

Abstract
Despite antiretroviral therapy (ART), HIV-1 persists in a stable latent reservoir1, 2, primarily in resting memory CD4+ T cells3, 4. This reservoir presents a major barrier to the cure of HIV-1 infection. To purge the reservoir, pharmacological reactivation of latent HIV-1 has been proposed5 and tested both in vitro and in vivo6–8. A key remaining question is whether virus-specific immune mechanisms including cytolytic T lymphocytes (CTL) can clear infected cells in ART-treated patients after latency is reversed. Here we show that there is a striking all or none pattern for CTL escape mutations in HIV-1 Gag epitopes. Unless ART is started early, the vast majority (>98%) of latent viruses carry CTL escape mutations that render infected cells insensitive to CTLs directed at common epitopes. To solve this problem, we identified CTLs that could recognize epitopes from latent HIV-1 that were unmutated in every chronically infected patient tested. Upon stimulation, these CTLs eliminated target cells infected with autologous virus derived from the latent reservoir, both in vitro and in patient-derived humanized mice. The predominance of CTL-resistant viruses in the latent reservoir poses a major challenge to viral eradication. Our results demonstrate that chronically infected patients retain a broad spectrum viral-specific CTL response and that appropriate boosting of this response may be required for the elimination of the latent reservoir.
HIV-1 establishes latent infection in resting CD4+ T cells3, 4. Recent efforts to eradicate HIV-1 infection have focused on reversing latency without global T cell activation5. However, inducing HIV-1 gene expression in latently infected cells is not sufficient to cause the death of these cells if they remain in a resting state9. Boosting HIV-1-specific immune responses including CTL responses may be required for clearance of the latent reservoir9. CTLs play a significant role in suppressing HIV-1 replication in acute infection10–14. Because of this strong selective pressure, HIV-1 quickly acquires mutations to evade CTL recognition12, 13, 15–18. CTL escape has been studied primarily through the analysis of plasma virus12, 13, 16, 18–20, and CTL-based vaccines have been designed based on conserved epitopes21, 22. A systematic investigation of CTL escape in the latent reservoir will be of great importance to the ongoing CTL-based eradication efforts, because latent HIV-1 likely represents the major source of viral rebound after treatment interruption. Earlier studies have suggested the presence of CTL escape mutations in proviral DNA15, 17, but it still remains unclear to what extent the latent reservoir in resting CD4+ T cells is affected by CTL escape, whether mutations detected in proviral DNA are representative of the very small fraction of proviruses that are replication-competent, and most importantly, whether the CTL response can recognize and clear infected cells after latency is reversed.
To investigate CTL escape variants in the latent reservoir, we deep sequenced the proviral HIV-1 DNA in resting CD4+ T cells from 25 patients (Extended Data Table 1). Among them, 10 initiated ART during the acute phase (AP, within 3 months of infection) while the other 15 initiated ART during the chronic phase (CP) of infection. The sequencing was focused on Gag because it is an important target of the CTL response23 and is highly conserved, which facilitates detection of escape variants. Our data show that previously documented CTL escape variants completely dominate the viral reservoirs of nearly all CP-treated patients (Extended Data Fig. 1 and Supplementary Table 1). This trend is especially obvious for several well characterized CTL epitopes: the HLA-A2-restricted epitope SLYNTVATL (SL9), the HLA-A3-restricted epitope RLRPGGKKK (RK9) and the HLA-B57/58-restricted epitope TSTLQEQIGW (TW10) (Fig. 1a and Extended Data Fig. 1). In these epitopes, close to 100% of the sequences harbored escape mutations. Comparison of mutation frequencies between HLA allele-relevant and -irrelevant epitopes in CP-treated patients suggests these CTL escape mutations identified are specific to patient’s HLA type (Fig. 1b). By contrast, except for SL9 from AP01 and RK9 from AP08, few if any CTL escape mutations were archived in AP-treated patients (Fig. 1c and Extended Data Fig. 1). The striking difference between AP- and CP-treated patients (Fig. 1c) indicates that unless treatment is initiated within the first several months of infection, the latent reservoir becomes almost completely dominated by variants resistant to dominant CTL responses.
Figure 1. CTL escape variants dominate the latent reservoir of CP-treated but not AP-treated patients.
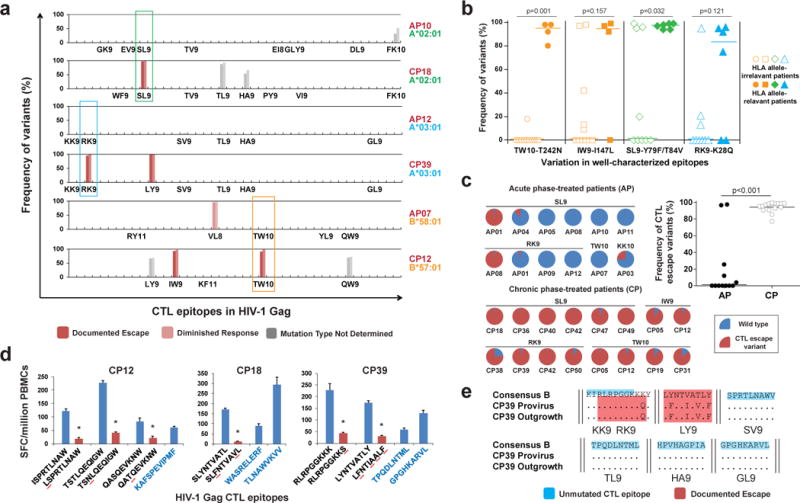
a, Frequency of variants in Gag CTL epitopes in proviruses from resting CD4+ T cells. Representative results of 6 patients are shown. Only optimal CTL epitopes relevant to each patient’s HLA type are listed. Results from both Pacbio (left bar) and MiSeq (right bar) sequencing are shown. The effect on CTL recognition (denoted by color) is determined from information in the Los Alamos National Laboratory (LANL) HIV Molecular Immunology Database. b, CTL escape variants identified by sequencing are specific to HLA type. Frequencies of documented escape-associated variants in four well-characterized epitopes are shown for all 15 CP-treated patients. Median and P value from Mann-Whitney test are shown. c, Comparison of CTL escape variant frequency in proviruses between CP- and AP-treated patients. Only well-characterized epitopes are shown. Median and P value from Mann-Whitney test are shown. d, Characterization of CTL responses against HIV-1 Gag epitopes by interferon-γ ELISpot. The peptides tested are listed below the x-axis (black type, epitopes in which sequence variation was detected; blue type, no variation). The observed mutation is underlined in red, and CTL escape (defined by the lost of positive response) is denoted by * above the bar. The peptide concentration was 10 μg/ml. Error bars represent s.e.m., n = 3. e, Sequences in Gag CTL epitopes for proviral DNA and outgrowth virus from resting CD4+ T cells in CP39. CTL epitopes with no observed variation are highlighted in blue. Epitopes with documented escape mutations are shaded in red.
