

Abstract
Inflammation in the tumor microenvironment has many tumor-promoting effects. In particular, tumor-associated macrophages (TAMs) produce many cytokines which can support tumor growth by promoting survival of malignant cells, angiogenesis, and metastasis. Enhanced cytokine production by TAMs is tightly coupled with protein synthesis. In turn, translation of proteins depends on tRNAs, short abundant transcripts that are made by RNA polymerase III (Pol III). Here, we connect these facts by showing that stimulation of mouse macrophages with lipopolysaccharides (LPS) from the bacterial cell wall causes transcriptional upregulation of tRNA genes. The transcription factor NF-κB is a key transcription factor mediating inflammatory signals, and we report that LPS treatment causes an increased association of the NF-κB subunit p65 with tRNA genes. In addition, we show that p65 can directly associate with the Pol III transcription factor TFIIIB and that overexpression of p65 induces Pol III-dependent transcription. As a consequence of these effects, we show that inhibition of Pol III activity in macrophages restrains cytokine secretion and suppresses phagocytosis, two key functional characteristics of these cells. These findings therefore identify a radical new function for Pol III in the regulation of macrophage function which may be important for the immune responses associated with both normal and malignant cells.
INTRODUCTION
Chronic inflammation caused by microbial infection, autoimmune diseases, or other pathologies increases the risk of tumorigenesis. Failure to clear the infection during chronic inflammation is a major source of tissue damage. During this process, reactive oxygen species lead to DNA damage and mutation (1). Furthermore, to sustain tissue homeostasis, cells continuously proliferate, and this can be a major driving force for the initial transformation of tumor cells (2). Tumor-infiltrating immune cells produce cytokines that activate transcription factors (e.g., nuclear factor κB [NF-κB], STAT3, or AP-1) in premalignant cells to control numerous protumorigenic processes, including survival, proliferation, angiogenesis, and invasion (3).
Macrophages are professional phagocytic cells of the innate immune system. They are a major line of host defense, being responsible for pathogen killing and for triggering inflammation. In addition, macrophages are also responsible for maintaining tissue homeostasis and repair, mainly through extracellular matrix remodeling and scavenging apoptotic cells and cellular debris (4).
Microenvironmental cues can polarize macrophages to exhibit either proinflammatory (M1, classically activated macrophages) or anti-inflammatory (M2, alternatively activated macrophages) phenotypes. Classically activated macrophages secrete a large variety of factors, including interleukins, chemokines, interferons, reactive oxygen species, and complement components (5).
Growing evidence indicates that macrophages present in tumors (called tumor-associated macrophages [TAMs]), instead of being effective in host defense, actually contribute to cancer progression by stimulating cancer cell proliferation, angiogenesis, metastasis, and suppression of adaptive immunity. Given the important role of macrophages in tumor promotion, TAMs are considered a potential target for anticancer therapy (6).
NF-κB is a key transcription factor mediating inflammatory signals and has also been suggested to have a role in tumor progression (7). There are five members of the NF-κB family of transcription factors: RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100). NF-κB may consist of many possible homo- and heterodimers; however, p50/RelA heterodimers are most commonly observed (8). In resting cells, NF-κB is bound (and kept in the cytoplasm) by IκB proteins (IκBα, IκBβ, and IκBε). Following an inflammatory stimulus, IκB is phosphorylated and undergoes proteasomal degradation, which leads to liberation of NF-κB and its translocation to the nucleus, where it activates target genes. The kinase responsible for the phosphorylation of IκB is the IκB kinase (IKK) complex, which consists of two kinases (IKKα and IKKβ) and a regulatory subunit, NEMO/IKKγ (reviewed in reference 9). NF-κB regulates the transcription of a wide variety of target genes, including inflammation-related genes (e.g., those encoding cytokines and chemokines, Cox-2, and those encoding tumor necrosis factor alpha [TNF-α]) (10). NF-κB is activated by a variety of inflammatory stimuli, such as interleukin 1 (IL-1) and TNF-α, and inhibited by IL-10 and nonsteroidal anti-inflammatory drugs (9, 11, 12). Moreover, aberrant NF-κB activity is frequently involved in diseases, and the function of RelA is altered in many cancers (13).
RNA polymerase III (Pol III) is responsible for the synthesis of many noncoding RNAs involved in protein biosynthesis and the maturation of other RNA molecules (14). In mammalian cells, Pol III transcription is directly activated by c-Myc and extracellular signal-regulated kinase (ERK) and repressed by RB, p53, and Maf1 (15, 16). Deregulation of Pol III transcription has been implicated in a variety of human diseases. In particular, elevated Pol III activity is a recurring feature of mouse and human tumors (14). Interestingly, NF-κB subunit RelA/p65 has been shown to associate with Pol III-transcribed genes in human lymphoblastoid cells (17). However, its role in Pol III transcriptional regulation has not yet been determined.
In this study, we show that stimulation of macrophages with the bacterial cell wall component lipopolysaccharides (LPS) upregulates transcription of tRNA genes. This effect occurs concomitantly with an increased association of p65 with tRNA genes, and we show that direct activation of the NF-κB pathway induces Pol III-dependent transcription. Ultimately and most importantly, we also show that inhibition of Pol III activity severely compromises macrophage function by delaying phagocytosis and restraining cytokine secretion, underscoring a novel and critical role for Pol III activity in the innate immune response in these cells.
MATERIALS AND METHODS
Cell culture.
Cells were cultured in a humidified incubator with 5% CO2 at 37°C. Murine leukemia virus transformed macrophages RAW264.7 and human U2OS cells (ATCC) were grown in DMEM supplemented with 2 mM l-glutamine, penicillin (100 U/ml), streptomycin (100 U/ml), and 10% fetal bovine serum (FBS), unless otherwise stated. THP-1 and murine bone marrow-derived macrophages were grown in RPMI 1640 supplemented with 2 mM l-glutamine, penicillin (100 U/ml), streptomycin (100 U/ml), and 10% FBS, unless otherwise stated. When indicated, cells were treated with LPS (Sigma) at 1 μg/ml, mouse recombinant macrophage colony-stimulating factor (M-CSF) (MACS Miltenyi Biotec) at 10 ng/ml, RNA polymerase III inhibitor ML-60218 (Merck) at 100 μM, phorbol-12-myristate-13-acetate (PMA) (Sigma) at 5 ng/ml, and CPI-203 (Cambridge Bioscience) at 10 μM.
THP-1 cell differentiation.
THP-1 monocytes were differentiated with PMA essentially as described previously (18). Briefly, one million cells were plated in 6-cm dishes in 3 ml medium containing 5 ng/ml PMA. Upon incubation of THP-1 monocytes with PMA, the cells become adherent to culture dishes. After 48 h, dishes were washed twice with phosphate-buffered saline (PBS) to remove nonadherent cells. Then, medium was replaced with fresh RPMI 1640 with supplements except PMA. After another 48 h, cells were used for experiments.
Bone marrow-derived macrophages (BMDM).
Wild-type male and female mice of a mixed genetic background were sacrificed at 3 to 6 months of age with CO2 and subsequent cervical dislocation. Bone marrow was flushed from femurs and tibias with RPMI 1640 medium using a 5-ml syringe and a 23-gauge needle. Cells were passed through a cell strainer, spun, resuspended in RPMI 1640 medium supplemented with 10 ng/ml M-CSF, and plated on 10-cm non-tissue culture sterile petri dishes. After 72 h, dishes were washed with PBS to remove nonadherent cells, and fresh RPMI 1640 medium supplemented with 10 ng/ml M-CSF was added. Forty-eight hours later, dishes were washed with PBS, and cells were flushed off the culture dish with 7 ml RPMI 1640 medium supplemented with 10 ng/ml M-CSF using a 10-ml syringe and a 23-gauge needle. Cells were counted and plated on 6-well (phagocytosis) or 12-well (enzyme-linked immunosorbent assay [ELISA] and RNA isolation) tissue culture plates. After another 24 h, cells were used for experiments.
Detection of de novo protein synthesis.
Levels of newly synthesized protein were estimated using a nonisotopic labeling method as described previously (19), with a Click-it HPG Alexa Fluor 488 protein synthesis assay kit (Life Technologies). Briefly, primary bone marrow-derived macrophages were grown for 30 min in RPMI 1640 medium without methionine (Life Technologies) and supplemented with 50 μM l-homopropargylglycine (HPG). Then, cells were washed with ice-cold PBS, harvested, and fixed with 50% methanol. The subsequent steps were performed according to the manufacturer's instructions. Labeled cells were subjected to flow cytometry on a FACSCalibur instrument (BD Biosciences) and analyzed with the Cell Quest Pro software. Data are represented as percentages of total fluorescent signal relative to the signal of the nontreated sample (NT), which was set to 100%.
