

Abstract
Left ventricular (LV) function in rheumatic mitral stenosis (MS) remains an issue of controversy, due to load dependency of previously employed assessment methods. We investigated LV performance in MS employing relatively load-independent indices robust to the altered loading state. We studied 106 subjects (32 ± 8 years, 72% female) with severe MS (0.8 ± 0.2 cm2) and 40 age-matched controls. MS subjects underwent simultaneous bi-ventricular catheterization and transthoracic echocardiography (TTE) before and immediately after percutaneous transvenous mitral commisurotomy (PTMC). Sphygmomanometric brachial artery pressures and TTE recordings were simultaneously acquired in controls. Single-beat LV elastance (Ees) was employed for LV contractility measurements. Effective arterial elastance (Ea) and LV diastolic stiffness were measured. MS patients demonstrated significantly elevated afterload (Ea: 3.0 ± 1.3 vs. 1.5 ± 0.3 mmHg ml−1; P < 0.001) and LV contractility (Ees: 4.1 ± 1.6 vs. 2.4 ± 0.5 mmHg ml−1; P < 0.001) as compared to controls, with higher Ea in subjects with smaller mitral valve area (≤ 0.8 cm2) and pronounced subvalvular fusion. Stroke volume (49 ± 16 to 57 ± 17 ml; P < 0.001) and indexed LV end-diastolic volume (LVEDVindex: 57 ± 16 to 64 ± 16 ml m−2; P < 0.001) increased following PTMC while Ees and Ea returned to more normal levels. Elevated LV stiffness was demonstrated at baseline and increased further following PTMC. Our findings provide evidence of elevated LV contractility, increased arterial load and increased diastolic stiffness in severe MS. Following PTMC, both LV contractility and afterload tend to normalize.
Key points
A hallmark of mitral stenosis (MS) is the markedly altered left ventricular (LV) loading.
As most of the methods used to determine LV performance in MS patients are influenced by loading conditions, previous studies have shown conflicting results.
The present study calculated LV elastance, which is a robust method to quantify LV function. We demonstrate that LV loading in MS patients is elevated but normalizes after valve repair and might be a result of reflex pathways.
Additionally, we show that the LV in MS is less compliant than normal due to a combination of right ventricular loading and the valvular disease itself. Immediately after valve dilatation the increase in blood inflow into the LV results in even greater LV stiffness.
Our findings enrich our understanding of heart function in MS patients and provide a simple reproducible way of assessing LV performance in MS.
Introduction
Despite numerous attempts to characterize left ventricular (LV) systolic function in the setting of mitral stenosis (MS), current evidence remains largely conflicting. Early studies demonstrated impaired LV systolic performance, ascribing this to mechanisms such as myocardial fibrosis (Sunamori et al. 1983), impaired inter-ventricular interaction (Curry et al. 1972) and chronic LV under-filling (Kaku et al. 1988). However, later investigations challenged this notion, revealing normal LV contractility in pure MS (Gash et al. 1983; Liu et al. 1992). These discrepancies may at least in part be attributed to the fact that the majority of the conventional methods for LV function assessment are influenced by the considerably altered loading conditions prevailing in the setting of MS. Thus, a more robust approach would comprise the use of indices that are less dependent on changes in LV loading and provide further insight into the ventricular and arterial interaction.
The instantaneous relationship between pressure and volume in the human heart is an expression of the integration of arterial pressure, preload, heart rate and inotropic state of the myocardium. Sunagawa et al. (1984) proposed a comprehensive model by which LV energetics, myocardial function and ventricular performance can be investigated taking into account their interaction with the vascular system. Briefly, the framework of arterio-ventricular coupling allows the characterization of heart function in terms of effective arterial elastance (Ea), a ‘lumped index’ denoting the LV afterload in the time domain, and LV elastance (Ees) representing the slope of the end-systolic pressure–volume relationship (ESPVR) and expressing the contractile force of the LV. A number of validating studies have provided evidence that ESPVR is relatively insensitive to afterload alterations, rendering Ees the gold standard for LV contractility (Suga et al. 1973; Weber et al. 1976). As this approach largely overcomes the limitations associated with haemodynamic loading, it is of particular value in the setting of MS.
Based on the above reasoning, we undertook this study to (1) evaluate LV performance in a large cohort of patients with pure MS using methods that are less susceptible to the altered haemodynamic state, (2) investigate the features of ventricular–arterial interaction in the setting of MS and (3) interrogate the possible alterations in LV diastolic and systolic function following the acute preload increase secondary to valve dilatation.
Methods
Study population
Symptomatic MS patients referred for percutaneous transvenous mitral commisurotomy (PTMC) to the Sri Sathya Sai Institute were enrolled prospectively between January and June 2012. Subjects were excluded if they presented with more than mild (grade > 1) mitral regurgitation (MR), concomitant aortic valve disease, ischaemic heart disease, atrial fibrillation or hypertension. All patients were on low dose β-blockers (atenolol 25 mg), and a combined regime of diuretics (amiloride + furosemide 40 mg). The control group comprised 40 healthy, age-matched subjects free of any medications. The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institutional review board. All subjects provided written informed consent.
Echocardiographic data
All MS subjects underwent transthoracic echocardiogram (TTE) using a GE Vivid E9 system (GE Ultrasound, Horten, Norway) and a 2.5 MHz matrix array transducer in keeping with current recommendations (Lang et al. 2005). LV elastance measurements were derived from simultaneously acquired LV volumes by echocardiography and invasive pressures just prior the PTMC. The echocardiographic and invasive recordings were then repeated within 5 min following PTMC.
LV end-systolic volume (LVESV), end-diastolic volume (LVEDV) and ejection fraction (EF) were measured according to current recommendations employing the Simpsons biplane method from two-dimensional TTE four- and two-chamber apical recordings (Lang et al. 2005). Stroke volume (SVDoppler) was calculated by multiplying the cross-sectional area of the LV outflow tract (LVOT) with the Doppler-derived velocity time integral (VTILVOT). Mitral valve area (MVA) was measured by planimetry and MR was graded semi-quantitatively. Continuous (CW) and pulsed wave (PW) recordings of the inflow mitral velocities (E and A wave) were performed. The mean transmitral gradient (MVGmean) was measured using the CW recordings according to current recommendations (Lang et al. 2005). Spectral tissue velocities were recorded in the septal and lateral mitral annulus in the patient cohort and in controls using a 5 mm PW sample volume and the early myocardial relaxation velocity (E′) as well as the annular tissue velocity during atrial contraction (A′) were recorded. The Wilkins score (WS) was employed to assess valve suitability for the procedure (Wilkins et al. 1988). All analyses were performed offline (EchoPac PC, GE Ultrasound, Waukesha, WI, USA).
