

Abstract
GATA1 is a key transcription factor for erythropoiesis. GATA1 gene expression is strictly regulated at the transcriptional level. While the regulatory mechanisms governing mouse Gata1 (mGata1) gene expression have been studied extensively, how expression of the human GATA1 (hGATA1) gene is regulated remains to be elucidated. To address this issue, we generated hGATA1 bacterial artificial chromosome (BAC) transgenic mouse lines harboring a 183-kb hGATA1 locus covering the hGATA1 exons and distal flanking sequences. Transgenic hGATA1 expression coincides with endogenous mGata1 expression and fully rescues hematopoietic deficiency in mGata1 knockdown mice. The transgene exhibited copy number-dependent and integration position-independent expression of hGATA1, indicating the presence of chromatin insulator activity within the transgene. We found a novel insulator element at 29 kb 5′ to the hGATA1 gene and refer to this element as the 5′ CCCTC-binding factor (CTCF) site. Substitution mutation of the 5′ CTCF site in the hGATA1 BAC disrupted the chromatin architecture and led to a reduction of hGATA1 expression in splenic erythroblasts under conditions of stress erythropoiesis. Our results demonstrate that expression of the hGATA1 gene is regulated through the chromatin architecture organized by 5′ CTCF site-mediated intrachromosomal interactions in the hGATA1 locus.
INTRODUCTION
The GATA1 protein is a founding member of the GATA family of zinc finger transcription factors. GATA1 plays a crucial role in the differentiation of several hematopoietic lineages, including the erythroid lineage (reviewed in references 1 to 3). GATA1 is a representative of the transcription factors that act in lineage-specific gene expression. Regulation of mouse Gata1 gene expression has been extensively studied under the concept of the regulation of the regulator. Genes encoding GATA1 usually consist of five coding exons and one or two noncoding first exons. For instance, mGata1 contains two noncoding first exons, IT and IE, which are differentially utilized in distinct cell lineages (4). The IT exon primarily directs mGata1 expression in Sertoli cells of the testis (5). The IT exon also resides in the rat Gata1 gene (6) (but not in the human GATA1 (hGATA1) gene (4). In contrast, the proximal IE exon (and promoter) is utilized in mGata1 expression in hematopoietic cells (4). It has been shown that mGata1 expression from the IE exon/promoter is strictly regulated in each differentiation stage, as homeostasis of GATA1 expression levels is essential for hematopoiesis. Indeed, forced transgenic expression of GATA1 in relatively differentiated erythroid cells leads to maturation arrest of the cells (7).
Through a series of studies on the structure and regulation of the mGata1 gene, we have found that a 3.9-kb mGata1 upstream region including the IE promoter plus 4.2 kb of the first intron sequence harbors sufficient regulatory information to recapitulate mGata1 gene expression in yolk sac primitive and fetal liver definitive erythroid cells (6). We now refer to these regions as the GATA1 hematopoietic regulatory domain (G1HRD) (8–10).
Extensive transgenic LacZ reporter mouse analyses utilizing the G1HRD-based transgene revealed multiple cis-acting elements in the 5′ flanking region of the IE exon of the mGata1 gene. Those include a GATA-binding motif in the Gata1 hematopoietic enhancer (G1HE/HS1; 3.9 kb upstream) and a proximal palindromic double GATA (dbGATA) motif located 680 bp upstream of the IE exon (11–14). An element in the first intron which contains multiple GATA motifs is also required for Gata1 gene expression in fetal liver definitive erythropoiesis and adult bone marrow progenitors (15). All these regulatory elements are evolutionarily conserved between humans and mice and appear to be essential for erythroid lineage-specific G1HRD reporter expression (12, 14, 16, 17).
While the G1HRD-based transgenic reporter mouse system represents significant progress and provides new insights into mouse Gata1 gene regulation, we have noticed that G1HRD is susceptible to positional effect variegation (PEV), as often occurs in short transgenic constructs (17). Therefore, we have exploited and examined transgenic green fluorescent protein reporter expression under the regulatory influence of 196-kb mGata1 bacterial artificial chromosome (BAC) DNA. The 196-kb mGata1 BAC shows resistance to PEV and displays transgene expression in a copy number-dependent manner (12). This implies that the distal flanking sequences beyond the evolutionarily conserved G1HRD region in mGata1 BAC DNA harbors potential insulator activity or locus control region (LCR) activity that protects mGata1 gene expression from regulatory influences around the transgenic integration site.
In contrast to the intensive analyses of the mGata1 gene, there exists only limited information regarding the regulatory mechanism of the hGATA1 gene. For instance, it has been suggested that the expression of murine and human GATA1 genes is regulated in a species-specific manner, since genomic sequences in the distal flanking region of these genes are not evolutionarily conserved (18). Epigenomic analyses of flanking regions in the hGATA1 and mGata1 genes by mapping of DNase I-hypersensitive sites and histone modification patterns suggest that the chromatin structures of these genes are quite different from each other (18, 19). Therefore, we decided to elucidate the regulatory mechanisms of the hGATA1 gene, knowledge of which is crucial for our understanding of human hematopoiesis and the diseases arising from perturbation of the process.
To this end, we have generated multiple transgenic mouse lines by using a 183-kb hGATA1 BAC (hG1B) DNA clone that harbors the hGATA1 genomic locus plus extensive flanking sequences. The regulatory activity of hG1B was compared with that of the endogenous mGata1 gene. The hG1B-directed hGATA1 expression profile coincides nicely with the endogenous mGata1 expression profile, and hG1B-directed hGATA1 expression fully rescues the hematopoietic deficiency in Gata1.05 knockdown mouse embryos. Importantly, hG1B shows a copy number-dependent expression of hGATA1, indicating that hG1B retains an enhancer activity coupled with an insulator activity that protects the hGATA1 locus from the influences of neighboring regulatory sequences. By employing chromosome conformation capture (3C) analysis in combination with the hG1B transgenic system, we found a binding site for the CCCTC-binding factor (CTCF; a factor eliciting insulator activity) 29 kb 5′ to the hGATA1 gene which is essential for maintenance of the higher-order chromatin architecture in the hGATA1 locus and hGATA1 expression in splenic erythroblasts under hemolytic conditions. Our present results reveal for the first time that the chromatin insulator sequences contribute to regulation of hGATA1 gene expression in hematopoietic lineages through maintenance of the correct chromatin architecture.
MATERIALS AND METHODS
BAC modification and generation of transgenic mice.
hGATA1 BAC transgenic mice were generated utilizing hG1B clone RP11-416B14 (Fig. 1A). For construction of the 5′ CTCF site-targeting vector, a 1,910-bp 5′ homologous region was amplified by PCR using sense primer AACTGTATGCGGCCGCAAACACCAGAATCTGACCCCAGT and antisense primer TAAATTCGCGGCCGCAAAATTGAAAGCTTAAAGTGGATG. A 2,983-bp 3′ homologous region was amplified by PCR using sense primer TAGACCAGAGGCATCACACG and antisense primer GGTAATGCCTGTCTCCCTGA. Substitution of the 5′ CTCF site was introduced by overlap extension PCR (see Fig. 7A). These fragments were cloned into a vector containing a neomycin resistance-conferring gene (neo) cassette to generate the targeting construct. The copy number and the integrity of the transgenes were determined by quantitative genomic PCR (qGPCR) using primer pairs amplifying the hGATA1 and mGata1 gene loci (Table 1) (20). Induction of hemolytic anemia with phenylhydrazine (PHZ) was performed as previously described (21). Gata1.05 knockdown mutant mice were described previously (22). All mice were handled according to the regulations of the Standards for Human Care and Use of Laboratory Animals of Tohoku University and the guidelines for the proper conduct of animal experiments of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
FIG 1.
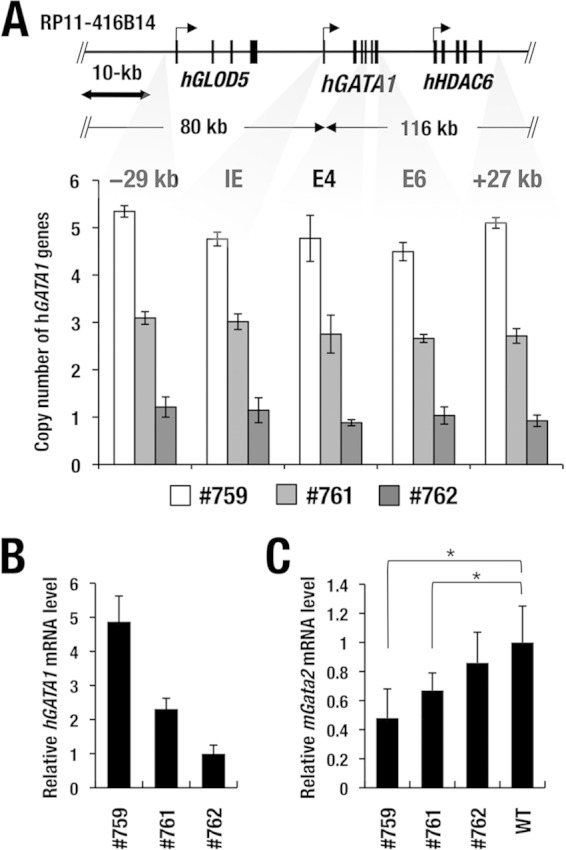
Generation of hGATA1 BAC transgenic mouse. (A) Structure of hGATA1 BAC clone RP11-416B14. Analyses of the copy numbers at the five different regions of the hGATA1 BAC transgene show 5 copies, 3 copies, and 1 copy in hG1B transgenic mouse lines 759, 761, and 762, respectively. Data represent the average ± SD for three mice of each line. (B) Relative expression of transgenic hGATA1 mRNA. Expression of transgenic hGATA1 mRNA was monitored. Note that the hG1B transgenic mouse lines (lines 759, 761, and 762; n = 4 for each line) express graded levels of the hGATA1 transcript in the bone marrow. (C) Relative expression of mouse Gata2 mRNA. Note that the increase in the level of hGATA1 mRNA expression correlates with the decrease in the level of endogenous Gata2 mRNA expression in each line of hG1B transgenic mice. mRNA levels are normalized to GAPDH levels. Statistically significant differences between wild-type mice (n = 4) and hG1B mice (n = 4 for each line) are depicted (*, P < 0.05, Student's unpaired t test).
FIG 7.
