

Abstract Abstract
In experimental animals and in patients with pulmonary arterial hypertension (PAH), a wide spectrum of structural and functional conditions is known that may be responsible for the switch of a state of “compensated” right ventricular (RV) hypertrophy to a state of RV failure. In recent years, therapy with differentiated cells, endothelial progenitor cells, and mesenchymal stem cells has been shown to cause partial or complete reversal of pathological characteristics of PAH. The therapeutic effects of stem or progenitor cell therapy are considered to be (1) paracrine effects from stem or progenitor cells that had engrafted in the myocardium (or elsewhere), by compounds that have anti-inflammatory, antiapoptotic, and proangiogenic actions and (2) unloading effects on the right ventricle due to stem or progenitor cell–induced decrease in pulmonary vascular resistance and decrease in pulmonary artery pressure.
Keywords: pulmonary arterial hypertension, right ventricular hypertrophy, right ventricular failure, cell therapy, stem cells, mesenchymal stem cells, progenitor cells, endothelial progenitor cells, homing, engraftment, differentiation, paracrine factors
Pulmonary arterial hypertension (PAH) is associated with right ventricular (RV) hypertrophy (RVH) or RV failure (RVF), considered to be the result of RV pressure overload. Pharmacotherapy, gene therapy, stem and progenitor cell therapy, and combinations of these treatment modalities have been shown to treat PAH, by lowering pulmonary artery pressure, lowering RV weight, and relieving RVF, thereby improving exercise capacity and life span.1 Here we review the evidence that, in animals with PAH, stem and progenitor cell therapy leads to lower RV weight and relief of RVF by mechanisms that can be ascribed to (1) lowering of RV afterload and (2) direct effects of the stem and progenitor cells on RV myocardial structure and function.
RVH due to PAH-induced RV overload: pathological or physiological hypertrophy?
Originally, left ventricular (LV) hypertrophy (LVH) secondary to pressure overload was considered to be “compensatory,”2 but several studies have clearly demonstrated that LVH is a risk factor and has unfavorable prognostic implications.3-5 On the other hand, the athlete’s heart often implies the presence of LVH, but this LVH is considered an adaptation to volume overload that occurs during long-lasting rowing, running, and cycling.6 To date, the healthy aspects of exercise training have been documented firmly and are not restricted to healthy individuals, such as athletes, but even to patients with heart failure.7,8
The two main forms of myocardial hypertrophy are often termed “pathological” hypertrophy, as occurs secondary to pressure overload, and “physiological” hypertrophy, as occurs in the athlete’s heart. Pathological and physiological hypertrophy have been characterized in detail,9-13 and the most significant differences are found in the membrane receptor and signaling pathways that convey the message to the nucleus, where hypertrophy is effectuated with or without changes in gene expression. In pathological hypertrophy, Ca2+-dependent calcineurin activation and calcineurin-induced dephosphorylation of nuclear factor of activated T cells (NFAT) trigger gene transcription in the nucleus, leading to multiple changes in gene expression, including expression of “fetal” genes such as atrial natriuretic peptide (ANP), 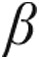 -myosin heavy chain (
-myosin heavy chain (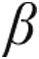 -MHC), and
-MHC), and  -skeletal-actin. The
-skeletal-actin. The  -adrenergic receptor density is decreased, and this receptor is desensitized.14 In addition, during the development of cardiac hypertrophy, a mismatch between the number of capillaries and the size of cardiomyocytes can lead to ischemia and contractile dysfunction.15 In physiological hypertrophy, signalling through Akt, mTOR, p70S6 kinase, and S6 activates gene transcription in the nucleus, which is hardly associated with changes in gene expression. The
-adrenergic receptor density is decreased, and this receptor is desensitized.14 In addition, during the development of cardiac hypertrophy, a mismatch between the number of capillaries and the size of cardiomyocytes can lead to ischemia and contractile dysfunction.15 In physiological hypertrophy, signalling through Akt, mTOR, p70S6 kinase, and S6 activates gene transcription in the nucleus, which is hardly associated with changes in gene expression. The  -adrenergic receptor density and capillary density in the myocardium remain unchanged, compared to those in controls.16
-adrenergic receptor density and capillary density in the myocardium remain unchanged, compared to those in controls.16
The transition from RVH to RVF is dependent on a large number of known and unknown conditions and factors. A new player in this field is the family of microRNAs of which the expressions are changing during the process of RV remodeling.17 MicroRNAs are small, noncoding RNAs that are emerging as crucial regulators of cardiac remodeling in LVH and LV failure.18,19
Administration of exogenous stem or progenitor cells for lung repair
Cytotherapy with stem and progenitor cell types is a novel approach to treat several lung diseases. This approach is based on the concept that stem and progenitor cells may help to regenerate and repair pulmonary vasculature.20 Stem cells are undifferentiated cells that display self-renewal, clonogenicity, and multipotency. Mesenchymal stem cells (MSCs) are cells that adhere to plastic, express surface cell markers such as CD73, CD90, and CD105; lack surface expression of CD11b, CD14, CD19, CD34, CD45, CD79 , and HLA-DR; and are able to differentiate into multiple cell types, such as adipocytes, osteocytes, and chondrocytes, in vitro.21 MSCs have the ability and tendency to preferentially migrate to injured lung tissue, where they secrete angiogenic (e.g., vascular endothelial growth factor [VEGF], Ang-1), antiapoptotic (Bcl-2), and anti-inflammatory factors (e.g., interferon-
, and HLA-DR; and are able to differentiate into multiple cell types, such as adipocytes, osteocytes, and chondrocytes, in vitro.21 MSCs have the ability and tendency to preferentially migrate to injured lung tissue, where they secrete angiogenic (e.g., vascular endothelial growth factor [VEGF], Ang-1), antiapoptotic (Bcl-2), and anti-inflammatory factors (e.g., interferon-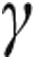 , interleukin-10, VEGF, hepatocyte growth factor [HGF]).22 They also exert immunomodulatory effects by direct cell-to-cell contact.23,24 These properties, in combination with their low immunogenicity and their ability to evade clearance by the immune system of the host,25 make MSCs an ideal candidate in therapies for a variety of lung diseases.