Catheterization data
Right heart catheterization was performed using a 6F Swan-Ganz catheter in all MS patients. Right atrial mean pressure (RAPm), right ventricular systolic pressure (RVPs), pulmonary artery systolic and mean pressure (PAPs; PAPm) and mean pulmonary capillary wedge pressure (PCWPm) were measured under fluoroscopy after careful calibration with the zero level set at the mid-thoracic line. Concurrently, a 6F pigtail catheter was advanced through the femoral artery to measure systolic, mean and diastolic arterial pressures (Ps, Pm, Pd) with subsequent LV end-diastolic pressure (LVEDP) and end-systolic pressure (LVESP) recordings before and after PTMC. Trans-septal puncture was performed with an 8F Mullins sheath, dilator and a Brockenbrough needle. Left atrial pressure (LAP) was subsequently recorded. Pressure tracings were stored (WITT Series III, Witt Biomedical Corp., Melbourne, FL, USA) and analysed off-line.
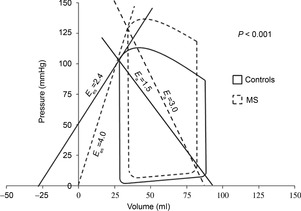
PTMC was performed using a 24 to 28 mm Accura balloon catheter (Vascular Concepts, Halstead, UK) by experts (P.K.D., B.B.) who have individually performed >4000 procedures. The procedure was considered successful if the resultant MVA was >1.5 cm2 with less than +1 grade increase in MR. Cardiac output (CO) and vascular resistances were measured before and after PTMC in conjunction with pressure–volume measurements. CO was calculated employing the estimated Fick's method with the oxygen consumption 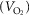 obtained from a standard nomogram.
obtained from a standard nomogram.
Measurements of LV and effective arterial elastance
Ea constitutes a ‘lumped index’ of LV afterload in the time domain and was calculated as
| 1 |
For the study's purposes the calculation of Ea was performed based on estimated LVESP values as derived from the equation
| 2 |
as this accurately approximates LVESP in pressure–volume loop measurements and has been widely used to evaluate ventriculo-arterial coupling (Kelly et al. 1992). More specifically, in MS patients Ea was calculated invasively (Ea INV) using the Ps recorded from the femoral artery. Additionally, non-invasive estimated Ea (Ea NI) was calculated using the regression equation derived from the validation group in order for the measurement to correspond to the Ea NI assessment in controls.
Ees was calculated using the single-beat approach developed by Chen et al. (2001). Importantly, this method does not assume that the volume axis intercept of ESPVR is at the origin of the diagram (0; 0) but can be extrapolated to intersect the volume axis at the point V0; 0 (Chen et al. 2001). Briefly, Ees was calculated as:
| 3 |
where ENd(est) represents group-averaged normalized Ees values as a function of EF and the ratio of diastolic (Pd fem) and systolic (Ps fem) arterial pressure at the level of the femoral artery as described by the equation:
| 4 |
In this equation, ENd (avg) is given by a seven-term polynomial function:
where summation is performed for i = 0 to 7, using values for ai of [0.35695; −7.2266; 74.249; −307.39; 684.54; −856.92; 571.95; −159.1], respectively.
The tNd value was determined as the ratio of the pre-ejection (R-wave to flow onset) to the total systolic period (R-wave to flow termination), with the time at onset and termination of flow obtained from pulsed Doppler in LVOT. LVESP in eqn (3) was estimated as stated above in eqn (2), i.e. LVESP = 0.9 × Ps fem.
LV end-diastolic chamber stiffness was estimated from the ratio of LVEDP and LVEDVi as described by Kass (2000). Furthermore, the LV end-diastolic pressure–volume relationship (EDPVR) was investigated employing the single-beat method described by Klotz et al. (2006).
Validation study
As provided above, Ees measurements in MS patients were based on invasive pressure measurements at the femoral artery level. However, invasive pressure measurements were not performed in controls and the Ees in that group was calculated based on non-invasive sphygmomanometric measurements in the brachial artery. To investigate the relationship between the two different approaches we performed a validation study on 14 MS patients referred for PTMC in whom simultaneous pressure measurements were performed sphygmonanometrically in the brachial artery and invasively in the femoral artery.
Stratification of subjects by severity of MS
MS subjects were dichotomized post-hoc based on MVA of ≤ 0.8 and > 0.8 cm2. Additionally, the MS group was stratified based upon WS (low: ≤9; high: >9).
Echocardiographic and haemodynamic measurements in controls
The 40 subjects constituting the control arm of the study underwent TTE and simultaneous sphygmomanometric measurements for pressure recordings at the left arm. Systolic (Ps brach) and diastolic brachial artery pressures (Pd brach) were recorded. With regard to echocardiographic data, volumetric and quantitative two-dimensional and Doppler measurements were performed and analysed as assessed in the patient cohort. For measurements of Ea and Ees, LVESP was estimated using the systolic brachial artery pressure as recorded sphygmomanometrically and derived from the equation: LVESP = 0.9 × Ps brach. Similarly, for Ees measurements eqns (3) and (4) were modified for controls using the non-invasive brachial artery pressures, i.e.:
| 3a |
and
| 4a |
In controls, PCWP was calculated according to the equation PCWP = 1.24 [E/E′] + 1.9, as proposed by Nagueh et al. (1997). In this equation, E denotes the peak early transmitral inflow velocity (E wave) and the E′ is the early myocardial tissue Doppler velocity at the lateral mitral annulus (Nagueh et al. 1997).
Based on the assumption that PCWP equals LVEDP in healthy individuals, LV end-diastolic chamber stiffness was estimated in controls using the two aforementioned methods, i.e. the end diastolic pressure–volume ratio (Kass, 2000) and the single beat approach (Klotz et al. 2006).
Statistical analysis
Statistical analysis was performed using SPSS version 16.0 (SPSS Inc., Chicago, IL, USA). Continuous variables were expressed as mean ± SD and categorical variables in absolute values and percentage. The Shapiro–Wilk test was used to check normality. Continuous variables were compared using the paired Student t test or the Wilcoxon test. Controls were compared with study subjects using the Mann–Whitney test. Correlations between variables were tested by the Pearson two-tailed correlation. Multiple regression analysis was used to identify independent confounders of end-diastolic LV stiffness. All tests were performed at 95% confidence intervals, and a P-value of < 0.05 was considered statistically significant. Mann–Whitney U test was performed for analysis of the difference between the predicted elastance values derived from the validation study and the corresponding non-invasive values for controls. Analysis of inter- and intraobserver variability was performed for Ees in 10 patients by two observers. Methodological error (Err) in a single measurement estimated from double measurements was calculated according to formula: Err = (SDdiff × 100%)/(total mean × √2), where SDdiff is the SD of the difference between the measurements (Dahlberg, 1940)
Results
Study population
Of the 120 patients enrolled, 14 were excluded due to severe MR following leaflet tear during PTMC (n = 6), tamponade (n = 2), unsuccessful PTMC (n = 1) and incomplete oximetry data (n = 5). In effect, 106 subjects (age 32 ± 8 years, 72% female) were analysed. Table1 summarizes the population characteristics. In total, 46% demonstrated markedly narrowed MVA (≤0.8 cm2). Despite lower EF as compared to controls, LV contractility was significantly higher in MS patients (Ees: 4.1 ± 1.6 vs. 2.4 ± 0.6 mmHg ml−1; P < 0.001).