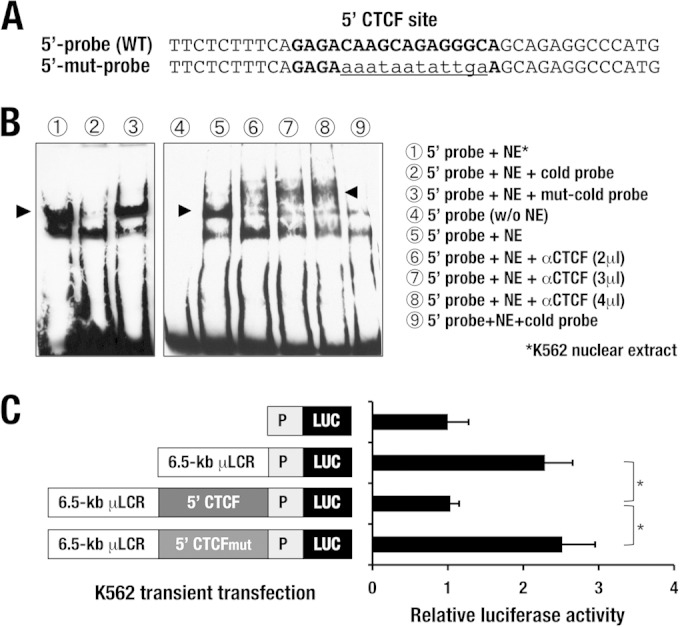
The sequence of the 5′ CTCF site retains enhancer-blocking activity. (A) Wild-type (upper) and mutated (mut; lower) sequences of the probe for the 5′ CTCF site. The boldface nucleotides represent the predicted CTCF-binding sequences, and the lowercase and underlined nucleotides represent the mutated sequences. (B) Electrophoretic gel mobility shift assay of CTCF binding. The probe for the 5′ CTCF site yields a band shift of a specific DNA-protein complex (arrowhead, lane 1), which is eliminated by adding 100-fold more wild-type cold probe (lane 2). The 5′ mutant probe fails to compete for the DNA-protein complex (lane 3). The band shift of the specific DNA-protein complex (arrowhead, lane 5) is supershifted by incubation with incrementally increased amounts of anti-CTCF antibody (arrowheads, lanes 6 to 8). (C) Results of an enhancer-blocking assay for the 5′ CTCF-binding site. A 1,259-bp fragment containing the sequence of the 5′ CTCF site exerts enhancer-blocking activity when placed between the 6.5-kb β-globin μLCR and the SV40 promoter-luciferase cassette. Note that the substitution mutation of the CTCF-binding site diminished the enhancer-blocking activity. Data represent the mean ± SD. Statistically significant differences from three independent experiments are depicted (*, P < 0.05, Student's unpaired t test).
TABLE 1.
Sequences of primers used in qGPCR, RT-qPCR, and genotyping
| Primer specificity | Primer sequence |
Purpose | |
|---|---|---|---|
| Forward | Reverse | ||
| mGata1 | TCTGGACAACCCAAGTCTCTG | GCTTTGAAGGTTCAAGCC | RT-qPCR |
| hGATA1 | TCTGGACAACCCAAGTCTCTG | GCTTTGAAGGTTCAAGCC | RT-qPCR |
| mGata2 | ACCTGTGCAATGCCTGTGGG | TTGCACAACAGGTGCCCGCT | RT-qPCR |
| hGLOD5 | GGGCAGGACTTTGGAGAAAC | CGATGTGGTCAAGTCTACGG | RT-qPCR |
| hHDAC6 | TATCTGCCCCAGTACCTTCG | GCAGCACCATTCAGAACCTC | RT-qPCR |
| Human CTCF | GGCTTGAGAGCTGGGTTCTA | CGACTGCATCACCTTCCAT | RT-qPCR |
| Mouse GAPDH | GTCGTGGAGTCTACTGGTGTCTT | GAGATGATGACCCTTTTGGC | RT-qPCR |
| Human GAPDH | CGAGATCCCTCCAAAATCAAGT | GGCAGAGATGATGACCCTTTTG | RT-qPCR |
| hGATA1 5′ CTCF | AAGGGTGTTGGCACTGAAAC | GCCCTCTGCTTGTCTCTGAA | qGPCR, ChIP-qPCR |
| hGATA1 kb −29 | CCTCCCATTCCTGCCCCTTG | CTGGCTCAGCGCCTGGAGAT | qGPCR, ChIP-qPCR |
| hGATA1 dbGATA | CCCCAAGACAGCCTGTTACT | CTGGGGCAGCAGATAAGTCT | qGPCR, ChIP-qPCR |
| hGATA1 E4 | CGGAGGGACAGGACAGG | CTTCTTGGGCCGGATGAG | qGPCR, ChIP-qPCR |
| hGATA1 E6 | AACCGCAAGGCATCTGG | CCACCTCCCCACAATTCC | qGPCR, ChIP-qPCR |
| hGATA1 3′ CTCF | GGATTTGGTGTGGCTACTGC | AACCCCCTGGTCAAATTAGG | qGPCR, ChIP-qPCR |
| hGATA1 kb +27 | CCCATGGGGGATGAGGGTGCCAG | TCAGCATCACCCATGCGGGGCCC | qGPCR, ChIP-qPCR |
| mGata2-2.8 | GCCCTGTACAACCCCATTCTC | TTGTTCCCGGCGAAGATAAT | qGPCR, ChIP-qPCR |
| Mouse HS5 | ATGAGGCGTTTTCACCAC | AAGGGGTCTTTTCACCGT | qGPCR, ChIP-qPCR |
| Human HS5 | TAGCTGAAGCTGCTGTTATGACCAC | CCAGATGTCCTGTCCCTGTAAGGT | qGPCR, ChIP-qPCR |
Fluorescence-activated cell sorter analysis and cell sorting.
For separation of late erythroid progenitors (LEPs) and erythroblasts, bone marrow mononucleated cells were stained with anti-c-Kit, anti-CD71, and anti-Ter119 antibodies (12, 23). For separation of the common myeloid progenitor (CMP) and the megakaryoerythroid progenitor (MEP), lineage marker-negative cells were stained with anti-c-Kit, anti-CD34, and anti-phycoerythrin-FcγRII/III antibodies (24). All antibodies were purchased from BD Pharmingen. Sorting and analysis of cells were performed using FACSAria II and FACSCalibur flow cytometers (BD Biosciences).
Western blotting.
Whole bone marrow cells were lysed with standard 2× SDS buffer. The whole-cell lysates were separated by 10% SDS-PAGE. Western blot analyses were performed using anti-GATA1 (N6 and C20; catalog numbers sc-265 and sc-1233, respectively; Santa Cruz) and anti-GATA2 (catalog number sc-1235; Santa Cruz) antibodies as described previously (25, 26). The Western blot film was scanned, and the band intensities were quantified using ImageJ software (NIH).
Quantitative ChIP analysis.
Chromatin immunoprecipitation (ChIP) analysis for detection of CTCF in the hGATA1 locus was performed using the K562 erythroid cell line and PHZ-treated splenic erythroblasts as previously described (20). Briefly, cells were fixed with 1.0% formaldehyde for 5 min at room temperature. After fixation, the formaldehyde was neutralized with 350 mM glycine. The cells were washed with ice-cold phosphate-buffered saline and resuspended in SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 1% SDS, 10 mM EDTA). The fixed cells were sonicated to fragment chromatin DNA into samples 300 to 1,000 bp in length. Sonicated chromatin DNA samples were centrifuged at 13,000 × g for 10 min, and the supernatant was diluted 5-fold in ChIP dilution buffer (50 mM Tris-HCl [pH 8.0], 167 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, protease inhibitors [Complete mini-EDTA-free; Roche]). Immunoprecipitation was carried out with anti-CTCF (Active Motif) and control rabbit IgG (catalog number sc-2027; Santa Cruz) antibodies. The immunoprecipitated materials were eluted in ChIP elution buffer (10 mM Tris-HCl [pH 8.0], 300 mM NaCl, 5 mM EDTA, 0.5% SDS). The cross-links were reversed by incubation with 1.5 μg/ml proteinase K at 55°C for 3 h, followed by 65°C for 8 h. Quantitative PCR (qPCR) was carried out using 2× SYBR green PCR master mix (PE Applied Biosystems). The primer pairs used for amplification of each point in the hGATA1 locus are listed in Table 1. Relative enrichment compared to the input was calculated.
Silencing of CTCF expression by siRNA.
K562 cells were transfected with small interfering RNA (siRNA) against CTCF or control siRNA (Stealth RNAi siRNA negative-control Hi GC; Invitrogen) using a Neon transfection system (Invitrogen). At 48 h after transfection, the cells were subjected to analysis. The siRNA sequence for human CTCF was 5′-UCACCCUCCUGAGGAAUCACCUUAA-3′.
RT-qPCR.
Total RNA was purified from cells using RNeasy (Qiagen) and reverse transcribed by SuperScript III (Invitrogen) with random hexamers. Quantitative real-time reverse transcription (RT-qPCR) was conducted using an ABI Prism 7300 sequence detector system (PE Applied Biosystems) and 2× SYBR green PCR master mix (Invitrogen). The mRNA level of each gene was normalized to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA level. The primer sequences are listed in Table 1.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were conducted as previously described (27). For supershift assays, the reaction mixture was incubated with anti-CTCF antibody (catalog number 61311; Active Motif). The sense-strand sequences of the wild-type and the mutated 5′ CTCF probes are depicted in Fig. 7A.
Enhancer blocking assay.
A 1,259-bp DNA fragment containing the 5′ CTCF site was amplified by PCR using sense primer CACTGCAACCTCCGCCTCCCGGATTCACGCGATTC and antisense primer AATTAGCTGGGCATGGTGACGTGCACCTGTAATCC. A nucleotide substitution was introduced into the 5′ CTCF site by overlapping PCR-based mutagenesis. The DNA fragments were inserted between the 6.5-kb human β-globin short recombined LCR (μLCR) and the simian virus 40 (SV40) promoter (7). Enhancer blocking assays using a dual-luciferase (Luc) reporter system were performed by a standard protocol in K562 cells (28).
3C analysis.