To confirm variants detected at high frequency in the latent reservoir represent functional CTL escape mutants, cells from 7 CP-treated subjects were tested for reactivity to synthetic peptides representing wild-type and mutant versions of the relevant epitopes. As expected, there were only minimal responses to previously documented CTL escape mutants by patient CD8+ T cells, and no de novo response was detected (Fig. 1d and Extended Data Fig. 2). In contrast, all tested subjects retained a strong response to peptides representing the wild-type epitopes, suggesting wild-type virus was initially transmitted, with subsequent evolution of CTL escape variants. Most HIV-1 proviruses detected in patients are defective24. Therefore, to determine whether these CTL escape variants can be reactivated and lead to viral rebound if therapy is stopped, we isolated replication-competent viruses from the latent reservoirs of 9 CP-treated patients. We found all dominant CTL escape mutations identified in proviruses in resting CD4+ T cells were also present in the replication-competent viruses that grew out after T cell activation (Fig. 1e and Extended Data Fig. 3), indicating that these CTL escape variants not only dominate the population of proviruses, but can also be released and replicate once latency is reversed.
We next asked whether the host CTL response could recognize and eliminate the cells infected with these escape variants. We infected activated CD4+ T cells from these patients with autologous, replication-competent virus derived from the latent reservoir (Extended Data Fig. 4a). The infected cells were then co-cultured with autologous CD8+ T cells, either unstimulated or pre-stimulated, to assess HIV-1-specific cytolytic activity. Non-specific activation of CD8+ T cells was not observed after co-cultured with PHA-activated CD4+ T cells (Extended Data Fig. 4b). From all 13 CP-treated subjects tested, CD8+ T cells pre-stimulated by a Gag peptide mixture efficiently killed autologous infected CD4+ T cells (median 61% elimination), while unstimulated CD8+ T cells from most subjects had significantly less effect (median 23% elimination) (Fig. 2a and Extended Data Fig. 4c and d). CD8+ T cells from 7/7 healthy donors completely failed to eliminate autologous infected cells (Fig. 2a), confirming the observed killing was HIV-1-specific. The killing effect was enhanced by increasing the effector to target (E:T) ratio (Extended Data Fig. 5a), and was cell-cell contact dependent (Extended Data Fig. 5b). When the co-culture was maintained over time in the absence of ART, viral replication was significantly reduced, but not completely inhibited by pre-stimulated CD8+ T cells (Fig. 2b). We found that peptide mixtures from other HIV-1 proteins (Nef, Tat, Rev, and Env) could also boost CTL responses and facilitate the elimination of infected cells (Fig. 2c) and that CTLs pre-stimulated by Gag peptides generally had the highest activity. Together, these results demonstrate that chronically infected patients retain CTL clones that can recognize and eliminate autologous infected CD4+ T cells, despite the presence of CTL escape mutations in dominant epitopes. However, these clones require stimulation with antigen for optimal activity.
Figure 2. CD8+ T cells pre-stimulated with a mixture of Gag peptides eliminate autologous CD4+ T cells infected with autologous HIV-1 from resting CD4+ T cells.
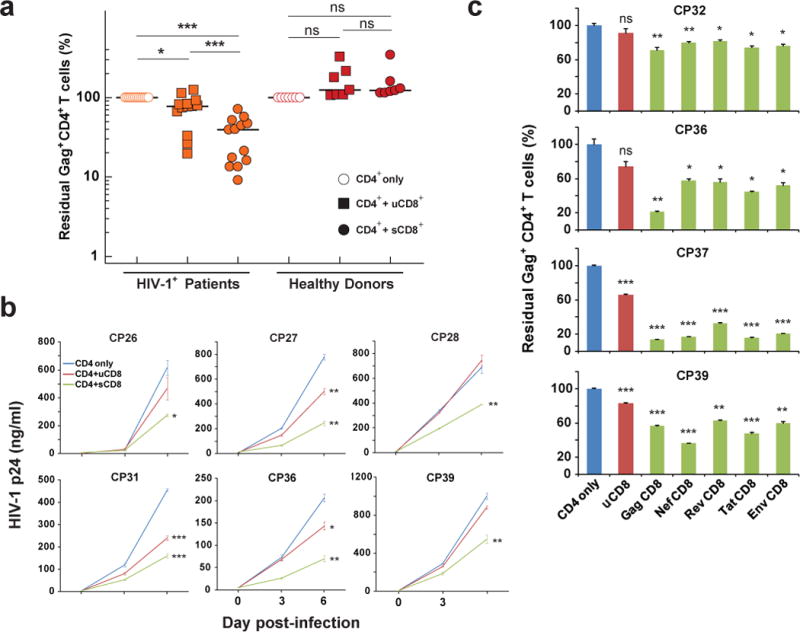
a, Pre-stimulated CD8+ T cells (sCD8) eliminate autologous infected CD4+ T cells more efficiently than unstimulated CD8+ T cells (uCD8). Each symbol represents the mean of 3 replicates. Median and P value from Mann-Whitney test are shown. b, sCD8 inhibit viral growth in autologous infected CD4+ T cells with higher efficacy than uCD8. c, sCD8 pre-stimulated by different viral peptides eliminate autologous CD4+ T cells infected with viruses derived from resting CD4+ T cells. For b and c, results are compared with CD4 only using paired t tests. Error bars represent s.e.m., n = 3. *: p<0.05; **: p<0.01; ***: p<0.001; ns: p>0.05.