Phagocytosis assay.
Fluorescent latex beads 2 μm in diameter (Life Technologies) were preopsonized by 10× dilution in macrophage medium (RPMI 1640 with all supplements) for 1 h at 4°C. Beads were added to macrophage culture plates in 10:1 ratio (beads to cells) and incubated for 3 h. Plates were then placed on ice, and macrophages were washed twice with ice-cold PBS and scraped into 0.25 ml PBS using a cell scraper. Cells were then subjected to flow cytometry. A control sample without fluorescent beads was used to assess the autofluorescence of cells.
Transfections.
U2OS cells were plated in 6-well plates at a density of 7.5 × 104 per well. Cells were transfected using Lipofectamine 2000 according to the manufacturer's recommendations. A 250-ng amount of each plasmid was used per well. pRSV, pRSV-p65, pRSV-p65(RHD), and pCMV-p50 were kindly provided by Neil Perkins (Institute for Cell and Molecular Biosciences, Newcastle University). For Brf1 knockdown, murine primary bone marrow-derived macrophages were transfected with 20 nM small interfering RNA (siRNA) oligonucleotides using Lipofectamine 2000. Transfections were done six times with 1-day intervals after every two transfections. The treatment experiments were performed on the day following the last transfection. The siRNAs used were Brf1 ON-TARGETplus SMARTpool (L-046566-01; Dharmacon) and AllStars negative-control siRNA (1027280; Qiagen).
Construction of stable RAW264 cell lines.
Stable RAW264 cell lines were generated by retroviral transduction. Ecotropic HEK293 packaging cells were transfected with retroviral pBabe-EMPTY vector, pBabe vector encoding a mutant version (A32/36) of IκBα, or pBabe vector encoding enhanced green fluorescent protein (EGFP)-tagged Brf1. RAW264 cells were infected three times using HEK293 conditioned medium and subsequently selected in medium containing puromycin at 2 μg/ml until there were no live cells on the control plate (usually 6 to 7 days). Pools of cells were used for experiments.
Protein extracts and Western blotting.
Cells were washed with ice-cold PBS and harvested by scraping directly into microextraction buffer (450 mM NaCl, 20 mM HEPES [pH 7.9], 0.2 mM EDTA, 25% glycerol, 0.5% Triton X-100). Extracts were sonicated for 30 s in Bioruptor (Diagenode) with maximum energy setting and spun for 10 min at 14,000 × g at 4°C. Supernatants were collected, and protein concentration was assessed using Bio-Rad protein assay. Proteins were precipitated from a volume of extract corresponding to 30 μg protein with an equal volume of 10% trichloroacetic acid (TCA). Samples were spun for 10 min at 14,000 × g at 4°C, and the pellet was washed with cold acetone, dried, and resuspended in Laemmli buffer (pH 8.8), followed by incubation for 15 min at 55°C with shaking. A 15-μg amount of protein was resolved on a 4-to-20% gradient gel (Bio-Rad), transferred to a nitrocellulose membrane, and incubated with antibodies in 5% milk in Tris-buffered saline–Tween (TBST). Antibodies to the following proteins were used: p65 (SC-392/SC-109; for Rel homology domain [RHD] Western blotting), p50 (SC-7178), IκBα (SC-847), actin (SC-1615), and c-Myc (SC-764; Santa Cruz Biotechnology); TBP (ab818), hemagglutinin (HA) tag (ab1424), and GFP tag (ab290; Abcam); and Brf1 (A301-228A; Bethyl Laboratories).
RNA isolation and cDNA synthesis.
Total RNA was isolated from cells using TRIzol (Life Technologies) according to the manufacturer's instructions. Fifty nanograms of RNA was used for cDNA synthesis using a QuantiTect reverse transcriptase kit (Qiagen). To increase the efficiency of tRNAs' cDNA synthesis, oligonucleotides specific to the 3′ end of tRNA were added to the reaction mixture, each at the final concentration 0.6 μM. The oligonucleotides sequences are listed in Table S1 in the supplemental material.
ChIP.
For each time point, one 15-cm culture dish of cells at around 80% percent confluence was used. Cell cross-linking was done by adding 37% formaldehyde to a final concentration of 1% for 8 min at room temperature, followed by the addition of glycine to a final concentration of 125 mM. Cells were lysed in 0.5 ml chromatin immunoprecipitation (ChIP) buffer (150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, 0.5% SDS, 1 mM sodium butyrate, 50 mM Tris-HCl [pH 8.0]) containing 1.5× protease and phosphatase inhibitor cocktail (Thermo Scientific). The lysate was sonicated 6 times for 15 s with 30-s intervals in a Diagenode Bioruptor set to maximum power. For each IP, 125 μl of extract was used, and 50 μl of extract was used as input. For IPs, extract was diluted five times with ChIP buffer with no SDS, and 3 μg of p65 (SC-372) antibody was added. Following overnight incubation at 4°C with rotation, 50 μl of protein G magnetic beads (Dynal Life Technologies) was added, and the mixtures were further incubated for 6 h. Beads were washed once with ChIP buffer (no SDS), twice with high-salt buffer (600 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, 50 mM Tris-HCl [pH 8.0]), twice with LiCl buffer (250 mM LiCl, 1 mM EDTA, 0.5% sodium deoxycholate, 0.5% NP-40, 10 mM Tris-HCl [pH 8.0]), one more time with ChIP buffer (no SDS), and once with final wash buffer (0.1% NP-40, 10 mM Tris-HCl [pH 7.5]). All the washes were performed on a rotating wheel at room temperature for 4 min. DNA recovery and purification were performed using Chelex 100 resin (Bio-Rad), as described previously (20).
Quantitative PCR.
Quantitative PCR was performed on a Bio-Rad CFX 96 unit using a 3-min incubation at 95°C, followed by 40 cycles of 20 s at 95°C, 30 s at 57°C, and 30 s at 72°C (with a plate read after each cycle). A melting curve analysis was performed for each sample after PCR amplification to ensure that a single product with the expected melting curve characteristics was obtained. Each sample was loaded in triplicate. Each plate contained cDNA dilutions for the standard curve, a non-reverse transcriptase control, and a nontemplate control. PCR efficiencies were between 90% and 100%. The primer sequences used are described in Table S2 in the supplemental material. Data were processed in Bio-Rad CFX Manager 3.0 and then analyzed in Excel (Microsoft). Data are expressed in arbitrary units calculated from standard curve where the highest cDNA concentration was set to 1.
Coimmunoprecipitation.
U2OS cells were transfected with EGFP-Brf1 and HA-p65 and harvested after 48 h. Total protein was isolated with extraction buffer (30 mM Tris-HCl [pH 8], 150 mM NaCl, 1% Triton X-100, 1% Igepal CA-630, 10% glycerol) and mechanical disruption by freezing and thawing cells three times. After insoluble debris had been spun down, extracts were diluted twice with dilution buffer (30 mM Tris-HCl [pH 8], 150 mM, 10% glycerol) to bring the concentration of Triton and Igepal to 0.5%. All buffers were supplemented with complete protease inhibitor cocktail (Roche). Protein concentration was estimated by Bradford assay. A suspension of Dynabeads (protein G magnetic beads) (50 μl; Invitrogen) was washed twice with 0.5 ml PBS containing 0.5% bovine serum albumin (BSA), and then 5 μl antibody against GFP (ab290; Abcam) and 1.25 μl of normal rabbit IgG (X0903; Dako) were added and incubated for 6 h. After that time, beads were washed with IP buffer (30 mM Tris-HCl [pH 8], 150 mM NaCl, 0.5% Triton X-100, 0.5% Igepal CA-630, 10% glycerol), and 250 μg of protein extract was added and incubated overnight with rotation at 4°C. For coimmunoprecipitation experiments using RAW264 cells stably expressing EGFP-Brf1, 750 μg of protein extract was incubated overnight with 5 μg of either antibody against HA tag (ab1424; Abcam) or GFP tag (A11120; Life Technologies). Then, 50 μl of a suspension of Dynabeads was added, and extracts were incubated for another 6 h. Following 3 washes with IP buffer, immunoprecipitated proteins were eluted by boiling for 3 min in the SDS gel electrophoresis loading buffer and analyzed by SDS-PAGE followed by Western blotting.
ELISA.