Table 1.
Baseline and echocardiographic characteristics of the study population
| Variable | Controls (n = 40) | MS (n = 106) | P |
|---|---|---|---|
| Female | 28 (70%) | 77 (72%) | |
| BSA (m2) | 1.5 ± 0.1 | 1.4 ± 0.1 | |
| HR (beats min–1) | 76 ± 10 | 75 ± 10 | |
| SBP (mmHg) | 116 ± 9 | 108 ± 10 | <0.001 |
| NYHA Class | |||
| II | 59 (56%) | ||
| III | 47 (44%) | ||
| Prior PTMC | 34 (32%) | ||
| Medication (%) | |||
| Atenolol 25 mg | 100% | ||
| Amiloride + furosemide 40 mg | 100% | ||
| MR severity | |||
| No MR | 34 (32%) | ||
| Grade I | 72 (68%) | ||
| Wilkins score | |||
| ≤8 | 47 (44%) | ||
| >8 | 59 (56%) | ||
| MVA (cm2) | 4.7 ± 0.9 | 0.8 ± 0.2 | <0.001 |
| MVGmean (mmHg) | 1 ± 0.3 | 19 ± 9 | <0.001 |
| LVEDV (ml) | 88 ± 18 | 82 ± 24 | n.s. |
| LVEDVindex (ml m−2) | 57 ± 9 | 57 ± 16 | n.s. |
| LVESV (ml) | 28 ± 7 | 34 ± 12 | <0.05 |
| LVESVindex (ml m−2) | 19 ± 4 | 23 ± 9 | <0.001 |
| SVDoppler (ml) | 71 ± 13 | 49 ± 16 | <0.001 |
| EF (%) | 66 ± 5 | 60 ± 8 | <0.001 |
PTMC, percutaneous transvenous mitral commisurotomy; BSA, body surface area; HR, heart rate; MR, mitral regurgitation; LVID, left ventricular internal diameter; d, diastolic; s, systolic; LVEDV, end-diastolic volume; LVESV, end-systolic volume; SVDoppler, Doppler-derived stroke volume; EF, ejection fraction; MVA, mitral valve area; MVGmean, mean mitral valve gradient; RVSP, right ventricular systolic pressure.
LV and arterial elastance in MS
Ees was inversely associated with SV in the MS group (r = −0.66; P < 0.001). LVEDV did not differ significantly, but LVESV was larger among MS subjects as was afterload (Ea: 3.0 ± 1.3 vs. 1.5 ± 0.3 mmHg ml−1; P < 0.001). Figure 1 illustrates ESPVR in controls and MS subjects. Controls demonstrated a positive association between Ea and LVEDV (r = 0.60; P < 0.001), whereas an inverse relationship was seen in the MS group (r = −0.73; P < 0.001) with a strong positive correlation between Ea and Ees (r = 0.74; P < 0.001). Ea in the MS group was inversely related to EF (r = −0.54; P < 0.001), exhibiting a weak association with heart rate (r = 0.34; P = 0.005).
Figure 1. Left ventricular end-systolic elastance (Ees) and arterial elastance (Ea) in severe rheumatic mitral stenosis (MS) as compared to normal subjects.
MS patients demonstrated significantly elevated Ees and Ea as compared to controls. Values shown here represent the mean for the two study groups.
MS severity and elastance
Patients with MVA ≤ 0.8 cm2 (n = 47) displayed similar LV contractility (Ees: 4.3 ± 1.6 vs. 3.8 ± 1.7 mmHg ml−1; P > 0.05) but considerably higher Ea (3.3 ± 1.3 vs. 2.8 ± 1.3 mmHg ml−1; P = 0.03) compared to the rest of the MS group. Subjects with high WS (n = 17) had elevated arterial load compared to those with low WS (n = 85) (Ea: 3.7 ± 1.7 vs. 3.0 ± 1.2 mmHg ml−1; P = 0.04), while Ees between the two groups did not differ significantly (Ees: 4.0 ± 1.5 vs. 4.6 ± 2.2 mmHg ml−1; P > 0.05).
Normal vs. reduced EF
Twenty-five MS patients (24%) exhibited reduced EF (≤ 55%). They showed significantly higher Ees (5.5 ± 2.9 vs. 3.9 ± 1.4 mmHg ml−1; P = 0.01) and Ea (5.1 ± 2.3 vs. 2.9 ± 1 mmHg ml−1; P < 0.001) as well as more extensive subvalvular fusion (SVF: 3 ± 0.4 vs. 3.4 ± 0.4, P < 0.05) compared to those with normal EF (n = 81).
Invasive measurements
As shown in Table2, all MS subjects demonstrated elevated intracardiac pressures and reduced CO. Immediately after commissurotomy, LVEDP (12 ± 4 to 16 ± 4 mmHg; P < 0.001) and cardiac index (CI; 2.5 ± 0.6 to 3.2 ± 0.8 l min m−2; P < 0.001) rose significantly, with concomitant SVR (27 ± 8 to 21 ± 8 Wood Units; P < 0.001) and PVR reduction (4.5 ± 4 to 3.4 ± 3 Wood Units; P < 0.001); no significant changes in arterial pressures were noted.
Table 2.