To further characterize which CTL population contributed to the elimination of cells infected by CTL escape variants, we compared the killing activity of two specific CTL populations: the one that targets epitopes in which escape has been identified and the one that targets unmutated epitopes (Fig. 3a). CD8+ T cells from CP36 and CP39 were pre-stimulated with IL-2 and different synthetic peptides representing the wild-type forms of the relevant epitopes. After incubation for 6 days, each CTL population exhibited significant proliferation compared to no treatment or IL-2 alone (Fig. 3b and c). Pentamer staining for three available epitopes revealed that the number of epitope-specific CD8+ T cells increased dramatically after stimulation with wild-type peptides (Fig. 3b). After co-culture with autologous target cells infected with latent reservoir-derived viruses, CTLs targeting unmutated epitopes clearly showed stronger cytolytic activity than the IL-2 only controls, while CTLs targeting epitopes with identified escaped mutations showed no significant killing (Fig. 3d). CTLs pre-stimulated by the Gag peptide mixture exhibited stronger killing than all single peptide-stimulated populations (Fig. 3d).
Figure 3. CD8+ T cells targeting unmutated epitopes, not epitopes with identified escaped mutations, eliminate CTL escape variants.
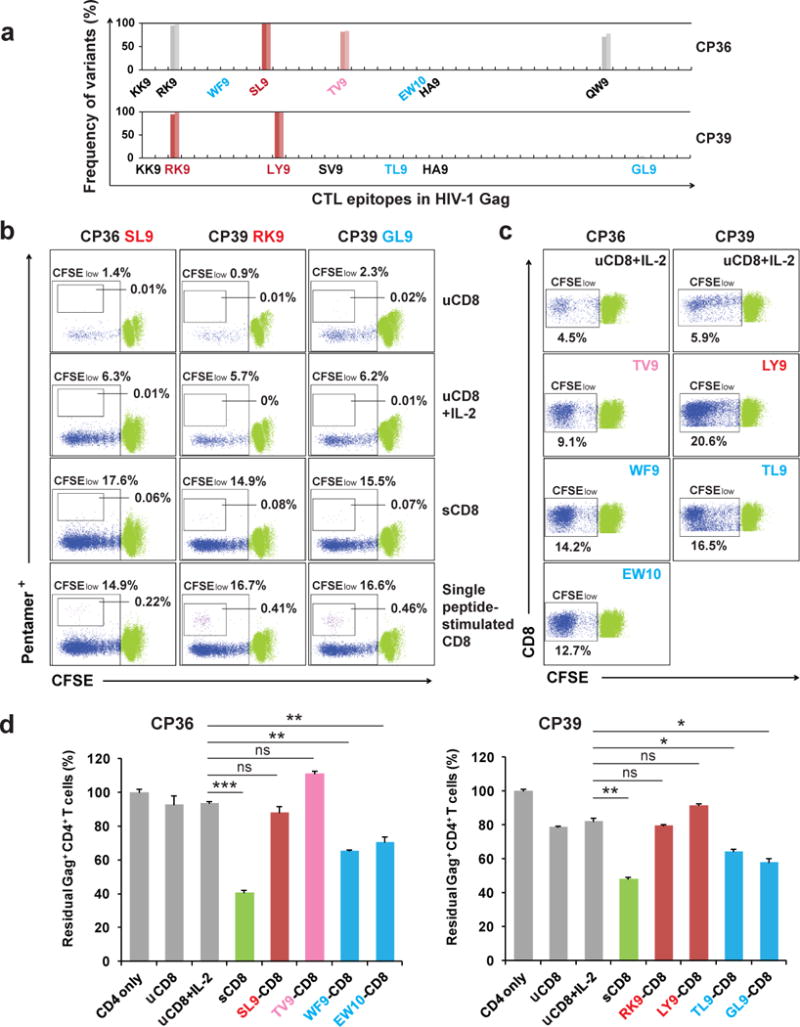
a, Frequency of variants in Gag CTL epitopes in proviruses from resting CD4+ T cells of CP36 and CP39. Epitopes tested with single peptide stimulation herein are denoted in colors (red or pink, epitopes with escape observed; blue, no escape observed). b, Epitope-specific CD8+ T cells proliferate significantly after single peptide stimulation. Only CD8+ cells are shown. Percentages of CFSElow, pentamer-positive cells are indicated for unstimulated cultures (uCD8) with or without IL2, for cultures stimulated with Gag peptide mixture (sCD8) and IL2, and for cultures stimulated with the indicated single peptides and IL2. Wild type versions of peptides were used for all single peptide stimulations. c, CD8+ T cell proliferative responses after single peptide stimulation. Only CD8+ cells are shown. Percentages of CFSElow cells are indicated. d, CD8+ T cells targeting unmutated epitopes, not epitopes with identified escaped mutations, eliminate autologous CD4+ T cells infected with CTL escape variants. uCD8: unstimulated CD8+ T cells; sCD8: Gag peptide mixture stimulated CD8+ T cells. Error bars represent s.e.m., n = 3. *: p<0.05; **: p<0.01; ***: p<0.001; ns: p>0.05, paired t test.
