

Abstract
The mechanisms triggering disuse muscle atrophy remain of debate. It is becoming evident that mitochondrial dysfunction may regulate pathways controlling muscle mass. We have recently shown that mitochondrial dysfunction plays a major role in disuse atrophy of soleus, a slow, oxidative muscle. Here we tested the hypothesis that hindlimb unloading-induced atrophy could be due to mitochondrial dysfunction in fast muscles too, notwithstanding their much lower mitochondrial content. Gastrocnemius displayed atrophy following both 3 and 7 days of unloading. SOD1 and catalase up-regulation, no H2O2 accumulation and no increase of protein carbonylation suggest the antioxidant defence system efficiently reacted to redox imbalance in the early phases of disuse. A defective mitochondrial fusion (Mfn1, Mfn2 and OPA1 down-regulation) occurred together with an impairment of OXPHOS capacity. Furthermore, at 3 days of unloading higher acetyl-CoA carboxylase (ACC) phosphorylation was found, suggesting AMP-activated protein kinase (AMPK) pathway activation. To test the role of mitochondrial alterations we used Tg-mice overexpressing PGC-1α because of the known effect of PGC-1α on stimulation of Mfn2 expression. PGC-α overexpression was sufficient to prevent (i) the decrease of pro-fusion proteins (Mfn1, Mfn2 and OPA1), (ii) activation of the AMPK pathway, (iii) the inducible expression of MuRF1 and atrogin1 and of authopagic factors, and (iv) any muscle mass loss in response to disuse. As the effects of increased PGC-1α activity were sustained throughout disuse, compounds inducing PGC-1α expression could be useful to treat and prevent muscle atrophy also in fast muscles.
Key points
Skeletal muscle atrophy occurs as a result of disuse. Although several studies have established that a decrease in protein synthesis and increase in protein degradation lead to muscle atrophy, little is known about the triggers underlying such processes.
A growing body of evidence challenges oxidative stress as a trigger of disuse atrophy; furthermore, it is also becoming evident that mitochondrial dysfunction may play a causative role in determining muscle atrophy.
Mitochondrial fusion and fission have emerged as important processes that govern mitochondrial function and PGC-1α may regulate fusion/fission events.
Although most studies on mice have focused on the anti-gravitary slow soleus muscle as it is preferentially affected by disuse atrophy, several fast muscles (including gastrocnemius) go through a significant loss of mass following unloading.
Here we found that in fast muscles an early down-regulation of pro-fusion proteins, through concomitant AMP-activated protein kinase (AMPK) activation, can activate catabolic systems, and ultimately cause muscle mass loss in disuse. Elevated muscle PGC-1α completely preserves muscle mass by preventing the fall in pro-fusion protein expression, AMPK and catabolic system activation, suggesting that compounds inducing PGC-1α expression could be useful to treat and prevent muscle atrophy.
Introduction
The molecular mechanisms that govern the imbalance between pathways controlling protein synthesis and degradation in disuse muscle atrophy remain unclear, notwithstanding the large amount of work done (Bonaldo & Sandri, 2013). Investigations of multiple models of disuse in animals have shown an up-regulation of genes related to protein degradation and stress response as well as a down-regulation of those related to transcription and translation, and energy metabolism (Lecker et al. 2004; Bodine, 2013).
Debate is ongoing regarding whether and to what extent redox signalling can actually be the trigger of such phenomena (Pellegrino et al. 2011a,b; Powers et al. 2012). A growing body of evidence challenges oxidative stress as a trigger of disuse atrophy (Ikemoto et al. 2002; Koesterer et al. 2002; Servais et al. 2007; Brocca et al. 2010; Desaphy et al. 2010; Glover et al. 2010; Kuwahara et al. 2010; Pellegrino et al. 2011b; Powers et al. 2012; Cannavino et al. 2014). We have recently reported that, in soleus muscle, notwithstanding an early persistent redox imbalance, a metabolic programme controlling muscle mass, rather than oxidative stress, played a major role in triggering hindlimb unloading (HU) muscle atrophy. A down-regulation of peroxisome proliferative activated receptor-γ coactivator 1α (PGC-1α) expression and protein content was, in fact, shown to activate catabolic systems by promoting ubiquitin ligase and autophagy gene expression through FoxO3 disinhibition (Cannavino et al. 2014).
Although most studies on mice have focused on the anti-gravitary, slow soleus muscle as it is preferentially affected by disuse atrophy, several fast muscles (including gastrocnemius) go through a significant loss of mass following unloading (Kyparos et al. 2005; Brocca et al. 2010; Desaphy et al. 2010). As fast muscles in mice have mostly a glycolytic metabolism and express low levels of PGC-1 (Lin et al. 2002), the metabolic programme supporting disuse-induced muscle mass loss in soleus might not be relevant in fast muscles.
Data regarding PGC-1α during disuse in fast muscles are inconsistent: increases (Wagatsuma et al. 2011), decreases (Mazzatti et al. 2008) as well as no change (Nagatomo et al. 2011) of PGC-1α levels have been reported. However, alterations of mitochondrial biogenesis (Liu et al. 2012b), mitochondrial respiration (Yajid et al. 1998) and mitochondrial dynamics (Wagatsuma et al. 2011; Liu et al. 2012b) have been shown after prolonged unloading in fast muscles. Moreover, in our previous study we demonstrated that antioxidant treatment during unloading did not prevent gastrocnemius atrophy, suggesting that oxidative stress is not the major cause of disuse atrophy also in fast muscle (Brocca et al. 2010).
A recent report has highlighted the importance of mitochondrial dynamics in cell and animal physiology. Mitochondria constantly fuse and divide, and an imbalance of these two processes dramatically alters overall mitochondrial morphology and function (Chen & Chan, 2005). Mitochondrial fusion and fission have emerged as important processes that govern mitochondrial function (Detmer & Chan, 2007; Hoppins et al. 2007). The mitofusins, Mfn1 and Mfn2, are located on the mitochondrial outer membrane and are involved in early steps in membrane fusion (Koshiba et al. 2004; Meeusen et al. 2006; Song et al. 2009). The dynamin-related protein optic atrophy 1 (OPA1) is associated with the inner membrane and is essential for inner membrane fusion (Meeusen et al. 2006; Song et al. 2009). In addition to its well-recognized function of controlling mitochondrial morphology, mitochondrial fusion clearly protects mitochondrial function (Detmer & Chan, 2007). Mice lacking Mfn1/2 function in skeletal muscle exhibit both mitochondrial dysfunction and profound muscle atrophy (Chen et al. 2010).
Mfn2 is a key target of the nuclear coactivator PGC-1α. Maintenance of a normal expression of Mfn2 is critical for the stimulatory effect of PGC-1α on mitochondrial membrane potential (Soriano et al. 2006) and PGC-1α may regulate mitochondrial fusion/fission events and hence mitochondrial function (Soriano et al. 2006). The latter phenomena could reduce ATP generating capacity, which in turn could shift signalling pathways toward catabolism through activation of 5’AMP-activated protein kinase (AMPK). AMPK, a cellular kinase, senses the energy level of the cell and causes atrogin-1 and muscle-specific ring finger protein-1 (MuRF-1) induction via a FoxO3-dependent mechanism (Tong et al. 2009) determining muscle loss.
