

Abstract Abstract
Decreased synthesis of nitric oxide (NO) by NO synthases (NOS) is believed to play an important role in the pathogenesis of pulmonary arterial hypertension (PAH). Multiple factors may contribute to decreased NO bioavailability, including increased activity of arginase, the enzyme that converts arginine to ornithine and urea, which may compete with NOS for arginine; inadequate de novo arginine production from citrulline; and increased concentration of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NOS. We hypothesized that PAH patients with the lowest arginine availability secondary to increased arginase activity and/or inadequate de novo arginine synthesis might have a slower rate of NO synthesis and greater pulmonary vascular resistance. Nine patients with group 1 PAH and 10 healthy controls were given primed, constant intravenous infusions of 15N2-arginine, 13C,2H4-citrulline, 15N2-ornithine, and 13C-urea in the postabsorptive state. The results showed that, compared with healthy controls, PAH patients had a tendency toward increased arginine clearance and ornithine flux but no difference in arginine and citrulline flux, de novo arginine synthesis, or NO synthesis. Arginine-to-ADMA ratio was increased in PAH patients. Two endotypes of patients with low and high arginase activity were identified; compared with the low-arginase group, the patients with high arginase had increased arginine flux, slower NO synthesis, and lower plasma concentrations of ADMA. These results demonstrate that increased breakdown of arginine by arginase occurs in PAH and affects NO synthesis. Furthermore, there is no compensatory increase in de novo arginine synthesis to overcome this increased utilization of arginine by arginase.
Keywords: nitric oxide, arginase, asymmetric dimethylarginine, ornithine
Introduction
Pulmonary arterial hypertension (PAH) is a disease of unknown etiology characterized by an increase in pulmonary vascular resistance (PVR) secondary to pulmonary arterial vasoconstriction, vascular remodeling, and thrombosis. Decreased bioavailability of the vasodilatory molecule nitric oxide (NO) is believed to play an important role in the pathogenesis of PAH.1-3 In support of this, intrapulmonary NO, biochemical reaction products of NO in bronchoalveolar fluid, and exhaled NO have been found to be decreased in PAH patients compared with controls.3,4 NO is produced in the body by a family of enzymes known as NO synthases (NOS). In a reaction that consumes molecular oxygen and cofactors such as nicotinamide adenine dinucleotide phosphate and tetrahydrobiopterin (BH4), NOS converts l-arginine to l-citrulline while simultaneously producing NO and water.5 Both reduced and increased expression of NOS have been reported in the pulmonary arterial endothelium of PAH patients compared with controls,1,6 suggesting that NOS expression does not necessarily reflect activity of the enzyme.
The synthesis of NO via NOS depends on the availability of substrate and cofactors as well as the presence of endogenous inhibitors of the enzyme. In humans, the major source of arginine is whole-body protein breakdown. De novo arginine synthesis occurs in the kidney via a two-step reaction starting with citrulline and with argininosuccinate as an intermediate7 and constitutes 5%–15% of endogenous arginine flux in the fasting state.8 Under conditions of stress or disease states, rates of endogenous arginine synthesis may be insufficient to meet the body’s demands.9,10 Utilization of arginine by other enzymes, particularly arginase, may also decrease its availability for reaction with NOS. Arginases, which exist in two isoforms, convert arginine to ornithine and urea and compete with NOS for arginine.8,11 Arginase I is expressed in the liver and contributes to the majority of the body’s total arginase activity, whereas arginase II is present in most tissues, including the lungs.12 Increased arginase I expression has been implicated in the development of PAH in sickle cell disease,13 while increased arginase II expression has been found in pulmonary endothelial cells of patients with World Health Organization (WHO) class I pulmonary hypertension.12 Endogenous inhibitors of NOS may also affect NO production. The dimethylarginines, including asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA), are synthesized by the methylation of arginine residues in protein, which are released by proteolysis.14 ADMA is the most potent endogenous inhibitor of NOS and competes with l-arginine for active sites on endothelial NOS (eNOS). Both ADMA and SDMA interfere with y+-mediated transport of arginine into cells.14
The relationship among arginases, dimethylarginases, and eNOS is complex and provides a potential explanation for the “arginine paradox.” The arginine paradox describes the phenomenon that increased extracellular arginine is needed to produce maximal NO production despite the fact that both extracellular and intracellular concentrations of arginine far exceed the theoretical Km of the enzyme.15 The ratios of arginine to its catabolic products and to the dimethylarginines may better reflect systemic arginine availability, and thus arginine supplementation under conditions where these ratios are low could improve arginine availability for NO production (Fig. 1).
Figure 1.

Arginine metabolic pathways. Extracellular arginine is transported intracellularly by y+ cationic amino acid transporters. Intracellular arginine is derived from protein breakdown and de novo synthesis from citrulline via argininosuccinate. Nitric oxide synthases (NOS) converts arginine to NO and citrulline. Arginase hydrolyzes arginine to ornithine and urea. Endogenous methylarginines (asymmetric dimethylarginine [ADMA], symmetric dimethylarginine [SDMA]) are produced after proteolysis from methylated arginine residues in protein and inhibit NOS as well as arginine transport. Dotted lines indicate inhibition.