To test whether CTL that recognize unmutated viral epitopes can inhibit HIV-1 replication and clear infected cells in vivo, we generated patient-derived humanized mice using an improved version of a recently reported mouse system named MISTRG25. Whereas the previously reported MISTRG mice bear a BAC transgene encoding human SIRPα, the newly generated MIS(KI)TRG mice harbor a knock-in replacement of the endogenous mouse Sirpa gene with a humanized version. With humanization by knock-in replacement of the Csf1, Csf2, Il3, Tpo and Sirpa genes in the Rag2−/− Il2rg−/− genetic background, MIS(KI)TRG mice are highly permissive for human hematopoiesis and support the reconstitution of robust human lymphoid and myelomonocytic systems. With the demonstrated development of functional T-lymphocytes and monocytes/macrophages, MIS(KI)TRG mice provide a useful humanized mouse host for HIV-1 infection studies. Bone marrow biopsies were obtained from study participants and purified CD34+ cells were used to reconstitute the MIS(KI)TRG mice. We infected these patient-derived humanized mice with primary HIV-1 isolates grown from resting CD4+ T cells of the same patient and then evaluated antiviral effect of autologous CD8+ T cells (Fig. 4a). MIS(KI)TRG mice engrafted with CP18 bone marrow CD34+ cells successfully developed human T-lymphocyte and monocyte/macrophage subsets (Fig. 4b and c), which were sufficient to support HIV-1 infection (Fig. 4d). Plasma HIV-1 RNA levels peaked 20–30 days after infection (Extended Data Fig. 6a). Depletion of CD4+ T cells was clearly evident 12 days after infection in peripheral blood and spleen (Fig. 4e and Extended Data Fig. 6b and 6c). Cell-associated HIV-1 RNA was detected in both T cells and macrophages/monocytes (Extended Data Fig. 6d). Viral infection was also observed in various tissues where a large number of memory CD4+ T cells were detected (Extended Data Fig. 7). In control mice or mice that received autologous patient CD8+ T cells pre-stimulated with a peptide representing the unmutated dominant SL9 epitope, levels of plasma HIV-1 RNA and proviral DNA in peripheral blood continued to increase from day 14 to day 29 after infection (Fig. 4f). In sharp contrast, mice that received CD8+ T cells pre-stimulated with unmutated epitopes (Gag mix or WF9) had a significantly lower level of viral replication (Fig. 4g and h). Dramatic decreases in plasma HIV-1 RNA of 100- to 1,000-fold were observed in all three mice that received CD8+ T cells pre-stimulated with the mixture of Gag peptides including dominant and subdominant epitopes. Two of three mice had undetectable levels of plasma HIV-1 RNA and proviral DNA in peripheral blood measured at three time points (Fig. 4g). We performed the same experiments using patient CP36-derived humanized mice and reduction of peripheral HIV-1 RNA and DNA levels was also observed in mice that received CP36 CD8+ T cells pre-stimulated with the mixture of Gag peptides (Extended Data Fig. 8). Since the post-engraftment lifespan of MIS(KI)TRG mice is only 10–12 weeks33, we were only able to investigate the acute phase of HIV-1infection and demonstrate the in vivo functionality of patient CD8+ T cells. Future developments of the MIS(KI)TRG model will prolong the post-engraftment lifespan of the mice and will allow the studies of establishment and clearance of HIV-1 latent reservoir in vivo. Together, our in vitro and in vivo experiments demonstrate that only the CTL clones targeting unmutated epitopes are effective against cells infected with the viral variants that are likely to represent the major source of rebound HIV-1 after reversal of latency.
Figure 4. Broad-spectrum cytotoxic T lymphocytes suppress in vivo replication of HIV-1 from the latent reservoir of the same patients in patient-derived humanized mice.
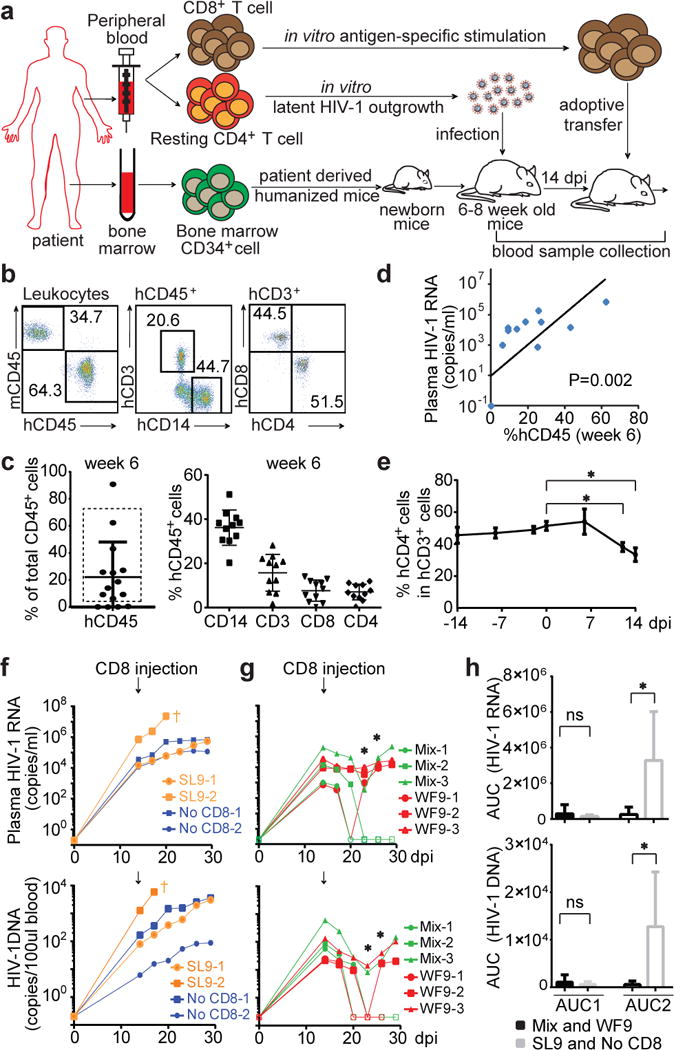
a, Experimental design. b and c, Efficient engraftment of patient CP18-derived hematopoietic cells in MIS(KI)TRG mice at week 6. Representative flow cytometry analysis (b) and summary (c) of human CD45+ cells, human T-lymphocyte and monocyte subsets. 11 out of 15 mice (enclosed in the rectangle) were used for HIV-1 infection. d, Correlation between frequency of peripheral human CD45+ cells (6 weeks after engraftment) and plasma HIV-1 RNA levels (14 days after infection). e, Depletion of peripheral CD4+ T cells after HIV-1 infection. *: p<0.05, paired t-test, n=11. f and g, Reduction of levels of plasma HIV-1 RNA and copies of peripheral blood HIV-1 DNA after injection of viral-specific CTLs. Filled: above detection limit; open: below detection limit. *: p<0.05, unpaired t test. h, Effect of CTL on the level of viral replication in vivo. The area under the curve of the viremia vs. time plot for each mouse in f and g before (AUC1) or after (AUC2) injection of CD8+ T cells was calculated to quantitatively represent viral replication over time. *: p<0.05, unpaired t test.