, interleukin-10, VEGF, hepatocyte growth factor [HGF]).22 They also exert immunomodulatory effects by direct cell-to-cell contact.23,24 These properties, in combination with their low immunogenicity and their ability to evade clearance by the immune system of the host,25 make MSCs an ideal candidate in therapies for a variety of lung diseases.
Progenitor cells are committed to a lineage and differentiate into a specific cell or cell types. Compared to stem cells, progenitor cells have a more limited capacity for self-renewal and represent an “intermediate” between stem cells and terminally differentiated ones. Endothelial progenitor cells (EPCs) are circulating, bone marrow–derived cells that possess the ability to differentiate and mature into endothelial cells that may repair and regenerate damaged vessels.26,27 A large number of studies have demonstrated that systemic administration of EPCs can treat experimentally induced lung injuries. Whether this includes structural contributions of the administered cells, paracrine stimulation of endogenous vascular progenitor cells, or other paracrine immunomodulatory actions remains unclear.28
In animal models of lung disease, engraftment of lung epithelium, vasculature, or interstitium by exogenously administered stem or progenitor cells (of bone marrow or other nonlung origin) is believed to occur rarely and with uncertain physiologic relevance.20,25,29,30 The therapeutic effects of stem or progenitor cells are exerted through a combination of mechanisms, including transdifferentiation of a small subset of cells, direct cell-to-cell connections with transfer of cell components, release of angiogenic, antiapoptotic, and anti-inflammatory factors, and release of other cellular components, such as microvesicles or exosomes.25
PAH-induced RVF and interstitial myocardial fibrosis
Pressure overload–induced RVH is associated with differentiation of fibroblasts to myofibroblasts that proliferate rapidly and secrete large quantities of collagen.31 The normally prevalent collagen III, which confers elastic properties, is replaced by collagen I that is inelastic, thereby impairing diastolic relaxation.32 In rats with RVF, by ≈4 weeks after monocrotaline (MCT) injection (60 mg/kg), RV fibrosis had occurred in the epicardial layers (17% ± 2% vs. 1.5% ± 0.4%; P < 0.01) and in the endocardial layers (18% ± 3% vs. 1.4% ± 0.3%; P < 0.01). The LVs of these hearts did not develop fibrosis.33 Yen et al.34 found that 3 weeks after MCT injection, the rats suffered from RV fibrosis, had high expression of fibrotic biomarkers (transforming growth factor 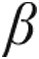 [TGF
[TGF ], p-SMAD3) in the RV, and low expression of antifibrotic biomarkers (bone morphogenetic protein 2 [BMP2], p-SMAD1/5). Treatment with a combination of sildenafil and bone marrow–derived EPCs was able to reverse these changes to levels almost equal to those of healthy rats, and this combination was more potent than either of the two therapies alone.34 Hessel et al.35 demonstrated that in the RV of MCT-treated (80 mg/kg) hearts, at 4 weeks after MCT injection, tenascin-C (TNC) was expressed at the messenger RNA level and at the protein level (from 0.0 to 0.62 ± 0.64 ng/mg). In the heart, TNC is an extracellular matrix glycoprotein that is expressed during cardiogenesis but disappears in the myocardium after birth. TNC may reappear in the heart under various pathological conditions, including acute myocardial infarction (MI), myocarditis, and dilated cardiomyopathy.36 In rats with MCT-induced RVF, Umar et al.37 demonstrated that the high TNC levels in RV myocardium at 4 weeks after MCT injection were almost absent if treated with MSCs at day 14. These studies demonstrate that PAH is associated with pathological RVH and interstitial RV fibrosis that both reverse upon cell therapy with stem and progenitor cells.