Invasive haemodynamic variables in the study population before and immediately after PTMC
| Variable | Controls (n) | Pre-PTMC (n) | Post-PTMC (n) | P |
|---|---|---|---|---|
| RAPm (mmHg) | 6 ± 3 | 5.7 ± 3 | n.s. | |
| PAPs (mmHg) | 58 ± 24 (105) | 49 ± 17 (105) | <0.001 | |
| PAPm (mmHg) | 39 ± 14 (105) | 32 ± 11 (105) | <0.001 | |
| PCWPm (mmHg) | 9.5 ± 1.2 (40) | 25 ± 7 (104) | 18 ± 5 (104) | <0.001 |
| LAPm (mmHg) | 26 ± 7 (104) | 18 ± 5 (104) | <0.001 | |
| LVESP (mmHg) | 135 ± 18 (104) | 132 ± 18 (104) | n.s. | |
| LVEDP (mmHg) | 12 ± 4 (104) | 16 ± 4 (104) | <0.001 | |
| RVEDP (mmHg) | 7.8 ± 4.2 (104) | 7.6 ± 4.1 (104) | n.s. | |
| Arterial systolic pressure (mmHg) | 134 ± 22 (98) | 131 ± 19 (98) | n.s. | |
| Arterial mean pressure (mmHg) | 97 ± 13 (98) | 97± 14 (98) | n.s. | |
| Arterial diastolic pressure (mmHg) | 74 ± 12 (98) | 75 ± 12 (98) | n.s. | |
| Arterial pulse pressure (mmHg) | 59 ± 17 (98) | 56 ± 15 (98) | n.s. | |
| CI (l min–1 m−2) | 2.5 ± 0.6 (104) | 3.2 ± 0.8 (104) | <0.001 | |
| PVR (Wood Units) | 4.5 ± 4 (103) | 3.4 ± 3 (103) | <0.001 | |
| SVR (Wood Units) | 27 ± 8 (99) | 21 ± 8 (99) | <0.001 | |
| Ees INV (mmHg ml−1) | 4.1 ± 1.6* (95) | 3.5 ± 1.3* (91) | <0.001 | |
| Ees NI (mmHg ml−1) | 2.4 ± 0.6 (40) | 3.7 ± 1.4* (95) | 3.2 ± 1.2* (91) | <0.001 |
| Ea INV (mmHg ml−1) | 3.1 ± 1.3* (101) | 2.6 ± 1.1* (102) | <0.001 | |
| Ea NI (mmHg ml−1) | 1.5 ± 0.3 (40) | 2.7 ± 1.2* (101) | 2.3 ± 1.0* (102) | <0.001 |
| LV stiffness (mmHg ml−1) | 0.17 ± 0.04 (40) | 0.23 ± 0.1* (99) | 0.26 ± 0.1* (99) | <0.001 |
| Beta (EDPVR) | 5.84 ± 0.03 (40) | 5.95 ± 0.17* (99) | 6.2± 0.52* (99) | <0.001 |
| Alpha (EDPVR) × 10−11 (mmHg) | 7.6 ± 7.7 (40) | 35.2 ± 143 (99) | 9.1 ± 2.52* (99) | <0.001 |
PAP, pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure, LAPm, left atrial mean pressure; LVEDP, left ventricular end diastolic pressure; LVESP; left ventricular end systolic pressure; RVEDP, right ventricular end diastolic pressure CI, cardiac index; PVR, pulmonary vascular resistance; SVR, systemic vascular resistance; Ees INV, invasively derived LV elastance; Ees NI, non-invasively derived estimated LV elastance; Ea INV, invasively derived arterial elastance; Ea NI, non-invasively derived estimated arterial elastance; EDPVR, end diastolic pressure–volume relation. P values indicate the significance of differences between pre- and post-PTMC data in the MS group. Significant differences between the control and pre- or post PTMC MS group values are indicated by asterisks.
Diastolic changes
In MS patients, LVEDP correlated significantly with RVEDP (r = 0.43, P < 0.001) and CI (r = 0.24, P < 0.05) and was elevated (>16 mmHg) in 24 cases (23%). Following PTMC, LVEDP further increased (>16 mmHg in 52% of the cases). LV stiffness was significantly higher in the MS group compared to controls both when using the EDPVR of the operant LV stiffness and when estimating the beta value derived from the single beat approach (Fig. 2). In the patient cohort, a reduction in LV stiffness occurred in only 11 patients. When comparing the two groups, patients with a reduction in chamber stiffness following PTMC had lower MVGmean prior to PTMC as compared to the corresponding group with increased LV stiffness (MVGmean 15.7 ± 10.8 vs. 20.8 ± 9.1 mmHg) and lower RVPs (44 ± 10.1 vs. 63.5 ± 24 mmHg). Multiple regression analysis identified RVEDP, Ea and MVAi as constituting the only independent predictors of LV stiffness both before and after PTMC as described by the regression equation: LV stiffness = 0.165 + 0.1 × RVEDP + 0.03 × Ea −0.147 × MVAi, with an overall model fit of r2 = 0.41, F1,94 = 21, P < 0.001. Furthermore, RVEDP at baseline acted as the sole independent predictor of the magnitude of augmentation of the beta value following dilatation of the MV (r = 0.41, P < 0.001). On the other hand, no correlation between LV stiffness and WS, SVF or age was found.
Figure 2. Left ventricular (LV) diastolic stiffness as expressed by the ratio of LV end-diastolic pressure to LV end-diastolic volume.
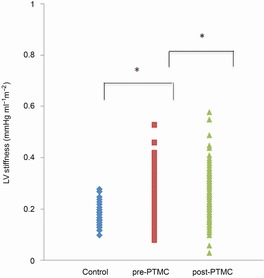
Patients with MS demonstrated elevated LV stiffness compared to controls. Following percutaneous transvenous mitral commissurotomy (PTMC), a further significant increase in LV stiffness was documented.
Haemodynamic alterations following PTMC
MVA increased in all cases following PTMC (0.8 ± 0.2 to 1.6 ± 0.2 cm2; P < 0.001) with a corresponding reduction in the transmitral gradient (19 ± 9 to 5 ± 2 mmHg; P < 0.001) (Table3). Immediately after PTMC, both Ees (4.1 ± 1.6 to 3.5 ± 1.3 mmHg ml−1; P < 0.001) and Ea (3.0 ± 1.6 to 2.6 ± 1.1 mmHg ml−1; P < 0.001) returned to more normal values with a concomitant increase in preload (LVEDV: 82 ± 2 to 90 ± 24 ml; P < 0.001), EF (60 ± 8 to 64 ± 8%; P < 0.001) and SVDoppler (49 ± 16 to 57 ± 17 ml; P < 0.001). Importantly, both methods for assessing LV stiffness revealed a significant reduction in chamber compliance following PTMC, with higher beta and lower alpha values following PTMC, indicating right and upward change in EDPVR following intervention.
Table 3.
Echocardiographic variables in the study population before and immediately after PTMC
| Variable | Pre-PTMC | Post-PTMC | P |
|---|---|---|---|
| HR (beats min–1) | 75 ± 10 | 77 ± 15 | n.s. |
| LVEDV (ml) | 82 ± 24 | 90 ± 24 | <0.001 |
| LVEDVindex (ml/m2) | 57 ± 16 | 64 ± 16 | <0.001 |
| LVESV (ml) | 34 ± 12 | 33 ± 12 | n.s. |
| LVESVindex (ml/m2) | 23 ± 9 | 23 ± 9 | n.s. |
| SVDoppler (ml) | 49 ± 16 | 57 ± 17 | <0.001 |
| EF (%) | 60 ± 8 | 64 ± 8 | <0.001 |
| MVA (cm2) | 0.8 ± 0.2 | 1.6 ± 0.2 | <0.001 |
| MVGmean (mmHg) | 19 ± 9 | 5 ± 2 | <0.001 |
HR, heart rate; LVEDV, left ventricular end diastolic volume; LVESV, left ventricular end systolic volume; SVDoppler, Doppler-derived stroke volume; EF, ejection fraction; MVA, mitral valve area; MVG, mitral valve gradient.