The seeding of the HIV-1 latent reservoir starts just a few days after infection26, prior to the development of robust CTL response14. This is consistent with our finding that patients who initiated treatment early in acute infection have few if any CTL escape variants archived in the latent reservoir. However, if treatment was initiated in chronic infection, CTL escape variants became dominant in the latent reservoir, indicating a complete replacement of the initially established ‘wild-type’ reservoir. The mechanism behind this replacement warrants further investigation, but likely reflects the dynamic nature of the reservoir in untreated infection. In any event, the overwhelming presence of escape variants in the latent reservoir of chronic patients certainly presents an additional barrier to eradication efforts. The striking difference between AP- and CP-treated patients presents another argument for early treatment of HIV-1 infection; early treatment not only reduces the size of the latent reservoir27, but also alters the composition of the reservoir, as shown here, in a way that may enhance the efficacy of potential CTL-based eradication therapies.
The hierarchy of HIV-1-specific CTL response in acute infection appears to play a significant role in initial viral suppression as demonstrated by the fact that certain immunodominant CTL populations are frequently linked to lower set point viremia later in infection17, 28. These immunodominant responses in acute infection have been identified as the major selection force driving the development of CTL escape mutations13, 20. Here we show that these immunodominant response-driven mutations are not only archived in the latent reservoir, but also in fact dominate the latent provirus population in CP-treated patients. Therefore, directing CTL responses to unmutated viral epitopes is essential to clear latent HIV-1. Due to bias in antigen presentation or recognition29, common vaccination strategies will likely re-stimulate immunodominant CTL clones which do not kill infected cells after reversal of latency. Stimulation of CTL responses with viral peptides circumvents antigen processing and is able to elicit broad-spectrum CTL responses against unmutated regions of viral proteins. Our study suggests that latent HIV-1 can be eliminated in chronically infected patients despite the overwhelming presence of CTL escape variants. Future directions in therapeutic vaccine design need to focus on boosting broad CTL responses as also reported elsewhere30 and/or manipulating immunodominance.
Methods
Human subjects
Peripheral blood or bone marrow for the isolation of primary CD4+, CD8+ or CD34+ T cells was obtained from 30 HIV-1-infected patients (Extended Data Table 1) and 7 healthy adult volunteers. All patients had been on antiretroviral therapy (ART) for at least 2 years and had maintained undetectable plasma HIV-1 RNA levels (<50 copies/ml) for at least 1 year prior to study. 10 AP-treated patients were recruited from the OPTIONS cohort at the University of California San Francisco (UCSF). This study was approved by the Johns Hopkins Internal Review Board and by the UCSF Committee on Human Research. Written informed consent was provided by all study participants. HLA typing for each patient was performed by the Johns Hopkins University Immunogenetics Laboratory.
Sample preparation for deep sequencing
Peripheral blood mononuculear cells (PBMCs) were isolated from whole blood by Ficoll gradient separation. CD4+ T cells were purified from PBMCs by negative selection using CD4+ Isolation Kit II (Miltenyi). Resting CD4+ T cells were then purified from CD4+ T cells by negative selection using CD25, CD69 and HLA-DR microbeads (Miltenyi). Genomic DNA was extracted from 5 million resting CD4+ T cells from each patient using QIAamp DNA Mini Kit (Qiagen). The gag gene was amplified from genomic DNA by a two-round nested PCR using these primers: 5′ outer primer (5′-TTGACTAGCGGAGGCTAGAAGG-3′); 3′ outer primer (5′-GATAAAACCTCCAATTCCCCCTATC-3′); 5′ inner primer (5′-GAGAGATGGGTGCGAGAGCGTC-3′); 3′ inner primer (5′-CTGCTCCTGTATCTAATAGAGC-3′). For each patient, the entire genomic DNA from 5 million of resting CD4+ T cells was evenly distributed as template into 80 PCR reactions. The reactions were performed by using High Fidelity Plantinum Taq Polymerase (Life Technologies) following manufacturer’s instruction. PCR amplicons were purified by gel extraction after gel electrophoresis.
Deep sequencing
For PacBio RS single molecule sequencing, amplicons were barcoded with a group of 10 bp indexes and then multiple samples were pooled together to generate smrtbell sequencing library following Pacific Biosciences template preparation and sequencing-C2 user guide for 2 kb insert size and using Pacific Bioscience DNA template preparation kit. For MiSeq Sequencing, the pooled amplicon DNA was end repaired, adenylated, and ligated to Illumina TruSeq adaptors and PCR enriched for 10 cycles. The resulting library was then run on bioanalyzer high sensitivity DNA chip for size and concentration determination. The library was then sequenced on MiSeq for paired end 250 bp reads. The sequence reads from PacBio and MiSeq were demultiplexed using Fastx-Toolkit.