Despite the evidence supporting derangement of arginine and NO metabolism as an important mechanism underlying the development of PAH, in vivo studies comprehensively dissecting the relevant metabolic pathways are lacking. One study found decreased whole-body NO production, as measured by conversion of 15N2-arginine to urinary 15N-nitrite and 15N-nitrate, in 4 patients with primary pulmonary hypertension.16 We hypothesized that PAH patients with the lowest arginine availability, secondary to increased arginase activity and/or inadequate de novo arginine synthesis, might have a slower rate of NO synthesis and greater PVR. We tested this hypothesis by using stable-isotope tracer techniques and biochemical methods to determine the metabolic fates of arginine in PAH patients in comparison with healthy controls and relative to clinical parameters of pulmonary hypertension.
Methods
Subjects
Nine patients with group I pulmonary hypertension as defined by the WHO17 were enrolled from the pulmonary clinics of Baylor College of Medicine and the Cleveland Clinic. For comparison, 10 healthy volunteers without chronic illnesses matched as closely as possible for age and sex were also enrolled. All participants provided written informed consent, and the study protocol and consents were approved by the institutional review boards at both institutions. All PAH patients were on stable medications and had undergone right heart catheterization as part of their standard care. On the day of isotope infusion, a subgroup of participants also had fractional exhaled NO (FeNO) measured by an online method at a constant flow rate of 50 mL s−1 according to the standards published by the American Thoracic Society.18
Isotope infusions
Tracer infusions were performed in all subjects at the Clinical Research Unit of the Cleveland Clinic, the General Clinical Research Center of Baylor College of Medicine, and the Metabolic Research Unit of the Children’s Nutrition Research Center. Sterile solutions of guanidino-15N2-arginine, 13C,2H4-citrulline, 15N-citrulline, 13C-urea, and 15N2-ornithine (Cambridge Isotope Laboratories, Tewksbury, MA) were prepared using strict aseptic techniques and were tested for sterility and lack of pyrogens prior to infusion.
After an 8-hour overnight fast, all participants had an intravenous catheter placed in the antecubital vein for isotope infusions and in a hand vein of the contralateral arm for blood sampling. The hand was heated to arterialize blood samples.19,20 After a baseline blood sample was obtained, primed, continuous, intravenous infusions of 15N2-arginine (prime = 8 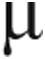 mol kg−1, infusion = 8
mol kg−1, infusion = 8  mol kg−1 h−1), 13C,2H4-citrulline (prime = 1.5
mol kg−1 h−1), 13C,2H4-citrulline (prime = 1.5  mol kg−1, infusion = 1
mol kg−1, infusion = 1  mol kg−1 h−1), 13C-urea (prime = 80
mol kg−1 h−1), 13C-urea (prime = 80  mol kg−1, infusion = 8
mol kg−1, infusion = 8  mol kg−1 h−1), and 15N2-ornithine (prime = 2
mol kg−1 h−1), and 15N2-ornithine (prime = 2  mol kg−1, infusion = 2
mol kg−1, infusion = 2  mol kg−1 h−1) were started and maintained for 6 hours. In addition, a prime of 15N-citrulline (0.16
mol kg−1 h−1) were started and maintained for 6 hours. In addition, a prime of 15N-citrulline (0.16  mol kg−1) was administered to prime the secondary 15N-citrulline pool. Blood samples were obtained every 30 minutes between 4.5 and 6 hours (4 samples) of the isotope infusions.
mol kg−1) was administered to prime the secondary 15N-citrulline pool. Blood samples were obtained every 30 minutes between 4.5 and 6 hours (4 samples) of the isotope infusions.
Sample analyses
Blood samples were drawn into prechilled tubes containing sodium heparin or EDTA. Tubes were centrifuged immediately at 4°C, and the plasma was separated and stored immediately at −70°C for later analysis.
The plasma isotope enrichments of arginine, citrulline, and ornithine were measured by tandem liquid chromatography mass spectrometry. The amino acids were converted into their 5-dimethylamino-1-napthalene sulfonamide derivatives, and ions were analyzed by selected reaction monitoring on a triple-quadrupole mass spectrometer. The transitions observed were precursor ion m/z 410 to product ion 392 for arginine, precursor ion m/z 414 to product ion 397 for citrulline, and precursor ion m/z 601 to product ion 170 for ornithine. The plasma isotope enrichments of urea were measured by gas chromatography mass spectrometry of its n-propyl ester heptafluorobutyramide derivative. The isotope enrichments of plasma NO metabolites, nitrites and nitrates (NOx), were determined by negative chemical ionization mass spectrometry as described elsewhere,21 after nitrate was reduced to nitrite and converted to its pentaflourobenzyl derivative.
Plasma amino acid concentrations were measured by ultraperformance liquid chromatography using precolumn derivitization with 6-amino-quinolyl-N-hydroxysuccinimidyl carbamate. Plasma concentrations of ADMA and NO metabolites were determined by stable isotope dilution with 2H7-ADMA and Na15NO3 as internal standards, as described elsewhere.21,22
Arginase activity
Plasma arginase activity was determined with aliquots of 50  L of plasma using a modification of the radioisotope method.23 Arginase activity was measured by the conversion of 15N2-arginine to 15N2-urea. Concentrations of 15N2-urea at baseline and after reaction were measured using isotope dilution with 13C,15N2-arginine as an internal standard. The unit of arginase activity is defined as microliters of urea per minute.