Validation measurements
In the validation group comprising 14 MS patients, simultaneous pressures were acquired sphygmomanom-etrically at the brachial artery level and invasively at the femoral artery level (Table4). The non-invasive Ees measurements (Ees NI) were highly correlated to the invasively derived Ees (Ees INV) (r2 = 0.94, Ees NI = 0.16 + 0.86 × Ees INV) although significantly lower (P = 0.005). Using the regression equation derived from the validation study, we calculated the predicted Ees NI in our patient cohort to verify that the observed difference between the Ees INV and non-invasive measurements in controls was valid. As shown in Table4, the predicted Ees NI values in MS patients both before (Ees NI pre = 3.7 ± 1.4 mm Hg ml−1) and after PTMC (Ees NI post = 3.2 ± 1.1 mm Hg ml−1) were higher as compared to the controls (Ees NI controls = 2.4 ± 0.6) (z = −4.4 and z = −2.2, respectively) with the mean Ees NI pre and Ees NI post being 38 and 28% higher than the corresponding values in controls.
Table 4.
Simultaneous invasive and non-invasive pressure and elastance measurements in the validation group
| Variable | Invasive | Non-invasive PTMC | P |
|---|---|---|---|
| Systolic arterial pressure (mmHg) | 123 ± 18 (14) | 107 ± 11 (14) | <0.001 |
| Diastolic arterial pressure (mmHg) | 68 ± 9 (14) | 62 ± 7 (14) | 0.047 |
| Ees (mmHg ml–1) | 4.15 ± 1.8 (14) | 3.74± 1.6 (14) | <0.001 |
| Ea (mmHg ml–1) | 3.1 ± 1.4 (14) | 2.7 ± 1.2 (14) | <0.001 |
Ees, LV elastance; Ea, effective arterial elastance.
Similarly, we calculated the predicted Ea values based on the validation study. An excellent correlation between invasive (Ea INV) and non-invasively derived Ea NI (r2 = 0.97, P < 0.001) was found in the validation group. As shown in Table4, the Ea NI was lower as compared to Ea INV (Ea NI = 0.04+0.87 × Ea INV, P < 0.001). Based on that equation, the predicted Ea NI was calculated in our patient group both before and following PTMC (Table3). We also inferred that MS patients showed significantly higher Ea values compared to controls, and this was valid for the measurements both before and after PTMC.
Inter- and intraobserver variability analysis showed relatively low error for repeated measurements of Ees (7.7 and 12.8% for intra- and interobserver measurements, respectively).
Discussion
To our knowledge, this is the largest invasive study evaluating LV performance in patients with pure rheumatic MS. Contrary to some previous reports, we demonstrate augmented LV contractility along with reduced LV compliance in severe MS. Our data indicate that EF poorly describes the inherent LV performance in this patient population. The elevated arterial load is strongly associated with LV stiffness and MS severity. Finally, the elevated LV contractility and afterload at baseline returned to more normal levels immediately following PTMC, along with a further increase in LV stiffness.
LV performance in rheumatic MS has been an issue of debate with conflicting observations that might partly be attributed to the load susceptibility of the various measurements employed for the quantification of LV function (Ahmed et al. 1977; Kaku et al. 1988). Indeed, based on the analysis of indices such as EF, stroke work as well as ESPVR and wall motion scoring, earlier studies reported evidence of impaired LV function in MS patients (Heller & Carleton 1970; Curry et al. 1972; Hildner et al. 1972). Reduced SV in relation to LV end-diastolic pressures at rest and during exercise as well as depressed EF have been previously interpreted as indicative of impaired LV function in these patients (Horwitz et al. 1973). Regardless, given the limited preload recruitment secondary to MS, lower LV output per se may not imply impaired ventricular performance. Similarly, circumferential fibre shortening rate (Vcf) was found to be reduced, thus arguing for myocardial dysfunction in this patient population (Holzer et al. 1973). However, experimental studies demonstrated that Vcf varies inversely with afterload alterations (Covell et al. 1966; Urschel et al. 1968). In contrast, Ahmed and colleagues (Ahmed et al. 1977) assessed dP/dt/Pmax and reported preserved LV contractile function in MS patients, although later studies have shown that even this approach is subjected to load dependency, thus providing unreliable results in the setting of MS (Schmidt and Scheer, 1981).
In the present report, LV function was assessed by ESPVR, a relatively load-independent approach well suited for MS (Suga et al. 1973; Suga and Sagawa, 1974). Our findings refute the notion of impaired LV contractile performance in severe MS advocated in previous studies. Instead, they indicate a state of elevated LV contractility, as demonstrated by a roughly 40% higher Ees in MS patients compared to that of age-matched controls. A direct comparison of the single beat Ees with other than EF LV performance indices was not performed in the current study, and thus a detailed physiological explanation of the discrepancy compared to previous findings is not appropriate. However, in an attempt to approach a constructive appreciation of the current results we suggest that they might reflect a less pronounced load sensitivity of Ees as compared to other previously employed LV function measurements. Increased sympathetic activity has been demonstrated in patients with MS and has been ascribed to the decreased SV secondary to the reduced space of the mitral valve (Ashino et al. 1997). The depressed LV ventricular output would yield a reduction in afferent activity of the baroreceptors, which has been considered as a possible cause of sympathetic activation in these patients (Ashino et al. 1997). Apart from the elevated Ees, we demonstrate increased arterial load and systemic peripheral vascular resistance in MS patients as compared to controls, a constellation of findings that might advocate increased sympathetic activity in our patient cohort. A significant association between sympathetic activity and systemic vascular resistance has been previously demonstrated (Ashino et al. 1997). The elevated LV elastance in MS patients as compared to controls in the present study stands in contrast to the findings of Liu et al. (1992) showing similar Ees values in MS patients compared to controls. A plausible explanation for this disparity might lie in the discrepancy of haemodynamic findings between the two cohorts; in our study, MS subjects demonstrated lower CO (3.0 ± 1.0 vs. 3.7 ± 0.9 l min−1), higher LAP (26 ± 7 vs. 18 ± 7 mmHg) and higher PAPm (58 ± 24 vs. 41 ±13 mmHg). These haemodynamic discrepancies suggest lower baroreceptor sensitivity (Ferguson et al. 1990) and increased atrial stretch (Koizumi et al. 1977; Ashino et al. 1997), which in turn imply increased sympathetic tone and hence elevated contractility.