The 3C assay was performed as described previously (29). Briefly, after PHZ-induced hemolytic anemia, formaldehyde cross-linked chromatin from K562 cells or whole spleen cells (107 cells) was digested with EcoRI at 37°C overnight. Religation of the restriction fragments was performed with T4 DNA ligase at 16°C for 4 h. To prepare control templates for standard curves, a BAC clone (RP11-416B14) harboring the hGATA1 locus was treated by use of the same protocol; restriction digestion with EcoRI was followed by religation with T4 DNA ligase. After reversing the cross-links, genomic DNA was purified by phenol extraction and subsequent ethanol precipitation. Assessment of the religated products was performed by real-time PCR with a TaqMan probe using an ABI Prism 7500 system (PE Applied Biosystems). All PCR products were cloned and sequenced to confirm the sequences of the ligated products. 3C-quantitative PCR data were normalized to the data for a loading control, using internal primers located in the excision repair cross-complementation group 3 gene (ERCC3). Statistical analysis of the data from three independent experiments was performed by Student's t test. The sequences of the primers and the TaqMan probes are listed in Table 2.
TABLE 2.
3C primers designed for EcoRI restriction sites
| Position no. or gene | Primer sequence | TaqMan probe (anchor fragment) | Purpose |
|---|---|---|---|
| 0 | CCATGTGGAGAGAGGCTAGG | CTCCTAAAGCTCAAGGTCAGCGCTGTGTTT | 3C assay |
| 1 | CTGAGGAGGGAGGATTGCTT | 3C assay | |
| 2 | GGCCTCCTCTGTGTCTTGTC | 3C assay | |
| 4 | CTGCCTAGGTTCGAATTCCTC | 3C assay | |
| 5 | TCTCACTCTCAGGGTATTTAGCA | CTTTGATCTACTAGATCCAAAGGTAGAAA | 3C assay |
| 6 | CACCAGCAGGAGCTCAATAA | 3C assay | |
| 7 | GCCGACCACTTTCCCTAGTT | 3C assay | |
| 8 | GAGGGGTGGTGTCCTTCTC | 3C assay | |
| 9 | TTGGTGTTGGATGAGCAGTT | 3C assay | |
| 10 | TGCATCTTACAAACAGTGACATCT | 3C assay | |
| 11 | ATCTGCCAAGGGTCTGTCAT | 3C assay | |
| ERCC3 | GTAGCACGTGTCTAGCCTTGAACATTAGGCCGGAGTAGC | Normalization |
Public ChIP-seq data.
ChIP sequencing (ChIP-seq) data for CTCF, Rad21, monomethylated H3K4 (H3K4me1), trimethylated H3K4 (H3K4me3), and acetylated H3K27 (H3K27Ac) in K562 and MEL cells were obtained from the University of Washington transcription factor binding site database (accession numbers GSE30263 and GSE29218). The Pennsylvania State University Bioinformatics Group created the vertebrate conservation data (http://www.bx.psu.edu/miller_lab/). All the data are part of the mouse/human ENCODE project (30).
RESULTS
Copy number-dependent mRNA expression of hGATA1 in hGATA1 BAC transgenic mouse lines.
To investigate the regulatory mechanisms and to assess the functional boundaries of the human GATA1 (hGATA1) gene, we examined hGATA1 BAC (hG1B) clone RP11-416B14, which contains a 183-kb genomic region spanning the sequence flanking from approximately 80 kb 5′ to 116 kb 3′ of the hGATA1 gene (Fig. 1A). We generated three lines of hG1B transgenic mice (lines 759, 761, and 762) by using the BAC clone. After breeding the three established lines of transgenic mice to a C57BL/6J mouse background for more than five generations, we quantified the BAC copy number at five different regions encompassing the region from kb −29 to kb + 27 in the transgenic hGATA1 locus by quantitative genomic PCR (qGPCR) analysis. The 759, 761, and 762 lines harbor 5 copies, 3 copies, and 1 copy of the hG1B transgene, respectively (Fig. 1A).
To examine whether transgene copy numbers correlate with the level of hGATA1 mRNA expression, hGATA1 transcript levels in whole bone marrow cells were examined by quantitative real-time reverse transcription-PCR (RT-qPCR) analysis with a specific hGATA1 primer pair. We found that among the three transgenic lines, the 762 line with a single copy of the hG1B transgene showed the lowest level of the hGATA1 transcript, while the 759 line with 5 copies of the hG1B transgene showed the highest level of hGATA1 mRNA expression, which was approximately 4.9-fold higher than that of the 762 line (Fig. 1B). The line 761 mice with 3 copies of the transgene exhibited a medium level of hGATA1 expression that was approximately 2.3-fold higher than that of line 762 mice. All three lines of transgenic mice were free of hematopoietic abnormalities and were indistinguishable in appearance from their wild-type littermates (data not shown). The increase in the level of hGATA1 mRNA expression correlated nicely with the decrease in the level of endogenous Gata2 mRNA expression (Fig. 1C), in agreement with the report that GATA1 negatively regulates Gata2 expression during erythroid differentiation (31). These results thus indicate that hG1B confers a copy number-dependent expression of hGATA1.
Transgenic hGATA1 expression shows hematopoietic lineage and stage specificity.
To verify hematopoietic lineage-specific hGATA1 expression in the hG1B transgenic mice, we separated Ter119-positive erythroblasts (BM-EBs), Mac1-positive macrophages, CD41-positive megakaryocytes, and thymocytes from the transgenic mice and examined transgene-derived hGATA1 and endogenous mGata1 mRNA expression. We found that hGATA1 and mGata1 were expressed abundantly in the Ter119-positive erythroid cells and CD41-positive megakaryocytes, while both mRNAs were expressed at low levels in Mac1-positive macrophages and thymocytes (Fig. 2A), suggesting that, in this mouse milieu, the hG1B transgene recapitulates the original regulatory activity exerted in human erythroid lineage cells, and the specificity showed a good resemblance to endogenous mGata1 expression.
FIG 2.
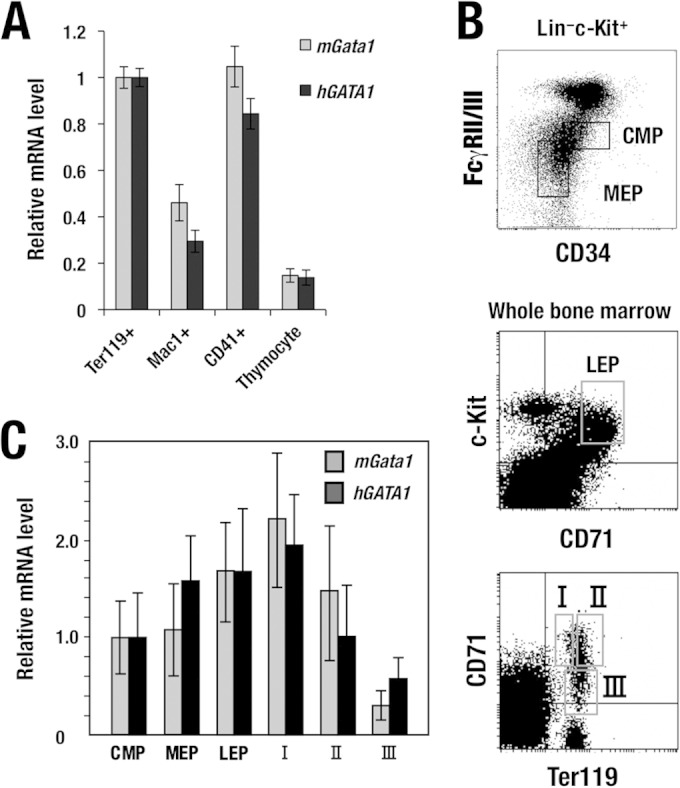
Hematopoietic lineage and differentiation stage-specific hGATA1 expression from the hGATA1 BAC transgene. (A) Relative expression of hGATA1 and mGata1 mRNAs in Ter119-positive erythroid cells, CD41-positive megakaryocytes, Mac1-positive macrophages, and thymocytes. Note that hGATA1 and mGata1 mRNAs are expressed at comparable levels and abundantly in both erythroid cells and megakaryocytes, while macrophages and thymocytes showed comparable levels of expression of both mRNAs, but the levels were lower than those in erythroid cells and megakaryocytes. The level of hGATA1 and mGata1 mRNA expression in Ter119-positive erythroid cells was set equal to 1. (B) Flow cytometric separation of hematopoietic progenitor cells (top), late erythroid progenitor cells (middle), and erythroblasts (bottom). Hematopoietic progenitors are separated from the Lin− c-Kit+ fraction of bone marrow cells, while late erythroid progenitors and erythroblasts are separated from whole bone marrow cells. (C) Both hGATA1 and mGata1 mRNAs showed similar expression profiles during erythroid differentiation through the CMP, MEP, LEP, fraction I (ProEB), fraction II (basophilic erythroblast), and fraction III (poly- and orthochromatic erythroblast) stages. The level of hGATA1 and mGata1 mRNA expression in the CMP stage was set equal to 1. mRNA levels were normalized to GAPDH levels. Data represent the average ± SD for four hGATA1 BAC transgenic mice (line 762). The remaining two lines of hG1B transgenic mice (the 759 and 761 lines) exhibited patterns of lineage- and stage-specific hGATA1 expression similar to those of the 762 line.
To address how hGATA1 expression changes during the erythroid differentiation process, we separated hematopoietic progenitor fractions, including the common myeloid progenitor (CMP), the megakaryoerythroid progenitor (MEP), and the c-Kit+ CD71+ late erythroid progenitor (LEP) (32), from hG1B transgenic mice by flow cytometry (Fig. 2B, top and middle). It has been reported that mGata1 expression is detected at a low level in the CMP stage and reaches a peak in the LEP and proerythroblast (ProEB) stages; thereafter, the level of expression gradually decreases toward terminal erythroblast maturation (32, 33). The levels of expression of endogenous mGata1 and transgene-derived hGATA1 mRNAs in these fractions were separately quantified. Showing very good agreement with the mGata1 mRNA expression profile, hG1B-derived hGATA1 mRNA became detectable in the CMP stage, and the level of expression increased in the MEP and LEP stages (Fig. 2C).
Utilizing a protocol with Ter119 and CD71 antibodies (23), we separated erythroid lineage cells (Fig. 2B, bottom). We examined hGATA1 expression in each stage of erythroblasts and compared that with endogenous mGata1 expression. The hGATA1 expression profile during erythroblast differentiation coincided with that of endogenous mGata1 (Fig. 2C). These results demonstrate that the 186-kb hG1B harbors a comprehensive set of regulatory sequences that directs lineage- and stage-specific expression of the hGATA1 gene in a mouse hematopoietic milieu in vivo.