L of plasma using a modification of the radioisotope method.23 Arginase activity was measured by the conversion of 15N2-arginine to 15N2-urea. Concentrations of 15N2-urea at baseline and after reaction were measured using isotope dilution with 13C,15N2-arginine as an internal standard. The unit of arginase activity is defined as microliters of urea per minute.
Calculations
The rate of appearance or total flux (Q) of arginine, citrulline, ornithine, and urea were calculated from the steady state equation
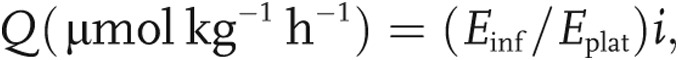 |
where Einf is the isotopic enrichment in the infusate, Eplat is the isotopic enrichment in plasma at isotopic steady state, and i is the infusion rate of the tracer ( mol kg−1 h−1).
mol kg−1 h−1).
Under steady state conditions, the rate of appearance of arginine equals the rate of disappearance. Therefore, arginine clearance is
Because arginine is converted to citrulline and NO at a 1∶1 ratio, the rate of conversion of 15N2-arginine to 15N-citrulline is an index of NO synthesis. Similarly, the conversion of 13C,2H4-citrulline to 13C,2H4-arginine is a measure of de novo arginine synthesis, and the conversion of 15N2-arginine to 15N2-urea is an index of arginase activity. The conversion of precursor to product is calculated by
 |
where Qprod is the flux of the product (citrulline, arginine, or urea), Eprod is the plasma enrichment of the product (m+1 citrulline, m+5 arginine, or m+2 urea), and Epre is the plasma enrichment of the precursor (m+2 arginine, m+5 citrulline, or m+2 arginine).
The fractional synthesis rate (FSR) of NO was calculated according to the precursor-product equation as described elsewhere,21 using the following equation:
 |
where  is the increase in isotopic enrichment of NOx over the period of hours 4.5 and 6 (
is the increase in isotopic enrichment of NOx over the period of hours 4.5 and 6 ( ) of the infusion and
) of the infusion and  is the plateau isotopic enrichment of plasma arginine. In this calculation, the plateau enrichment of arginine in plasma is assumed to represent the enrichment of all the arginine pools from which NO—and, hence, nitrite and nitrate—are synthesized. The absolute synthesis rate (ASR) of plasma NOx in the intravascular compartment was calculated as the product of the plasma NOx concentration and the FSR (
is the plateau isotopic enrichment of plasma arginine. In this calculation, the plateau enrichment of arginine in plasma is assumed to represent the enrichment of all the arginine pools from which NO—and, hence, nitrite and nitrate—are synthesized. The absolute synthesis rate (ASR) of plasma NOx in the intravascular compartment was calculated as the product of the plasma NOx concentration and the FSR ( mol L plasma−1 h−1).
mol L plasma−1 h−1).
Additional cohort
A second cohort of 9 healthy individuals and 9 patients diagnosed with class I PAH between the ages of 18 and 60 years was used to explore the relationship between plasma arginase activity and cardiac function. Plasma arginase activity and plasma amino acid concentrations were measured as described above. Echocardiograms were performed on the PAH patients by a single experienced sonographer, and multiple parameters were measured as described elsewhere.24
Statistical analysis
Continuous variables were summarized by group as means ± SE, unless otherwise indicated. Normality of data was confirmed using the Kolmogorov-Smirnov test. Differences between groups of subjects were assessed by the unpaired Student’s t test. Correlations were performed using Pearson’s correlation (r). Tests were considered statistically significant if P < 0.05. Data analysis was performed using STATA software (ver. 11).
Results
Subject characteristics
PAH patients and controls were similar in age, sex, ethnicity, weight, and body mass index (all P > 0.05; Table 1). All PAH was class 1, and all but 1 patient had idiopathic PAH. PAH patients had high pulmonary arterial pressures (PAPs), PVR, and normal pulmonary arterial occlusion pressures (Table 2). All patients were receiving treatment for PAH at the time of the isotope infusions, and 3 were receiving multiple drug therapy. These therapies included inhaled and intravenous prostanoids (treprostinil and epoprostenol), endothelin antagonists (bosentan and ambrisentan), phosphodiesterase inhibitors (sildenafil), and calcium channel blockers (diltiazem).
Table 1.
Study population
| Controls (n = 10) | PAH (n = 9) | |
|---|---|---|
| Age, years | 37 ± 10 | 43 ± 13 |
| Sex | ||
| Male | 1 | 1 |
| Female | 9 | 8 |
| Race | ||
| White | 6 | 6 |
| Black | 4 | 3 |
| Height, inches | 64.5 ± 3.0 | 63.5 ± 2.6 |
| Weight, kg | 66.0 ± 10.7 | 77.5 ± 19.4 |
| Body mass index | 24.5 ± 2.8 | 29.9 ± 7.6 |
| Mean arterial blood pressure, mmHg | 86 ± 9 | 81 ± 10 |
| Heart rate, beats min−1 | 70 ± 12 | 82 ± 10 |
| Respiratory rate, breaths min−1 | 18 ± 3 | 19 ± 3 |
| O2 saturation, % | 98 ± 1 | 96 ± 4 |
Data are mean ± SD. All P > 0.05. PAH: pulmonary arterial hypertension.