The goal of the present study was to clarify whether mytochondrial dysfunction, recently indicated to play a major role in soleus (Cannavino et al. 2014), could trigger disuse-induced muscle atrophy also in a fast muscle, the gastrocnemius, which has different metabolism, much lower mitochondrial content (Jackman & Willis, 1996; Berchtold et al. 2000; Lin et al. 2002), low PGC-1α expression (Lin et al. 2002) and different redox balance response to disuse (Brocca et al. 2010). We show that: (1) HU resulted in the early down-regulation of pro-fusion protein expression and mitochondrial dysfunction; (2) energy stress activated the protein degradation pathway through AMPK activation; and (3) gastrocnemius atrophy was totally prevented by muscle-specific over-expression of PGC-1α through stimulation of Mfn2 expression.
Methods
Ethical approval
Experiments were approved by the Italian Health Department and complied with the Italian guidelines for the use of laboratory animals, which conform to the European Community Directive published in 1986 (86/609/ECC).
Animal care and hindlimb unloading
Six-month-old male C57BL/6 mice (Charles River Laboratories, Wilmington, MA, USA) and transgenic mice overexpressing PGC-1α (TgPGC-1α) in skeletal muscle, as previously described (Lin et al. 2002), were used. Mice were unloaded for 3, 7 and 14 days as previously described (Brocca et al. 2010). Briefly, animals were suspended individually in special cages by thin string tied at one end to the tail and at the other end to the top of the cage; the length of the string was adjusted to allow the animals moving freely on the forelimbs, while the body was inclined at 30–40 deg from the horizontal plane. All mice had water and food ad libitum. The ground C57BL/6 and TgPGC-1α mice were maintained free in single cages and killed after 3, 7 or 14 days. The animals of all experimental groups were killed at 10.00 h after 2 h without food by cervical dislocation to allow removal of gastrocnemius muscle. Muscles were immediately frozen in liquid nitrogen and stored at −80°C.
Experimental plan
Not all analyses were performed on both WT and TgPGC-1α mice at all times (3, 7 and 14 days), but the timing of HU, the strain of the animals and the analyses performed were chosen according to a precise rationale. WT mice were given 3 and 7 days of HU to analyse the time course of adaptations of muscle mass, intracellular pathways controlling muscle mass, redox status, mitochondrial impairment and AMPK pathway. As the experiment on WT mice suggested that mitochondrial dysfunction could be involved in causing gastrocnemius atrophy, to test such a conclusion and study the mechanisms underlying the potential prevention of muscle mass loss, TgPGC-1α mice were subjected to HU for 3 days. The time was chosen based on the observations that intracellular signalling pathways controlling muscle mass were previously shown to go through clear adaptations at that time in soleus (Cannavino et al. 2014) and in gastrocnemius (experiments on WT mice of this study), and that gastrocnemius was atrophic following 3 days of HU (Fig.1). TgPGC-1α mice underwent 14 days of HU because we wanted to check that PGC-1 α protein levels were maintained during chronic disuse in transgenic mice and that the positive effect on muscle size found at 3 days of HU was maintained also after prolonged disuse.
Figure 1. CSA is reduced in gastrocnemius in the early phases of HU.
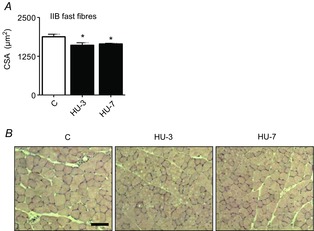
A, quantification of average fibre area from haematoxylin and eosin-stained gastrocnemius muscle from control mice (C) and mice unloaded for 3 days (HU-3) and 7 days (HU-7). B, representative haematoxylin and eosin stainings of the gastrocnemius muscle of control mice (C), and mice unloaded for 3 days (HU-3) and 7 days (HU-7). *Significantly different from C, P < 0.05. Scale bar: 100 μm. Data are presented as means ± SEM.
Cross-sectional area (CSA) analysis
CSA of individual muscle fibres was determined in the mid-belly region of gastrocnemius muscles as previously described (Brocca et al. 2010). Briefly, muscle serial transverse sections (10 μm thick) were stained with haematoxylin-eosin. Images of the stained sections were captured from a light microscope (Leica DMLS) and transferred to a personal computer using a video camera (Leica DFC 280). Fibre CSA was measured with Image J analysis software (NIH, Bethesda, MD, USA) and expressed in micrometres squared.
Analysis of myosin heavy chain (MHC) isoform content
MHC isoform content was determined using an electrophoretic approach previously described in detail (Brocca et al. 2010). Briefly, about 6 μg of each muscle sample were dissolved in lysis buffer (20 mm Tris-HCl, 1% Triton X100, 10% glycerol, 150 mm NaCl, 5 mm EDTA, 100 mm NaF, 2 mm NaPPi, 1× inhibitors protease phosphatase (Protease Inhibitor Cocktail, Sigma-Aldrich, St. Louis, MO, USA) and 1 mm PMSF). The lysates were loaded onto 8% polyacrylamide SDS-PAGE gels. Electrophoresis was run for 2 h at 200 V and then for 24 h at 250 V; the gels were stained with Coomassie Blue. Densitometric analysis of MHC bands was performed to assess the relative proportion of the MHC isoforms (Pellegrino et al. 2003)
Western blot analysis
Frozen muscle samples were pulverized and immediately re-suspended in a lysis buffer (20 mm Tris-HCl, 1% Triton X100, 10% glycerol, 150 mm NaCl, 5 mm EDTA, 100 mm NaF and 2 mm NaPPi supplemented with 1× protease, phosphatase inhibitors (Sigma-Aldrich) and 1 mm PMSF). The homogenate obtained was centrifuged at 18000 g for 20 min at 4°C and the supernatant stored at −80°C until ready to use. Equal amounts of muscle samples were loaded on gradient precast gels purchased from Bio-Rad (AnyKd; Hercules, CA, USA). Proteins were electro-transferred to PVDF membranes at 35 mA overnight. The membranes were probed with specific primary antibodies (see below). Thereafter, the membranes were incubated in HRP-conjugated secondary antibody. The protein bands were visualized by an enhanced chemiluminescence method. The content of each protein investigated was assessed by determining the brightness–area product of the protein band as previously described (Gondin et al. 2011).
OxyBlot analysis
Muscle samples previously stored at −80°C were pulverized and homogenized at 4°C in an antioxidant buffer containing protease inhibitors, 25 mm imidazole and 5 mm EDTA, pH 7.2, adjusted with NaOH as previously described in detail (Brocca et al. 2010). The lysate was left for 20 min on ice and the homogenate obtained was centrifuged at 18000 g for 20 min at 4°C. Protein concentration was determined on the supernatant using the RC DCTM protein assay kit (BioRad). The supernatant was stored at −80°C until ready to use.
Protein carbonylation level was detected using an OxyBlot kit (AbNova, Taipei City, Taiwan), which provides reagents for sensitive immunodetection of these carbonyl groups. The carbonyl groups in the protein side chains are derivatized to 2,4-dinitrophenylhydrazone (DNP hydrazone) by reaction with 2,4-dinitrophenylhydrazine (DNPH); 6 μg of the DNP-derivatized protein samples were separated by PAGE (15% SDS polyacrylamide gels) and then blotted for 2 h at 100 V to a nitrocellulose membrane. Membranes obtained were stained with Ponceau Red and then scanned. Membranes were incubated with primary antibody, specific to the DNP moiety of the proteins, and subsequently with an HRP–antibody conjugate directed against the primary antibody (secondary antibody: goat anti-rabbit IgG). Blots were developed by using an enhanced chemiluminescence method. Positive bands emitting light were detected by short exposure to photographic films. Protein oxidation was quantified by defining the oxidative index (OI), i.e. the ratio between densitometric values of the OxyBlot bands and those stained with Ponceau Red.