The hG1B transgene rescues Gata1.05 knockdown mice from embryonic lethality.
To address whether the hG1B transgene directs levels of hGATA1 expression physiologically sufficient to sustain mouse survival, we tried to rescue Gata1.05 knockdown mice from lethal hematopoietic deficiency. For this complementation rescue analysis, we bred Gata1.05/X heterozygous female mice with hG1B transgenic male mice and examined whether the hG1B transgene could rescue the Gata1.05/Y male mice from embryonic lethality. We found that Gata1.05/Y male mice were successfully rescued from embryonic lethality and sustained their life until adulthood when mated with mice from these three lines of hG1B transgenic mice (Table 3). The numbers of hG1B-rescued Gata1.05/Y mice, which we refer to as hG1R mice, followed a Mendelian transmission rate when the mice were genotyped at 2 to 3 months after birth. The appearance of hG1R mice was normal, and we did not notice any signs of anemia throughout embryonic development and adult stages (data not shown). The peripheral blood of hG1R mice indeed exhibited normal hematopoietic indices (Table 4). This result was quite reproducible, and all three rescued mouse lines showed similar results.
TABLE 3.
Transgenic rescue of Gata1.05/Y knockdown mice from embryonic lethality by crossing with hG1B transgenic micea
| Line | Total no. of mice | No. of Gata1.05/Y::hGATA1 BAC transgenic mice |
|---|---|---|
| 759 | 104 | 12 (14) |
| 761 | 82 | 7 (10) |
| 762 | 96 | 10 (12) |
Genotyping was conducted 8 to 12 weeks after birth. Numbers in parentheses are the expected number of transgene-rescued Gata1.05/Y mice.
TABLE 4.
Hematopoietic indices of hG1B transgenic rescued adult micea
| Genotype (no. of mice) | WBC count (102/μl) | RBC count (104/μl) | Hb (g/dl) | Ht (%) | PLT count (104/μl) |
|---|---|---|---|---|---|
| Wild type (6) | 58.3 ± 12.2 | 932 ± 118 | 14.3 ± 1.1 | 44.8 ± 4.7 | 95.6 ± 32.8 |
| hG1R (7) | 55.0 ± 23.0 | 972 ± 162 | 13.9 ± 0.5 | 46.0 ± 3.2 | 96.5 ± 21.9 |
Peripheral blood from 2- to 3-month-old mice of line 762 was examined. WBC, leukocyte; RBC, erythrocyte; Hb, hemoglobin; Ht, hematocrit; PLT, platelet.
We also examined the differentiation of erythroid lineage cells in bone marrow. We found that LEPs reside in hG1R mice at levels comparable to those in wild-type control mice (Fig. 3A). The levels of three erythroblast fractions separated by CD71 and Ter119 (fractions I, II, and III) (23) were also comparable between hG1R mice and wild-type mice (Fig. 3B). Furthermore, CD41+ CD61+ megakaryocytes were normally developed in hG1R mouse bone marrow (Fig. 3C). While in Fig. 3 we show the results for line 762 of mice expressing 1 copy of the hG1B transgene, these results were reproducible in all three lines of rescued mice. We have therefore demonstrated that the hG1B transgene sustains adequate erythropoiesis and megakaryopoiesis in the mouse in vivo.
FIG 3.
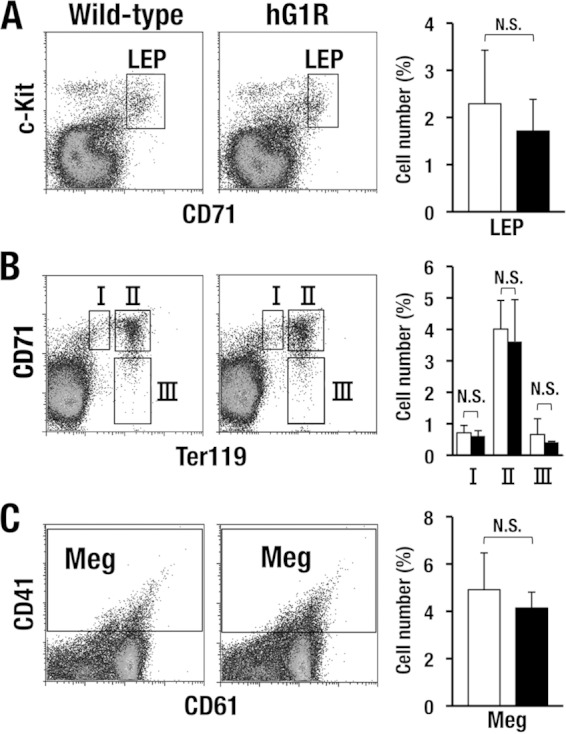
Erythroid lineage cells in the bone marrow of hG1R mice are within the normal range. (A and B) hG1B-rescued Gata1.05/Y (hG1R) mice show normal populations of erythroid lineage cells. hG1R mice of the 762 line were used in this analysis, and LEPs and three different stages of erythroblasts were analyzed and compared with those from wild-type mice. (C) CD41+ CD61+ megakaryocytes (Meg) are normally developed in the hG1R mice. Quantification data for each fraction (right column) represent the average ± SD derived for hG1R (black bars; n = 7) and wild-type (white bars; n = 6) male mice. N.S., not significant.
hG1B-directed hGATA1 protein expression in transgenic rescued Gata1.05 mice.
We assessed the hG1B transgene-directed expression of the hGATA1 protein quantitatively in whole bone marrow cells from the lines of hG1R mice expressing high (line 759) and low (line 762) levels of the hG1B transgene. Western blotting using a goat antibody raised against the C terminus of the hGATA1 protein (C20) detected high (line 759) and low (line 762) levels of hGATA1 protein expression in hG1R mice (Fig. 4A). The bands were scanned and the intensities were quantified, and we found that the expression of hGATA1 in line 762 was almost comparable to that of mGATA1 in wild-type mice and that in line 759 it was approximately 8-fold higher than the level of endogenous mGATA1 expression (Fig. 4B).
FIG 4.
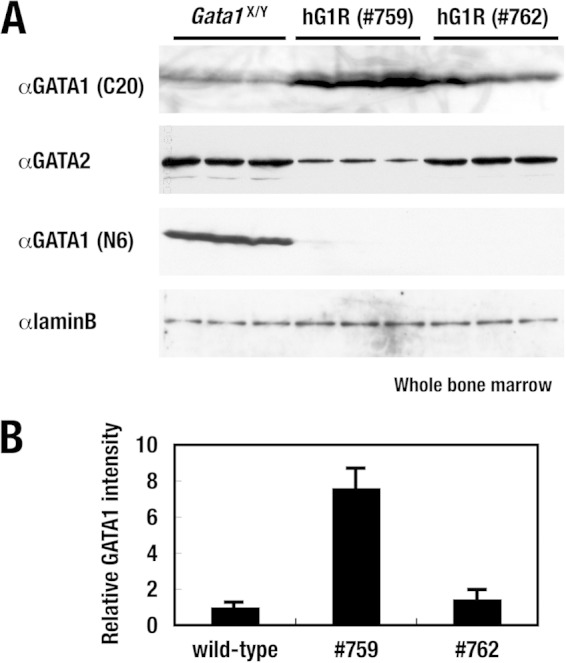
hGATA1 protein expression in hG1R mice. (A) Western blotting analyses of the GATA1 and GATA2 proteins in the bone marrow of transgenic rescued mice. A goat antibody raised against the C terminus of the hGATA1 protein (C20) detects high (line 759) and low (line 762) levels of abundance of the hGATA1 protein in hG1R mice (top row). The GATA2 protein level was decreased significantly in hG1R mice expressing high levels of the hGATA1 transgene (line 759) and only marginally in hG1R mice expressing low levels of the hGATA1 transgene (line 762) compared with the GATA2 protein level in wild-type control mice (second row). A rat monoclonal antibody against the N terminus of the mGATA1 protein (N6) detects only endogenous mGATA1 protein in the wild-type control mice (third row). In hG1R mice, endogenous mGATA1 gene expression is knocked down 5% compared to the normal level and mGATA1 protein expression is usually not detectable. Lamin B served as a loading control. (B) The levels of mouse or human GATA1 protein expression detected by the C20 antibody were quantified and normalized to the lamin B level. Data represent the average ± SD for three mice of each genotype.
Of note, in hG1R mice expressing a high level of the hG1B transgene (line 759), the GATA2 protein level fell to almost half of that in wild-type mice, while the level in hG1R mice expressing a low level of the hG1B transgene (line 762) was only marginally decreased in comparison with that in wild-type control mice (Fig. 4A). Thus, high-level hGATA1 expression is able to suppress Gata2 gene expression in hematopoietic progenitor cells in vivo, as is the case for high-level expression of mGATA1 (31, 33).
We also exploited our homemade rat monoclonal antibody against the N terminus of the mouse GATA1 protein (N6) (4). Whereas N6 detected endogenous mGATA1 protein in the wild-type control mice, the antibody did not react with hGATA1 in the 759 and 762 lines of hG1R mice (Fig. 4A). Although Gata1.05 knockdown mice retained approximately 5% of the level of mGATA1 expression as wild-type mice (22), such a level of mGATA1 expression was rarely detected under the experimental conditions used in the present study. Thus, the hG1B transgene directs sufficient (or more than sufficient) hGATA1 expression to reconstitute normal erythroid and megakaryocytic lineages in Gata1.05 knockdown mutant mice.
The hGATA1 locus contains evolutionarily unique CTCF-binding sites.
The copy number-dependent expression of the hG1B transgene led us to surmise that the 183-kb human GATA1 locus corresponding to the hG1B transgene harbors a locus control region with activity that is constituted of both insulator and enhancer activities. The insulator activity eliminates the positional effect variegation (PEV) of transgenes by generating gene boundaries and by blocking enhancer influences (28). As vertebrate insulators that exhibit enhancer-blocking activity are shown to bind to the CCCTC-binding factor (CTCF) (34, 35), we searched for CTCF-binding sequences in the hGATA1 locus and found several high CTCF-binding peaks within the hGATA1 locus in the ChIP-seq database with human erythroid cell line K562 (http://genome.ucsc.edu/). Particularly of note, high and sharp peaks were located 29 kb upstream and 21, 33, and 50 kb downstream of the hGATA1 gene locus (Fig. 5A).
FIG 5.