Supernatants from the macrophage treatment experiments were collected, the volume was measured, and cytokine concentration was estimated using inflammatory cytokine multianalyte ELISA or custom ELISA (IL-6, IL-10, and TNF-α) kits from Qiagen (MEM-004A and CMEM-0608A, respectively). Samples were treated as described in the manufacturer's instructions. Samples for TNF-α detection were diluted twice in macrophage RPMI 1640 medium. The optical density (OD) values were corrected for the volume of the medium harvested and total RNA content in the cells. To reduce the variation between replicates, the largest amount of cytokine within a replicate was set to 100%.
Statistics.
P values were calculated using a paired Student t test or a ratio Student t test in situations where data were presented as fold change. All the calculations were performed in Excel (Microsoft). If no statistical value is shown, there was no significant difference between data sets.
RESULTS
LPS stimulates protein synthesis and tRNA expression in macrophages.
LPS is a major component of the outer membranes of most Gram-negative bacteria and is a strong proinflammatory mediator, potently activating immune system cells (21). LPS treatment leads to activation of multiple signaling pathways, including NF-κB, interferon regulatory factor 3 (IRF3), and members of the mitogen-activated protein kinase (MAPK) family (ERK/c-Jun N-terminal kinase [JNK]/p38) (22). LPS triggers cell cycle arrest (23–25) and together with interferon gamma (IFN-γ) polarizes macrophages toward an M1 phenotype (classically activated macrophages). M1 macrophages secrete high levels of proinflammatory cytokines (such as IL-1, TNF-α, and IL-6) and exhibit antibacterial capacity (3).
It has been shown previously that the activation of macrophages is associated with dynamic changes in transcriptional and protein synthesis profiles (26). The authors of that study showed that almost five thousand different proteins are newly synthesized upon LPS treatment in RAW264 macrophages. Other studies reported a 20% increase in total protein content in murine primary bone marrow-derived macrophages after 27 h of treatment with LPS (27) and also de novo protein synthesis after 22 h of treatment with LPS, as measured by radioactive leucine incorporation (25). Because the reported experiments were performed with quite long LPS exposure times, we were interested to see whether the response is more rapid. To test this, we monitored incorporation of the amino acid analog l-homopropargylglycine (HPG) into newly synthesized proteins in primary murine bone marrow-derived macrophages treated with LPS for 1, 2, 3, and 5 h. As shown in Fig. 1, the LPS treatment induced a gradual increase of HPG incorporation. After 3 and 5 h of LPS treatment, the rate of protein synthesis was about 80% higher than the rate in nontreated control cells. This confirms the published observations and also shows that de novo protein synthesis is rapidly induced within hours of LPS treatment.
FIG 1.
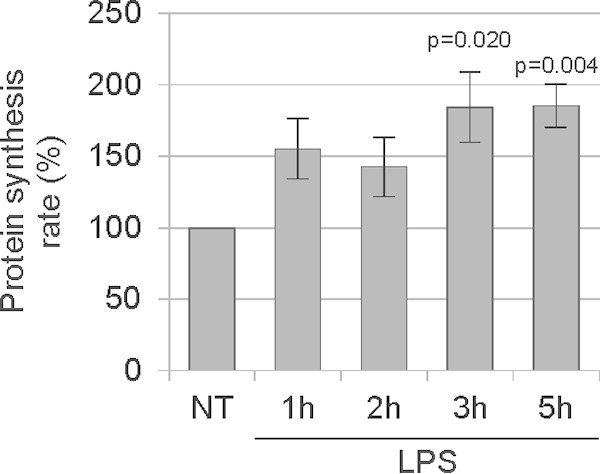
LPS treatment enhances de novo protein synthesis in macrophages. Mouse primary bone marrow-derived macrophages were treated with 1 μg/ml LPS for the indicated periods of time. The medium was replaced with methionine-free medium containing the methionine analogue HPG, and cells were incubated for 30 min. Cells were harvested and subjected to flow cytometry to assess HPG incorporation. The value for the NT sample was set to 100%. The experiment was performed on macrophages derived from three different mice (n = 3). Error bars represent standard errors of the means (SEM).
Since Pol III products, such as tRNAs, are crucial for translation and a rapid increase in protein synthesis is associated with LPS treatment, we sought to determine whether Pol III activity is also upregulated in response to LPS. To test this, RAW264 macrophages were treated with LPS for 1, 2, 3, 5, and 8 h. Cells were harvested and total RNA was isolated. Reverse transcription-quantitative PCR (RT-qPCR) analysis revealed that upon LPS treatment, tRNATyr and tRNAIle levels increase, reaching a peak at 5 h (Fig. 2A). The response is more pronounced if RAW264 macrophages are deprived of serum for 16 h before LPS addition (see Fig. S1 in the supplemental material). Similar effects were observed in primary murine bone marrow-derived macrophages and human acute monocytic leukemia THP-1 cells, which had been differentiated into macrophages (dTHP-1). RT-qPCR analysis revealed that tRNATyr and tRNAIle levels are upregulated in bone marrow-derived macrophages treated with LPS for 5 and 8 h (Fig. 2B) and in dTHP-1 cells treated with LPS for 6 h (Fig. 2C). These collective data show that tRNA expression is upregulated by LPS.
FIG 2.
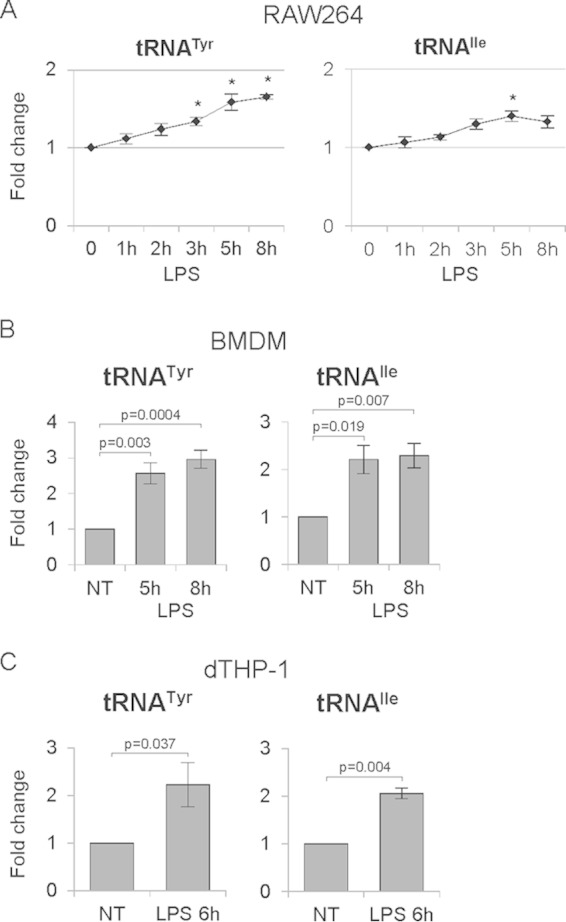
LPS upregulates RNA polymerase III activity. Mouse RAW264 macrophages (A), murine primary bone marrow-derived macrophages (BMDM) (B), and human THP-1 cells differentiated into macrophages (dTHP-1) (C) were treated with 1 μg/ml LPS and harvested at the indicated time points. RNA was isolated from cells and transcript levels of the indicated genes were assessed using RT-qPCR. RNA levels were normalized to GAPDH (A and B) or RPLP0 mRNA (C), and the value for the zero time point or the NT (nontreated) cells was set to 1. An asterisk indicates a P value of <0.05. Error bars represent SEM. The experiments were performed three times each in duplicate (A), five times (B), and four times (C).
NF-κB is involved in the regulation of Pol III activity by LPS.
Because of its key role in responses to inflammatory stimuli, we investigated whether NF-κB is involved in regulation of Pol III activity upon LPS treatment. To test this, we took advantage of the fact that inhibition of the IKK kinase complex blocks activation of NF-κB by preventing its nuclear localization (9). RAW264 macrophages and THP-1 cells differentiated into macrophages were therefore pretreated for 1 h with IKK inhibitor PS1145 or dimethyl sulfoxide (DMSO) as a control and then treated with LPS for 5 or 6 h, respectively. Cells were harvested, and total RNA was isolated. As a marker of NF-κB activity, we examined IκBα (NFKBIA) mRNA levels, which are highly responsive to changes in NF-κB activity (28, 29). As shown in Fig. 3A and B, IKK inhibitor PS1145 reduced the upregulation of IκBα mRNA after LPS treatment in both RAW264 and dTHP-1 cells, proving the effectiveness of the IKK inhibitor. Similarly, after LPS treatment the Pol III product tRNATyr was upregulated in DMSO-treated RAW264 and dTHP-1 cells (Fig. 3A and B). However, IKK inhibition significantly reduced this LPS-induced upregulation of tRNATyr.