Gene expression analysis
Total RNA, from skeletal samples, was extracted using the Promega SV Total RNA isolation kit; the concentration of RNA was evaluated by using a NanoDrop instrument (Thermo Scientific, Waltham, MA, USA) and 300 ng was reverse-transcribed with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) to obtain cDNA. The cDNA was analysed by real-time PCR (RT-PCR; AB 7500) with the SYBR Green or TaqMan PCR kit (Applied Biosystems, Foster City, CA, USA) and the data were normalized to hypoxanthine-guanine phosphoribosyl transferase rRNA. Oligonucleotide primers used for RT-PCR are listed below.
Primers
Forward (FP) and reverse (RP) primers used for RT-PCR are listed below:
MuRF1 FP: ACCTGCTGGTGGAAAACATC; RP: CTTCGTGTTCCTTGCACATC
Atrogin-1 FP: GCAAACACTGCCACATTCTCTC; RP: CTTGAGGGGAAAGTGAGACG
p62 FP: CCCAGTGTCTTGGCATTCTT; RP: AGGGAAAGCAGAGGAAGCTC
Beclin1 FP: GCTCCTGAGGCATGGAGGGGTCT; RP: GGTTTCGCCTGGGCTGTGGTAA
PGC-1α FP: ACCCCAGAGTCACCAAATGA; RP: CGAAGCCTTGAAAGGGTTATC
Mfn-1, Mfn-2 and OPA1 TaqMan gene expression assay (Applied Biosystems)
HPRT Quanti Tect Primer Assay (Qiagen, Valencia, CA, USA)
Antibodies
Anti-rabbit superoxide dismutase 1 (Abcam, Cambridge, MA, USA); anti-rabbit catalase (Abcam); anti-rabbit α tubulin (Sigma-Aldrich); anti-mouse OXPHOS complexes (Abcam); anti-rabbit PGC-1α (Abcam); anti-rabbit DRP1 (Cell Signaling, Danvers, MA, USA); anti-rabbit p-AKT(ser473) (Cell Signaling); anti-rabbit AKT (Cell Signaling); anti-rabbit p-S6Rp(ser 235/236) (Cell Signaling); anti-rabbit S6Rp (Cell Signaling); anti-rabbit p-4EBP1(thr 37/46) (Cell Signaling); anti-rabbit 4EBP1 (Cell Signaling); anti-rabbit p-AMPK(thr 172) (Cell Signaling); anti-rabbit AMPK (Cell Signaling); anti-rabbit p-ACC(ser79) (Cell Signaling); anti-rabbit ACC (Cell Signaling); anti-rabbit LC3B (Sigma Aldrich); anti-rabbit citrate synthase (Abcam); anti-mouse IgG (Dako, Glostrup, Denmark); anti-rabbit IgG (Cell Signaling).
Hydrogen peroxide quantification
Frozen muscle samples were pulverized in a steel mortar with liquid nitrogen to obtain a powder that was immediately re-suspended in the assay buffer provided with the kit (Abcam). After protein quantification, samples were removed of all proteins. A standard curve with known H2O2 concentration was generated following the supplied protocol. Samples and H2O2 standards were incubated at room temperature for 10 min in HRP and OxiRed Probe that reacted with H2O2 to produce red fluorescence (Ex/Em = 535/587 nm) and measured with a micro-plate reader (Tecan Infinite 200 Pro) (Cannavino et al. 2014).
OXPHOS capacity
Maximal ADP-stimulated mitochondrial respiration (‘OXPHOS capacity’) was determined in permeabilized muscle fibres by high-resolution respirometry (Oxygraph 2-K; Oroboros Instruments, Innsbrück, Austria) (Salvadego et al. 2013). Fibre bundles were separated using sharp forceps in ice-cold preservation solution (BIOPS; Oroboros Instruments). Muscle samples in solution plus 10% (w/v) fatty acid-free BSA and 30% (v/v) DMSO were quickly frozen in liquid nitrogen and stored at −80°C until analysis within 1 month. For analysis, muscle samples were quickly thawed by immersion in the preservation solution at room temperature, washed and permeabilized in saponin (50 μg ml–1) for 30 min at 4°C. Fibres were subsequently washed in a mitochondrial respiration solution (MiR05; Oroboros Instruments) and weighed before adding to a 3 ml chamber in the high-resolution respirometer and incubated with MiR05 at 37°C. The O2 concentration in the chamber was kept above 270 μm throughout the experiment, to avoid O2 diffusion limitation. Intermittent reoxygenation steps were performed by adding a 200 mm H2O2 solution into the medium containing catalase.
O2 consumption in the chamber was measured polarographycally following a sequential administration of substrates. Maximal ADP-stimulated mitochondrial respiration (state 3 respiration) was measured after the addition of ADP (5.0 mm) as phosphate acceptor with malate (4 mm), glutamate (10 mm) and succinate (10 mm) as substrates. The addition of cytochrome c (10 μm) had no additive effects (<10%) on respiration, thereby confirming the integrity of the outer mitochondrial membrane. Mitochondrial respiration values were then normalized per citrate synthase (CS) content, taken as an estimate of mitochondrial mass.
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance of the differences between means was assessed by one-way ANOVA followed by Student–Newman–Keuls test. A probability of less than 5% was considered significant (P < 0.05).
Results
CSA was reduced in gastrocnemius in the early phases of HU
CSA of skeletal muscle fibres was analysed in the mid-belly region of gastrocnemius in control and unloaded mice. The measurements were performed on cryosections stained with haematoxylin–eosin. Gastrocnemius muscle fibres went through 14 and 12% of atrophy, respectively, at 3 and 7 days of HU (Fig.1). As the portion of muscle analysed contained almost solely MHC isoform IIB (Control MHC-IIB 91 ± 1.5%, MHC-IIX 5.5 ± 1.1% MHC-IIA 3.5 ± 0.8%), all fibres analysed were considered type IIB.
The antioxidant defence system efficiently reacted to the initial reactive oxygen species (ROS) increase
To examine the effects of HU on redox imbalance, we studied the expression of SOD1 and catalase, the two major enzymes of ROS scavenging, H2O2 content and protein carbonylation. Gastrocnemius showed an early SOD1 and catalase up-regulation evident at 3 and 7 days of HU compared with control (Fig.2D). H2O2 concentration (Fig.2A) and protein carbonylation (Fig.2B) levels were not different from control at any of the experimental times analysed.
Figure 2. The antioxidant defence system efficiently reacts to the initial ROS increase.
