suppress mHtt aggregation. DUBs are isopeptidases regulating the ubiquitination state of target proteins (22). The catalytic activity of DUBs is tightly controlled by posttranslational modifications such as acetylation, sumoylation, ubiquitination, and phosphorylation. These modifications have been reported to affect DUBs in several ways, including localization, adaptor protein binding, protein stability, and catalytic activity (37). Posttranslational modifications of USP25 also regulate its DUB functions, which impair binding of the ubiquitin chain by sumoylation (26) and protein degradation by SYK-mediated phosphorylation (38). Furthermore, we have shown that VRK2-mediated phosphorylation of USP25 causes impairment of DUB activity for CCT4 deubiquitination.
FIG 6.
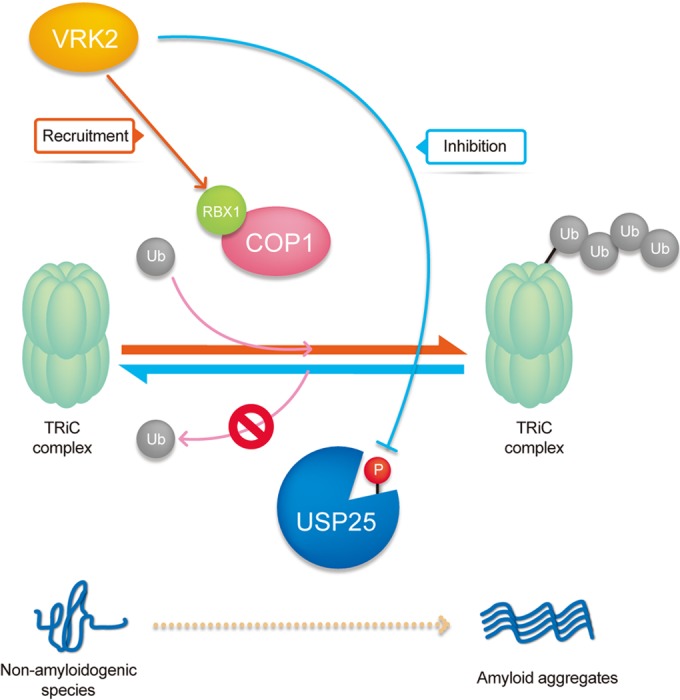
Diagram of VRK2 function in the negative regulation of TRiC via inhibition of USP25 deubiquitinating activity. TRiC functions to properly fold abnormally folded proteins. If VRK2 levels are increased or its enzymatic activity is activated in cells, TRiC ubiquitination is induced by inhibition of USP25 and E3 ligase COP1 is recruited. Subsequently, TRiC protein levels are decreased. Therefore, chaperonin-mediated proper folding does not occur, and aggregate formation increases.
Because the levels of TRiC represent the chaperone capacity for ameliorating toxicity resulting from misfolded proteins, VRK2-mediated downregulation of TRiC expression may be critical for the onset and progression of misfolded protein-associated neurodegenerative diseases. VRK2 expression is reported to be maintained at low levels in the brain (39). However, in neurological disorders, VRK2 mRNA levels were upregulated in the brains from patients with schizophrenia compared to normal controls (20). Moreover, polyQ aggregation was increased in active VRK2-expressing SH-SY5Y neuroblastoma cells (14). Further studies that determine whether VRK2 levels and activity are elevated in brains from HD patients will provide promising evidence to help us understand VRK2 function in HD pathogenesis. Although the physiological role of VRK2 in the brain remains to be investigated, our results raise the possibility that inhibition of VRK2 enzymatic activity would be a potential therapeutic target for the treatment of many neurodegenerative diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Frydman and J. Yoo for providing valuable plasmids.
This study was supported by grants from the Basic Research Laboratory Program (no. 2014054324), the Brain Korea 21 Plus Program ( 10Z20130012243) funded by the Korean Ministry of Science, and the Next-Generation BioGreen 21 Program (no. PJ00950302) funded by the Korean Rural Development Administration.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01325-14.