Data analysis for deep sequencing results
For the paired MiSeq reads, the two reads were first merged using FLASH31. MiSeq and PacBio reads from each individual were then aligned to the reference HIV-1 consensus B Gag sequence using Bowtie232. Custom program was written using perl scripts to identify and compute the frequency of all sequence variants that caused non-synonymous amino acid changes in each individual’s relevant optimal Gag epitopes (based on reported information in the HIV Molecular Immunology Database, Los Alamos National Laboratory, http://www.hiv.lanl.gov/content/immunology/index.html) according to their HLA type. For each individual, variants that occurred at a frequency > 3% were retained. Additional for PacBio reads, sequences with identified premature stop codons were eliminated from the analyzed results. For each identified variation, the mutation type regarding CTL recognition was determined by matching with the information in the above-mentioned database. The five mutation types adopted in this paper are: Documented Escape (no CTL response when patient cells are challenged with the variant peptide), Inferred Escape (variant is predicted to be an escape mutant by longitudinal study or transmission study, but the reactivity of the variant is not tested experimentally), Diminished Response (experimental data suggest partial escape as evidenced by decreased CTL response), Susceptible Form (CTL response is elicited when patient cells are challenged with the variant peptide) and Mutation Type Not Determined (no experimental data on CTL recognition of this variant).
Elispot essays
The Elispot assays were performed using Human IFN-γ ELISpot PLUS kit (Mabtech) according to previously described33 and manufacturer’s instructions. PBMCs were added at 200,000 cells per well and synthetic peptides were added in a final concentration of 0.1, 1 or 10 μg/ml. A response is considered positive if it was 3 fold higher than the mean background (cell only control) and greater than 55 SFC/million cells. The number of specific T cells was calculated by subtracting the mean background values.
Recovery and sequencing of patient viruses from resting CD4+ T cells
Co-culture assays were performed to recover and amplify replication-competent viruses as previously described34. The viruses were recovered from five to ten million resting CD4+ T cells. The concentration of outgrowth viruses was determined by p24 ELISA (PerkinElmer). Total RNA of outgrowth viruses was extracted using Trizol LS reagent (Life Technologies). Residual DNA was then removed by TURBO DNase (Life Technologies) treatment. First strand cDNA was synthesized using SuperScript III Reverse Transcriptase (Life Technologies) and the gag gene was amplified from cDNA using the gag outer primer pair mentioned above. The PCR amplicons were then purified by gel extraction and sequenced by regular Sanger sequencing.
In vitro HIV-1 infection
PBMCs from HIV-1-infected patients and healthy donors were stimulated by adding 0.5μg/ml Phytohaemagglutinin (PHA) and IL-2 (100U/ml) to basal media (RPMI with 10% heat-inactivated fetal bovine serum and antibiotics) for three days prior to isolation of CD4+ T cells. Each patient’s activated CD4+ T cells were infected with viruses recovered from the same patient’s resting CD4+ T cells. Healthy donor’s CD4+ cells were infected with a lab strain virus, BaL. Virus concentration used in infection was equivalent to the p24 concentration of 200 ng/ml. All infections were performed by centrifugation of target cells with virus at 1,200g for 2 hours.
Stimulation of CD8+ T cells
PBMCs from CP-treated patients were cultured in the presence of IL-2 (100U/ml) with a mixture of consensus B Gag (or Nef, Rev, Tat, Env) peptides (800 ng/ml for each) (NIH AIDS Reagent Program), or with individual synthetic peptide (0.5μg/ml) (Genemed Synthesis). CD8+ T cells were purified after six days of incubation by positive selection using human CD8 microbeads (Miltenyi). To monitor CTL proliferation, PBMCs were stained with CFSE (Life Technologies) prior to incubation and with relevant pentamer (Proimmune) after incubation. PBMCs were then stained with CD8-APC (Becton Dickson, BD) and analyzed by flow cytometry using FACS Canto II (BD).
Co-culture of autologous CD4+ and CD8+ T cells
Three hours after infection, CD4+ T cells were mixed with autologous unstimulated or stimulated CD8+ T cells at 1:1 ratio in basal media at 5 million cells per ml. Two days after co-culture, enfuvirtide (T-20, Roche) was added into the culture at 10 μM to prevent further infection events except if the measurement was p24 ELISA. Three days after co-culture, cells were stained with CD8-APC (BD) first, fixed and permeabilized with Cytoperm/Cytofix (BD pharmingen), then stained for intracellular p24 Gag (PE, Coulter). Cells were analyzed by flow cytometry using FACS Canto II (BD). For measurement of viral growth, 5 μl of supernatant was taken from the co-culture at Days 0, 3 and 6, and subjected to p24 ELISA. For analysis of cell contact dependence, CD4+ and CD8+ T cells were placed in separate chambers of trans-well plates (0.4 μm, Costar).