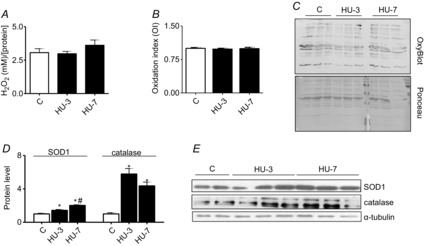
A, determination of H2O2 concentration. B, quantification of protein carbonylation level. C, representative OxyBlot and Red Ponceau staining. D, quantification of protein levels of SOD1 and catalase by Western blot. E, representative Western blot of SOD1, catalase and α-tubulin. *Significantly different from C, P < 0.05; #significantly different from HU-3, P < 0.05, n = 6 for each group. Data are presented as means ± SEM.
Mitochondrial fusion was impaired in gastrocnemius muscle in the early phases of HU
To define the adaptations of oxidative metabolism to muscle disuse we used RT-PCR and Western blot analysis.
To examine the effects of HU on expression of mitochondrial biogenesis-related genes, the mRNA transcript and protein level of PGC-1α, playing a central role in mitochondrial biogenesis, were studied. PGC-1α mRNA and protein expression was unchanged both at 3 and at 7 days of HU (Fig.3). Similarly, no changes in levels of protein in mitochondrial complexes (Fig.3D) (Complexes I–V) at either time analysed were found.
Figure 3. Mitochondrial fusion alteration is established early during HU in gastrocnemius muscle.
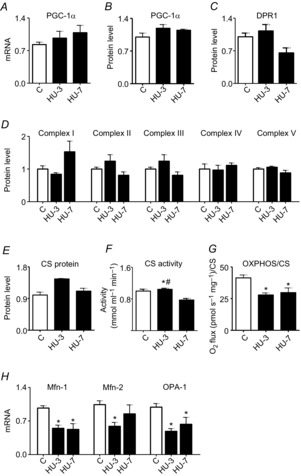
A, quantification of mRNA levels of PGC-1α by RT-PCR. B, quantification of protein levels of PGC-1α by Western blot. C, quantification of protein levels of DRP1 involved in fission machinery by Western blot. D, quantification of protein levels of mitochondrial complexes by Western blot. E, quantification of protein levels of citrate synthase by Western blot. F, determination of citrate synthase activity in skeletal muscle. G, determination of OXPHOS capacity, normalized per CS activity. H, quantification of mRNA levels of pro-fusion proteins by RT-PCR. C, control; HU-3, 3 days of hindlimb unloading; HU-7, 7 days of hindlimb unloading. *Significantly different from C, P < 0.05; #significantly different from HU-3, P < 0.05. Data are presented as means ± SEM.
As the mitochondrial fission machinery is able to cause muscle wasting in mice (Romanello et al. 2010), the expression of DRP1, one of its major components, was analysed. Levels of DRP1 protein did not change at either time analysed (Fig.3C).
However, a mismatch between the increase in CS protein level and its activity was observed on day 3 of HU (Fig.3E,F). CS protein and activity levels dropped significantly on day 7 of HU. Moreover, OXPHOS capacity in permeabilized fibres was impaired both at 3 and at 7 days (Fig.3G), suggesting an alteration of mitochondrial function. OXPHOS capacity was assessed based on maximal ADP-stimulated state 3 mitochondrial respiration (normalized per CS content), taken as an estimate of mitochondrial mass. As mitochondrial fusion protects mitochondrial function (Detmer & Chan, 2007), we also analysed expression of the pro-fusion proteins Mfn1, Mfn2 and OPA1. Importantly, a significantly lower level of such proteins was detected in HU animals compared to controls (Fig.3H).
The latter results indicate alteration to the mitochondrial network, which could potentially lead to energy stress. Because when mitochondria are fragmented FoxO3 is activated via AMPK in myofibres and induces the expression of atrogin-1 and MuRF-1 (Romanello & Sandri, 2010), we studied the ratio of the active (phosphorylated) and total form of AMPK and its downstream acetyl-CoA carboxylase (ACC) by Western blotting. At 3 days of HU, no significant changes were observed in AMPK activation whereas a significant increase of ACC activation was found (Fig.4).
Figure 4. The AMPK pathway is activated early during HU in gastrocnemius muscle.
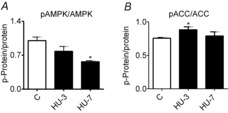
Determination of the activation level of AMP-kinase (A) and of ACC (B) by Western blot measuring the ratio between the phosphorylated (p) and total forms. C, control; HU-3, 3 days of hindlimb unloading; HU-7, 7 days of hindlimb unloading. *Significantly different from C, P < 0.05, n = 6 for each group. Data are presented as means ± SEM.
Collectively, the results indicate that the mitochondrial network was compromised in gastrocnemius muscle and that this sustained an impairment of function and activation of the AMPK catabolic system.
PGC-1α levels are maintained in transgenic mice during HU
To elucidate the role of Mfn2 expression in disuse atrophy, muscle-specific transgenic mice for the peroxisome PGC-1α were used as PGC-1α is known to enhance expression of pro-fusion proteins (Soriano et al. 2006; Zorzano, 2009).
First, to ensure muscle specificity of expression, PGC-1α expression in the gastrocnemius of Tg-PGC-1α mice using the WT samples as reference was assessed. Then, to check that protein levels were maintained during chronic disuse, Tg-PGC-1α mice were unloaded for 14 days.
As expected, both PGC-1α mRNA and protein levels were higher in Tg-PGC-1α gastrocnemius than in the WT (Fig.5). Furthermore, the PGC-1α overexpression was maintained during disuse, i.e. both mRNA and protein levels were significantly up-regulated in Tg-PGC-1α after 14 days of HU in comparison to control Tg-PGC-1α (Fig. 5).
Figure 5. In transgenic mice, PGC-1α levels are maintained during HU.
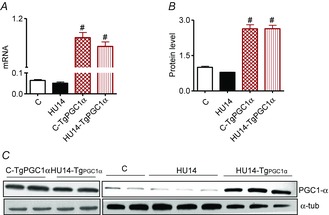
A, quantification of mRNA levels of PGC-1α in WT and transgenic mice. B, quantification of protein levels of PGC-1α by Western blot in WT and transgenic mice. C, representative Western blot of PGC-1α. *Significantly different from C; #significantly different from C and HU-14 at P < 0.05, n = 5 for each group. Data are presented as means ± SEM.
Elevated muscle PGC-1α completely preserved muscle mass by preventing the activation of catabolic systems
As gastrocnemius atrophy occurs early into disuse (3 days, Fig.1) and we have previously found that the catabolic systems are also active early (Cannavino et al. 2014), Tg-PGC-1α mice were unloaded for 3 days. The TgPGC-1α mice showed complete resistance to HU muscle atrophy (Fig.6). The CSA of gastrocnemius fibres of Tg-PGC-1α mice was not different from that of C-Tg-PGC-1α. To assess whether the protection against muscle atrophy might fade with time, Tg-PGC-1α mice were unloaded for 14 days following HU. No atrophy was observed following 14 days of HU (Fig.6).
Figure 6. Increased PGC-1α expression in muscle prevents muscle atrophy during HU.
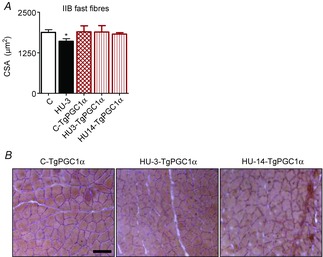
A, CSA of gastrocnemius fibres stained with haematoxylin and eosin. B, Scale bar: 100 μm. C-TgPGC-1α, control, transgenic PGC-1α; HU3-TgPGC-1α, 3 days of hindlimb unloading, transgenic PGC-1α; HU14-TgPGC-1α, 14 days of hindlimb unloading, transgenic PGC-1α. *Significantly different from C, P < 0.05. Data are presented as means ± SEM.