REFERENCES
- 1.Zoghbi HY, Orr HT. 2000. Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA. 2002. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35:819–822. doi: 10.1016/S0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 3.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. 2003. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 4.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. 2000. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A 97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. 2002. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 6.Vashishtha M, Ng CW, Yildirim F, Gipson TA, Kratter IH, Bodai L, Song W, Lau A, Labadorf A, Vogel-Ciernia A, Troncosco J, Ross CA, Bates GP, Krainc D, Sadri-Vakili G, Finkbeiner S, Marsh JL, Housman DE, Fraenkel E, Thompson LM. 2013. Targeting H3K4 trimethylation in Huntington disease. Proc Natl Acad Sci U S A 110:E3027–E3036. doi: 10.1073/pnas.1311323110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. 2004. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. 2006. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Tyedmers J, Mogk A, Bukau B. 2010. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, Takahashi Y, Kimura H, Kinjo M, Morimoto RI, Nagata K. 2006. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol 8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- 11.Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, Hartl FU. 2006. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell 23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. 1998. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480. doi: 10.1016/S0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 13.Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. 2008. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, Park DY, Lee D, Kim W, Jeong YH, Lee J, Chung SK, Ha H, Choi BH, Kim KT. 2014. Vaccinia-related kinase 2 mediates accumulation of polyglutamine aggregates via negative regulation of the chaperonin TRiC. Mol Cell Biol 34:643–652. doi: 10.1128/MCB.00756-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanco S, Santos C, Lazo PA. 2007. Vaccinia-related kinase 2 modulates the stress response to hypoxia mediated by TAK1. Mol Cell Biol 27:7273–7283. doi: 10.1128/MCB.00025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez IF, Blanco S, Lozano J, Lazo PA. 2010. VRK2 inhibits mitogen-activated protein kinase signaling and inversely correlates with ErbB2 in human breast cancer. Mol Cell Biol 30:4687–4697. doi: 10.1128/MCB.01581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vazquez-Cedeira M, Lazo PA. 2012. Human VRK2 (vaccinia-related kinase 2) modulates tumor cell invasion by hyperactivation of NFAT1 and expression of cyclooxygenase-2. J Biol Chem 287:42739–42750. doi: 10.1074/jbc.M112.404285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monsalve DM, Merced T, Fernandez IF, Blanco S, Vazquez-Cedeira M, Lazo PA. 2013. Human VRK2 modulates apoptosis by interaction with Bcl-xL and regulation of BAX gene expression. Cell Death Dis 4:e513. doi: 10.1038/cddis.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinberg S, de Jong S, Irish Schizophrenia Genomics Consortium, Andreassen OA, Werge T, Borglum AD, Mors O, Mortensen PB, Gustafsson O, Costas J, Pietilainen OP, Demontis D, Papiol S, Huttenlocher J, Mattheisen M, Breuer R, Vassos E, Giegling I, Fraser G, Walker N, Tuulio-Henriksson A, Suvisaari J, Lonnqvist J, Paunio T, Agartz I, Melle I, Djurovic S, Strengman E, Jurgens G, Glenthoj B, Terenius L, Hougaard DM, Orntoft T, Wiuf C, Didriksen M, Hollegaard MV, Nordentoft M, van Winkel R, Kenis G, Abramova L, Kaleda V, Arrojo M, Sanjuan J, Arango C, Sperling S, Rossner M, Ribolsi M, Magni V, Siracusano A, et al. 2011. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum Mol Genet 20:4076–4081. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Wang Y, Zheng XB, Ikeda M, Iwata N, Luo XJ, Chong SA, Lee J, Rietschel M, Zhang F, Muller-Myhsok B, Cichon S, Weinberger DR, Mattheisen M, Schulze TG, Martin NG, Mitchell PB, Schofield PR, Liu JJ, Su B, Moo DSC. 2012. Meta-analysis and brain imaging data support the involvement of VRK2 (rs2312147) in schizophrenia susceptibility. Schizophr Res 142:200–205. doi: 10.1016/j.schres.