FIG 3.
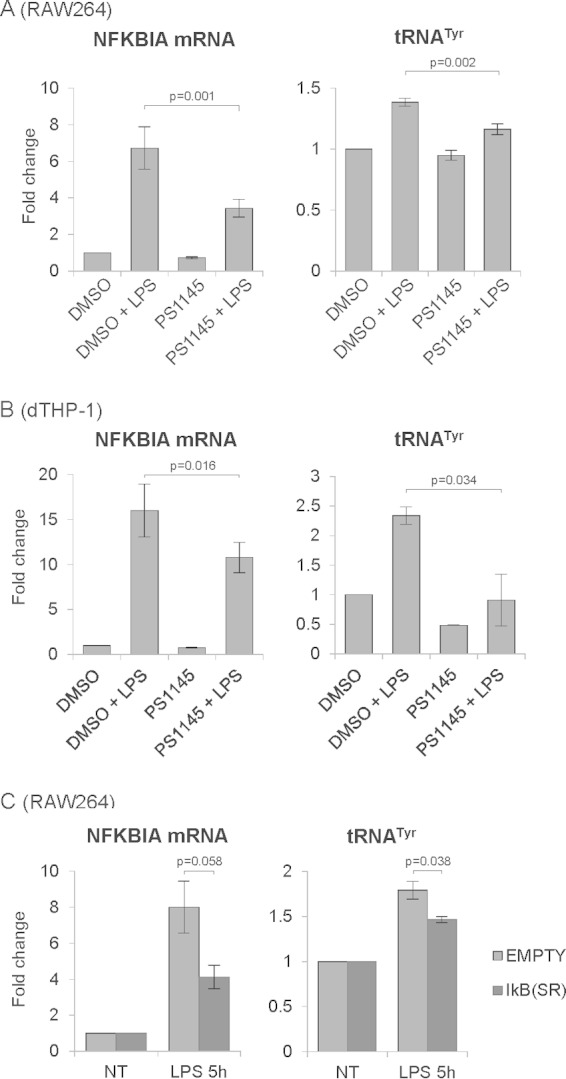
Inhibition of the NF-κB pathway partially precludes Pol III activation upon LPS treatment. Mouse RAW264 macrophages (A) and human THP-1 cells differentiated into macrophages (dTHP-1) (B) were pretreated for 1 h with 10 μM IKK inhibitor PS1145 or with DMSO (0.2%) as a control and then treated with 1 μg/ml LPS for 5 (RAW264) or 6 (dTHP-1) hours. (C) RAW264 control cells or cells stably expressing the superrepressor IkB were treated with 1 μg/ml LPS for 5 h. RNA was isolated from cells, and transcript levels of the indicated genes were assessed using RT-qPCR. RNA levels were normalized to GAPDH mRNA (A and C) or RPLP0 mRNA (B), and the value for DMSO-treated or NT (nontreated) cells was set to 1. Error bars represent SEM (n = 4 [A and C] or n = 3 [B]).
To further assess the role of NF-κB in Pol III regulation, we constructed a RAW264 cell line stably expressing a mutant form of NF-κB repressor IκBα. This phosphorylation-defective IκBα (S32A/S36A), called a superrepressor, is resistant to degradation and thus continuously sequesters NF-κB in the cytoplasm, preventing its activation (30, 31). As shown in Fig. 3C, expression of the superrepressor IκBα partially prevented the activation of its own gene (NFKBIA) upon LPS treatment. Superrepressor IκBα also significantly reduced LPS-induced upregulation of tRNATyr (Fig. 3C). Moreover, the same effect was observed in serum-deprived RAW264 macrophages (see Fig. S2 in the supplemental material). Overall, these data show that the NF-κB pathway is involved in regulation of Pol III activity in response to LPS.
p65 binds to tRNA genes.
The NF-κB family of transcription factors consists of five proteins, namely, RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100) (13). Kasowski and colleagues performed chromatin precipitation followed by deep sequencing to look at genome-wide binding of p65 protein in human lymphoblastoid cells (17). We took advantage of these data and manually inspected the presence of p65 on tRNA genes using the ENCODE transcription factor ChIP track in the UCSC genome browser (http://genome.ucsc.edu) (32, 33). This revealed that p65 is present at many Pol III-transcribed genes, including tRNAs, 7SL RNA, 7SK RNA, and RMRP (RNase MRP) genes. As shown in Fig. S3 in the supplemental material, p65 binding peaks overlap tRNA genes and Pol III peaks. The ChIP sequencing (ChIP-seq) data revealed that in GM12878 lymphoblastoid cells, p65 is present at 151 out of a total 631 tRNA genes in the hg18 human genome compilation (see Fig. S4 in the supplemental material).
We also looked at overlap between p65 binding and Pol III binding in three cell lines from which four sets of ChIP-seq data were available for Pol III, that is, the GM12878, K562 (two antibodies against different subunits, RPC155 and RPC32) and HeLa-S3 cell lines (see Fig. S4 in the supplemental material). It should be clearly noted here that none of those data are straightforwardly comparable, since p65 data are from cells treated with TNF-α and Pol III data are from untreated cells. However, it is noteworthy that in all four data sets, p65 binding overlaps Pol III binding. This suggests that p65 may preferentially bind to tRNA genes that are already occupied by Pol III. A minority of p65-positive, Pol III-negative tRNA genes also appear in this comparison, but this should be viewed with caution, as the actual Pol III binding is likely to be different upon TNF-α treatment.
Given our finding that Pol III targets are upregulated upon LPS treatment in an IKK-dependent manner, we sought to determine whether LPS treatment induces p65 binding to tRNA genes in mouse macrophages. To test this, RAW264 cells were treated with LPS for 1, 2, and 3 h prior to analysis by ChIP assay. Under these conditions, p65 protein showed time-dependent binding to the promoter of its known target gene NFKBIA and also to tRNATyr and tRNAIle genes (Fig. 4). In all cases, the binding peak occurred after 1 h of LPS treatment. The p65 binding efficiency to tRNA genes was much lower than its binding to the NFKBIA promoter (∼1% versus ∼7% IP/input, respectively). This is probably because p65 binds directly to its DNA motif in the NFKBIA promoter, whereas p65 may be recruited to tRNA genes by protein/protein interactions, rather than binding DNA directly (see below).
FIG 4.
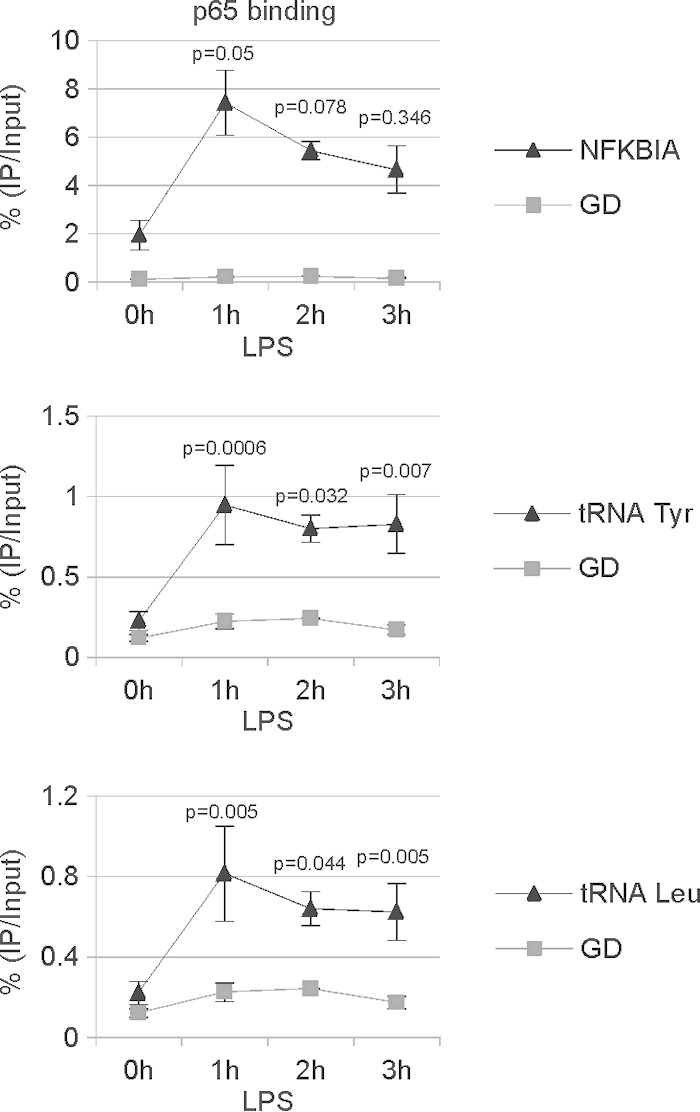
p65 binds tRNA genes upon LPS treatment. Mouse RAW264 macrophages were treated with 1 μg/ml LPS and harvested at the indicated time points. p65 binding to the NFKBIA promoter, the gene desert (GD), and tRNA genes was assessed by ChIP assay followed by RT-qPCR. Error bars represent SEM (n = 3).