LV systolic function and arterial load
Consistent with previous results, our MS patients demonstrated significantly increased afterload (Liu et al. 1992) with 24% of them showing reduced EF (< 55%) (Kennedy et al. 1970; Gash et al. 1983). Ees, however, was not significantly different in these patients, whereas arterial load was higher (Ea: 4.1 ± 1.9 vs. 2.8 ± 0.9, P < 0.001) compared to those with preserved EF. The concept of ventriculo-arterial coupling posits that EF is determined by the interaction between LV contractility and afterload (Sunagawa et al. 1985). Providing there are no alterations in contractility, PV loop analysis suggests that a 33% afterload reduction (representing the measured difference between mean Ea in the two MS groups) yields an EF increase of roughly 20%, consistent with our results (Kass et al. 1990). Furthermore, with the reduction of Ea observed following PTMC (from 4.1 ± 1.9 to 3.5 ± 1.7 mmHg ml−1), EF normalized (EF: 60±9%) in all cases with depressed LV performance at baseline. This, together with the significant inverse correlation between Ea and EF, suggests that employing EF to describe LV performance in severe MS can be misleading.
MS subjects demonstrated significantly higher LVESV, but not LVEDV, suggesting afterload mismatch (Ea vs. LVEDV, r = −0.75; P < 0.001) only partially compensated for by an increase in contractility (Ees vs. LVEDV, r = 0.61; P < 0.001). In normal hearts, afterload elevation is countered by preload increase to prevent SV reduction. However, MS hinders adequate preload recruitment (MVA vs. LVEDV, r = 0.4; P < 0.001), thereby limiting preload reserve. Hence, despite elevated Ees, the raised arterial load cannot be overcome owing to a hampered Frank–Starling mechanism, yielding lower SV. Although in normal hearts increased LVESV often indicates reduced inotropy, a more applicable explanation in MS could be a state of exhausted contractile reserve, or the LV's inability to further increase Ees. This ‘ceiling effect’ in contractility may also be attributed to the inhibiting impact of β-blockers, partially preventing further increases in LV contractility.
LV performance and MS severity
Previous studies have demonstrated a rapid rise in tension in the subvalvular apparatus (Salisbury et al. 1963; Semafuko & Bowie, 1975) during isovolumetric LV contraction, and a considerable reduction in Ees when the chordae were severed (Hansen et al. 1986). To investigate the influence of mitral apparatus on LV performance, we sub-grouped our patients based on WS, degree of chordal fusion and length separately. Although LV contractility did not differ between these groups, subjects with EF < 55% displayed a significantly higher degree of subvalvular fusion. Our results also suggest that LV afterload is related to the degree of valvular deformation, as reflected by increased Ea in patients with higher valve scores.
LV diastolic function in MS
Previous studies have shown reduced LV compliance in MS patients (Liu et al. 1992; Mayer et al. 1999). A number of possible mechanisms for this observation have been proposed, including a mechanically mediated increase in LV stiffness by the rigid mitral apparatus (Heller & Carleton 1970; Curry et al. 1972), inherent myocardial alterations due to rheumatic disease, as well as altered RV loading and inter-ventricular interaction with this condition (Mayer et al. 1999). Our results provide important details regarding the possible underlying mechanisms of the reduced end-diastolic LV compliance in MS patients. First, we show that the degree of MV stenosis could independently predict the degree of LV compliance. In that sense a more rigid and immobile valvular apparatus could act in a constraining manner reducing the distensibility of the LV during diastolic filling, as suggested by Liu et al. (1992). More importantly, RVEDP was identified as a strong predictor of end-diastolic LV stiffness. The role of inter-ventricular interaction in the setting of EDPVR has been suggested by Curry et al. (1972). In their study, MS patients with enlarged RV, reflecting increased RV preload, displayed impaired anterolateral wall motion and LV function; this observation was ascribed to the mechanical influence of the RV. Another group of investigators showed that RVEDP was associated with LV diastolic conditions in MS patients (Mayer et al. 1999). It has been suggested that a right to left interaction in MS might be secondary to alterations in anterolateral wall motion due to RV pressure overload (Nagel et al. 1996).
Acute haemodynamic alterations following PTMC
Following PTMC, SV and LVEDV rose significantly, while Ees returned to more normal values. SV elevation was directly related to the effect of the Frank–Starling mechanism (ΔLVEDV vs. ΔSV; r = 0.73, P < 0.001), suggesting that following MV dilatation, the LV counteracts afterload mismatch by recruiting preload reserve. Hence, the fall of LV elastance following PTMC can be assigned partly to SV increase after the intervention (ΔSV vs. ΔEes; r = −0.37, P < 0.001) as the increased SV is expected to yield afferent parasympathetic baroreceptor stimulation. This in turn would inhibit sympathetic systemic output, resulting in lower arterial tonus (Ea reduction) and less pronounced LV contractility. Normalization of the baroreceptor reflex function has been attributed to CI increase and occurs within 1 week following PTMC (Ashino et al. 1997). Our findings imply that the arterial pressor reflexes may be reactivated immediately following valve dilatation, supporting the notion that their function is impaired due to haemodynamic rather than structural alterations (Ferguson et al. 1989). An intriguing finding of the present study is that LV end-diastolic stiffness showed a statistically significant albeit slight increase immediately after dilatation of the MV when employing two separate non-invasive measurements of chamber stiffness. In fact, an elevation of chamber stiffness was noted in roughly 90% of the patients. Our data provide a plausible explanation for this finding as we have identified RV preload as the only independent predictor of the increases in EDPVR slope following PTMC. The presence of a non-distensible pericardium surrounding the two ventricles and the common septal wall shared by the two chambers contribute to the haemodynamic interaction between the two ventricles. In particular, the diastolic interaction between the two chambers has been increasingly recognized recently. In the setting of MS, the increased RVEDP occurring in MS due to pulmonary hypertension might be a plausible explanation for the increased LV stiffness. The observation that RVEDP did not change significantly following intervention, despite a significant fall in systolic RV pressures, adds weight to this hypothesis as after opening of the MV the LV has to accommodate larger volumes (increase in LVEDV). This would add a further constraint and thus lead to elevated LV stiffness. Although the present study did not investigate possible changes in myocardial stiffness following PTMC, it appears not to provide a plausible explanation for the altered LV chamber stiffness observed after MV dilatation.
Clinical implications
EF is misleading when studying LV performance in patients with MS as a result of the elevated LV afterload. The single-beat approach to measuring Ees provides a feasible, more comprehensive evaluation of LV function. Optimal pharmacological inhibition of the adrenergic activation might have a beneficial effect in MS patients.