Table 2.
Clinical characteristics of pulmonary arterial hypertension (PAH) patients
| Value | |
|---|---|
| Etiology of group I PAH | |
| Idiopathic | 8 |
| Systemic lupus erythematosus | 1 |
| Central venous pressure, mmHg | 4.8 ± 1.6 |
| Mean pulmonary arterial pressure, mmHg | 42.5 ± 3.0 |
| Pulmonary arterial systolic pressure, mmHg | 67.5 ± 5.7 |
| Pulmonary arterial diastolic pressure, mmHg | 28.8 ± 2.5 |
| Pulmonary arterial opening pressure, mmHg | 10.5 ± 1.6 |
| Cardiac output (thermodilution), L min−1 | 5.1 ± 0.4 |
| Cardiac index (thermodilution), L min−1 | 2.7 ± 0.1 |
| Pulmonary vascular resistance, dyn s cm−5 | 721.6 ± 132.5 |
| Treatmenta | |
| Prostanoids | |
| Injectable | 3/9 |
| Inhaled | 1/9 |
| Endothelin antagonists | 3/9 |
| Phosphodiesterase inhibitors | 3/9 |
| Calcium channel blockers | 3/9 |
Some patients were receiving combination therapy.
Arginine, citrulline, and NO kinetics
Arginine and citrulline kinetics and plasma concentrations were similar among PAH patients and controls (Table 3). There was no difference in arginine or citrulline flux between controls and PAH patients. De novo arginine production and conversion of 15N2-arginine to 15N-citrulline, an index of NO synthesis (Fig. 2), were also similar between PAH patients and controls. Citrulline flux was negatively correlated with mean PAP (mPAP; Table 4), although this did not meet statistical significance. There was also a significant negative correlation between de novo arginine production and cardiac output (Table 4).
Table 3.
Parameters of arginine production
| Controls (n = 10) | PAH (n = 9) | |
|---|---|---|
Arginine flux, 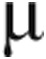 mol kg−1 h−1 mol kg−1 h−1
|
48.6 ± 2.4 | 52.6 ± 4.1 |
Citrulline flux,  mol kg−1 h−1 mol kg−1 h−1
|
8.2 ± 0.5 | 8.7 ± 0.7 |
De novo arginine production,  mol kg−1 h−1 mol kg−1 h−1
|
4.8 ± 0.5 | 5.5 ± 0.6 |
P > 0.05 for all.
Figure 2.

Parameters of nitric oxide (NO) synthesis. The conversion of 15N2-arginine to 15N-citrulline, an index of NO synthesis, was similar in pulmonary arterial hypertension (PAH) patients and controls (P = 0.36; A), as were fractional synthesis rates (FSRs) of 15NOx from 15N2-arginine (P = 0.36; B), but absolute synthesis rates (ASRs) were higher in PAH patients than in controls (P = 0.06; C). NOx: nitrites and nitrates.
Table 4.
Significant correlations between metabolic/kinetic and clinical parameters
| Parameter | |||
|---|---|---|---|
| Metabolic/kinetic | Clinical | r | P |
| Citrulline flux | Mean pulmonary arterial pressure | −0.632 | 0.07 |
| De novo arginine production | Cardiac output | −0.744 | 0.03 |
| NOx absolute synthesis rate | Mean pulmonary arterial pressure | 0.665 | 0.05 |
| Ornithine flux | FeNO | −0.600 | 0.07 |
| Conversion of arginine to urea | Mean pulmonary arterial pressure | −0.674 | 0.047 |
NOx: nitrites and nitrates; FeNO: fractional exhaled nitric oxide.
Plasma NOx concentration was not different between PAH patients and controls (PAH = 32.0 ± 4.6  M, controls = 30.8 ± 4.0
M, controls = 30.8 ± 4.0 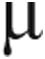 M; P = 0.84). The FSR of 15NOx from 15N2-arginine was also similar between the groups, but the ASR was higher in PAH patients than in controls, although this did not meet statistical significance (Fig. 3). There was a significant positive correlation between ASR and mPAP (Table 4).
M; P = 0.84). The FSR of 15NOx from 15N2-arginine was also similar between the groups, but the ASR was higher in PAH patients than in controls, although this did not meet statistical significance (Fig. 3). There was a significant positive correlation between ASR and mPAP (Table 4).
Figure 3.

Arginine clearance and catabolism. Compared with controls, pulmonary arterial hypertension (PAH) patients had a tendency toward greater arginine clearance (P = 0.07; A) and a tendency toward increased arginine catabolism by arginase as indicated by ornithine flux (P = 0.08; B). Although there was no difference in arginase activity as measured by conversion of 15N2-arginine to 15N2-urea (P = 0.78; C), high and low arginine metabolic endotypes of PAH are suggested by the data.