As expected on the basis of muscle atrophy, catabolic systems were significantly induced at 3 days of HU compared to control animals, whereas high levels of PGC-1α completely blunted the up regulation of MuRF-1, atrogin-1, beclin1 and p62 genes (Fig.7A).
Figure 7. Increased PGC-1α expression in muscle prevents activation of the catabolic systems.
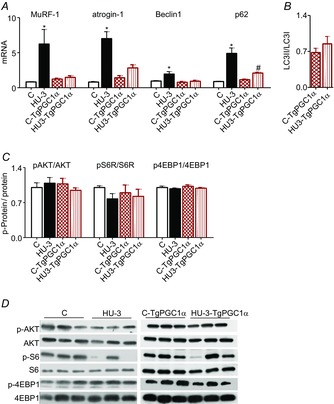
A, quantification of mRNA levels of MuRF-1 and atrogin-1 (ubiquitin proteasome system) and of Beclin1 and p62 (autophagy system) by RT-PCR. B, quantification of protein levels of LC3-II based on the ratio between the content in forms II and I of LC3 by Western blotting. C, determination of activity levels of AKT, S6R and 4EBP1 by Western blot analysis of the ratio between the content in the phosphorylated (p) and total forms. D, representative Western blot of synthetic factors. *Significantly different from C, P < 0.05; #significantly different from C-TgPGC-1α, P < 0.05. Data are presented as means ± SEM.
To check whether the slight increase of p62 in the unloaded gastrocnemius muscle of TgPGC-1α animals resulted in increased autophagy, the level of lipidated microtubule-associated protein light chain 3 (LC3) were studied. The cytosolic form of LC3-I, when recruited on autophagosomes, covalently binds phospholipids, resulting in a higher molecular weight protein named LC3-II. Therefore, the protein level of LC3-II is often used as a marker of autophagy (Kabeya et al. 2004). No significant changes in LC3-II/LC3-I ratio in unloaded TgPGC-1α samples compared to controls were found (Fig.7B). Therefore, the increased PGC-1α levels completely prevented activation of the catabolic systems in gastrocnemius.
Hind limb suspension did not significantly affect the anabolic pathway in gastrocnemius muscle of WT and TgPGC-1α mice (Fig.7C). In fact, no differences were found in pAKT, pS6R and p4EBP1 activation.
Elevated muscle PGC-1α completely prevented pro-fusion protein down-regulation and ACC activation
To assess whether PGC-1α over-expression actually elicited its effect through a modulation of mitochondrial dynamics, Mfn1, Mfn2, OPA1 and ACC were studied. PGC-1α overexpression completely prevented the decrease of Mfn1, Mfn2 and OPA1 (Fig.8A) and also the increase of ACC (Fig.8B) in HU muscles. PGC-1α therefore appeared to be sufficient to preserve mitochondrial fusion proteins, to block activation of the energy-stress pathway and muscle atrophy.
Figure 8. Increased PGC-1α expression in muscle prevents the reduction of pro-fusion proteins.
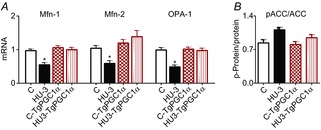
A, quantification of mRNA levels of pro-fusion proteins (Mfn-1, Mfn-2 and OPA-1) by RT-PCR. B, determination of the activation level of ACC by Western blot measuring the ratio between the phosphorylated (p) and total forms. *Significantly different from C, P < 0.05. Data are presented as means ± SEM.
Discussion
Although several studies have attempted to elucidate the signalling pathways involved in triggering atrophy during disuse of limb muscles, the exact mechanisms responsible for disuse-induced muscular atrophy are currently debated and there is still no therapy to treat muscle atrophy. Our previous findings have indicated that a metabolic programme might be the major phenomenon triggering muscle atrophy in HU mice (Cannavino et al. 2014). We demonstrated, in fact, that a fall in the levels of PGC-1α can be responsible for the activation of proteolytic pathways through FoxO3 disinhibition, leading to the conclusion that deregulation in PGC-1α is the molecular switch that triggers protein breakdown. The latter work was performed on soleus muscle, a slow muscle with high oxidative metabolism, mitochondrial content and PGC-1α expression. As fast muscles rely mainly on glycolytic metabolisms, have much lower mitochondrial content (Jackman & Willis, 1996; Berchtold et al. 2000; Lin et al. 2002), and express PGC-1α at low levels (Lin et al. 2002), a metabolic programme might not be as relevant in gastrocnemius as in soleus. In the present study, we tested the hypothesis that also in fast muscles disuse-induced muscle atrophy could be due to a metabolic programme.
Gastrocnemius muscle went through atrophy following both 3 and 7 days of HU, confirming, and extending to earlier times, the results of previous studies, which showed significant gastrocnemius mass loss with long-term disuse (Kyparos et al. 2005; Brocca et al. 2010; Desaphy et al. 2010). Although the loss of CSA appeared to plateau between 3 and 7 days, note that, following 14 days of HU, CSA of type IIB fibres from gastrocnemius was 25% lower than in controls, indicating that disuse atrophy in gastrocnemius actually progresses beyond 7 days. However, the observation of a plateau is intriguing. It is consistent with some previous observations (Isfort et al. 2002) and at variance with others (Bodine, 2013). The rate of muscle mass loss might vary according to the time course of the adaptations of the intracellular signalling pathways controlling muscle mass, which in turn might vary from pathway to pathway.
Oxidative stress and disuse atrophy in a fast muscle
The present study supports the idea that even in fast muscle oxidative stress might not be the main phenomenon in triggering disuse muscle atrophy (Ikemoto et al. 2002; Koesterer et al. 2002; Servais et al. 2007; Brocca et al. 2010; Desaphy et al. 2010; Glover et al. 2010; Kuwahara et al. 2010; Pellegrino et al. 2011b; Powers et al. 2012; Cannavino et al. 2014). Redox imbalance occurred early into HU in gastrocnemius consistently with the up-regulation of SOD1 and catalase (Fig.2). The lack of H2O2 accumulation together with the absence of carbonylation both at 3 and at 7 days of HU (Fig.2) indicates that, in the early phase of disuse, the antioxidant defence system efficiently reacts to the initial ROS increase. This compensatory increase in antioxidant activity is maintained over time, in agreement with the absence of carbonylation found at 14 days of HU (Brocca et al. 2010). Given that we have previously shown that antioxidant treatment during unloading did not prevent gastrocnemius atrophy, the data suggest that oxidative stress might not be the major phenomenon of disuse atrophy in fast muscle (Brocca et al. 2010).