The response is not limited to LPS stimulus, as we could see similar effects in RAW264 macrophages treated with TNF-α (see Fig. S5A in the supplemental material). Moreover, TNF-α upregulated tRNA gene transcription in murine primary bone marrow-derived macrophages (see Fig. S5B in the supplemental material). Overall, these data suggest a direct role of p65 in regulating tRNA expression.
Overexpression of p65 activates several Pol III-transcribed genes.
To further investigate the role of NF-κB in Pol III activation, we overexpressed its p65 and p50 subunits. U2OS cells were transfected with plasmids encoding p65 or p50 or empty vectors as controls. At 24 h posttransfection, cells were serum deprived (0.2% FBS) for another 24 h and then harvested. Western blotting revealed that overexpression of p65 led to increased levels of the known NF-κB targets (28, 34, 35) IκBα and p50 (Fig. 5A), whereas overexpression of p50 had no effect on IκBα levels. Importantly, we also found that overexpression of p65 also increased the levels of tRNATyr, tRNAThr, tRNAIle, and 7SL RNA (Fig. 5B). Overexpression of p50 had no effect on IκBα, p65, or Pol III activity, suggesting that p65 is a limiting factor and p50 may be in relative excess in these cells.
FIG 5.

Overexpression of NF-κB subunit p65 increases tRNA expression. U2OS cells were transfected with empty vectors and/or vectors encoding p65 or p50, as indicated. After 24 h, cells were serum deprived (0.2% FBS), cultured for another 24 h, and harvested. (A) Western blot analysis of p65, p50, IκBα, and actin. (B) RT-qPCR analysis of indicated Pol III transcripts (n = 3). (C and D) Transactivation domain of p65 is necessary for upregulation of Pol III activity. U2OS cells were transfected with empty vector or vector encoding full-length p65 or truncated version of p65 containing only the Rel homology domain (RHD). After 24 h, cells were serum deprived (0.2% FBS), cultured for another 24 h, and harvested. (C) Western blot analysis of p65 and tubulin. (D) RT-qPCR analysis to monitor levels of the indicated Pol III transcripts (n = 2). RNA levels were normalized to RPLP0 mRNA, and the value for empty vector samples was set to 1. Error bars represent SEM.
p65 belongs to the family of Rel proteins, characterized by the presence of a Rel homology domain (RHD). Additionally, p65, c-Rel, and RelB proteins also contain transactivation domains (TAD), which are required for activation of NF-κB target genes (7). We were interested to know whether this TAD is also required for activation of Pol III-transcribed genes. To test this, U2OS cells were transfected with either empty plasmid or plasmids encoding full-length p65 or a truncated version of p65, containing only the Rel homology domain. At 24 h posttransfection, cells were serum deprived (0.2% FBS) for another 24 h and then harvested. As shown by Western blotting (Fig. 5C), we were able to overexpress both full-length p65 and the truncated version containing only the RHD. Although overexpression of p65 increased tRNA and 7SL RNA levels, the truncated version did not, despite its robust expression (Fig. 5D). These results show that the TAD required for activation of Pol II-transcribed p65 target genes is also necessary for stimulating Pol III activity.
It has been shown that c-myc is an NF-κB target gene (36), and we consistently observed increased levels of c-Myc in cells overexpressing p65 (see Fig. S6A in the supplemental material). Moreover, c-Myc is known to positively regulate Pol III activity (37). It was possible, therefore, that the effect of p65 overexpression on Pol III activity is indirect and mediated by c-Myc. To examine this possibility, we transfected U2OS cell with either a plasmid encoding p65 or empty vector as a control. At 24 h posttransfection, cells were serum deprived (0.2% FBS), cultured for 24 h, and harvested. At 6 h before harvesting, cells were treated with 10 μM CPI-203, a potent bromodomain-containing protein (BET) inhibitor which inhibits c-myc transcription (38). As expected, inhibitor treatment decreased both c-myc transcript and protein levels (see Fig. S6A and B in the supplemental material). Although c-myc induction was blocked, the Pol III products tRNATyr and 7SL RNA were still induced by p65 (see Fig. S6B in the supplemental material). This indicates that c-Myc is not required for p65-dependent upregulation of Pol III activity, and these data further support the direct role of p65 in Pol III activation.
p65 associates with TFIIIB.
Transcription of tRNA genes requires transcription factor TFIIIB, comprising subunits TBP, Bdp1, and Brf1 (39). Brf1 protein belongs to the family of TFIIB-related transcription factors, which are characterized by the presence of a zinc ribbon domain and two internal cyclin repeats (39). TFIIB and TBP have independently been shown to interact with p65. Moreover, the transactivation domain of p65 and one of the cyclin repeat domains of TFIIB were crucial for this interaction (40, 41). Since Brf1 is homologous to TFIIB, we asked whether p65 is able to associate with Brf1. In order to test this, U2OS cells were cotransfected with plasmids encoding EGFP-tagged Brf1 and HA-tagged p65. Protein extracts were then prepared, coimmunoprecipitated, and analyzed by Western blotting. This revealed that Brf1 was associated with TBP protein, a known Brf1 binding partner. Furthermore, we also found that p65 protein coimmunoprecipitated with Brf1 (Fig. 6A).
FIG 6.

p65 interacts with TFIIIB. (A) U2OS cells were transfected with EGFP-Brf1 and HA-p65 and harvested after 48 h. (B) RAW264 cells stably expressing EGFP-tagged Brf1were treated with 1 μg/ml LPS for the indicated periods of time. Cellular extracts were incubated with magnetic beads coated with rabbit anti-GFP antibody or normal rabbit IgG as a control (A) or with mouse anti-GFP or mouse anti-HA antibody as a control (B). After extensive washes, immunoprecipitated proteins were eluted and analyzed by Western blotting with antibodies against GFP, HA tag, and TBP (A) or Brf1, p65, and TBP (B).
To assess whether endogenous p65 in macrophages is able to interact with TFIIIB, we generated a RAW264 cell line stably expressing EGFP-tagged Brf1. The cells were treated with LPS for 1 and 2 h, and protein extracts were prepared, coimmunoprecipitated, and analyzed by Western blotting. As shown in Fig. 6B, we could again see the association of TBP with Brf1 in both treated and untreated cells. Furthermore, we could also see that LPS induced association of p65 with Brf1. These data indicate that p65 can associate with TFIIIB through interactions with its components Brf1 and/or TBP.
Pol III inhibition restrains cytokine secretion by primary macrophages.
Macrophages are one of the principal cell types involved in innate immune responses. Upon treatment with LPS, macrophages secrete a large variety of cytokines, including TNF-α, IL-6, and IL-10 (42), which modulate the immune response. We were therefore interested to know whether inhibition of Pol III affects cytokine secretion by macrophages. To test this, bone marrow-derived macrophages were pretreated with DMSO or the Pol III inhibitor ML-60218 for 6 h. Macrophages were then treated with 1 μg/ml LPS for 5 h and 8 h. Cells and culture medium were collected for RNA extraction and ELISA analysis, respectively. As shown in Fig. 7A, RT-qPCR analysis revealed that after 5 or 8 h of LPS treatment, tRNATyr and tRNAIle levels were upregulated, and this response was diminished in the cells treated Pol III inhibitor. Furthermore, ELISA analysis revealed that TNF-α, IL-6, and IL-10 were secreted into the medium upon LPS treatment (Fig. 7B). Reducing Pol III activity significantly decreased levels of secreted IL-6 and IL-10 but had a mild or no effect on TNF-α. As shown in Fig. 7C, TNF-α, IL-6, and IL-10 mRNA levels were upregulated upon LPS treatment, which is consistent with the previously published data (43–45). Moreover, after 8 h of LPS treatment, all three transcript levels were lower than those in corresponding samples at 5 h of treatment, which suggests that at the time points tested, the cellular response to LPS was decreasing. Pol III inhibition had no effect on TNF-α mRNA levels, whereas IL-6 mRNA levels were modestly elevated (Fig. 7C). The effect of Pol III inhibition on secretion of these cytokines can therefore not be explained by changes in their mRNA expression. In contrast, IL-10 mRNA expression showed a markedly different pattern: Pol III inhibition resulted in lower IL-10 mRNA levels than in DMSO-treated cells at both time points tested (Fig. 7C). The changes in IL-10 mRNA expression most likely reflect indirect effects of Pol III inhibition.