FeNO
In the subgroup of participants who underwent FeNO testing, there was no difference in FeNO between PAH patients and controls (PAH = 14.4 ± 2.8 ppb, controls = 17.7 ± 4.7 ppb; P = 0.56). There were positive correlations between FeNO and arginine plasma concentration (r = 0.74, P = 0.02) and between FeNO and the global arginine availability ratio (r = 0.778, P = 0.008). There was also a positive correlation between FeNO and cardiac index (r = 0.959, P = 0.04) and a negative correlation between FeNO and ornithine flux, although this did not meet statistical significance (Table 4). There was also a significant correlation between FeNO and the rate of NO synthesis as measured by conversion of 15N2-arginine to 15N-citrulline (r = 0.772, P = 0.009).
The arginine metabolome: arginine, ornithine, citrulline, and ADMA
Plasma concentrations of arginine, citrulline, and ornithine were similar among healthy controls and PAH patients (Table 5). When arginine availability was evaluated using the ratio of arginine to products generated from catabolism (Arg/[Cit + Orn]), arginine availability was 30% lower in PAH patients, although the difference was not statistically significant (P = 0.12). Plasma ADMA also tended to be higher in PAH patients (P = 0.07), and arginine availability as determined by Arg/ADMA was significantly higher in controls than in PAH patients (P = 0.02). Citrulline concentration was inversely related to the pulmonary arterial diastolic pressure and mPAP (Table 4).
Table 5.
Plasma concentrations of arginine, citrulline, ornithine, and methylated arginine (ADMA) and relative arginine availability
| Controls (n = 10) | PAH (n = 9) | |
|---|---|---|
Arginine, 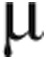 M M |
80.0 ± 6.4 | 76.2 ± 5.3 |
Citrulline,  M M |
31.0 ± 2.0 | 37.2 ± 2.2 |
Ornithine,  M M |
58.8 ± 4.7 | 65.6 ± 5.7 |
| Arg/(Cit + Orn) | 0.92 ± 0.08 | 0.71 ± 0.09 |
ADMA,  M M |
0.48 ± 0.04 | 0.61 ± 0.06 |
| Arg/ADMA | 174.1 ± 16.7 | 119.7 ± 12.3a |
P < 0.05 compared with controls.
Arginase activity
Because ornithine and urea are products of arginine hydrolysis by arginase, their rates of production reflect arginase activity. The rate of conversion of 15N2-arginine to 15N2-urea is also an index of arginase activity. There was a trend toward increased ornithine flux in PAH patients (P = 0.08), but there was no such trend for the conversion of arginine to urea (P = 0.78; Fig. 3). Arginine clearance reflects the rate of removal of arginine from the plasma compartment, and there was a trend toward increased arginine clearance in PAH patients (P = 0.07). Plasma arginase activity measured in vitro was not significantly different between the two groups (PAH = 0.435  mol urea min−1, controls = 0.464
mol urea min−1, controls = 0.464  mol urea min−1; P = 0.73).
mol urea min−1; P = 0.73).
There was a strong correlation between the different measurements of arginase pathway activity (Fig. 4). There was a significant negative correlation between conversion of arginine to urea and mPAP (Table 4). On further inspection of the data, the conversion of arginine to urea was clearly clustered into two groups among PAH patients: 4 patients had low conversion (less than mean −1 SD of controls), and 5 patients had high conversion, that is, low and high arginase activity.
Figure 4.

Correlations among measures of arginine catabolism. Arginine clearance and catabolism are strongly related to measures of ornithine flux. Circles indicate controls, and triangles indicate pulmonary arterial hypertension patients.
Hemodynamic and kinetic parameters by arginase activity
Because PAH patients appeared to be divided into those with low and those with high arginase activity, hemodynamic and kinetic parameters were analyzed in these two endotypes. PAH patients with high arginase activity had higher arginine flux, lower NO synthesis, lower ASR of NOx, and lower plasma concentration of ADMA (Fig. 5). There was no difference in PAPs (mean [P = 0.15], systolic [P = 0.29], or diastolic [P = 0.19]), PVR (P = 0.44), cardiac index (P = 0.54), or cardiac output (P = 0.88) between the two groups.
Figure 5.

Arginine metabolic endotypes of pulmonary arterial hypertension (PAH). A subgroup of PAH patients with high arginase activity had greater arginine flux (P = 0.08; A), less nitric oxide (NO) synthesis measured by conversion of 15N2-arginine to 15N-citrulline (P = 0.05; B), less NO synthesis measured by conversion of 15N2-arginine to 15NOx (P = 0.02; C), and lower levels of asymmetric dimethylarginine (ADMA; P = 0.04; D). Asterisks indicate P ≤ 0.05 compared with the low-arginase group. NOx: nitrites and nitrates; ASR: absolute synthesis rate.
Additional cohort
In the additional cohort of 9 PAH patients and 9 controls, there was a trend toward higher plasma arginase activity in PAH patients than in controls (PAH = 0.78 ± 0.05  mol urea min−1, controls = 0.65 ± 0.05
mol urea min−1, controls = 0.65 ± 0.05 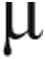 mol urea min−1; P = 0.10). There were no differences in plasma concentrations of arginine, citrulline, or ornithine between the groups, but there was a significantly lower global arginine availability ratio in PAH patients than in controls (PAH = 0.62 ± 0.8, controls = 0.88 ± 0.09; P = 0.04). There were no significant correlations between plasma amino acid levels or arginase activity with echocardiographic measurements, including cardiac output, cardiac index, right atrial pressure, right ventricular systolic pressure, tricuspid regurgitant velocity, or PVR.
mol urea min−1; P = 0.10). There were no differences in plasma concentrations of arginine, citrulline, or ornithine between the groups, but there was a significantly lower global arginine availability ratio in PAH patients than in controls (PAH = 0.62 ± 0.8, controls = 0.88 ± 0.09; P = 0.04). There were no significant correlations between plasma amino acid levels or arginase activity with echocardiographic measurements, including cardiac output, cardiac index, right atrial pressure, right ventricular systolic pressure, tricuspid regurgitant velocity, or PVR.