Imbalance between mitochondrial fission and fusion and AMPK activation in fast muscles following HU
In contrast to what was observed in soleus (Cannavino et al. 2014), unloaded gastrocnemius showed normal PGC-1α levels (Fig.2) and no alterations in mitochondrial complex expression, suggesting different mechanisms for fast muscle atrophy. Previous findings regarding PGC-1α during disuse in fast muscle are contradictory. In fact, higher (Wagatsuma et al. 2011), lower (Mazzatti et al. 2008) and unchanged (Nagatomo et al. 2011) PGC-1α levels have been reported. Furthermore, mitochondrial complexes did not drastically change with disuse in fast muscle, and only a reduction of complex I expression was found after 28 days of unloading in gastrocnemius muscle (Liu et al. 2012b). This notwithstanding, several lines of evidence suggest that mitochondrial alterations could be good candidates in triggering atrophy even in fast muscle. In fact, alteration in mitochondrial biogenesis (Liu et al. 2012b) and mitochondrial respiration (Yajid et al. 1998) have been found after chronic disuse in fast skeletal muscle.
Mitochondria play critical roles in the life and death of cells. Mitochondrial dynamics are regulated by coordinated fusion and fission cycles performed by a complex molecular machinery. Mitochondrial dynamics maintain normal mitochondrial function by degrading damaged mitochondria and generating new mitochondria. The mitochondrion is vital to cellular energy and metabolism, being the site of generation of most ATP. Derangements in fusion–fission have been proposed as a mechanism underlying the formation of dysfunctional aberrant mitochondria (Yoon et al. 2006) and causing muscle atrophy through activation of AMPK (Romanello et al. 2010).
Fusion–fission derangement can be due to an excessive activation of fission. In contrast to observations in soleus (Cannavino et al. 2014), unchanged DRP1 mRNA levels (Fig.3) suggest that in the early phases of gastrocnemius atrophy there is no excessive fission activation.
Mitochondrial fusion was assessed based on the expression of three large GTPases that require the coordinated fusion of the outer and inner membranes: Mfn1, Mfn2 and OPA1. A defective mitochondrial fusion, revealed by a down-regulation of Mfn1, Mfn2 and OPA1, occurred (Fig.3), in agreement with previous reports in fast muscle during early (Wagatsuma et al. 2011) and chronic disuse (Liu et al. 2012b). Mitochondrial fusion can protect mitochondrial function (Detmer & Chan, 2007). Consistently, we found an impairment of OXPHOS capacity assessed as the maximal ADP-stimulated state 3 mitochondrial respiration (Fig.3). Together with a mismatch between the increase in CS protein level and its activity, the latter suggests a compensatory mechanism to an intrinsic oxidative defect in fast twitch muscle.
The decrease in mitochondrial biogenesis and impaired mitochondrial function could lower ATP production, generate energy imbalance and activate protein catabolism. The energy level of the cell is, in fact, sensed by AMPK, a cellular kinase, which can cause atrogin-1 and MuRF-1 induction via a FoxO3-dependent mechanism (Tong et al. 2009). Interestingly, an early up-regulation of ACC, a kinase downstream of AMPK, was observed (Fig.4), indicating activation of AMPK. The latter results suggest that mitochondrial alteration could be important in determining muscle size during disuse in fast muscles.
Several signalling kinases, including AMPK, have been implicated in activating PGC-1α transcription in response to various stimuli (Irrcher et al. 2008). This might explain why, following HU, PGC-1α level is unchanged.
PGC-1α over-expression prevents HU-induced muscle atrophy and imbalance in mitochondrial fusion and fission
To test the role of mitochondrial alterations we used transgenic mice overexpressing PGC-1α. In fact, it has been shown that the Mfn2 gene is controlled by PGC-1α and PGC-1β in conjunction with oestrogen-related receptor-α (Soriano et al. 2006; Liesa et al. 2008) and PGC-1α stimulates Mfn2 expression (Soriano et al. 2006; Zorzano, 2009). We subjected transgenic mice overexpressing PGC-1α to 3 days of HU. The duration of HU was chosen based on the observation that in soleus maximal induction of catabolic genes occurred 3 days after unloading (Cannavino et al. 2014). Skeletal muscle overexpressing PGC-α displayed complete resistance to loss of mass following 3 days of HU and this positive effect on muscle size was fully maintained also after prolonged disuse (14 days) (Fig.6), consistent with the high and steady protein levels of PGC-1α (Fig.5).
To test the mechanism underlying this effect on muscle size, we analysed the main pathways controlling muscle mass. The ubiquitine proteasome system (UPS) and autophagy are the two major proteolytic systems responsible for muscle wasting. Among several markers of UPS, two E3 ubiquitin ligases, MAFbx/Atrogin-1 and MuRF1 (also known as atrogenes), have been widely used as they are highly expressed in many conditions of muscle wasting (Bodine et al. 2001a; Cohen et al. 2009).
MuRF1 and atrogin-1 mRNA expression at 3 days (Fig.7) was dramatically increased, indicating UPS activation in the early phases of disuse atrophy. Disuse also induced expression of genes involved in autophagy (Beclin1 and p62) (Fig.7). It has been recently understood that an increase in activity of the autophagy system can have a role in muscle atrophy (Sandri, 2010). The latter findings indicate that, at least in the early stages of disuse, gastrocnemius mass loss is supported by activation of both the UPS and autophagy systems, consistent with previous observations (Bodine et al. 2001a; Ikemoto et al. 2002; Haddad et al. 2006; Kline et al. 2007).
Signalling through the phosphatidyl-inositol 3-kinase (PI3K/AKY/mTOR) pathway controls protein synthesis and muscle hypertrophy (Rommel et al. 2001). In gastrocnemius muscle, HU did not result in any significant alterations in the phosphorylation states of the anabolic pathway after 3 days (Fig.7). Little and contradictory information is available on the impact of disuse on activation of the protein synthesis signalling pathway in fast muscles. Although our results are not in agreement with those of Bodine et al. (2001b), who found a decrease in the protein synthesis signalling pathway in atrophied gastrocnemius muscle at 14 days of unloading, they are in line with the concept that protein synthesis regulation differs among muscle types (Hornberger et al. 2001). Indeed, several lines of evidence indicate an increase or no changes in protein synthesis in fast muscles in different disuse models, including HU. An increase in translation factors (p70S6K, eEF-2 and eIF-2a) has been reported in the atrophied fast EDL muscle at 7 days of denervation (Hornberger et al. 2001). Loss of gastrocnemius mass independent of a decrease in protein synthesis has been reported after unilateral hindlimb immobilization (Krawiec et al. 2005). Finally, in line with our results, Liu et al. (2012a) showed a biphasic protein translational signalling response of gastrocnemius to HU. Specifically, the authors found that the initial unloading phase was associated with increased repression of 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1) on translation initiation factor eIF4E without alteration of the Akt/mTOR/p70S6K/S6 pathway, while prolonged disuse (14 days) was associated with activation of translational signalling, including activation of the Akt/mTOR/p70S6K/S6 pathway and diminished repression of 4EBP1 on eIF4E.
The findings discussed so far prompted the hypothesis that in fast muscles an early down-regulation of pro-fusion proteins at 3 days of HU, through concomitant AMPK activation, can activate the ubiquitin proteasome pathway and autophagy, and ultimately cause muscle mass loss in disuse.
The latter hypothesis was confirmed by the observation that overexpression of PGC-α was sufficient to prevent (i) the decrease of pro-fusion proteins (Mfn1, Mfn2 and OPA1) (Fig.8), (ii) activation of the AMPK pathway (Fig.8), (iii) the inducible expression of MuRF1 and atrogin1 and the autophagic factors (Fig.7); and (iv) any muscle mass loss in response to disuse.