FIG 7.

Inhibition of RNA polymerase III activity restrains cytokine secretion by mouse bone marrow-derived macrophages. Bone marrow-derived macrophages were pretreated with DMSO (0.2%) or 100 μM RNA polymerase III inhibitor ML-60218 for 6 h, and then cells were stimulated with 1 μg/ml LPS for the indicated times. Cells and culture medium were harvested for RNA extraction and ELISA analysis, respectively. (A) RT-qPCR measurements of the levels of indicated tRNAs. RNA levels were normalized to GAPDH mRNA, and the value for the nontreated (NT) sample was set to 1. (B) Levels of the indicated interleukins secreted into the culture medium, as determined by ELISA. Results are expressed as corrected OD values as described in Materials and Methods. (C) RT-qPCR measurements of mRNA levels of the indicated interleukins, shown as fold change relative to GAPDH mRNA levels. Error bars represent SEM (n = 3).
To further assess the impact of abolishing Pol III activity on cytokine secretion, we knocked down Brf1 in bone marrow-derived macrophages using RNA interference. The effectiveness of Brf1 depletion is shown by the Western blots in Fig. 8A. Macrophages were treated with 1 μg/ml LPS for 2.5 h or 5 h. As shown in Fig. 8B, Brf1 knockdown efficiently abolished LPS-induced upregulation of tRNATyr and tRNAIle. ELISA analysis revealed that, similar to the ML-60218 inhibitor, Brf1 knockdown significantly decreased levels of secreted IL-6 and IL-10 but had no or a modest effect on TNF-α (Fig. 8C). Abolishing Pol III activity by Brf1 knockdown had a minor or no effect on TNF-α and IL-6 mRNA levels, respectively. IL-10 mRNA levels were consistently lower than those in control cells after LPS treatment, although after 5 h of treatment, the values did not reach statistical significance (Fig. 8D). Altogether, these data suggest that Pol III activity is required for efficient cytokine secretion by macrophages. Moreover, given the similarities between the effects of Brf1 knockdown and the inhibitor ML-60218, the results also confirm the specificity of the latter.
FIG 8.

Brf1 knockdown reduces cytokine secretion by mouse bone marrow-derived macrophages. Bone marrow-derived macrophages were transfected with a siRNA pool targeting Brf1 or nontargeting negative (Neg) siRNA. Cells were stimulated with 1 μg/ml LPS for the indicated time. Cells and culture medium were harvested for protein and RNA extraction and ELISA analysis. (A) Western blot showing Brf1 knockdown efficiency. (B) RT-qPCR measurements of the levels of indicated tRNAs. RNA levels were normalized to GAPDH mRNA, and the value for the nontreated (NT) Neg siRNA sample was set to 1. (C) Levels of the indicated interleukins secreted to the culture medium, as determined by ELISA. Results are expressed as corrected OD values as described in Materials and Methods. (D) RT-qPCR measurements of mRNA levels of the indicated interleukins, shown as fold change relaive to GAPDH mRNA. Error bars represent SEM (n = 3).
Pol III inhibition impairs phagocytosis by macrophages.
Phagocytosis is one of the primary functions of macrophages (4). Because Pol III inhibition affects cytokine secretion, another important macrophage function, we were interested to know if Pol III activity is also required for phagocytosis.
In order to test this, first we used THP-1 cells differentiated into macrophages (dTHP-1), which were treated with DMSO (0.2%) or the Pol III inhibitor ML-60218 (100 μM) for 4 h. The efficacy of the inhibitor was confirmed by RT-qPCR, which revealed a 2-fold decrease in tRNATyr and 4-fold decrease in tRNAIle expression after normalization to RPLP0 mRNA (see Fig. S7 in the supplemental material). Serum-opsonized fluorescent beads were then added, and after 3 h, cells were harvested and analyzed by flow cytometry.
Phagocytic capacity was assessed by estimating the total number of cells that engulfed at least one bead, by measuring total mean fluorescence of engulfed beads, and also by estimating the number of cells that engulfed one, two, three, four or more beads. As shown in Fig. 9A, approximately 70% of both nontreated and DMSO-treated dTHP-1 cells engulfed at least one bead. However, upon Pol III inhibitor treatment, phagocytosis was markedly reduced, with only 40% of cells having engulfed at least one bead. Moreover, we could also see a difference in the number of engulfed beads, as measured by the total fluorescent signal generated by those cells. As shown in Fig. 9B, the total fluorescence signal was reduced by 40% upon Pol III inhibition. These results suggested that phagocytosis is generally impaired in cells treated with Pol III inhibitor. The biggest difference was in the population of cells that engulfed four or more beads (Fig. 9C). The lower the number of engulfed beads, the smaller was the difference between Pol III inhibitor-treated and control cells, suggesting that the former cells are still able to engulf beads, although at a lower rate. Thus, partial Pol III inhibition restricts phagocytosis but does not block it completely.
FIG 9.
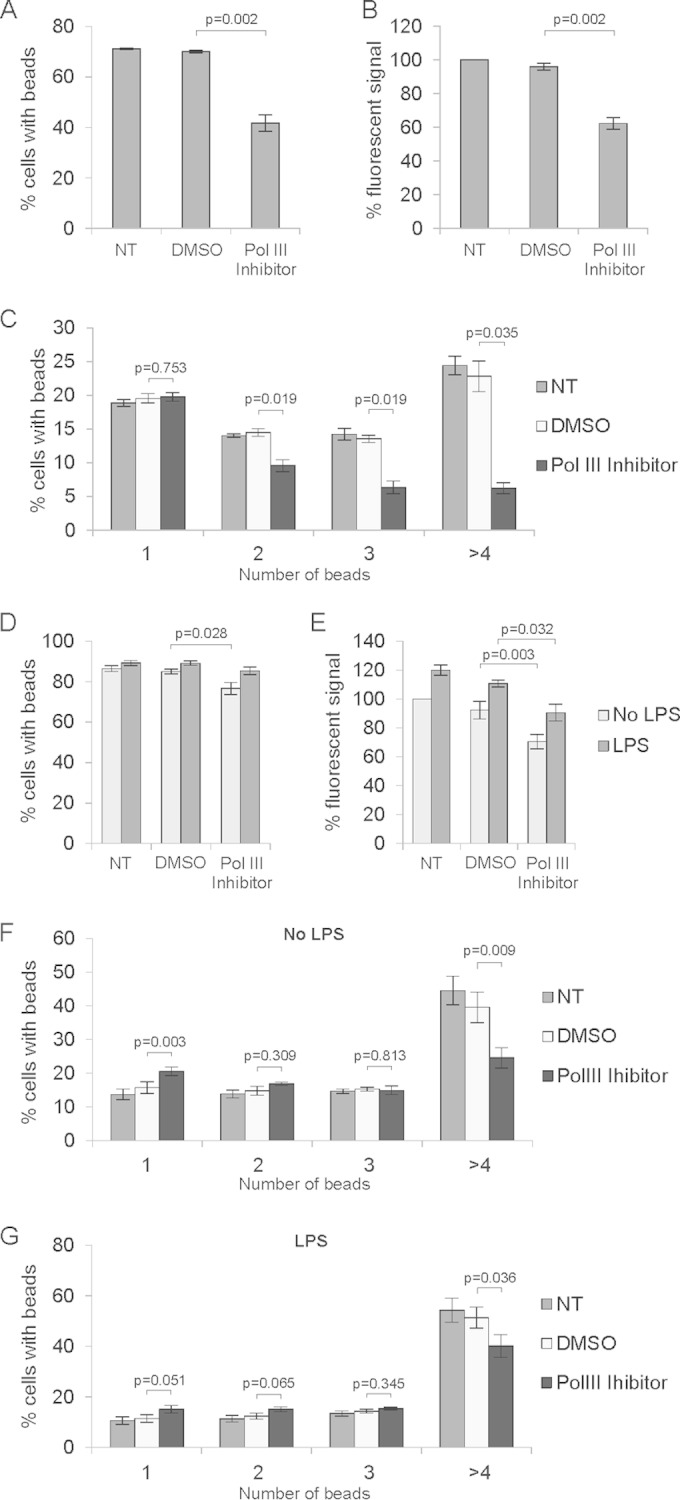
Inhibition of RNA polymerase III activity decreases phagocytosis by macrophages. PMA-differentiated THP-1 cells (n = 3) (A to C) and primary bone marrow-derived macrophages (n = 4) (D to G) were treated for 4 h with DMSO (0.2%) or 100 μM RNA polymerase III inhibitor ML-60218, as indicated, and then FBS-opsonized fluorescent latex beads were added. BMDMs were additionally treated with 1 μg/ml LPS 3 h or left untreated before addition of beads. After 3 h, cells were harvested and analyzed by flow cytometry. (A and D) Percentages of cells that engulfed at least one bead. (B and E) Percentage of total mean fluorescent signal of engulfed beads. (C, F, and G) Percentages of cells that engulfed the indicated numbers of beads. Error bars represent SEM.