Discussion
There is substantial evidence that NO deficiency is an important mechanism underlying the development of PAH, although the specific metabolic derangements contributing to decreased NO bioavailability are not completely understood.25 This is the first study, to our knowledge, to use stable isotope tracer techniques to characterize the production and catabolism of arginine and its relationship to NO synthesis in pulmonary hypertension, thereby increasing our knowledge of the role played by arginine metabolism in the disease process. The results demonstrate that there is decreased arginine bioavailability in patients with pulmonary hypertension as a result of increased catabolism by arginase and higher concentrations of ADMA. The global arginine bioavailability ratio (the ratio of arginine to ornithine plus citrulline) has been shown to be associated with mortality in patients with sickle cell disease and pulmonary hypertension.13 This study similarly found decreased arginine bioavailability measured by low global arginine availability ratio in two separate cohorts of PAH patients compared with controls but also substantiates this finding with kinetic measurements showing faster arginine clearance and ornithine production in PAH patients. Furthermore, there is decreased NO synthesis in the subgroup of patients with high compared with low arginase activity, demonstrating that arginase activity regulates arginine availability for NOS.
Alterations in arginase (specifically arginase II) expression and activity have been implicated in the pathophysiology of pulmonary hypertension. Rats with pulmonary hypertension induced by monocrotaline have significantly accelerated arginase activity in their pulmonary arterial endothelial cells, along with decreased NOS activity and expression.26 Similarly, human endothelial cells isolated from PAH patients have increased expression of arginase II and lower NO production than endothelial cells from controls.12 On a systemic level, these PAH patients also had higher serum arginase activity and a lower arginine-to-ornithine ratio. This is the first study to confirm these findings in vivo. A study by Castillo et al.27 first established the use of stable isotope techniques to characterize in vivo arginine catabolism and demonstrated the importance of arginine degradation in the regulation of arginine availability. When healthy adult men were given an arginine-free diet, there was a significant decline in ornithine flux as well as arginine conversion to ornithine in an effort to conserve body arginine. A study by the same authors in burn patients subsequently found that plasma arginine to ornithine conversion constitutes only 7% of total urea production,28 indicating that the majority of urea flux is derived from turnover of arginine via the urea cycle in the liver rather than plasma conversion of arginine to urea. Ornithine flux, on the other hand, is a measure of non-urea-cycle arginine metabolism and serves as a surrogate for arginine turnover and catabolism, since changes in arginine turnover and oxidation were paralleled by changes in ornithine flux. In this study, we found that PAH patients and controls had similar urea flux, and, in line with the prior tracer studies, plasma conversion of arginine to urea constituted 7.8% of urea production in the healthy controls. However, the PAH patients had a tendency toward increased ornithine flux and arginine clearance, two parameters that were highly correlated. Furthermore, ornithine flux was also significantly correlated with in vitro plasma arginase activity, and findings of increased in vitro arginase activity were confirmed in a second cohort. Taken together, these results indicate that there is increased catabolism of arginine by arginase in PAH. The finding that ornithine but not urea flux differs between PAH patients and controls suggests that this is specifically due to accelerated activity of arginase II.
Increased arginase catabolism is believed to be important in PAH due to decreased arginine availability for reaction with NOS and thus slower NO synthesis. However, contrary to our hypothesis, despite increased arginase activity PAH patients as a whole in this study did not have slower NO production; in fact, the results suggest that NO production may be increased in PAH patients. One potential explanation is that arginase contributes to the development of PAH via NO-independent pathways. For example, in a hypoxia model of pulmonary hypertension, arginase II was responsible for proliferation of pulmonary arterial smooth muscle cells, possibly via ornithine metabolism to polyamines, which are a key component needed for cellular growth.29 In addition, upregulation of arginase expression increases production of proline,30 an important precursor for synthesis of collagen. Thus, increased ornithine production in PAH patients could help sustain vascular remodeling via increased metabolism to polyamine and proline, although this was not directly measured in this study. We also found that there was a subgroup of patients with high arginase activity as measured by arginine to urea conversion, and, as predicted, this group had slower NO synthesis and faster arginine production compared with the group with low arginase activity. At the same time, there was no difference in de novo arginine production between the two groups. These findings suggest that, in PAH patients with high arginine catabolism by arginase, increased arginine production results from increased arginine release from protein breakdown and that there is an inadequate response in de novo arginine synthesis to compensate for increased arginine catabolism. This may be because de novo arginine synthesis from citrulline does not readily adapt to changes in arginine availability; in support of this, healthy men fed arginine-free diets also did not have compensatory increase in de novo arginine production.31 In this study, citrulline plasma concentration and flux were negatively correlated with PAPs and de novo arginine production was negatively correlated with cardiac output, suggesting that citrulline supply—and subsequently its conversion to arginine—are important for improving hemodynamic parameters, likely via NO production. Therefore, citrulline may be a potential treatment for PAH. Citrulline supplementation has been shown to be superior to arginine supplementation for increasing NO synthesis in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome as a result of marked increase in de novo arginine synthesis.32 Oral citrulline supplementation raised arginine plasma concentrations in patients with sickle cell disease33 and increased de novo arginine production and NO synthesis in piglets with hypoxia-induced PAH, thereby improving pulmonary arterial pressure, pulmonary vascular resistance, and NO production.34 Further studies are needed to determine the efficacy of citrulline supplementation in PAH.