The hGATA1 locus contains multiple CTCF-binding sites. (A) The chromosome locations of hGATA1 and flanking genes (hGLOD5 and hHDAC6) are depicted. The degree of sequence conservation in vertebrate and ChIP-seq data for CTCF, Rad21, H3K4me1, H3K4me3, and H3K27Ac in K562 cells was obtained from the University of California, Santa Cruz, genome browser (http://genome.ucsc.edu/). (B) Mouse ChIP-seq data for CTCF in MEL cells. All the ChIP-seq data were derived from the ENCODE project.
We also searched for binding sites for Rad21, which is a component of the CTCF/cohesin complex and is known to colocalize with the insulator (36). We found that the Rad21 site is localized at the 29-kb upstream and 21-kb downstream sites, as indicated by the gray lines in Fig. 5A (http://genome.ucsc.edu). In particular, the 29-kb upstream region showed the highest peak of Rad21 binding in the hGATA1 locus (Fig. 5A). We refer to these two sites as the 5′ CTCF and 3′ CTCF sites, respectively. Of note, among vertebrate species the genomic sequences around the CTCF-binding peaks are less evolutionally conserved than the body of the GATA1 gene locus (Fig. 5A). The distribution patterns of the CTCF-binding peaks in the mGata1 and hGATA1 loci are quite different from each other (http://genome.ucsc.edu/) (Fig. 5B).
We next examined the histone modification pattern in the hGATA1 locus using the ChIP-seq database with K562 cells (http://genome.ucsc.edu/) and found that active histone modifications, i.e., H3K4me1 and HeK27Ac, both of which mark active enhancer regions, predominantly accumulated between the 5′ and 3′ CTCF sites (Fig. 5A) (37). H3K4me3, which marks transcription start sites, was also detected mainly between the 5′ and 3′ CTCF sites (Fig. 5A). These data further support our contention that the two CTCF/Rad21 sites located at kb −29 (5′ CTCF site) and kb +21 (3′ CTCF site) in the flanking regions of the hGATA1 locus have insulator activity and protect the hGATA1 gene locus from PEV.
CTCF accumulates in the hGATA1 locus and exerts regulatory activity.
We next examined whether CTCF binds to these binding sites by means of chromatin immunoprecipitation (ChIP) analysis using an anti-CTCF antibody. CTCF indeed bound to the 5′ and 3′ CTCF sites but not around the IE promoter region of the hGATA1 gene in K562 cells (Fig. 6A). In addition, CTCF also bound to the 3′ end of the hGATA1 gene body (Fig. 6A, hGATA1 E6).
FIG 6.
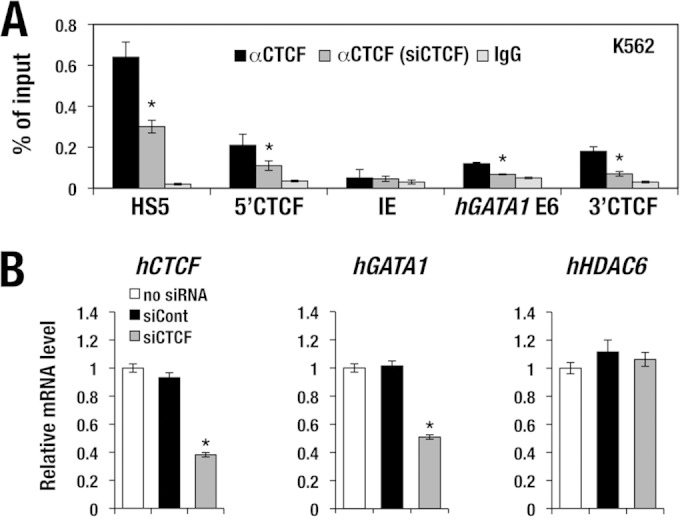
CTCF accumulation in the hGATA1 locus. (A) CTCF accumulates at the 5′ and 3′ CTCF sites in the hGATA1 locus of K562 cells. CTCF also binds to the 3′ end of the hGATA1 gene (E6) but not to the IE promoter region. Note that upon siRNA knockdown of human CTCF, CTCF binding in the hGATA1 locus was diminished. HS5, human β-globin LCR DNase I hypersensitive site 5, which served as a positive-control locus. (B) siRNA knockdown reduces the human CTCF mRNA level to 40% of the normal level. The hGATA1 expression level is decreased 50% upon knockdown of human CTCF. In contrast, the level of hHDAC6 expression is not changed regardless of the human CTCF expression level. mRNA levels are normalized to GAPDH levels. Data represent the mean ± SD. Statistically significant differences from three independent experiments are depicted (*, P < 0.05, Student's unpaired t test). siCont, control siRNA.
We also examined whether CTCF functionally contributes to hGATA1 gene regulation in erythroid cells. To this end, we employed an approach that used siRNA knockdown of CTCF and examined the consequences of reduced CTCF expression in K562 cells. The siRNA for CTCF (siCTCF) suppressed the CTCF mRNA expression level to 40% of the normal level (Fig. 6B, left). Consistently, the level of CTCF accumulation at the 5′ and 3′ CTCF sites and at the hGATA1 E6 site was decreased to less than approximately half of the normal level (Fig. 6A). Of note, hGATA1 mRNA expression was decreased to 40% of the normal level in K562 cells after the siRNA-mediated reduction of CTCF expression (Fig. 6B, center), while the expression level of the human HDAC6 (hHDAC6) genes, which are localized in the 3′ flanking region of hGATA1, was not changed (Fig. 6B, right). These results indicate that CTCF plays an important role for maintenance of hGATA1 gene expression in K562 cells, while the hHDAC6 gene is not under the regulatory influence of CTCF.
CTCF directly binds to the 5′ CTCF site in the hGATA1 gene.
To further characterize CTCF binding to the 5′ and 3′ CTCF sites, we employed an algorithm for prediction of the CTCF-binding site (http://insulatordb.uthsc.edu/) (38) and found one candidate CTCF-binding sequence in the 5′ CTCF site (Fig. 7A). We prepared double-strand oligonucleotide probes corresponding to the 5′ CTCF site and performed electrophoretic mobility shift assays (EMSAs) using K562 nuclear extracts to assess whether the CTCF protein directly binds to the sequence. The probe for the 5′ CTCF site yielded a specific DNA-protein complex on EMSA, which was competed out by adding an excess amount of cold probe (Fig. 7B, lane 2). The specific band was supershifted by incubating with anti-CTCF antibody. We have tested incremental amounts of the antibody, and the results were quite reproducible (Fig. 7B, lanes 6 to 8). These data thus demonstrate that, in vitro, CTCF binds to the 5′ CTCF sequences in the hGATA1 locus.
In contrast, while there is one consensus CTCF-binding sequence in the 3′ CTCF site, we could not detect specific DNA-protein complex formation with the probe for the 3′ CTCF site using EMSA. We tried the experiments five times; each time, nonspecific bands were visible, but the cold probe did not compete out these bands (data not shown). While there still remains the possibility that CTCF binds to the 3′ CTCF site in vivo in the chromatin context, in the following analyses we focused on the 5′ CTCF site.
The 5′ CTCF site exerts enhancer-blocking activity.
We next conducted an enhancer-blocking analysis of the 5′ CTCF site essentially by exploiting the method developed by Gaszner and Felsenfeld (28). For this purpose, we prepared a new enhancer-blocking assay plasmid by inserting the 1,259-bp-long 5′ fragment of the CTCF-binding site between a 6.5-kb human β-globin μLCR (7) and an SV40 promoter-directed luciferase (Luc) cassette (Fig. 7C). These plasmids were transfected into K562 cells, and Luc reporter activity was assessed. μLCR activated SV40 promoter-directed Luc expression 2.3-fold compared to the level of expression obtained with the enhancer-less Luc reporter (Fig. 7C). The μLCR activity was abrogated by the insertion of the 5′ CTCF site, but this blocking activity was cancelled by the substitution mutations within the CTCF site shown in Fig. 7A (Fig. 7C). These results demonstrate that the 5′ CTCF site of the hGATA1 locus exerts enhancer-blocking activity against the β-globin μLCR in K562 erythroid cells.
The 5′ CTCF site participates in the organization of the hGATA1 locus chromatin architecture.
CTCF-mediated chromatin insulator activity is often elicited through formation of a higher-order chromatin architecture around the gene locus. To test this possibility, we examined the long-range chromosomal interaction in the endogenous hGATA1 locus by means of chromosome conformation capture (3C) analysis with K562 cells. When we set an anchor fragment at the 5′ CTCF site, we found that a chromosomal interaction loop was formed between the 5′ CTCF site and the 3′ end of the human GLOD5 (hGLOD5) gene (Fig. 8B). The 5′ CTCF site also interacted with the 3′ end of the hGATA1 gene, but the anchor site did not show an interaction with the 3′ CTCF site (Fig. 8B). These results were somewhat surprising for us, as the strongest signals of CTCF (and Rad21) binding were seen in the 5′ and 3′ CTCF sites of this locus in the data from the public ChIP sequence database (Fig. 5A).
FIG 8.