We also performed the same experiments using primary murine bone marrow-derived macrophages (BMDM). As LPS treatment stimulates phagocytosis in primary macrophages (27), we tested whether Pol III inhibition affects LPS-induced phagocytosis.
BMDM were treated with DMSO (0.2%) or the Pol III inhibitor ML-60218 for 4 h. One hour later, cells were also treated with 1 μg/ml LPS. Serum-opsonized fluorescent beads were then added, and after 3 h, cells were harvested and analyzed by flow cytometry. Similar to dTHP-1 cells, BMDM showed a reduced capacity to engulf opsonized beads upon Pol III inhibition (Fig. 9D to F). There were 10% fewer cells that had engulfed at least one bead (Fig. 9D) and a 20% reduction in the total fluorescent signal generated by engulfed beads (Fig. 9E) compared to DMSO-treated cells. As for dTHP-1 cells, the greatest effect of the Pol III inhibitor was on the number of cells engulfing four or more beads. Moreover, as reported previously (27), LPS induced the rate of phagocytosis in macrophages (Fig. 9E to G). We did not see much difference in the number of cells that engulfed at least one bead compared to non-LPS samples, which may be explained by the fact that the engulfment rate was already high, reaching 90% of all cells. On the other hand, we did see an almost 20% increase in the total fluorescent signal, suggesting that LPS indeed increased the number of engulfed beads. Importantly, this increase was partially blocked by the inhibition of Pol III (Fig. 9E). These results show that Pol III activity is important for efficient phagocytosis in dTHP-1 cells and primary bone marrow-derived macrophages.
DISCUSSION
The study presented here demonstrates that activation of innate immune responses in mouse macrophages causes upregulation of transcription by RNA polymerase III. We show that the NF-κB pathway is involved in this process, since its inhibition precludes Pol III upregulation. Furthermore, overexpression of the NF-κB subunit p65 activates Pol III transcription. In macrophages treated with LPS, p65 binds to tRNA genes, which may allow direct interaction with the Pol III transcription apparatus. We also provide evidence that inhibition of RNA polymerase III activity abolishes the major macrophage functions of phagocytosis and cytokine secretion.
Macrophages have many biological functions, such as antigen presentation, target cell cytotoxicity, phagocytosis, tissue remodeling, and regulation of inflammation. They play an essential role in frontline defense, leading to the killing of microorganisms, parasites, and tumor cells (reviewed in reference 4). Activated macrophages (e.g., by LPS or IFN-γ) secrete a large amount of products, including enzymes, complement components, reactive oxygen intermediates, coagulation factors, cytokines, nitrite, and various other substances (46). All these compounds contribute to the microbicidal, cytotoxic (tumoricidal), or fungicidal potential of macrophages (47). Such a vast repertoire of products synthesized by macrophages requires efficient protein synthesis, which in turn tightly depends on RNA polymerase III products such as tRNAs. Indeed, our data show that concomitantly with increased de novo protein synthesis, tRNAs are upregulated during activation of macrophages by LPS.
The decreased cytokine secretion after Pol III inhibition could be partially explained by the reduced efficiency of the translation machinery. However, since secretion of TNF-α was only modestly affected by inhibition of Pol III activity, it is unlikely that the effect is the result of a general defect in protein synthesis. Pol III may influence some later stage of signal propagation that is initiated by TNF-α or IL-6 secreted into the medium. IL-10 gene transcription is primarily regulated by NF-κB (48), but it has also been shown that expression of this cytokine is regulated by IL-6 in a STAT3-dependent manner (49). It is possible therefore that a defect in IL-6 secretion abolishes IL-10 synthesis at the transcription level. Since we have shown that IL-6 mRNA levels are not changed upon Pol III inhibition, regulation appears to be posttranscriptional. Secretion is probably not impeded, as IL-6 and TNF-α use the same secretory pathway (50). However, previous studies have shown that IL-6 levels can be regulated posttranscriptionally by 3′ untranslated region (UTR)-driven mRNA decay (51). Additional control at the translational level has not been excluded.
The most striking effect of Pol III inhibition in macrophages is the reduced phagocytosis. It is widely appreciated that phagocytosis is a complex process which involves multiple stages, such as particle recognition, internalization, maturation and transport of phagosomes, and fusion of the phagosomes with lysosomes (52, 53). Each of these steps is highly regulated, thus creating the possibility of multiple points being affected by Pol III inhibition. Elucidation of the role of Pol III in phagocytosis is undoubtedly interesting but is beyond the scope of this work and requires extensive further investigation.
Regardless of the mechanisms underlying the observed phenotypes—restriction of cytokine secretion and phagocytosis—it is clear that Pol III inhibition has a great impact on these important functions of macrophages. Partial suppression of Pol III may therefore be worth consideration as an additional treatment in diseases such as sepsis or macrophage activation syndrome, where macrophages play an important role.
To understand how LPS regulate Pol III-dependent transcription, we considered downstream signaling. LPS engage the TLR4 receptor and induces various signaling pathways, including those involving MAPK, IRF3, and NF-κB (22). NF-κB is a key transcription factor implicated in inflammatory responses but has not previously been connected to Pol III. Besides its role in the transcription of genes encoding inflammatory cytokines, NF-κB also regulates gene expression related to cell survival, differentiation, and proliferation. The most extensively studied and most abundant form of NF-κB is a heterodimer consisting of p50 and p65. We therefore focused our attention on the NF-κB pathway, in particular on p65. We found that inhibition of the NF-κB pathway, using either IKK inhibitor or the degradation-resistant superrepressor IkBα, partially blocked Pol III induction upon LPS treatment and that overexpression of p65 protein increased Pol III-dependent transcription. Moreover, we found that p65 associates with TFIIIB and binds to tRNA genes upon LPS treatment. It seems also that p65 binding to tRNA genes is induced not only by LPS but also by another potent NF-κB activator, TNF-α. Furthermore, we show that the TAD of p65 is required for Pol III activation. This is reminiscent of c-Myc, which directly binds to TFIIIB and also requires its TAD to activate Pol III-dependent transcription (37). These data strongly support the idea of direct involvement of p65 in regulation of Pol III activity. However, since blocking the NF-κB pathway caused only partial reduction of LPS-induced tRNA upregulation, the involvement of other signaling pathways and/or transcription factors seems likely. Indeed, activation of MAPK signaling has also been shown to stimulate Pol III transcription directly (54).
Although ChIP-Seq data sets from different sources are not reliably comparable, the emerging picture is that p65 associates with tRNA genes which are already prebound by the Pol III machinery. Thus, it is unlikely that NF-κB serves as a pioneer transcription factor, and its role may rather be related to adjusting Pol III activity.
It has been shown that infection of macrophages with the parasitic protozoan Leishmania leads to decreased expression of certain Pol III products, including tRNA and 7SL RNA (55, 56). Downregulation of 7SL RNA suppresses vesicular trafficking and makes macrophages more susceptible to parasitism. Conversely, overexpressing 7SL RNA in macrophages protected them against Leishmania infection (55). This is consistent with our finding that Pol III activity contributes to immune cell function.
Taken together, our results support the notion that Pol III activity influences macrophage function. Given that macrophages play a crucial role in immunity, our data provide a link between Pol III-dependent transcription and the innate immune response. In addition, our study provides novel evidence that the transcription factor NF-κB is involved in the regulation of Pol III-dependent transcription, in addition to its well-established role in Pol II-dependent transcription, adding further complexity to the mechanisms which regulate RNA polymerase III.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Kirsteen Campbell, Joanna Birch, Louise Mitchell, and members of the Tumor Cell Death Laboratory for critical reading of the manuscript. We are also grateful to Neil Perkins for providing p65 constructs.
This work was supported by the Cancer Research UK core grant to the Beatson Institute (C596/A17196) and an EMBO long-term fellowship (ALTF 1435-2011), which was awarded to D.G.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00990-14.