], p-SMAD3) in the RV, and low expression of antifibrotic biomarkers (bone morphogenetic protein 2 [BMP2], p-SMAD1/5). Treatment with a combination of sildenafil and bone marrow–derived EPCs was able to reverse these changes to levels almost equal to those of healthy rats, and this combination was more potent than either of the two therapies alone.34 Hessel et al.35 demonstrated that in the RV of MCT-treated (80 mg/kg) hearts, at 4 weeks after MCT injection, tenascin-C (TNC) was expressed at the messenger RNA level and at the protein level (from 0.0 to 0.62 ± 0.64 ng/mg). In the heart, TNC is an extracellular matrix glycoprotein that is expressed during cardiogenesis but disappears in the myocardium after birth. TNC may reappear in the heart under various pathological conditions, including acute myocardial infarction (MI), myocarditis, and dilated cardiomyopathy.36 In rats with MCT-induced RVF, Umar et al.37 demonstrated that the high TNC levels in RV myocardium at 4 weeks after MCT injection were almost absent if treated with MSCs at day 14. These studies demonstrate that PAH is associated with pathological RVH and interstitial RV fibrosis that both reverse upon cell therapy with stem and progenitor cells.
PAH-induced RVF and arrhythmogenesis
At ≈4 weeks after MCT injection, cardiomyocytes isolated from hypertrophied RVs had prolonged their action potential duration (APD90) from 33 ± 6 ms (control) to 57 ± 8 ms (P < 0.001). Cardiomyocytes isolated from the LVs of these hearts did not show action potential prolongation. Cardiomyocytes isolated from the RVs of MCT-treated hearts manifested greater dispersion in APD90 than did those from controls: 30 ± 1.8 ms vs. 20 ± 1.5 ms (P < 0.01).33 The reduced repolarization reserve in the MCT-treated hearts was associated with downregulation of at least 2 K+ channels, Kv1.5 (protein expression reduced by 88%) and KCNE2 (protein expression reduced by ≈75%). In combination with an increased susceptibility to depolarization-induced repetitive activity, all conditions for precipitation of sudden death are present in cardiomyocytes isolated from RVs of MCT-treated hearts. Sudden death occurred ≈1 week after the drop in RV ejection fraction.33 As to cell therapy for RVF in animal models of PAH, a reduction of mortality has been reported in numerous studies,38-43 but the exact mechanisms by which cell therapy repaired the pathologic changes associated with sudden death are not yet known.
Cardiomyocytes are electrically coupled via gap junctions and are built up of several proteins, including connexins, mainly connexin-43 (Cx43). Hearts that fail because of pressure overload have diminished Cx43 density at the intercalated disks,44,45 which slows down conduction velocity through the myocardium. In RVs of rats with MCT-induced PAH, Cx43 staining decreased at the intercalated disks and intensified at the lateral cell membrane.46,47 After 3 weeks of treatment with bosentan, a dual endothelin receptor antagonist, the Cx43 staining had redistributed to the intercalated disks, as in controls.47 In a prevention study, administration of bone marrow–derived EPCs, combined with pharmacotherapy using cilostazol, a phosphodiesterase III inhibitor, to rats treated on day 3 after MCT administration was able to prevent the decrease in Cx43 expression observed in RVs of rats with MCT-induced PAH.48 However, as the development of PAH and RVH was also prevented by this therapy, a direct effect of therapy on RV myocardial Cx43 expression is unlikely.
PAH-induced RVF and cardiomyocyte apoptosis
An increased rate of cardiomyocyte apoptosis has been detected in failing human hearts,49,50 as well as in hearts of animals with experimentally induced hypertrophy and cardiomyopathy16 and in RVs of rats with SU5416 (a VEGF-receptor-2 antagonist)-induced and chronic hypoxia-induced PAH.51 Stem cells from human amniotic fluid (hAFS) or cells from rat adipose tissue stromal vascular fraction (rSVC), administered intravenously in rats with MCT-induced PAH, engrafted in the lungs, heart, and skeletal muscle and differentiated into endothelial cells and vascular smooth muscle cells (VSMCs).52 Cell therapy for PAH with hAFS or rSVC was successful in treating PAH and was associated with reduction in the number of apoptotic cells per cubic millimeter in lung, heart, and skeletal muscle toward levels found in corresponding organs of healthy rats.52 Successful therapy for PAH is likely to include antiapoptotic actions.
PAH-induced RVF: reversible or not?