PAH patients with high arginase activity also had lower plasma ADMA concentrations than patients with low arginase activity. The lungs are known to be a major source of ADMA,35 and increased ADMA has been previously reported in patients with pulmonary hypertension12,36,37 and is an independent predictor of mortality.37 Elevated ADMA concentrations are thought to result from reduced activity of dimethylaminohydrolase, the enzyme responsible for ADMA clearance.38 Although PAH patients had increased ADMA and a significant reduction in the arginine/ADMA ratio in the current study, this was not accompanied by the expected decrease in NO synthesis. In fact, whole-body NO synthesis was increased, and the rate of NO production as measured by conversion of 15N2-arginine to 15N-citrulline was correlated with exhaled NO, demonstrating an association between whole-body and pulmonary NO production. However, although the rate of NO production may be accelerated, not all the NO formed may participate in physiological processes. In some cases where eNOS expression is unexpectedly increased, it is likely uncoupled, leading to superoxide production instead of NO, and the NO produced may then be inactivated by scavenging reactions.25 In addition, the majority of PAH patients in this study were receiving treatment for PAH, and although none of the medications act directly on NO synthesis, they may have indirectly affected NO production. In fact, administration of prostacyclin to PAH patients has been shown to increase exhaled NO, and successful treatment of PAH has led to increases in FeNO.39 Finally, NO production was measured in the whole body by stable isotopes, which may not account for compartmentalization intracellularly.40 Nevertheless, the important finding that ADMA was significantly higher in the patients with low arginase activity suggests that different mechanisms may be responsible for alterations in arginine metabolism in different patients.
This study has several limitations. It was conducted in a small number of patients with group I PAH and therefore may not be applicable to all types of pulmonary hypertension. Patients were studied while receiving PAH treatment, as it would not have been feasible to stop the medications for the study, and therefore potential effects of therapy on arginine metabolism and NO synthesis could not be excluded. In addition, lung-specific in vivo measurements could not be obtained, as this would have required invasive procedures and sampling of the pulmonary circulation. However, this study provides a rationale for future potential therapies for PAH, including citrulline supplementation, inhibition of arginase II, and enhancing degradation of ADMA.
Acknowledgments
We are grateful to the nursing staff of the Clinical Research Unit, the General Clinical Research Center, and the Metabolic Research Unit for care of the participants and to Grace Tang for assistance in laboratory analyses.
Source of Support: This work was supported in part by the National Institutes of Health (UL1TR000439, HL60917, HL115008, M01-RR-00188, 1K23DK084115). The research was also supported with federal funds from the Agricultural Research Service, US Department of Agriculture, under cooperative agreement 58-6250-6001.
Conflict of Interest: None declared.
References
- 1.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995;333:214–221. [DOI] [PubMed]
- 2.Cooper CJ, Landzberg MJ, Anderson TJ, et al. Role of nitric oxide in the local regulation of pulmonary vascular resistance in humans. Circulation 1996;93:266–271. [DOI] [PubMed]
- 3.Kaneko FT, Arroliga AC, Dweik RA, et al. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med 1998;158:917–923. [DOI] [PubMed]
- 4.Girgis RE, Champion HC, Diette GB, Johns RA, Permutt S, Sylvester JT. Decreased exhaled nitric oxide in pulmonary arterial hypertension: response to bosentan therapy. Am J Respir Crit Care Med 2005;172:352–357. [DOI] [PubMed]
- 5.Bruckdorfer R. The basics about nitric oxide. Mol Aspects Med 2005;26:3–31. [DOI] [PubMed]
- 6.Mason NA, Springall DR, Burke M, et al. High expression of endothelial nitric oxide synthase in plexiform lesions of pulmonary hypertension. J Pathol 1998;185:313–318. [DOI] [PubMed]
- 7.Curis E, Nicolis I, Moinard C, et al. Almost all about citrulline in mammals. Amino Acids 2005;29:177–205. [DOI] [PubMed]
- 8.Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J 1998;336:1–17. [DOI] [PMC free article] [PubMed]
- 9.Morris SM Jr. Arginine metabolism: boundaries of our knowledge. J Nutr 2007;137:1602S–1609S. [DOI] [PubMed]
- 10.Luiking YC, Poeze M, Dejong CH, Ramsay G, Deutz NE. Sepsis: an arginine deficiency state? Crit Care Med 2004;32:2135–2145. [DOI] [PubMed]
- 11.Maarsingh H, Pera T, Meurs H. Arginase and pulmonary diseases. Naunyn Schmiedebergs Arch Pharmacol 2008;378:171–184. [DOI] [PMC free article] [PubMed]