REFERENCES
- 1.Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 3.Dale DC, Boxer L, Liles WC. 2008. The phagocytes: neutrophils and monocytes. Blood 112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 4.Verschoor CP, Puchta A, Bowdish DM. 2012. The macrophage. Methods Mol Biol 844:139–156. doi: 10.1007/978-1-61779-527-5_10. [DOI] [PubMed] [Google Scholar]
- 5.Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solinas G, Germano G, Mantovani A, Allavena P. 2009. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 7.Perkins ND. 2012. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer 12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin AS., Jr 1996. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 9.Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Oeckinghaus A, Ghosh S. 2009. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fredriksson L, Herpers B, Benedetti G, Matadin Q, Puigvert JC, de Bont H, Dragovic S, Vermeulen NP, Commandeur JN, Danen E, de Graauw M, van de Water B. 2011. Diclofenac inhibits tumor necrosis factor-alpha-induced nuclear factor-kappaB activation causing synergistic hepatocyte apoptosis. Hepatology 53:2027–2041. doi: 10.1002/hep.24314. [DOI] [PubMed] [Google Scholar]
- 12.Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr. 1999. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem 274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 13.Perkins ND. 2000. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci 25:434–440. doi: 10.1016/S0968-0004(00)01617-0. [DOI] [PubMed] [Google Scholar]
- 14.White RJ. 2005. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol 6:69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 15.Felton-Edkins ZA, Kenneth NS, Brown TR, Daly NL, Gomez-Roman N, Grandori C, Eisenman RN, White RJ. 2003. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle 2:181–184. doi: 10.4161/cc.2.3.375. [DOI] [PubMed] [Google Scholar]
- 16.Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gerard M, Lefebvre O, White RJ. 2008. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J Mol Biol 378:481–491. doi: 10.1016/j.jmb.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 17.Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, Snyder M. 2010. Variation in transcription factor binding among humans. Science 328:232–235. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. 2007. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res 56:45–50. doi: 10.1007/s00011-007-6115-5. [DOI] [PubMed] [Google Scholar]
- 19.Tsai YC, Greco TM, Cristea IM. 2014. Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol Cell Proteomics 13:73–83. doi: 10.1074/mcp.M113.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 21.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F, et al. . 1994. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J 8:217–225. [DOI] [PubMed] [Google Scholar]
- 22.Lu YC, Yeh WC, Ohashi PS. 2008. LPS/TLR4 signal transduction pathway. Cytokine 42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Vadiveloo PK. 1999. Macrophages—proliferation, activation, and cell cycle proteins. J Leukoc Biol 66:579–582. [DOI] [PubMed] [Google Scholar]
- 24.Vairo G, Argyriou S, Knight KR, Hamilton JA. 1991. Inhibition of colony-stimulating factor-stimulated macrophage proliferation by tumor necrosis factor-alpha, IFN-gamma, and lipopolysaccharide is not due to a general loss of responsiveness to growth factor. J Immunol 146:3469–3477. [PubMed] [Google Scholar]
- 25.Vairo G, Royston AK, Hamilton JA. 1992. Biochemical events accompanying macrophage activation and the inhibition of colony-stimulating factor-1-induced macrophage proliferation by tumor necrosis factor-alpha, interferon-gamma, and lipopolysaccharide. J Cell Physiol 151:630–641. doi: 10.1002/jcp.1041510324. [DOI] [PubMed] [Google Scholar]
- 26.Eichelbaum K, Krijgsveld J. 2014. Rapid temporal dynamics of transcription, protein synthesis, and secretion during macrophage activation. Mol Cell Proteomics 13:792–810. doi: 10.1074/mcp.M113.030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper PH, Mayer P, Baggiolini M. 1984. Stimulation of phagocytosis in bone marrow-derived mouse macrophages by bacterial lipopolysaccharide: correlation with biochemical and functional parameters. J Immunol 133:913–922. [PubMed] [Google Scholar]
- 28.Sun SC, Ganchi PA, Ballard DW, Greene WC. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 29.Hao S, Baltimore D. 2009. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10:281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. 1996. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol 16:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu ZG, Hsu H, Goeddel DV, Karin M. 1996. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87:565–576. doi: 10.1016/S0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 32.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, Lee BT, Barber GP, Harte RA, Diekhans M, Long JC, Wilder SP, Zweig AS, Karolchik D, Kuhn RM, Haussler D, Kent WJ. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paya CV, Ten RM, Bessia C, Alcami J, Hay RT, Virelizier JL. 1992. NF-kappa B-dependent induction of the NF-kappa B p50 subunit gene promoter underlies self-perpetuation of human immunodeficiency virus transcription in monocytic cells. Proc Natl Acad Sci U S A 89:7826–7830. doi: 10.1073/pnas.89.16.7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ten RM, Paya CV, Israel N, Le Bail O, Mattei MG, Virelizier JL, Kourilsky P, Israel A. 1992. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappa B indicates that it participates in its own regulation. EMBO J 11:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duyao MP, Buckler AJ, Sonenshein GE. 1990. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc Natl Acad Sci U S A 87:4727–4731. doi: 10.1073/pnas.87.12.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomez-Roman N, Grandori C, Eisenman RN, White RJ. 2003. Direct activation of RNA polymerase III transcription by c-Myc. Nature 421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- 38.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ III, Singer DS. 2012. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A 109:6927–6932. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schramm L, Hernandez N. 2002. Recruitment of RNA polymerase III to its target promoters. Genes Dev 16:2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- 40.Schmitz ML, Stelzer G, Altmann H, Meisterernst M, Baeuerle PA. 1995. Interaction of the COOH-terminal transactivation domain of p65 NF-kappa B with TATA-binding protein, transcription factor IIB, and coactivators. J Biol Chem 270:7219–7226. doi: 10.1074/jbc.270.13.7219. [DOI] [PubMed] [Google Scholar]
- 41.Paal K, Baeuerle PA, Schmitz ML. 1997. Basal transcription factors TBP and TFIIB and the viral coactivator E1A 13S bind with distinct affinities and kinetics to the transactivation domain of NF-kappaB p65. Nucleic Acids Res 25:1050–1055. doi: 10.1093/nar/25.5.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweet MJ, Hume DA. 1996. Endotoxin signal transduction in macrophages. J Leukoc Biol 60:8–26. [DOI] [PubMed] [Google Scholar]
- 43.Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. 1990. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med 171:35–47. doi: 10.1084/jem.171.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Libermann TA, Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10:2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu LG, Shu HB. 2002. TNFR-associated factor-3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kappa B activation and IL-10 production. J Immunol 169:6883–6889. doi: 10.4049/jimmunol.169.12.6883. [DOI] [PubMed] [Google Scholar]
- 46.Mosser DM. 2003. The many faces of macrophage activation. J Leukoc Biol 73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 47.Adams DO, Hamilton TA. 1984. The cell biology of macrophage activation. Annu Rev Immunol 2:283–318. doi: 10.1146/annurev.iy.02.040184.001435. [DOI] [PubMed] [Google Scholar]
- 48.Saraiva M, Christensen JR, Tsytsykova AV, Goldfeld AE, Ley SC, Kioussis D, O'Garra A. 2005. Identification of a macrophage-specific chromatin signature in the IL-10 locus. J Immunol 175:1041–1046. doi: 10.4049/jimmunol.175.2.1041. [DOI] [PubMed] [Google Scholar]
- 49.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O'Shea JJ, Hunter CA. 2007. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol 8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 50.Stanley AC, Lacy P. 2010. Pathways for cytokine secretion. Physiology (Bethesda) 25:218–229. doi: 10.1152/physiol.00017.2010. [DOI] [PubMed] [Google Scholar]
- 51.Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. 2002. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem 277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- 52.Aderem A, Underhill DM. 1999. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 53.Stuart LM, Ezekowitz RA. 2005. Phagocytosis: elegant complexity. Immunity 22:539–550. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Felton-Edkins ZA, Fairley JA, Graham EL, Johnston IM, White RJ, Scott PH. 2003. The mitogen-activated protein (MAP) kinase ERK induces tRNA synthesis by phosphorylating TFIIIB. EMBO J 22:2422–2432. doi: 10.1093/emboj/cdg240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Misra S, Tripathi MK, Chaudhuri G. 2005. Down-regulation of 7SL RNA expression and impairment of vesicular protein transport pathways by Leishmania infection of macrophages. J Biol Chem 280:29364–29373. doi: 10.1074/jbc.M504162200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rana T, Misra S, Mittal MK, Farrow AL, Wilson KT, Linton MF, Fazio S, Willis IM, Chaudhuri G. 2011. Mechanism of down-regulation of RNA polymerase III-transcribed noncoding RNA genes in macrophages by Leishmania. J Biol Chem 286:6614–6626. doi: 10.1074/jbc.M110.181735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.