The structural and functional changes associated with the state of RVF are potentially reversible, given the many reports about successful therapy for MCT-induced PAH, hypoxia-induced PAH, and high flow–induced (by shunting) PAH in experimental animals, including (1) a decrease in pulmonary vascular resistance, (2) a decrease in pulmonary artery pressure, (3) a decrease in RV mass, and (4) an increase in RV ejection fraction. Other convincing evidence of the reversible nature of PAH-induced RVF comes from studies that have addressed RV function after lung transplantation for end-stage PAH53-55 and after successful pulmonary endarterectomy in patients with chronic thromboembolic PH.56 Following these surgical interventions, RV function slowly recovered to normal, and this recovery was sustained in long-term follow-up. Improvements included a return of the leftward-bowing interventricular septum (IVS) to its physiological position and a disappearance of high-grade tricuspid insufficiency.55,56 Whether there is a point of no return is to be seen, but so far numerous data on therapies for PAH, including cell therapies, indicate reversibility of PAH-induced RVF, albeit slow and maybe incomplete.
Engraftment of bone marrow stem or progenitor cells in the heart
To investigate whether circulating bone marrow–derived cells are capable of de novo cardiomyocyte formation, Deb et al.57 analyzed hearts of 4 female patients who had undergone sex-mismatched bone marrow transplantation. The time interval between transplantation and death was 35, 480, 510, and 600 days. The mean percentage of Y chromosome–positive cardiomyocytes was 0.23% ± 0.06%, without evidence that these chimeric cardiomyocytes were the result of cell fusion.57 Thiele et al.58 found higher numbers of chimeric cardiomyocytes in the heart after bone marrow–derived (stem) cell transplantation. In hearts of 7 patients who received sex-mismatched bone marrow cells, 5.4% ± 1.1% of the cardiomyocytes were sex mismatched, and in the coronary vessels 5.5% ± 2.2% of the endothelial cells and 3.8% ± 0.6% of the VSMCs were sex mismatched. Four other patients received sex-mismatched peripheral blood stem cells that contained up to 2% CD34+ progenitor cells. In this group 1.7% ± 0.5% of the cardiomyocytes were sex mismatched, and in the coronary vessels 4.6% ± 1.1% of the endothelial cells and 3.7% ± 0.5% of the VSMCs were sex mismatched.58 Spees et al.59 investigated the effects of MCT on homing, engraftment, and differentiation of bone marrow–derived cells in lungs and heart. To that purpose, these authors transplanted bone marrow from green fluorescent protein (GFP)-transgenic male rats into irradiated GFP-negative female rats. Three weeks after MCT injection into chimeric rats, the GFP-positive bone marrow–derived cells were found engrafted in the lungs and being differentiated into interstitial fibroblasts and pulmonary epithelial cells (Clara cells), vascular endothelial cells, and VSMCs. At the same time, the GFP-positive bone marrow–derived cells were found engrafted in the hypertrophied hearts of MCT-injected rats, with significantly more GFP-positive cells in the RV than in the LV (1.5∶1), whereas in the control chimeric rats (no MCT), the LV contained more male DNA than the RV (1∶0.5). In the RVs of rats with PAH, GFP-positive cells were mainly vascular cells and cardiomyocytes. Only a few GFP-positive cardiomyocytes appeared as binucleated cardiomyocytes. None of the GFP-positive vascular cells in the RV were fused cells.59 Transplantation of EPCs in the lungs of dogs with dehydroMCT-induced PAH by injection into the lung parenchyma decreased the pulmonary artery pressure and pulmonary vascular resistance and increased cardiac output.60
In a prevention study, bone marrow–derived EPCs, combined with cilostazol, a phosphodiesterase III inhibitor, were administered intravenously to rats on day 3 after MCT administration.48 Combination therapy (cells + drug) prevented (1) the development of PAH and RVH, (2) the decrease in pulmonary expression of Bcl-2 and endothelial nitric oxide synthase (eNOS), (3) the increase in pulmonary expression of caspase-3, matrix metalloproteinase 9 (MMP9), and tumor necrosis factor 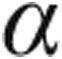 (TNF
(TNF ), (4) the decrease in alveolar sacs, (5) the decrease in pulmonary arterioles, and (6) the decrease in RV expression of Cx43. Combination therapy appeared superior to either EPCs or cilostazol alone in preventing MCT-induced PAH, including prevention of apoptotic and inflammatory actions in the lung and prevention of arrhythmogenesis in the RV.48
), (4) the decrease in alveolar sacs, (5) the decrease in pulmonary arterioles, and (6) the decrease in RV expression of Cx43. Combination therapy appeared superior to either EPCs or cilostazol alone in preventing MCT-induced PAH, including prevention of apoptotic and inflammatory actions in the lung and prevention of arrhythmogenesis in the RV.48
In PAH, the pulmonary resistance arteries are remodeled as a result of proliferation of endothelial cells and VSMCs. This vascular remodeling is considered to stimulate homing of MSCs. Takemiya et al.42 demonstrated that more intravenously administered MSCs engrafted in lungs of rats with MCT-induced PAH at days 1 and 14 after cell administration than in lungs of healthy rats, by 2.5- and 6.5-fold, respectively. Cell therapy was given 2 weeks after MCT injection. Two weeks after cell administration, the MSCs, transduced with GFP, had not lowered the pulmonary artery pressure or the RV/(LV + IVS) weight ratio, compared to those in rats with PAH 4 weeks after MCT injection. However, if the MSCs had been transduced with prostacyclin synthase, pulmonary artery pressure and the RV/(LV + IVS) weight ratio were significantly lower than corresponding measurements in rats with PAH 4 weeks after MCT injection.42
Bone marrow–derived MSCs genetically modified with HGF administered intravenously 3 weeks after MCT injection in rats caused lower pulmonary artery systolic pressure, lower RV weight, less interstitial fibrosis in the RV myocardium, and increased staining of RV myocardial Cx43 at the intercalated disks, compared with rats with MCT-induced PAH without cell therapy.61 The therapeutic effects of MSCs without genetic modification were less pronounced.