Conclusions
The present results and our recent previous study (Cannavino et al. 2014) strongly support the idea that a metabolic programme can play a major role in triggering disuse muscle atrophy both in slow and in fast muscles. However, the metabolic programme would exert its effects through different pathways in slow, soleus and fast, gastrocnemius muscles. In soleus the primary phenomenon would be a fall of PGC-1α expression and content, which would cause induction of atrogenes through FoxO3 disinhibition (Cannavino et al. 2014), whereas in gastrocnemius the primary phenomenon would be a down-regulation of pro-fusion proteins, which in turn would cause energy stress and induction of atrogenes through AMPK activation (Fig.9). It is known that during muscle wasting the enhanced FoxO activity results in increased transcription of Mul1, a mitochondrial E3 ligase, which ubiquitinates and targets Mfn2 for degradation through the UPS, resulting in mitochondrial dysfunction (Lokireddy et al. 2012). Interestingly, slow-twitch muscle has small amounts of Mul1 but high Mfn2 protein levels whereas fast-twitch muscles express higher levels of Mul1 and low levels of Mfn2 (Lokireddy et al. 2012). The latter phenomenon probably exposes fast muscles to a rapid degradation of Mfn2 with subsequent alteration of mitochondrial fusion, which precedes the fall of PGC-α levels that occurs in the late stages of disuse.
Figure 9. Scheme representing the effects of the early stages of disuse on gastrocnemius muscle.
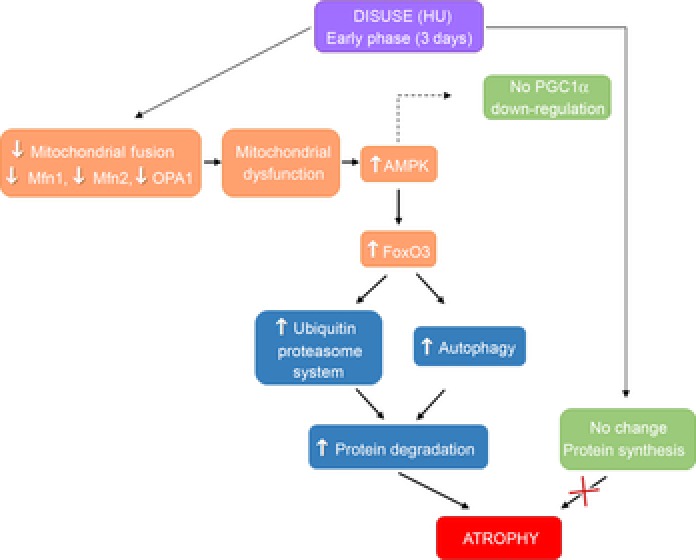
Skeletal muscle disuse would cause down-regulation of pro-fusion proteins leading to mitochondrial dysfunction and AMPK activation. AMPK activation leads to the activation of FoxO, which induces the transcription of genes encoding proteins involved in protein degradation via both the autophagy and the ubiquitin proteasome pathways. The increase in the rate of protein degradation causes gastrocnemius atrophy. AMPK might also prevent the down-regulation of PGC-1α in gastrocnemius in the early stages of HU. Changes of protein synthesis would not be responsible for gastrocnemius atrophy at this stage of disuse.
Moreover, our results show that PGC-1α levels higher than basal are necessary to increase pro-fusion proteins, thus preventing gastrocnemius muscle atrophy during disuse. Interestingly, the higher efficacy of resistance training in preventing muscle mass loss compared to endurance training could be due to the fact that although both types of exercise increase PGC-1α expression, at a given exercise volume the higher the intensity the higher the effect (Egan et al. 2010). As the effects of increased PGC-1α activity were sustained throughout disuse (Fig.6), compounds inducing PGC-1α expression could be useful to treat and prevent muscle atrophy.
Acknowledgments
We thank Professor Francesco Zorzato who kindly donated the TgPGC-1α mice and Mr Luigi Guidotti for excellent technical help during all experiments related to muscle disuse.
Glossary
- 4EBP1
eukaryotic translation initiation factor 4E-binding protein 1
- ACC
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- CS
citrate synthase
- CSA
cross-sectional area
- DNP
dinitrophenylhydrazone
- HU
hindlimb unloading
- LC3
microtubule-associated protein light chain 3
- Mfn1, Mfn2
mitofusin1 and mitofusin2
- MuRF-1
muscle-specific ring finger protein-1
- MHC
myosin heavy chain
- OPA1
optic atrophy 1
- PGC-1α
peroxisome proliferative activated receptor-γ coactivator 1α
- UPS
ubiquitine proteasome system
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
R.B. and M.A.P.: conception and design of the experiments; J.C., L.B. and B.G.: collection, analysis and interpretation of data; R.B. and M.A.P.: drafting the article or revising it critically for important intellectual content. All authors made comments on the manuscript and read and approved the final version. The OXPHOS experiments were performed at the Department of Medical and Biological Sciences, University of Udine, and all the others were performed at the Department of Molecular Medicine, University of Pavia, Italy.
Funding
This study was supported by Cariplo Foundation, Italy (grant no. 2010.0764), and the European Commission for the MYOAGE grant (no. 22 3576) funded under FP7. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- Berchtold MW, Brinkmeier H. Müntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000;80:1215–1265. doi: 10.1152/physrev.2000.80.3.1215. [DOI] [PubMed] [Google Scholar]
- Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol. 2013;45:2200–2208. doi: 10.1016/j.biocel.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD. Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001a;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ. Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001b;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- Bonaldo P. Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca L, Pellegrino MA, Desaphy JF, Pierno S, Camerino DC. Bottinelli R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp Physiol. 2010;95:331–350. doi: 10.1113/expphysiol.2009.050245. [DOI] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Bottinelli R. Pellegrino MA. PGC1-α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol. 2014;592:4575–4589. doi: 10.1113/jphysiol.2014.275545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14(Spec No. 2):R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM. Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E. Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaphy JF, Pierno S, Liantonio A, Giannuzzi V, Digennaro C, Dinardo MM, Camerino GM, Ricciuti P, Brocca L, Pellegrino MA, Bottinelli R. Camerino DC. Antioxidant treatment of hindlimb-unloaded mouse counteracts fiber type transition but not atrophy of disused muscles. Pharmacol Res. 2010;61:553–563. doi: 10.1016/j.phrs.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Detmer SA. Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Egan B, Carson BP, Garcia-Roves PM, Chibalin AV, Sarsfield FM, Barron N, McCaffrey N, Moyna NM, Zierath JR. O'Gorman DJ. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol. 2010;588:1779–1790. doi: 10.1113/jphysiol.2010.188011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Yasuda N, Tarnopolsky MA, Abadi A. Phillips SM. Little change in markers of protein breakdown and oxidative stress in humans in immobilization-induced skeletal muscle atrophy. Appl Physiol Nutr Metab. 2010;35:125–133. doi: 10.1139/H09-137. [DOI] [PubMed] [Google Scholar]
- Gondin J, Brocca L, Bellinzona E, D'Antona G, Maffiuletti NA, Miotti D, Pellegrino MA. Bottinelli R. Neuromuscular electrical stimulation training induces atypical adaptations of the human skeletal muscle phenotype: a functional and proteomic analysis. J Appl Physiol. 2011;110:433–450. doi: 10.1152/japplphysiol.00914.2010. [DOI] [PubMed] [Google Scholar]