- 12.Xu W, Kaneko FT, Zheng S, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004;18:1746–1748. [DOI] [PubMed]
- 13.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005;294:81–90. [DOI] [PMC free article] [PubMed]
- 14.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl 2003;4:33–40. [DOI] [PubMed]
- 15.Elms S, Chen F, Wang Y, et al. Insights into the arginine paradox: evidence against the importance of subcellular location of arginase and eNOS. Am J Physiol Heart Circ Physiol 2013;305:H651–H666. [DOI] [PMC free article] [PubMed]
- 16.Demoncheaux EA, Higenbottam TW, Kiely DG, et al. Decreased whole body endogenous nitric oxide production in patients with primary pulmonary hypertension. J Vasc Res 2005;42:133–136. [DOI] [PubMed]
- 17.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54:S43–S54. [DOI] [PubMed]
- 18.American Thoracic Society, European Respiratory Society. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am J Respir Crit Care Med 2005;171:912–930. [DOI] [PubMed]
- 19.Abumrad NN, Rabin D, Diamond MP, Lacy WW. Use of a heated superficial hand vein as an alternative site for the measurement of amino acid concentrations and for the study of glucose and alanine kinetics in man. Metabolism 1981;30:936–940. [DOI] [PubMed]
- 20.Copeland KC, Kenney FA, Nair KS. Heated dorsal hand vein sampling for metabolic studies: a reappraisal. Am J Physiol 1992;263:E1010–E1014. [DOI] [PubMed]
- 21.Villalpando S, Gopal J, Balasubramanyam A, Bandi VP, Guntupalli K, Jahoor F. In vivo arginine production and intravascular nitric oxide synthesis in hypotensive sepsis. Am J Clin Nutr 2006;84:197–203. [DOI] [PubMed]
- 22.Kao CC, Bandi V, Guntupalli KK, Wu M, Castillo L, Jahoor F. Arginine, citrulline and nitric oxide metabolism in sepsis. Clin Sci (Lond) 2009;117:23–30. [DOI] [PubMed]
- 23.Morris CR, Poljakovic M, Lavrisha L, Machado L, Kuypers FA, Morris SM Jr. Decreased arginine bioavailability and increased serum arginase activity in asthma. Am J Respir Crit Care Med 2004;170:148–153. [DOI] [PubMed]
- 24.Sharp J, Farha S, Park MM, et al. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol 2014;2:884–891. [DOI] [PMC free article] [PubMed]
- 25.Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;188:639–646. [DOI] [PubMed]
- 26.Sasaki A, Doi S, Mizutani S, Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol 2007;292:L1480–L1487. [DOI] [PubMed]
- 27.Castillo L, Sanchez M, Chapman TE, Ajami A, Burke JF, Young VR. The plasma flux and oxidation rate of ornithine adaptively decline with restricted arginine intake. Proc Natl Acad Sci USA 1994;91:6393–6397. [DOI] [PMC free article] [PubMed]
- 28.Yu YM, Ryan CM, Castillo L, et al. Arginine and ornithine kinetics in severely burned patients: increased rate of arginine disposal. Am J Physiol Endocrinol Metab 2001;280:E509–E517. [DOI] [PubMed]
- 29.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 2009;297:L1151–L1159. [DOI] [PubMed]
- 30.Li H, Meininger CJ, Hawker JR Jr., et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 2001;280:E75–E82. [DOI] [PubMed]
- 31.Castillo L, Chapman TE, Sanchez M, et al. Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets. Proc Natl Acad Sci USA 1993;90:7749–7753. [DOI] [PMC free article] [PubMed]
- 32.El-Hattab AW, Hsu JW, Emrick LT, et al. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab 2012;105:607–614. [DOI] [PMC free article] [PubMed]
- 33.Waugh WH, Daeschner CW 3rd, Files BA, McConnell ME, Strandjord SE. Oral citrulline as arginine precursor may be beneficial in sickle cell disease: early phase two results. J Natl Med Assoc 2001;93:363–371. [PMC free article] [PubMed]
- 34.Ananthakrishnan M, Barr FE, Summar ML, et al. l-Citrulline ameliorates chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 2009;297:L506–L511. [DOI] [PMC free article] [PubMed]
- 35.Bulau P, Zakrzewicz D, Kitowska K, et al. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am J Physiol Lung Cell Mol Physiol 2007;292:L18–L24. [DOI] [PubMed]
- 36.Pullamsetti S, Kiss L, Ghofrani HA, et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J 2005;19:1175–1177. [DOI] [PubMed]
- 37.Kielstein JT, Bode-Boger SM, Hesse G, et al. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2005;25:1414–1418. [DOI] [PubMed]
- 38.Bode-Boger SM, Scalera F, Ignarro LJ. The l-arginine paradox: importance of the l-arginine/asymmetrical dimethylarginine ratio. Pharmacol Ther 2007;114:295–306. [DOI] [PubMed]
- 39.Machado RF, Londhe Nerkar MV, Dweik RA, et al. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med 2004;37:1010–1017. [DOI] [PubMed]
- 40.Luiking YC, Deutz NE. Isotopic investigation of nitric oxide metabolism in disease. Curr Opin Clin Nutr Metab Care 2003;6:103–108. [DOI] [PubMed]