When administered intravenously in rats with MCT-induced PAH, hAFS and hSVC engrafted in the lungs (2%–4.5%), heart (0.4%–0.8%), and skeletal muscle (0.2%–0.4%) and differentiated into endothelial cells and VSMCs.52 These stem cells caused a decrease in pulmonary artery systolic pressure, RV weight, RV dilatation, plasma concentration of brain natriuretic peptide (BNP), and numbers of apoptotic cells in lung, heart, and skeletal muscle.52
These studies demonstrate that cell therapy with bone marrow–derived stem and progenitor cells, administered to treat PAH, may lead to engraftment of a small fraction of stem or progenitor cells in the lungs and an even smaller fraction in the RV myocardium, with subsequent differentiation into vascular and cardiac cell lineages, respectively.
Are the beneficial effects of cell therapy on RV structure and function indirect (secondary to a decrease in pulmonary vascular resistance) or direct, e.g., due to cells that had engrafted in the heart or to cells that had engrafted elsewhere? Human MSCs that were entrapped in the lungs of rats with MI had a therapeutic effect on the heart, as evidenced by production of anti-inflammatory substances, such as TNF-inducible gene-6 protein (TSG-6), that reduced inflammatory responses and even reduced infarct size.62 As a disease, PAH differs from MI, but a failing RV of an animal or individual with PAH may have become ischemic (or otherwise vulnerable) and may become a target for circulating cells administered as cell therapy. The so-called paracrine effects of engrafted cells on other cells, e.g., in lungs and/or heart, may be supplied by (1) soluble factors secreted by engrafted cells, as was shown by experiments in which the cells were replaced by conditioned medium,63,64 and/or (2) exosomes, being secreted membrane microvesicles that have been isolated from conditioned media of MSCs.65
An example that stem cells may be able to repair “myocardial injury” is presented by Gopinath et al.66 Human umbilical cord blood (hUCB)–derived stem cells were administered intravenously in immunoincompetent mice 14 days after the mice had been injected with doxorubicin, leading to pathological heart hypertrophy. The hUCB-derived stem cells migrated and integrated into the hearts and reversed the expression of pathological hypertrophic markers, such as ANP, 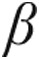 -MHC, and
-MHC, and 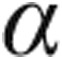 -skeletal actin, whereas the expression of physiological hypertrophic markers, such as sarcoplasmic reticulum Ca2+-ATPase (SERCA2), insulin-like growth factor 1 (IGF-1), and
-skeletal actin, whereas the expression of physiological hypertrophic markers, such as sarcoplasmic reticulum Ca2+-ATPase (SERCA2), insulin-like growth factor 1 (IGF-1), and 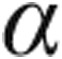 -MHC, increased, indicating that hUCB-derived stem cells caused a shift from pathological hypertrophy toward physiological hypertrophy in doxorubicin-challenged mice.66 Earlier, this shift from pathological to physiological hypertrophy was noticed in mice with angiotensin II–induced LVH that were treated with hUCB-derived stem cells.67 The hUCB-derived stem cell treatment in mice decreased the doxorubicin-induced increase in heart weight–to–body mass ratio, myocardial fibrosis, and number of apoptotic cardiomyocytes.66 These findings suggest that cell therapy with hUCB-derived stem cells is able to reverse heart failure, but whether this applies to doxorubicin-induced heart failure only is uncertain.
-MHC, increased, indicating that hUCB-derived stem cells caused a shift from pathological hypertrophy toward physiological hypertrophy in doxorubicin-challenged mice.66 Earlier, this shift from pathological to physiological hypertrophy was noticed in mice with angiotensin II–induced LVH that were treated with hUCB-derived stem cells.67 The hUCB-derived stem cell treatment in mice decreased the doxorubicin-induced increase in heart weight–to–body mass ratio, myocardial fibrosis, and number of apoptotic cardiomyocytes.66 These findings suggest that cell therapy with hUCB-derived stem cells is able to reverse heart failure, but whether this applies to doxorubicin-induced heart failure only is uncertain.