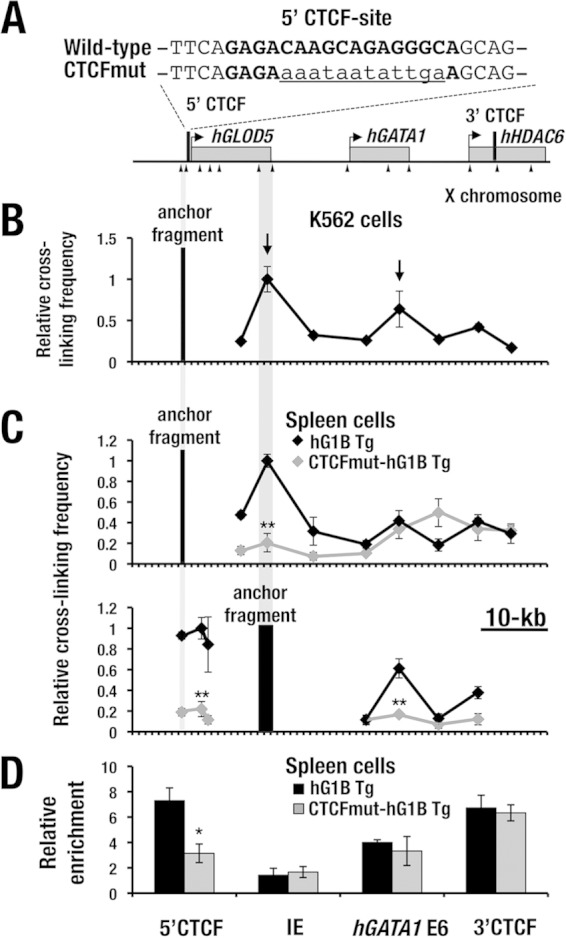
The 5′ CTCF site is important for the chromatin architecture in the hGATA1 locus. (A) Sequences of the 5′ CTCF site in the wild-type and mutant CTCF hG1B transgene. The boldface nucleotides represent the predicted CTCF-binding sequences, and the lowercase and underlined nucleotides represent the mutated sequences. (B) 3C analysis of the hGATA1 locus in K562 cells. Note that the 5′ CTCF site (anchor fragment) interacts with the 3′ end of the hGLOD5 gene and the 3′ end of the hGATA1 gene (arrows). The positions of the hGLOD5, hGATA1, and hHDAC6 genes are indicated by gray boxes. The positions of EcoRI sites (arrowheads) and 5′ and 3′ CTCF sites (black bars) are depicted. (C) (Top) 3C analysis of the hGATA1 locus in splenic erythroblasts from wild-type hG1B (black line) and CTCFmut-hG1B (gray line) transgenic (Tg) mice. The anchor fragment was changed to the 3′ end of the hGLOD5 gene (bottom). Each data point is an average from three independent experiments; error bars depict ±SD. (D) CTCF binds to the 5′ and 3′ CTCF sites and the 3′ end of the hGATA1 gene (E6) in the transgenic hGATA1 locus in splenic erythroblasts from hG1B transgenic mice. Note that CTCF binding is reduced at the 5′ CTCF site in CTCFmut-hG1B transgenic mice. The CTCF ChIP value is normalized against the values obtained for control IgG. All ChIP results represent the averages from three independent experiments. Statistically significant differences between the wild-type and CTCFmut-hG1B are depicted (**, P < 0.01; *, P < 0.05; Student's unpaired t test).
The interaction of the 5′ CTCF site with the 3′ end of the hGATA1 gene is consistent with CTCF accumulation at the 3′ end of the hGATA1 gene body (Fig. 5A and 6A, hGATA1 E6). However, we did not find the CTCF and Rad21 peaks at the 3′ end of the hGLOD5 gene in the data from the public ChIP sequence database (Fig. 5A). Thus, these results suggest that the 5′ CTCF site forms chromatin loops through an interaction with the 3′ boundary region of hGLOD5 and the 3′ end of the hGATA1 gene in K562 cells by using the CTCF-mediated mechanism and/or some other mechanisms.
Verification of the chromatin architecture utilizing spleen cells from hG1B transgenic mice with hemolytic anemia.
To examine whether the higher-order chromatin architecture is reproducible in mouse hematopoietic cells in vivo, we performed a 3C analysis similar to that described above utilizing spleen cells from transgenic mice with a single copy of hG1B (line 762). We injected phenylhydrazine (PHZ) into the mice, and at 5 days after the injection, whole spleen cells that mainly consisted of Ter119-positive erythroblasts (here referred to as PHZ-SP cells) were subjected to 3C analysis. When the 5′ CTCF site was used as the anchor fragment, the site interacted with both the 3′ end of the hGLOD5 gene and the 3′ end of the hGATA1 gene (Fig. 8C, top), showing very good agreement with the results obtained with K562 cells (Fig. 8B).
To further confirm the higher-order chromatin architecture of the hGATA1 transgene locus in the spleen cells, we changed the anchor fragment to the 3′ end of the hGLOD5 gene. The 3′ end of the hGLOD5 locus closely interacted with the 5′ CTCF site as well as with the 3′ end of the hGATA1 gene (Fig. 8C, bottom). Collectively, these results suggest that the 5′ CTCF site organizes the 3-dimensional structure of the hGATA1 locus by interacting with the 3′ end regions of the hGLOD5 and hGATA1 genes.
To verify the contribution of the 5′ CTCF site to the formation of the chromatin architecture, we generated two lines of hG1B transgenic mice in which the 5′ CTCF site was mutated by nucleotide substitution (CTCFmut-hG1B) (Fig. 8A, CTCFmut). We conducted qGPCR analyses for five different regions encompassing the region from kb −29 to kb +27 in the CTCFmut-hG1B locus, and the analyses demonstrated that one line harbors 1 copy of the CTCFmut-hG1B transgene, while the other line carries 3 copies of the CTCFmut-hG1B transgene (data not shown). CTCF ChIP-qPCR analysis using PHZ-SP cells from the wild-type hG1B mouse line (line 762 with 1 copy of the CTCFmut-hG1B transgene) indeed demonstrated CTCF binding to the 5′ and 3′ CTCF sites in the hGATA1 locus of the hG1B transgene, as in K562 cells (Fig. 8D). Furthermore, we found that CTCF binding to the mutated 5′ CTCF site was significantly lower in PHZ-SP cells from CTCFmut-hG1B transgenic mice (1 copy) than in those from wild-type hG1B transgenic mice (Fig. 8D). These results thus indicate that the substitution mutation of the 5′ CTCF site successfully eliminated CTCF binding to this region.
To assess whether the substitution mutation of the 5′ CTCF site alters the higher-order chromatin architecture around the hGATA1 locus in spleen cells, we also conducted the same set of 3C analyses using PHZ-SP cells from mice with 1 copy of the CTCFmut-hG1B transgene and examined the higher-order chromatin architecture around the hGATA1 locus. Of note, mutation of the 5′ CTCF site significantly reduced the chromatin interaction between the 5′ CTCF site and the 3′ end of the hGLOD5 gene (Fig. 8C, top, gray line), while reduction of the interaction between the anchor site and the 3′ end the hGATA1 gene was not apparent.
In the latter case, we rather observed an artificial rise in the 3′ adjacent primer region. Therefore, we changed the anchor fragment to the 3′ end of the hGLOD5 gene and found that the chromatin loops with the 5′ CTCF site and with the 3′ end of the hGATA1 gene were both significantly reduced (Fig. 8C, bottom, gray line). These results thus indicate that mutation of the 5′ CTCF site disrupts the chromosomal interactions between the 5′ CTCF site and the 3′ ends of both the hGLOD5 and hGATA1 genes.
The 5′ CTCF site-based chromatin architecture is important for hGATA1 expression in splenic erythroblasts under conditions of hemolytic anemia.
To assess whether the change in the higher-order chromatin architecture affects hGATA1 gene expression or not, we next examined the hGATA1 expression levels directed by the CTCFmut-hG1B transgene in PHZ-SP cells from CTCFmut-hG1B transgenic mice. We compared the results with those for wild-type hG1B transgenic mice harboring the equivalent copy number (line 762 for 1 copy and line 761 for 3 copies) and found that the PHZ-SP cells from the CTCFmut-hG1B transgenic mice with 1 copy of the hG1B transgene showed a reduced level of hGATA1 expression in comparison with that in wild-type mice with 1 copy of the hG1B transgene (Fig. 9A, left). The result was reproducible in the analyses of mice harboring 3 copies of the transgene (Fig. 9B, left).
FIG 9.
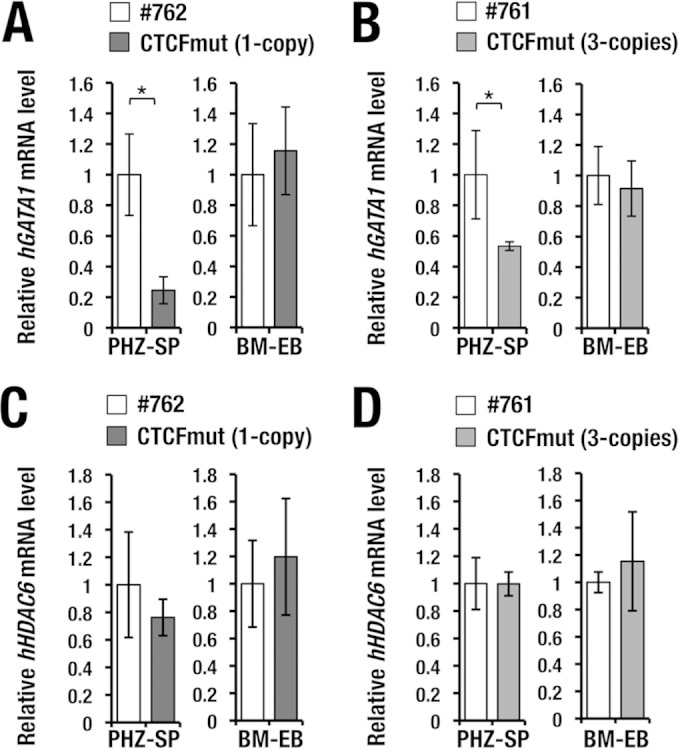
The 5′ CTCF site is important for hGATA1 expression in splenic erythroblasts. (A and B) Relative expression of hGATA1 mRNA in splenic erythroblasts under conditions of stress erythropoiesis and bone marrow cells under homeostatic conditions. Note that the level of hGATA1 mRNA expression is decreased in the splenic erythroblasts from CTCFmut-hG1B mice after PHZ treatment (PHZ-SP), while hGATA1 expression in the bone marrow erythroblasts (BM-EBs) is maintained in CTCFmut-hG1B mice. The result is reproducible in the analyses of mice harboring 1 copy (A) and 3 copies (B) of the CTCFmut-hG1B and wild-type transgenes. (C and D) Levels of hHDAC6 mRNA expression in BM-EBs and PHZ-SP cells. Note that the levels of hHDAC6 mRNA expression in BM-EBs and PHZ-SP cells are not changed in wild-type hG1B and CTCFmut-hG1B transgenic mice in the comparison of mice harboring 1 copy (C) or 3 copies (D) of the transgenes. mRNA levels were normalized to GAPDH levels. PHZ-SP cells and BM-EBs from 10 mice of each genotype were subjected to analysis. Statistically significant differences are depicted (*, P < 0.05, Student's unpaired t test).