Generation and infection of patient-derived humanized mice
The previously reported MISTRG mouse in the Rag2−/− Il2rg−/− 129×Balb/c (N2) genetic background harbors knock-in replacement of the endogenous mouse Csf1, Csf2, Il3 and Tpo genes with humanized version and a BAC transgene encoding human SIRPα25. We generate Sirpa(KI) mouse which harbors a knock-in replacement of the endogenous mouse Sirpa gene with a humanized version. Sirpa(KI) mouse will be thoroughly described elsewhere. The improved MIS(KI)TRG mouse was generated by breeding Sirpa(KI) mice to MITRG mice. All animal experimentations were performed in compliance with Yale Institutional Animal Care and Use Committee protocols. MIS(KI)TRG mice were maintained with continuous treatment with enrofloxacin in the drinking water (Baytril, 0.27 mg/ml). Patient bone marrow or fetal liver CD34+ cells were isolated by CD34 microbeads selection (miltenyi). Newborn mice (within first 3 days of life) were sublethally irradiated (X-ray irradiation with X-RAD 320 irradiator, PXi; 1 × 150 cGy) and 100,000 fetal liver or 250,000 patient CD34+ cells in 20 μl of PBS were injected into the liver with a 22-gauge needle (Hamilton Company). 6–8 weeks after engraftment, mice engrafted with patient CD34+ cells were infected by i.v. with HIV-1 (100ng p24) which was recovered and expanded from the resting CD4+ T cells of the same patient (CD34+ cell donor), as mentioned above. Mice engrafted with fetal liver CD34+ cells were infected by i.v. with HIV-1 BaL (100ng p24). Mice with less than 5% human CD45+ cells in the peripheral blood were excluded from the infection study. Mice with more than 70% human CD45+ cells in the peripheral blood were also excluded because they were unhealthy due to human macrophage/monocytes caused anemia33. 20 million autologous CD8+ T cells with or without pre-stimulation were injected by i.v. 9 or 14 days after infection. Group allocation was blinded. Peripheral blood samples were collected by retro-orbital bleeding at different time points before and after injection of CD8+ T cells. Engraftment of human CD45+ cells as well as lymphoid and myeloid subsets was determined by flow cytometry. Plasma HIV-1 RNA in peripheral blood was measured by one-step reverse transcriptase (Invitrogen) real-time PCR using the following primers and probe, described previously8: forward (5′ → 3′) ACATCAAGCAGCCATGCAAAT, reverse (5′ → 3′) TCTGGCCTGGTGCAATAGG and probe (5′ → 3′) VIC-CTATCCCATTCTGCAGCTTCCTCATTGATG-TAMRA. Assay sensitivity is 200 RNA copies per ml of plasma. HIV-1 DNA in peripheral blood was also measured by real time PCR using the same primers and probe mentioned above, with assay sensitivity at 5 copies per 100μl of blood. Total viral DNA in PBMCs was determined by measuring copies of viral DNA/100ul blood and blood volume per mouse (80ul blood/1g body weight). To quantitate total viral DNA in tissues, spleens, livers and lungs of infected mice were collected. For the spleen, single-cell suspensions were treated with ACK lysis buffer. Liver and lung leukocytes were isolated by digesting tissues with 100 U/ml collagenase IV and 0.02 mg/ml DNase I (Sigma), followed by density gradient centrifugation.
Statistical analysis
For comparison of HIV-1 variant frequency (Figure 1b and c) and viral infection in HIV-1 BaL-infected mice (Extended Data Figure 6 and 7), we applied Mann-Whitney tests. For comparison of inhibitory effect of autologous CTLs (Figure 2a), we applied a Wilcoxon matched pairs test. For comparison of viral replication in humanized mice (Figure 4f, g and h), we applied an unpaired t test. For all other comparisons, paired t tests were applied. All tests were calculated by the GraphPad Prism 6 software, and conducted as two-tailed tests with a type I error rate of 5%.
Extended Data
Extended Data Figure 1. CTL escape variants dominate the latent reservoir of CP-treated HIV-1-positive individuals, but not AP-treated individuals.

Frequency of variants in Gag CTL epitopes in proviruses from resting CD4+ T cells. Results of all 25 patients tested are shown. Only optimal CTL epitopes relevant to each patient’s HLA type are listed in linear positional order on the X axis. Results from both Pacbio (left bar) and MiSeq (right bar) sequencing platforms are shown for each epitope. The absence of bars above a listed epitope indicates that only wild type sequences were detected. For each mutation in a CTL epitope, information regarding the effect of the mutation on CTL recognition from the Los Alamos National Laboratory (LANL) HIV Molecular Immunology Database or from ELISpot assays described herein was used to assign the mutation to one of the categories indicated at the bottom. See Methods for definitions of these categories.
Extended Data Figure 2. Characterization of CTL responses against HIV-1 Gag epitopes by interferon-γ ELISpot.
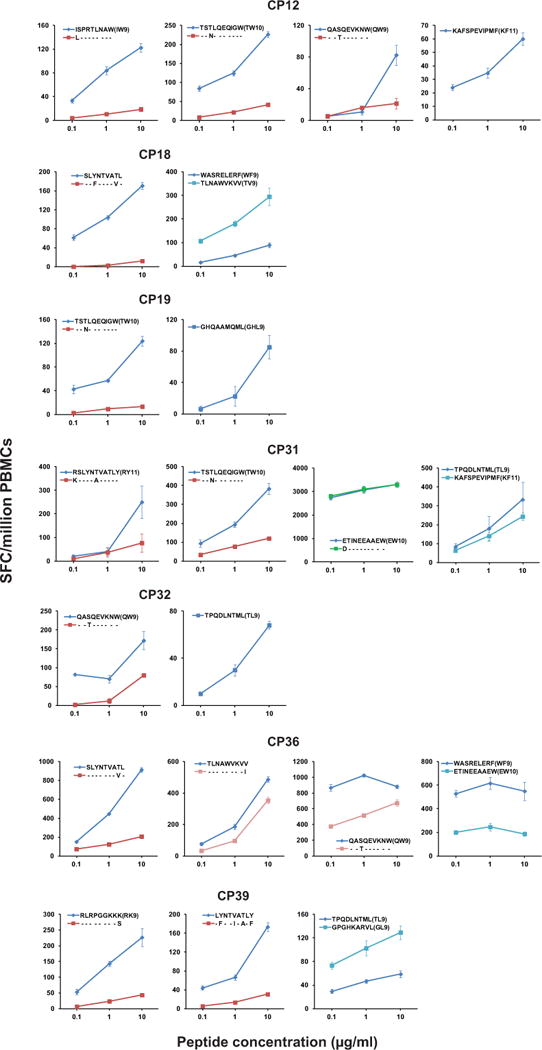
Results of 7 patients tested are shown. The peptides tested are listed for each patient in each graph. Error bars represent s.e.m., n = 3.
Extended Data Figure 3. Partial Gag sequences from proviral DNA and outgrowth virus from resting CD4+ T cells of 8 CP-treated patients (following Fig. 1e).