Conclusions
The conditions by which a state of “compensated” RVH switches to a state of RVF are partially known: (1) development of fibrosis, (2) “fetal” gene expression, (3) apoptosis, and (4) slowed conduction due to gap junction loss may determine whether the RV starts failing. We believe that these changes are potentially reversible, given (1) the many reports about successful therapy for MCT-induced PAH, hypoxia-induced PAH, and high flow–induced (by shunting) PAH in experimental animals and (2) the reports about reversed RV remodeling after lung transplantation in patients with end-stage PAH. The therapeutic effects of stem and progenitor cell therapy are considered to be (1) paracrine effects from stem and progenitor cells that had engrafted in the myocardium (or elsewhere), by compounds that have anti-inflammatory, antiapoptotic, and proangiogenic actions and (2) unloading effects on the RV due to stem or progenitor cell–induced decrease in pulmonary vascular resistance and decrease in pulmonary artery pressure. The therapeutic effects of stem and progenitor cell therapy are improved by genetic modification of the cells with genes encoding anti-inflammatory, antiapoptotic, and proangiogenic actions.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Umar S, Steendijk P, Ypey DL, Atsma DE, van der Wall EE, Schalij MJ, van der Laarse A. Novel approaches to treat experimental pulmonary arterial hypertension: a review. J Biomed Biotechnol 2010;2010:702836. doi:10.1155/2010/702836. [DOI] [PMC free article] [PubMed]
- 2.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 1975;56(1):56–64. [DOI] [PMC free article] [PubMed]
- 3.Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP. Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol 1987;59(9):956–960. [DOI] [PubMed]
- 4.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990;322(22):1561–1566. [DOI] [PubMed]
- 5.Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J 2001;141(3):334–341. [DOI] [PubMed]
- 6.Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete’s heart: a meta-analysis of cardiac structure and function. Circulation 2000;101(3):336–344. [DOI] [PubMed]
- 7.Piepoli MF, ExTraMATCH Collaborative. Exercise training meta-analysis of trials in patients with chronic heart failure (ExTraMATCH). BMJ 2004;328(7433):189. doi:10.1136/bmj.37938.645220.EE. [DOI] [PMC free article] [PubMed]
- 8.Smart N, Marwick TH. Exercise training for patients with heart failure: a systematic review of factors that improve mortality and morbidity. Am J Med 2004;116(10):693–706. [DOI] [PubMed]
- 9.Dorn GW II, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ Res 2003; 92(11):1171–1175. [DOI] [PubMed]
- 10.Dorn GW II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 2005;115(3):527–537. [DOI] [PMC free article] [PubMed]
- 11.Dorn GW II. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 2007;49(5):962–970. [DOI] [PubMed]
- 12.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol 2008;214(2):316–321. [DOI] [PubMed]
- 13.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 2010;128(1):191–227. [DOI] [PubMed]
-
14.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and /article/back/ref-list/ref/mixed-citation/inline-formula
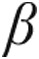 -adrenergic-receptor density in failing human hearts. N Engl J Med 1982;307(4):205–211. [DOI] [PubMed]
-adrenergic-receptor density in failing human hearts. N Engl J Med 1982;307(4):205–211. [DOI] [PubMed] - 15.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007;446(7134):444–448. [DOI] [PubMed]
- 16.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest 2006;116(6):1547–1560. [DOI] [PMC free article] [PubMed]
- 17.Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC, Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics 2012;44(10):562–575. [DOI] [PMC free article] [PubMed]
- 18.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 2006;103(48):18255–18260. [DOI] [PMC free article] [PubMed]
- 19.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 2007;116(3):258–267. [DOI] [PubMed]
- 20.Lau AN, Goodwin M, Kim CF, Weiss DJ. Stem cells and regenerative medicine in lung biology and diseases. Mol Ther 2012;20(6):1116–1130. [DOI] [PMC free article] [PubMed]
- 21.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans RJ, Keating A, Prockop DJ, Horwitz EM. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8(4):315–317. [DOI] [PubMed]
- 22.Antunes MA, Laffey JG, Pelosi P, Rocco PRM. Mesenchymal stem cell trials for pulmonary diseases. J Cell Biochem 2014;115(6):1023–1032. [DOI] [PubMed]