To assess whether erythroblasts show a similar difference under homeostatic conditions, we also examined the hGATA1 expression levels directed by the CTCFmut-hG1B transgene in bone marrow Ter119-positive erythroblasts (BM-EBs). Here, we found that the hGATA1 expression levels in BM-EBs from CTCFmut-hG1B mice were comparable to those in BM-EBs from wild-type hG1B transgenic mice harboring an equivalent number of copies of the transgene (Fig. 9A and B, right). Importantly, hHDAC6 expression levels were not changed in either BM-EBs or PHZ-SP cells from CTCFmut-hG1B transgenic mice (the lines with 1 copy and 3 copies of the hG1B transgene) compared with those in the same cell types from wild-type hG1B transgenic mice harboring the equivalent numbers of copies of the hG1B transgene (the 762 and 761 lines; Fig. 9C and D). We also examined hGLOD5 gene expression, but expression of this gene was very low or undetectable in the bone marrow and spleen, and the mutation did not affect the expression profile (data not shown). Thus, these results indicate that the mutation in the 5′ CTCF site specifically affects hGATA1 expression in splenic erythroblasts under conditions of anemia.
Based on these results, we propose that the 5′ CTCF site is required for generation of the correct chromatin architecture through chromosomal interaction with the 3′ ends of the hGLOD5 and hGATA1 genes. This 5′ CTCF site-mediated chromatin architecture in the hGATA1 locus appears to be important for induction of hGATA1 expression in splenic erythroblasts under conditions of stress erythropoiesis. It is not required for the expression of adjacent genes, nor does it affect hGATA1 expression in erythroblasts under homeostatic conditions.
DISCUSSION
In the present study, we found that the profiles of hGATA1 mRNA expression directed by a BAC-based hG1B transgene nicely coincide with those of endogenous mGATA1 mRNA expression during erythroid differentiation in a series of transgenic mice. The physiological level of hGATA1 expression in transgenic mice with a single copy of hG1B rescues the lethal erythroid deficiency in Gata1.05 knockdown mutant mice, indicating that the in vivo activity of hGATA1 is comparable to that of endogenous mGATA1. While the 8.5-kb orthologous G1HRD region is highly conserved between the hGATA1 and mGata1 genes (8–10), flanking regulatory sequences beyond the G1HRD region of these two genes show significant divergence. Therefore, the coincidence in the expression profiles of the hGATA1 transgene and the endogenous mGata1 gene in hG1B transgenic mice seems to be attributable to the conservation of the core cis-regulatory elements located in these orthologous G1HRD regions. In this study, we asked what the more distantly located cis-regulatory regions might contribute to hGATA1 gene expression.
In this regard, we have noticed in our previous studies that expression of the mGata1 BAC transgene tends to show resistance to influences from the integration site of the transgene or PEV (12). We surmised that this might be due to the presence of insulators or related cis-regulatory activity within the locus. When we examined CTCF-binding sites in both the hGATA1 and mGata1 genes, we found that there are multiple CTCF-binding sites in the mGata1 and hGATA1 loci. However, the distribution patterns of the CTCF-binding peaks are quite different from each other; the hGATA1 locus harbors high CTCF-binding peaks at kb −29, +21, +33, and +50 in the distal flanking region, whereas the mGata1 gene carries larger numbers of CTCF-binding peaks distributed throughout the gene body and flanking regions (Fig. 5). This observation led us to examine whether CTCF-based chromatin architectures are formed in the hGATA1 locus and whether the architectures contribute to the expression of the hGATA1 gene. Our present results unequivocally demonstrate that the hGATA1 BAC transgene harbors resistance against PEV in the transgenic mouse experiments. The surrounding genome sequences of the two obvious CTCF-binding sites (i.e., the 5′ and 3′ CTCF sites) in the hGATA1 locus are not evolutionarily conserved between the mouse and human genomes, suggesting that the chromatin architectures organized by the CTCF sites are significantly different between the hGATA1 and mGata1 loci.
The regulation of the mGata1 gene during mouse erythropoiesis has been extensively studied. Accumulating lines of evidence demonstrate that mGata1 is under elaborate regulation, organized by a GATA factor network that employs multiple cis-acting GATA motifs and epigenetic controls (reviewed in references 1, 9, and 15). The murine Gata1 gene also contains two first exons that are differentially utilized in distinct tissues (4, 6). In contrast, regulation of the hGATA1 gene has not been studied intensively. A comparative study of the genome sequences and epigenetic marks of the hGATA1 and mGata1 loci has been reported (18), but there remain a number of questions as to how the hGATA1 gene is regulated and how the regulation of the hGATA1 gene is diverged from or conserved with that of mGata1.
A recent series of integrative studies from the ENCODE Consortium has revealed that approximately 50% of transcription factor binding sites in cis-regulatory elements of the mouse genome are evolutionally diverged from the orthologous cis-regulatory elements in the human genome (39, 40). It has been shown that the patterns of erythroid-affiliated gene expression in erythroblasts are significantly diverged between humans and mice during genetic evolution (41). Thus, it seems reasonable that the regulatory mechanisms governing the hGATA1 gene are largely diverged from those governing the mGata1 gene. We need to place a high priority on investigation of the regulatory mechanisms governing the hGATA1 gene to better understand the development and differentiation of the human hematopoietic system and the pathogenesis arising from perturbation of the system. We believe that this study represents a first step in such an investigation.
We found that the 5′ CTCF site is crucial for the formation of the correct chromosomal architecture around the hGATA1 locus in splenic erythroblasts under stress erythropoiesis conditions (summarized in Fig. 10, left). In this regard, it is interesting to note that the mouse spleen has a distinct hematopoietic microenvironment that plays a crucial role in stress erythropoiesis (42). A recent report showed that mGata1 expression is more severely repressed in splenic CD71high erythroblasts than in the corresponding population in the bone marrow of Gata1low mutant mice in which G1HE/HS1, a potent cis-acting enhancer element in the Gata1 gene, is deleted from the endogenous locus (43). Showing very good agreement with these observations, the findings of our present study show that hGATA1 gene expression is significantly diminished in splenic erythroblasts upon the loss of the chromosome architecture in CTCFmut-hG1B transgenic mice under conditions of hemolytic anemia. This observation suggests that the compromised cis-regulatory network predominantly diminishes mGata1 expression in the spleen of anemic mice rather than steady-state mGata1 expression in the bone marrow. We surmise that the higher-order chromatin architecture plays a more crucial role for enhancement of hGATA1 expression driven by multiple enhancers in the spleen of anemic mice than for homeostatic hGATA1 expression in bone marrow at steady state in hG1B transgenic mice.
FIG 10.

Contribution of the 5′ CTCF site to the higher-order chromatin structure in the hGATA1 locus. (Left) CTCF binds to the 5′ CTCF site, localized 5′ to the hGLOD5 gene, and forms a chromatin hub through forming a looping structure with the 3′ end of the hGLOD5 gene and the 3′ end of the hGATA1 gene in the wild-type hGATA1 locus. (Right) When the 5′ CTCF site is mutated, the 5′ end of the hGATA1 gene is separated from the chromatin hub, and thereafter, hGATA1 expression is diminished in splenic erythroblasts. The hHDAC6 gene localized outside the loop is not under the direct regulatory influence of the 5′ CTCF site.
We suggest that in the hGATA1 locus, once the chromatin interaction between the 5′ CTCF site and the 3′ end of the hGLOD5 gene is disrupted by the substitution mutation of the 5′ CTCF site, the core cis-regulatory elements in the hGATA1 locus become open and can be interfered with by negative cis-regulatory influences outside the locus (Fig. 10, right). In this model, the hHDAC6 gene appears to be free from the regulatory influence of the 5′ CTCF site-mediated chromatin environment, as the hHDAC6 gene is localized beyond the major chromatin loop formed around the hGLOD5 and hGATA1 loci (Fig. 10, left). Indeed, our results indicate that hHDAC6 gene expression is not affected by the activity of the 5′ CTCF site.
A number of studies highlight the tight correlation between the altered activity or the diminished expression level of hGATA1 and clinical hematopoietic disorders, including inherited anemia, thrombocytopenia, and myelofibrosis (reviewed in references 1 and 44). To understand the underlying mechanisms, we have established a mouse model in which hematopoietic differentiation is promoted by the hGATA1 expressed from the transgenic hGATA1 gene. In this study, taking advantage of the hG1B transgenic mouse system, we demonstrate that the hGATA1 expression level is significantly reduced in the spleen of CTCFmut-hG1B transgenic mice under conditions of anemic stress. The result suggests that mutations in the chromatin insulator sequences may lead to alterations in hGATA1 expression levels in human. Studies on regulatory single nucleotide polymorphisms (rSNPs) have now come of age. rSNPs (or other types of polymorphisms) in the insulator sequences of the hGATA1 locus may result in variations in the expression level of hGATA1, leading to enhanced susceptibility to hematopoietic disorders.
In summary, we propose that studies of hGATA1 gene-specific regulatory mechanisms are important avenues to achieve further understanding of human hematopoiesis and its disorders. The hG1B transgenic mouse system will serve as an important model that enables us to examine lineage- and stage-specific hGATA1 gene expression in hematopoietic tissues. The hG1B-based transgenic rescue system of mGata1-deficient mice will provide a reliable platform for examining the functional importance of cis-regulatory sequences of the hGATA1 gene in vivo.
ACKNOWLEDGMENTS
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) (KAKENHI 22118001 and 24249015 to M.Y. and 24590371 to T.M.), the Core Research for Evolutionary Science and Technology (CREST) research program of the Japan Science and Technology Agency (to M.Y.), and the Naito Foundation, Mitsubishi Foundation, and Takeda Science Foundation (to M.Y.).
All the ChIP-seq data were derived from the ENCODE project. We thank the Biomedical Research Core of the Tohoku University Graduate School of Medicine for its technical support. We thank Maggie Walmsley Patient for critical review of the manuscript.