Limitations
In the present study, non-invasive indices of LV and arterial function were used. LV end-diastolic stiffness and LV elastance as well as arterial elastance are optimally recorded using conductance catheters. However, all the aforementioned non-invasive measurements are validated against gold standard invasive methods (Kelly et al. 1992; Kass, 2000; Chen et al. 2001). Additionally, in our study, we calculated Ees and Ea using the invasively derived pressures, whereas in the controls non-invasive pressure measurements were performed for the same reason. This limitation of the study was addressed by a validation study. The predicted values for the patient cohort, although different from the invasive data, did not alter the results of the study. Furthermore, in controls LV chamber stiffness was measured using PCWP values estimated using a previously proposed equation (Nagueh et al. 1997). As the authors in that study reported a standard error of approximately 4 mmHg, this would result in overlap in LV stiffness between MS patients and controls. To resolve this concern we proceeded by using a PCWP value of 11 mmHg for all healthy controls (the generally accepted upper normal value in young healthy subjects). Measurements of LV stiffness thus revealed that even in that extreme case LV stiffness in controls was significantly lower (0.2 ± 0.03 mmHg ml−1) as compared to MS patients (P < 0.001). Doppler and two-dimensional echocardiographic measurements entail an inherently larger variability compared to invasive measurements. On the other hand, measurements of intra- and interobserver variation performed for Ees show a rather low variation for repeated single measurements. Finally, patients in our study had advanced rheumatic MS (WS > 8 in 56% of cases) and may not represent the haemodynamic state in less severe degrees of stenosis. However, the study adds important physiological insight into severe rheumatic MS.
Conclusion
Subjects with severe MS exhibit a hypercontractile LV, most probably reflecting an increased sympathetic tone. With preload recruitment immediately following PTMC, LV afterload and contractility tend to normalize in most patients. Finally, we demonstrate that heightened arterial load is associated with MS severity.
Glossary
- BSA
body surface area
- CI
cardiac index
- CO
cardiac output
- Ea
effective arterial elastance
- Ea INV
invasive effective arterial elastance
- Ea NI
non-invasive effective arterial elastance
- Ees
LV elastance
- Ees NI
non-invasive LV elastance
- Ees INV
invasive LV elastance
- EF
ejection fraction
- E′lat
early diastolic mitral annular velocity of the lateral LV basal wall
- E′sept
early diastolic mitral annular velocity of the IV septum
- ESPVR
end-systolic pressure–volume relationship
- LA
left atrium
- LAP
left atrial pressure
- LV
left ventricle/ventricular
- LVEDP
left ventricular end-diastolic pressure
- LVESP
left ventricular end-systolic pressure
- LVEDVindex
left ventricular end-diastolic volume indexed to BSA
- LVESVindex
left ventricular end-systolic volume indexed to BSA
- LVOT
left ventricular outflow tract
- MS
mitral stenosis
- MVA
mitral valve area
- MVG
mean transmitral gradient
- PAPd
pulmonary arterial diastolic pressure
- PAPm
pulmonary arterial mean pressure
- PAPs
pulmonary arterial systolic pressure
- PCWP
pulmonary capillary wedge pressure
- Pd
diastolic systemic arterial pressure
- Pm
mean systemic arterial pressure
- Ps
systolic systemic arterial pressure
- PTMC
percutaneous transvenous mitral commissurotomy
- PVR
pulmonary vascular resistance
- RAP
right atrial mean pressure
- RHC
right heart catheterization
- RV
right ventricle/vetricular
- RVEDP
right ventricular end-diastolic pressure
- RVESP
right ventricular end-systolic pressure
- RVPs
RV systolic pressure
- SV
stroke volume
- SVi
stroke volume index
- SVR
systemic vascular resistance
- TTE
transthoracic echocardiography
Additional information
Competing interests
No extramural funding was used to support this work. None of the authors have any conflict of interest to disclose. The authors are solely responsible for the design and conduct of this study, all study analyses and drafting and editing of the paper. All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Author contributions
A.M., A.V., A.I.N., S.S. and R.W. designed the study; A.V., A.M., B.B. and P.K.D. performed invasive and echocardiographic measurements; A.V., A.M., A.I.N., K.S., S.C.G., A.S., R.W., B.B. and P.K.D. contributed to analysis and interpretation of the data; A.V., A.M. and A.I.N. wrote the manuscript. K.S., S.C.G., A.S., R.W., B.B. and P.K.D. revised the manuscript critically. All authors have read and approved the final manuscript.
References
- Ahmed SS, Regan TJ, Fiore JJ, Levinson GE. The state of the left ventricular myocardium in mital stenosis. Am Heart J. 1977;94:28–36. doi: 10.1016/s0002-8703(77)80340-2. [DOI] [PubMed] [Google Scholar]
- Ashino K, Gotoh E, Sumita S, Moriya A, Ishii M. Percutaneous transluminal mitral valvuloplasty normalizes baroreflex sensitivity and sympathetic activity in patients with mitral stenosis. Circulation. 1997;96:3443–3449. doi: 10.1161/01.cir.96.10.3443. [DOI] [PubMed] [Google Scholar]
- Chen CH, Fetics B, Nevo E, Rochitte CE, Chiou KR, Ding PA, Kawaguchi M, Kass DA. Noninvasive single-beat determination of left ventricular end-systolic elastance in humans. J Am Coll Cardiol. 2001;38:2028–2034. doi: 10.1016/s0735-1097(01)01651-5. [DOI] [PubMed] [Google Scholar]
- Covell JW, Ross J, Jr, Sonnenblick EH, Braunwald E. Comparison of the force–velocity relation and the ventricular function curve as measures of the contractile state of the intact heart. Circ Res. 1966;19:364–372. doi: 10.1161/01.res.19.2.364. [DOI] [PubMed] [Google Scholar]
- Curry GC, Elliott LP, Ramsey HW. Quantitative left ventricular angiocardiographic findings in mitral stenosis. Detailed analysis of the anterolateral wall of the left ventricle. Am J Cardiol. 1972;29:621–627. doi: 10.1016/0002-9149(72)90162-2. [DOI] [PubMed] [Google Scholar]
- Dahlberg G. Statistical Methods for Medical and Biological Students. London: G. Allen & Unwin; 1940. [Google Scholar]
- Ferguson DW, Berg WJ, Sanders JS. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct microneurographic recordings. J Am Coll Cardiol. 1990;16:1125–1134. doi: 10.1016/0735-1097(90)90544-y. [DOI] [PubMed] [Google Scholar]
- Ferguson DW, Berg WJ, Sanders JS, Roach PJ, Kempf JS, Kienzle MG. Sympathoinhibitory responses to digitalis glycosides in heart failure patients. Direct evidence from sympathetic neural recordings. Circulation. 1989;80:65–77. doi: 10.1161/01.cir.80.1.65. [DOI] [PubMed] [Google Scholar]