- 23.Gebler A, Zabel O, Seliger B. The immunomodulatory capacity of mesenchymal stem cells. Trends Mol Med 2012;18(2):128–134. [DOI] [PubMed]
- 24.Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005;105(7):2821–2827. [DOI] [PubMed]
- 25.Anversa P, Perrella MA, Kourembanas S, Choi AMK, Loscalzo J. Regenerative pulmonary medicine: potential and promise, pitfalls and challenges. Eur J Clin Invest 2012;42(8):900–913. [DOI] [PMC free article] [PubMed]
- 26.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997;275(5302):964–966. [DOI] [PubMed]
- 27.Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res 2008;78(3):413–421. [DOI] [PubMed]
- 28.Weiss DJ. Concise review: current status of stem cells and regenerative medicine in lung biology and diseases. Stem Cells 2014;32(1):16–25. [DOI] [PMC free article] [PubMed]
- 29.Kotton DN, Fabian AJ, Mulligan RC. Failure of bone marrow to reconstitute lung epithelium. Am J Respir Cell Mol Biol 2005;33(4):328–334. [DOI] [PMC free article] [PubMed]
- 30.Weiss DJ, Bertoncello I, Borok Z, Kim C, Panoskaltsis-Mortari A, Reynolds S, Rojas M, Stripp B, Warburton D, Prockop DJ. Stem cells and cell therapies in lung biology and lung diseases. Proc Am Thorac Soc 2011;8(3):223–272. [DOI] [PMC free article] [PubMed]
- 31.Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M, Low RB. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am J Pathol 1991;139(1):207–216. [PMC free article] [PubMed]
- 32.Yang CM, Kandaswamy V, Young D, Sen S. Changes in collagen phenotypes during progression and regression of cardiac hypertrophy. Cardiovasc Res 1997;36(2):236–245. [DOI] [PubMed]
- 33.Umar S, Lee JH, de Lange E, Iorga A, Partow-Navid R, Bapat A, van der Laarse A, et al. Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhyth Electrophysiol 2012;5(1):181–190. [DOI] [PMC free article] [PubMed]
- 34.Yen CH, Tsai TH, Leu S, Chen YL, Chang LT, Chai HT, Chung SY, et al. Sildenafil improves long-term effect of endothelial progenitor cell-based treatment for monocrotaline-induced rat pulmonary arterial hypertension. Cytotherapy 2013;15(2):209–223. [DOI] [PubMed]
- 35.Hessel M, Steendijk P, den Adel B, Schutte C, van der Laarse A. Pressure overload-induced right ventricular failure is associated with re-expression of myocardial tenascin-C and elevated plasma tenascin-C levels. Cell Physiol Biochem 2009;24(3–4):201–210. [DOI] [PubMed]
- 36.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res 2004;64(1):24–31. [DOI] [PubMed]
- 37.Umar S, de Visser YP, Steendijk P, Schutte CI, Laghmani EH, Wagenaar GTM, Bax WH, et al. Allogenic stem cell therapy improves right ventricular function by improving lung pathology in rats with pulmonary hypertension. Am J Physiol Heart Circ Physiol 2009;297(5):H1606–H1616. [DOI] [PubMed]
- 38.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res 2005;96(4):442–450. [DOI] [PubMed]
- 39.Zhao Q, Liu Z, Wang Z, Yang C, Liu J, Lu J. Effect of prepro-calcitonin gene-related peptide-expressing endothelial progenitor cells on pulmonary hypertension. Ann Thorac Surg 2007;84(2):544–552. [DOI] [PubMed]
- 40.Luan Y, Zhang ZH, Wei DE, Zhao JJ, Kong F, Cheng GH, Wang YB. Implantation of mesenchymal stem cells improves right ventricular impairments caused by experimental pulmonary hypertension. Am J Med Sci 2012;343(5):402–406. [DOI] [PubMed]
- 41.Kanki-Horimoto S, Horimoto H, Mieno S, Kishida K, Watanabe F, Katsumata T. Implantation of mesenchymal stem cells overexpressing endothelial nitric oxide synthase improves right ventricular impairments caused by pulmonary hypertension. Circulation 2006;114(suppl. 1):I-181–I-185. [DOI] [PubMed]
- 42.Takemiya K, Kai H, Yasukawa H, Tahara N, Kato S, Imaizumi T. Mesenchymal stem cell-based prostacyclin synthase gene therapy for pulmonary hypertension rats. Basic Res Cardiol 2010;105(3):409–417. [DOI] [PubMed]
- 43.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, et al. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 2010;182(5):652–660. [DOI] [PubMed]
- 44.Teunissen BE, Jongsma HJ, Bierhuizen MF. Regulation of myocardial connexins during hypertrophic remodelling. Eur Heart J 2004;25(22):1979–1989. [DOI] [PubMed]
- 45.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 2008;80(1):9–19. [DOI] [PMC free article] [PubMed]
- 46.Uzzaman M, Honjo H, Takagishi Y, Emdad L, Magee AI, Severs NJ, Kodama I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ Res 2000;86(8):871–878. [DOI] [PubMed]