- Haddad F, Adams GR, Bodell PW. Baldwin KM. Isometric resistance exercise fails to counteract skeletal muscle atrophy processes during the initial stages of unloading. J Appl Physiol. 2006;100:433–441. doi: 10.1152/japplphysiol.01203.2005. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Lackner L. Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Hornberger TA, Hunter RB, Kandarian SC. Esser KA. Regulation of translation factors during hindlimb unloading and denervation of skeletal muscle in rats. Am J Physiol Cell Physiol. 2001;281:C179–187. doi: 10.1152/ajpcell.2001.281.1.C179. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Okamura Y, Kano M, Hirasaka K, Tanaka R, Yamamoto T, Sasa T, Ogawa T, Sairyo K, Kishi K. Nikawa T. A relative high dose of vitamin E does not attenuate unweighting-induced oxidative stress and ubiquitination in rat skeletal muscle. J Physiol Anthropol Appl Human Sci. 2002;21:257–263. doi: 10.2114/jpa.21.257. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Kirwan AF. Hood DA. AMP-activated protein kinase-regulated activation of the PGC-1α promoter in skeletal muscle cells. PLoS One. 2008;3:e3614. doi: 10.1371/journal.pone.0003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isfort RJ, Wang F, Greis KD, Sun Y, Keough TW, Farrar RP, Bodine SC. Anderson NL. Proteomic analysis of rat soleus muscle undergoing hindlimb suspension-induced atrophy and reweighting hypertrophy. Proteomics. 2002;2:543–550. doi: 10.1002/1615-9861(200205)2:5<543::AID-PROT543>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Jackman MR. Willis WT. Characteristics of mitochondria isolated from type I and type IIb skeletal muscle. Am J Physiol. 1996;270:C673–678. doi: 10.1152/ajpcell.1996.270.2.C673. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y. Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kline WO, Panaro FJ, Yang H. Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol. 2007;102:740–747. doi: 10.1152/japplphysiol.00873.2006. [DOI] [PubMed] [Google Scholar]
- Koesterer TJ, Dodd SL. Powers S. Increased antioxidant capacity does not attenuate muscle atrophy caused by unweighting. J Appl Physiol. 2002;93:1959–1965. doi: 10.1152/japplphysiol.00511.2002. [DOI] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM. Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Krawiec BJ, Frost RA, Vary TC, Jefferson LS. Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab. 2005;289:E969–980. doi: 10.1152/ajpendo.00126.2005. [DOI] [PubMed] [Google Scholar]
- Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T. Shimizu T. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med. 2010;48:1252–1262. doi: 10.1016/j.freeradbiomed.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Kyparos A, Feeback DL, Layne CS, Martinez DA. Clarke MS. Mechanical stimulation of the plantar foot surface attenuates soleus muscle atrophy induced by hindlimb unloading in rats. J Appl Physiol (1985) 2005;99:739–746. doi: 10.1152/japplphysiol.00771.2004. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE. Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- Liesa M, Borda-d'Agua B, Medina-Gomez G, Lelliott CJ, Paz JC, Rojo M, Palacin M, Vidal-Puig A. Zorzano A. Mitochondrial fusion is increased by the nuclear coactivator PGC-1β. PLoS One. 2008;3:e3613. doi: 10.1371/journal.pone.0003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R. Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Liu H, Blough ER, Arvapalli R, Wang Y, Reiser PJ, Paturi S, Katta A, Harris R, Nepal N. Wu M. Regulation of contractile proteins and protein translational signaling in disused muscle. Cell Physiol Biochem. 2012a;30:1202–1214. doi: 10.1159/000343310. [DOI] [PubMed] [Google Scholar]
- Liu J, Peng Y, Cui Z, Wu Z, Qian A, Shang P, Qu L, Li Y. Long J. Depressed mitochondrial biogenesis and dynamic remodeling in mouse tibialis anterior and gastrocnemius induced by 4-week hindlimb unloading. IUBMB Life. 2012b;64:901–910. doi: 10.1002/iub.1087. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M. Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. doi: 10.1016/j.cmet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Mazzatti DJ, Smith MA, Oita RC, Lim FL, White AJ. Reid MB. Muscle unloading-induced metabolic remodeling is associated with acute alterations in PPARδ and UCP-3 expression. Physiol Genomics. 2008;34:149–161. doi: 10.1152/physiolgenomics.00281.2007. [DOI] [PubMed] [Google Scholar]
- Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM. Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Nagatomo F, Fujino H, Kondo H, Suzuki H, Kouzaki M, Takeda I. Ishihara A. PGC-1α and FOXO1 mRNA levels and fiber characteristics of the soleus and plantaris muscles in rats after hindlimb unloading. Histol Histopathol. 2011;26:1545–1553. doi: 10.14670/HH-26.1545. [DOI] [PubMed] [Google Scholar]
- Pellegrino MA, Canepari M, Rossi R, D'Antona G, Reggiani C. Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546:677–689. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC. Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. The Journal of physiology. 2011a;589:2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC. Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. J Physiol. 2011b;589:2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Smuder AJ. Judge AR. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr Opin Clin Nutr Metab Care. 2012;15:240–245. doi: 10.1097/MCO.0b013e328352b4c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R. Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V. Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep. 2010;12:433–439. doi: 10.1007/s11906-010-0157-8. [DOI] [PubMed] [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD. Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- Salvadego D, Domenis R, Lazzer S, Porcelli S, Rittweger J, Rizzo G, Mavelli I, Simunic B, Pisot R. Grassi B. Skeletal muscle oxidative function in vivo and ex vivo in athletes with marked hypertrophy from resistance training. J Appl Physiol (1985) 2013;114:1527–1535. doi: 10.1152/japplphysiol.00883.2012. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010;298:C1291–1297. doi: 10.1152/ajpcell.00531.2009. [DOI] [PubMed] [Google Scholar]
- Servais S, Letexier D, Favier R, Duchamp C. Desplanches D. Prevention of unloading-induced atrophy by vitamin E supplementation: links between oxidative stress and soleus muscle proteolysis? Free Radic Biol Med. 2007;42:627–635. doi: 10.1016/j.freeradbiomed.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG. Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Liesa M, Bach D, Chan DC, Palacin M. Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes. 2006;55:1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- Tong JF, Yan X, Zhu MJ. Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem. 2009;108:458–468. doi: 10.1002/jcb.22272. [DOI] [PubMed] [Google Scholar]
- Wagatsuma A, Kotake N, Kawachi T, Shiozuka M, Yamada S. Matsuda R. Mitochondrial adaptations in skeletal muscle to hindlimb unloading. Mol Cell Biochem. 2011;350:1–11. doi: 10.1007/s11010-010-0677-1. [DOI] [PubMed] [Google Scholar]
- Yajid F, Mercier JG, Mercier BM, Dubouchaud H. Prefaut C. Effects of 4 wk of hindlimb suspension on skeletal muscle mitochondrial respiration in rats. J Appl Physiol (1985) 1998;84:479–485. doi: 10.1152/jappl.1998.84.2.479. [DOI] [PubMed] [Google Scholar]
- Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou MJ, Martinou JC. Yoon G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- Zorzano A. Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab. 2009;34:433–439. doi: 10.1139/H09-049. [DOI] [PubMed] [Google Scholar]