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Denuc A, Bosch-Comas A, Gonzalez-Duarte R, Marfany G. 2009. The UBA-UIM domains of the USP25 regulate the enzyme ubiquitination state and modulate substrate recognition. PLoS One 4:e5571. doi: 10.1371/journal.pone.0005571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reyes-Turcu FE, Ventii KH, Wilkinson KD. 2009. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sontag EM, Joachimiak LA, Tan Z, Tomlinson A, Housman DE, Glabe CG, Potkin SG, Frydman J, Thompson LM. 2013. Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant huntingtin cellular phenotypes. Proc Natl Acad Sci U S A 110:3077–3082. doi: 10.1073/pnas.1222663110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tam S, Geller R, Spiess C, Frydman J. 2006. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol 8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco S, Klimcakova L, Vega FM, Lazo PA. 2006. The subcellular localization of vaccinia-related kinase-2 (VRK2) isoforms determines their different effect on p53 stability in tumour cell lines. FEBS J 273:2487–2504. doi: 10.1111/j.1742-4658.2006.05256.x. [DOI] [PubMed] [Google Scholar]
- 26.Meulmeester E, Kunze M, Hsiao HH, Urlaub H, Melchior F. 2008. Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Mol Cell 30:610–619. doi: 10.1016/j.molcel.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 27.Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, Wu J, Bezprozvanny I, Corey DR. 2009. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol 27:478–484. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. 2008. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet 17:170–178. [DOI] [PubMed] [Google Scholar]
- 29.Bauer PO, Wong HK, Oyama F, Goswami A, Okuno M, Kino Y, Miyazaki H, Nukina N. 2009. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J Biol Chem 284:13153–13164. doi: 10.1074/jbc.M809229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Im J, Kim S, Jeong YH, Kim W, Lee D, Lee WS, Chang YT, Kim KT, Chung SK. 2013. Preparation and evaluation of BBB-permeable trehalose derivatives as potential therapeutic agents for Huntington's disease. Med Chem Commun 4:310–316. doi: 10.1039/C2MD20112G. [DOI] [Google Scholar]
- 31.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. 2004. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 32.Bauer PO, Nukina N. 2009. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110:1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x. [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam S, Sixt KM, Barrow R, Snyder SH. 2009. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayatekin C, Matlack KE, Hesse WR, Guan Y, Chakrabortee S, Russ J, Wanker EE, Shah JV, Lindquist S. 2014. Prion-like proteins sequester and suppress the toxicity of huntingtin exon 1. Proc Natl Acad Sci U S A 111:12085–12090. doi: 10.1073/pnas.1412504111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neef DW, Jaeger AM, Gomez-Pastor R, Willmund F, Frydman J, Thiele DJ. 2014. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep 9:955–966. doi: 10.1016/j.celrep.2014.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao RF, Rubio V, Chen H, Bai L, Mansour OC, Shi ZZ. 2013. OLA1 protects cells in heat shock by stabilizing HSP70. Cell Death Dis 4:e491. doi: 10.1038/cddis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kessler BM, Edelmann MJ. 2011. PTMs in conversation: activity and function of deubiquitinating enzymes regulated via posttranslational modifications. Cell Biochem Biophys 60:21–38. doi: 10.1007/s12013-011-9176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cholay M, Reverdy C, Benarous R, Colland F, Daviet L. 2010. Functional interaction between the ubiquitin-specific protease 25 and the SYK tyrosine kinase. Exp Cell Res 316:667–675. doi: 10.1016/j.yexcr.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 39.Nezu J, Oku A, Jones MH, Shimane M. 1997. Identification of two novel human putative serine/threonine kinases, VRK1 and VRK2, with structural similarity to vaccinia virus B1R kinase. Genomics 45:327–331. doi: 10.1006/geno.1997.4938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.