- 47.Tan XY, He JG. The remodeling of connexin in the hypertrophied right ventricular in pulmonary arterial hypertension and the effect of a dual ET receptor antagonist (bosentan). Pathol Res Pract 2009;205(7):473–482. [DOI] [PubMed]
- 48.Sun CK, Lee FY, Sheu JJ, Yuen CM, Chua S, Chung SY, Chai HT, et al. Early combined treatment with cilostazol and bone marrow–derived endothelial progenitor cells markedly attenuates pulmonary arterial hypertension in rats. J Pharmacol Exp Ther 2009;330(3):718–726. [DOI] [PubMed]
- 49.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996;335(16):1182–1189. [DOI] [PubMed]
- 50.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, et al. Apoptosis in the failing human heart. N Engl J Med 1997;336(16):1131–1141. [DOI] [PubMed]
- 51.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120(20):1951–1960. [DOI] [PubMed]
- 52.Angelini A, Castellani C, Ravara B, Franzin C, Pozzobon M, Tavano R, Dalla Libera L, et al. Stem-cell therapy in an experimental model of pulmonary hypertension and right heart failure: role of paracrine and neurohormonal milieu in the remodeling process. J Heart Lung Transplant 2011;30(11):1281–1293. [DOI] [PubMed]
- 53.Ritchie M, Waggoner AD, Dávila-Román VG, Barzilai B, Trulock EP, Eisenberg PR. Echocardiographic characterization of the improvement in right ventricular function in patients with severe pulmonary hypertension after single-lung transplantation. J Am Coll Cardiol 1993;22(4):1170–1174. [DOI] [PubMed]
- 54.Schulman LL, Leibowitz DW, Anandarangam T, DiTullio MR, McGregor CC, Smith CR, Homma S. Variability of right ventricular functional recovery after lung transplantation. Transplantation 1996;62(5):622–625. [DOI] [PubMed]
- 55.Kasimir MT, Seebacher G, Jaksch P, Winkler G, Schmid K, Marta GM, Simon P, Klepetko W. Reverse cardiac remodelling in patients with primary pulmonary hypertension after isolated lung transplantation. Eur J Cardiothorac Surg 2004;26:(4)776–781. [DOI] [PubMed]
- 56.Reesink HJ, Marcus JT, Tulevski II, Jamieson S, Kloek JJ, Vonk Noordegraaf A, Bresser P. Reverse right ventricular remodeling after pulmonary endarterectomy in patients with chronic thromboembolic pulmonary hypertension: utility of magnetic resonance imaging to demonstrate restoration of the right ventricle. J Thorac Cardiovasc Surg 2007;133(1):58–64. [DOI] [PubMed]
- 57.Deb A, Wang S, Skelding KA, Miller D, Simper D, Caplice NM. Bone marrow–derived cardiomyocytes are present in adult human heart: a study of gender-mismatched bone marrow transplantation patients. Circulation 2003;107(9):1247–1249. [DOI] [PubMed]
- 58.Thiele J, Varus E, Wickenhauser C, Kvasnicka HM, Lorenzen J, Gramley F, Metz KA, Rivero F, Beelen DW. Mixed chimerism of cardiomyocytes and vessels after allogeneic bone marrow and stem-cell transplantation in comparison with cardiac allografts. Transplantation 2004;77(12):1902–1905. [DOI] [PubMed]
- 59.Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J, Prockop DJ. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J 2008;22(4):1226–1236. [DOI] [PubMed]
- 60.Takahashi M, Nakamura T, Toba T, Kajiwara N, Kato H, Shimizu Y. Transplantation of endothelial progenitor cells into the lung to alleviate pulmonary hypertension in dogs. Tissue Eng 2004;10(5–6):771–779. [DOI] [PubMed]
- 61.Su L, Guo Y, Guo N, Chang D, Xie L, Liu C. The efficacy of MSC-HGF in treating pulmonary arterial hypertension (PAH) and connexin remodelling. Cent Eur J Biol 2013;8(3):240–251.
- 62.Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009;5(1):54–63. [DOI] [PMC free article] [PubMed]
- 63.Di Santo S, Yang Z, Wyler von Ballmoos M, Voelzmann J, Diehm N, Baumgartner I, Kalka C. Novel cell-free strategy for therapeutic angiogenesis: in vitro generated conditioned medium can replace progenitor cell transplantation. PLoS ONE 2009;4:e5643. doi:10.1371/journal.pone.0005643. [DOI] [PMC free article] [PubMed]
- 64.Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol 2005;39(5):733–742. [DOI] [PubMed]
- 65.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012;126(22):2601–2611. [DOI] [PMC free article] [PubMed]
- 66.Gopinath S, Vanamala SK, Gondi CS, Rao JS. Human umbilical cord blood derived stem cells repair doxorubicin-induced pathological cardiac hypertrophy in mice. Biochem Biophys Res Commun 2010;395(3):367–372. [DOI] [PubMed]
- 67.Vanamala SK, Gopinath S, Gondi CS, Rao JS. Effect of human umbilical cord blood cells on Ang-II-induced hypertrophy in mice. Biochem Biophys Res Commun 2009;386(2):386–391. [DOI] [PMC free article] [PubMed]