- Gash AK, Carabello BA, Cepin D, Spann JF. Left ventricular ejection performance and systolic muscle function in patients with mitral stenosis. Circulation. 1983;67:148–154. doi: 10.1161/01.cir.67.1.148. [DOI] [PubMed] [Google Scholar]
- Hansen DE, Cahill PD, DeCampli WM, Harrison DC, Derby GC, Mitchell RS, Miller DC. Valvular–ventricular interaction: importance of the mitral apparatus in canine left ventricular systolic performance. Circulation. 1986;73:1310–1320. doi: 10.1161/01.cir.73.6.1310. [DOI] [PubMed] [Google Scholar]
- Heller SJ. Carleton RA. Abnormal left ventricular contraction in patients with mitral stenosis. Circulation. 1970;42:1099–1110. doi: 10.1161/01.cir.42.6.1099. [DOI] [PubMed] [Google Scholar]
- Hildner FJ, Javier RP, Cohen LS, Samet P, Nathan MJ, Yahr WZ, Greenberg JJ. Myocardial dysfunction associated with valvular heart disease. Am J Cardiol. 1972;30:319–326. doi: 10.1016/0002-9149(72)90559-0. [DOI] [PubMed] [Google Scholar]
- Holzer JA, Karliner JS, O'Rourke RA, Peterson KL. Quantitative angiographic analysis of the left ventricle in patients with isolated rheumatic mitral stenosis. Br Heart J. 1973;35:497–502. doi: 10.1136/hrt.35.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz LD, Mullins CB, Payne RM, Curry GC. Left ventricular function in mitral stenosis. Chest. 1973;64:609–614. doi: 10.1378/chest.64.5.609. [DOI] [PubMed] [Google Scholar]
- Kaku K, Hirota Y, Shimizu G, Kita Y, Saito T, Kawamura K. Depressed myocardial contractility in mitral stenosis–an analysis by force–length and stress-shortening relationships. Jpn Circ J. 1988;52:35–43. doi: 10.1253/jcj.52.35. [DOI] [PubMed] [Google Scholar]
- Kass DA. Assessment of diastolic dysfunction. Invasive modalities. Cardiol Clin. 2000;18:571–586. doi: 10.1016/s0733-8651(05)70162-4. [DOI] [PubMed] [Google Scholar]
- Kass DA, Grayson R, Marino P. Pressure–volume analysis as a method for quantifying simultaneous drug (amrinone) effects on arterial load and contractile state in vivo. J Am Coll Cardiol. 1990;16:726–732. doi: 10.1016/0735-1097(90)90366-w. [DOI] [PubMed] [Google Scholar]
- Kelly RP, Ting CT, Yang TM, Liu CP, Maughan WL, Chang MS, Kass DA. Effective arterial elastance as index of arterial vascular load in humans. Circulation. 1992;86:513–521. doi: 10.1161/01.cir.86.2.513. [DOI] [PubMed] [Google Scholar]
- Kennedy JW, Yarnall SR, Murray JA, Figley MM. Quantitative angiocardiography. IV. Relationships of left atrial and ventricular pressure and volume in mitral valve disease. Circulation. 1970;41:817–824. doi: 10.1161/01.cir.41.5.817. [DOI] [PubMed] [Google Scholar]
- Klotz S, Hay I, Dickstein ML, Yi GH, Wang J, Maurer MS, Kass DA, Burkhoff D. Single-beat estimation of end-diastolic pressure–volume relationship: a novel method with potential for noninvasive application. Am J Physiol Heart Circ Physiol. 2006;291:H403–412. doi: 10.1152/ajpheart.01240.2005. [DOI] [PubMed] [Google Scholar]
- Koizumi K, Nishino H, Brooks CM. Centers involved in the autonomic reflex reactions originating from stretching of the atria. Proc Natl Acad Sci U S A. 1977;74:2177–2181. doi: 10.1073/pnas.74.5.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ Chamber Quantification Writing Group; American Society of Echocardiography's Guidelines and Standards Committee; European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Liu CP, Ting CT, Yang TM, Chen JW, Chang MS, Maughan WL, Lawrence W, Kass DA. Reduced left ventricular compliance in human mitral stenosis. Role of reversible internal constraint. Circulation. 1992;85:1447–1456. doi: 10.1161/01.cir.85.4.1447. [DOI] [PubMed] [Google Scholar]
- Mayer IV, Fischer A, Jakob M, Mandinov L, Hug R, Vassalli G, Hess OM. Reversal of increased diastolic stiffness in mitral stenosis after successful balloon valvuloplasty. J Heart Valve Dis. 1999;8:47–56. [PubMed] [Google Scholar]
- Nagel E, Stuber M, Hess OM. Importance of the right ventricle in valvular heart disease. Eur Heart J. 1996;17:829–836. doi: 10.1093/oxfordjournals.eurheartj.a014963. [DOI] [PubMed] [Google Scholar]
- Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quiñones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527–1533. doi: 10.1016/s0735-1097(97)00344-6. [DOI] [PubMed] [Google Scholar]
- Salisbury PF, Cross CE, Rieben PA. Chorda tendinea tension. Am J Physiol. 1963;205:385–392. doi: 10.1152/ajplegacy.1963.205.2.385. [DOI] [PubMed] [Google Scholar]
- Schmidt HD. Scheer RD. Quantitative data on the afterload dependence of left ventricular dp/dtmax in isolated canine hearts. Basic Res Cardiol. 1981;76:89–105. doi: 10.1007/BF01908165. [DOI] [PubMed] [Google Scholar]
- Semafuko WE. Bowie WC. Papillary muscle dynamics: in situ function and responses of the papillary muscle. Am J Physiol. 1975;228:1800–1807. doi: 10.1152/ajplegacy.1975.228.6.1800. [DOI] [PubMed] [Google Scholar]
- Suga H. Sagawa K. Instantaneous pressure–volume relationships and their ratio in the excised, supported canine left ventricle. Circ Res. 1974;35:117–126. doi: 10.1161/01.res.35.1.117. [DOI] [PubMed] [Google Scholar]
- Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure–volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res. 1973;32:314–322. doi: 10.1161/01.res.32.3.314. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for the maximal stroke work studied in isolated canine left ventricle. Circ Res. 1985;56:586–595. doi: 10.1161/01.res.56.4.586. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Sagawa K, Maughan WL. Ventricular interaction with the loading system. Ann Biomed Eng. 1984;12:163–189. doi: 10.1007/BF02584229. [DOI] [PubMed] [Google Scholar]
- Sunamori M, Suzuki A, Harrison CE. Relationship between left ventricular morphology and postoperative cardiac function following valve replacement for mitral stenosis. J Thorac Cardiovasc Surg. 1983;85:727–732. [PubMed] [Google Scholar]
- Urschel CW, Covell JW, Sonnenblick EH, Ross J, Jr, Braunwald E. Effects of decreased aortic compliance on performance of the left ventricle. Am J Physiol. 1968;214:298–304. doi: 10.1152/ajplegacy.1968.214.2.298. [DOI] [PubMed] [Google Scholar]
- Weber KT, Janicki JS, Hefner LL. Left ventricular force–length relations of isovolumic and ejecting contractions. Am J Physiol. 1976;231:337–343. doi: 10.1152/ajplegacy.1976.231.2.337. [DOI] [PubMed] [Google Scholar]
- Wilkins GT, Weyman AE, Abascal VM, Block PC, Palacios IF. Percutaneous balloon dilatation of the mitral valve: an analysis of echocardiographic variables related to outcome and the mechanism of dilatation. Br Heart J. 1988;60:299–308. doi: 10.1136/hrt.60.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]