REFERENCES
- 1.Moriguchi T, Yamamoto M. 2014. A regulatory network governing Gata1 and Gata2 gene transcription orchestrates erythroid lineage differentiation. Int J Hematol 100:417–424. doi: 10.1007/s12185-014-1568-0. [DOI] [PubMed] [Google Scholar]
- 2.Kaneko H, Shimizu R, Yamamoto M. 2010. GATA factor switching during erythroid differentiation. Curr Opin Hematol 17:163–168. doi: 10.1097/MOH.0b013e32833800b8. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira R, Ohneda K, Yamamoto M, Philipsen S. 2005. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol 25:1215–1227. doi: 10.1128/MCB.25.4.1215-1227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito E, Toki T, Ishihara H, Ohtani H, Gu L, Yokoyama M, Engel JD, Yamamoto M. 1993. Erythroid transcription factor GATA-1 is abundantly transcribed in mouse testis. Nature 362:466–468. doi: 10.1038/362466a0. [DOI] [PubMed] [Google Scholar]
- 5.Yomogida K, Ohtani H, Harigae H, Ito E, Nishimune Y, Engel JD, Yokoyama M. 1994. Developmental stage- and spermatogenic cycle-specific expression of transcription factor GATA-1 in mouse Sertoli cells. Development 120:1759–1766. [DOI] [PubMed] [Google Scholar]
- 6.Onodera K, Yomogida K, Suwabe N, Takahashi S, Muraosa Y, Hayashi N, Ito E, Gu L, Rassoulzadegan M, Engel JD, Yamamoto M. 1997. Conserved structure, regulatory elements, and transcriptional regulation from the GATA-1 gene testis promoter. J Biochem 121:251–263. doi: 10.1093/oxfordjournals.jbchem.a021581. [DOI] [PubMed] [Google Scholar]
- 7.Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, de Bruijn M, Langeveld A, Gribnau J, Grosveld F, Philipsen S. 2000. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature 406:519–524. doi: 10.1038/35020086. [DOI] [PubMed] [Google Scholar]
- 8.Onodera K, Takahashi S, Nishimura S, Ohta J, Motohashi H, Yomogida K, Hayashi N, Engel JD, Yamamoto M. 1997. GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc Natl Acad Sci U S A 94:4487–4492. doi: 10.1073/pnas.94.9.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto M, Takahashi S, Onodera K, Muraosa Y, Engel JD. 1997. Upstream and downstream of erythroid transcription factor GATA-1. Genes Cells 2:107–115. doi: 10.1046/j.1365-2443.1997.1080305.x. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura S, Takahashi S, Kuroha T, Suwabe N, Nagasawa T, Trainor C, Yamamoto M. 2000. A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol Cell Biol 20:713–723. doi: 10.1128/MCB.20.2.713-723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trainor CD, Omichinski JG, Vandergon TL, Gronenborn AM, Clore GM, Felsenfeld G. 1996. A palindromic regulatory site within vertebrate GATA-1 promoters requires both zinc fingers of the GATA-1 DNA-binding domain for high-affinity interaction. Mol Cell Biol 16:2238–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki M, Moriguchi T, Ohneda K, Yamamoto M. 2009. Differential contribution of the Gata1 gene hematopoietic enhancer to erythroid differentiation. Mol Cell Biol 29:1163–1175. doi: 10.1128/MCB.01572-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu R, Hasegawa A, Ottolenghi S, Ronchi A, Yamamoto M. 2013. Verification of the in vivo activity of three distinct cis-acting elements within the Gata1 gene promoter-proximal enhancer in mice. Genes Cells 18:1032–1041. doi: 10.1111/gtc.12096. [DOI] [PubMed] [Google Scholar]
- 14.Moriguchi T, Suzuki M, Yu L, Takai J, Ohneda K, Yamamoto M. 2015. Progenitor stage-specific activity of a cis-acting double GATA motif for Gata1 gene expression. Mol Cell Biol 35:805–815. doi: 10.1128/MCB.01011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohneda K, Shimizu R, Nishimura S, Muraosa Y, Takahashi S, Engel JD, Yamamoto M. 2002. A minigene containing four discrete cis elements recapitulates GATA-1 gene expression in vivo. Genes Cells 7:1243–1254. doi: 10.1046/j.1365-2443.2002.00595.x. [DOI] [PubMed] [Google Scholar]
- 16.Vyas P, McDevitt MA, Cantor AB, Katz SG, Fujiwara Y, Orkin SH. 1999. Different sequence requirements for expression in erythroid and megakaryocytic cells within a regulatory element upstream of the GATA-1 gene. Development 126:2799–2811. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu R, Takahashi S, Ohneda K, Engel JD, Yamamoto M. 2001. In vivo requirements for GATA-1 functional domains during primitive and definitive erythropoiesis. EMBO J 20:5250–5260. doi: 10.1093/emboj/20.18.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valverde-Garduno V, Guyot B, Anguita E, Hamlett I, Porcher C, Vyas P. 2004. Differences in the chromatin structure and cis-element organization of the human and mouse GATA1 loci: implications for cis-element identification. Blood 104:3106–3116. doi: 10.1182/blood-2004-04-1333. [DOI] [PubMed] [Google Scholar]
- 19.Drissen R, Guyot B, Zhang L, Atzberger A, Sloane-Stanley J, Wood B, Porcher C, Vyas P. 2010. Lineage-specific combinatorial action of enhancers regulates mouse erythroid Gata1 expression. Blood 115:3463–3471. doi: 10.1182/blood-2009-07-232876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takai J, Moriguchi T, Suzuki M, Yu L, Ohneda K, Yamamoto M. 2013. The Gata1 5′ region harbors distinct cis-regulatory modules that direct gene activation in erythroid cells and gene inactivation in HSCs. Blood 122:3450–3460. doi: 10.1182/blood-2013-01-476911. [DOI] [PubMed] [Google Scholar]
- 21.Gutiérrez L, Tsukamoto S, Suzuki M, Yamamoto-Mukai H, Yamamoto M, Philipsen S, Ohneda K. 2008. Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood 111:4375–4385. doi: 10.1182/blood-2007-09-115121. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi S, Onodera K, Motohashi H, Suwabe N, Hayashi N, Yanai N, Nabesima Y, Yamamoto M. 1997. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J Biol Chem 272:12611–12615. doi: 10.1074/jbc.272.19.12611. [DOI] [PubMed] [Google Scholar]
- 23.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. 2001. Ineffective erythropoiesis in Stat5a(−/−) 5b(−/−) mice due to decreased survival of early erythroblasts. Blood 98:3261–3273. doi: 10.1182/blood.V98.12.3261. [DOI] [PubMed] [Google Scholar]
- 24.Akashi K, Traver D, Miyamoto T, Weissman IL. 2000. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 25.Minegishi N, Suzuki N, Kawatani Y, Shimizu R, Yamamoto M. 2005. Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes Cells 10:693–704. doi: 10.1111/j.1365-2443.2005.00864.x. [DOI] [PubMed] [Google Scholar]
- 26.Ainoya K, Moriguchi T, Ohmori S, Souma T, Takai J, Morita M, Chandler KJ, Mortlock DP, Shimizu R, Engel JD, Lim KC, Yamamoto M. 2012. UG4 enhancer-driven GATA-2 and BMP4 complementation remedies the CAKUT phenotype in Gata2 hypomorphic mutant mice. Mol Cell Biol 32:2312–2322. doi: 10.1128/MCB.06699-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriguchi T, Sakurai T, Takahashi S, Goto K, Yamamoto M. 2002. The human prepro-orexin gene regulatory region that activates gene expression in the lateral region and represses it in the medial regions of the hypothalamus. J Biol Chem 277:16985–16992. doi: 10.1074/jbc.M107962200. [DOI] [PubMed] [Google Scholar]
- 28.Gaszner M, Felsenfeld G. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet 7:703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- 29.Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forne T. 2007. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc 2:1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 30.ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohneda K, Yamamoto M. 2002. Roles of hematopoietic transcription factors GATA-1 and GATA-2 in the development of red blood cell lineage. Acta Haematol 108:237–245. doi: 10.1159/000065660. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki N, Suwabe N, Ohneda O, Obara N, Imagawa S, Pan X, Motohashi H, Yamamoto M. 2003. Identification and characterization of 2 types of erythroid progenitors that express GATA-1 at distinct levels. Blood 102:3575–3583. doi: 10.1182/blood-2003-04-1154. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki M, Kobayashi-Osaki M, Tsutsumi S, Pan X, Ohmori S, Takai J, Moriguchi T, Ohneda O, Ohneda K, Shimizu R, Kanki Y, Kodama T, Aburatani H, Yamamoto M. 2013. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells 18:921–933. doi: 10.1111/gtc.12086. [DOI] [PubMed] [Google Scholar]
- 34.Chung JH, Whiteley M, Felsenfeld G. 1993. A 5′-element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74:505–514. doi: 10.1016/0092-8674(93)80052-G. [DOI] [PubMed] [Google Scholar]
- 35.Bell AC, West AG, Felsenfeld G. 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98:387–396. doi: 10.1016/S0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 36.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 37.Ruthenburg AJ, Allis CD, Wysocka J. 2007. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Ziebarth JD, Bhattacharya A, Cui Y. 2013. CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res 41:D188–D194. doi: 10.1093/nar/gks1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vierstra J, Rynes E, Groudine M, Bender MA, Stamatoyannopoulos JA. 2014. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 346:1007–1012. doi: 10.1126/science.1246426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Y, Ma Z, Kim BH, Wu W, Cayting P, Boyle AP, Sundaram V, Xing X, Dogan N, Li J, Euskirchen G, Lin S, Lin Y, Visel A, Kawli T, Yang X, Patacsil D, Keller CA, Giardine B, Mouse ENCODE Consortium, Kundaje A, Wang T, Pennacchio LA, Weng Z, Hardison RC, Snyder MP. 2014. Principles of regulatory information conservation between mouse and human. Nature 515:371–375. doi: 10.1038/nature13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pishesha N, Thiru P, Shi J, Eng JC, Sankaran VG, Lodish HF. 2014. Transcriptional divergence and conservation of human and mouse erythropoiesis. Proc Natl Acad Sci U S A 111:4103–4108. doi: 10.1073/pnas.1401598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slayton WB, Georgelas A, Pierce LJ, Elenitoba-Johnson KS, Perry SS, Marx M, Spangrude GJ. 2002. The spleen is a major site of megakaryopoiesis following transplantation of murine hematopoietic stem cells. Blood 100:3975–3982. doi: 10.1182/blood-2002-02-0490. [DOI] [PubMed] [Google Scholar]
- 43.Migliaccio AR, Martelli F, Verrucci M, Sanchez M, Valeri M, Migliaccio G, Vannucchi AM, Zingariello M, Di Baldassarre A, Ghinassi B, Rana RA, van Hensbergen Y, Fibbe WE. 2009. Gata1 expression driven by the alternative HS2 enhancer in the spleen rescues the hematopoietic failure induced by the hypomorphic Gata1low mutation. Blood 114:2107–2120. doi: 10.1182/blood-2009-03-211680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu R, Engel JD, Yamamoto M. 2008. GATA1-related leukaemias. Nat Rev Cancer 8:279–287. doi: 10.1038/nrc2348. [DOI] [PubMed] [Google Scholar]