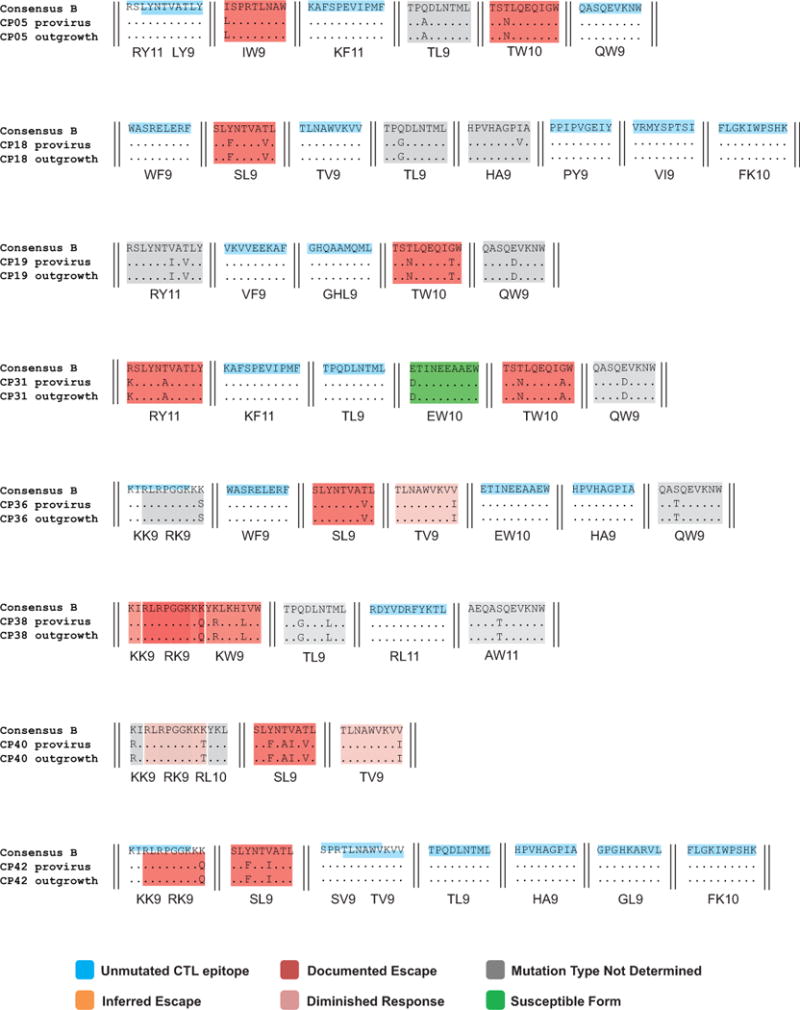
CTL epitopes with no observed variation are highlighted in blue. Documented escape mutations (red shading), inferred escape mutations (yellow shading), diminished response (pink shading), susceptible form (green shading) or undetermined variations (gray shading) in relevant optimal epitopes are indicated. See Methods for definitions of these types of mutations.
Extended Data Figure 4. CD8+ T cells pre-stimulated with a mixture of consensus B Gag peptides eliminate autologous CD4+ T cells infected with autologous HIV-1 from resting CD4+ T cells.
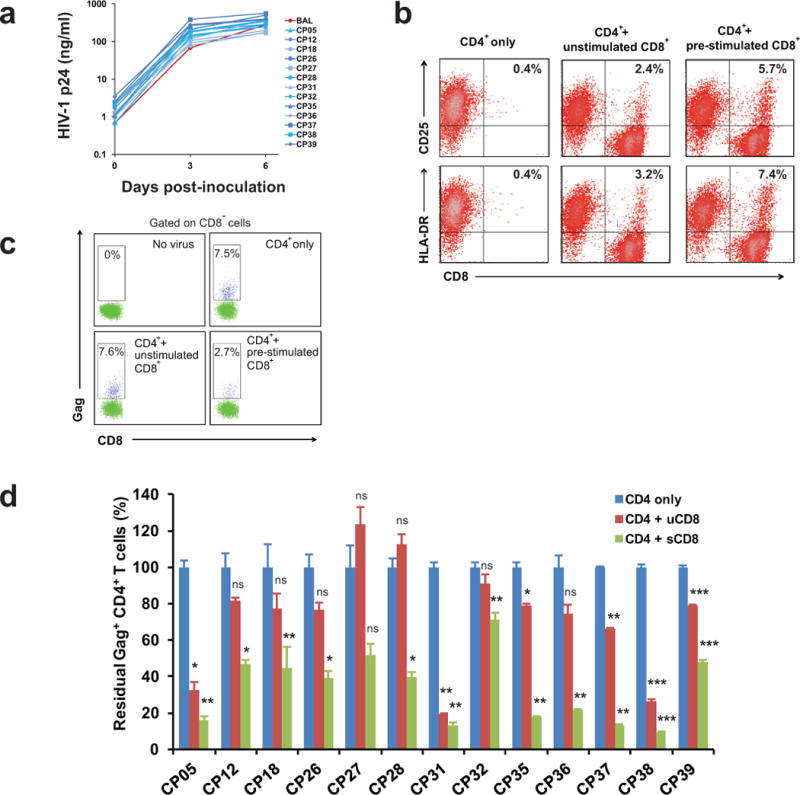
a, HIV-1 isolated from ART-treated individuals replicates as well as lab strain virus BaL. p24 values represent mean of 3 replicates. Error bars represent s.e.m., n = 3. b, CD8+ T cells are not stimulated after co-cultured with PHA-activated CD4+ T cells. c, A representative flow cytometric analysis of CTL-mediated killing after co-culture of infected CD4+ T cells with autologous CD8+ T cells. CTL activity is measured by the percentage of Gag-positive, CD8-negative cells after 3 days of co-culture relative to cultures without CD8+ T cells. d, Pre-stimulated CD8+ T cells eliminate autologous infected CD4+ T cells more efficiently than non-stimulated CD8+ T cells. All results were normalized to the CD4 only control group. Error bars represent s.e.m., n = 3. *: p<0.05; **: p<0.01; ***: p<0.001; ns: p>0.05, paired t test.
Extended Data Figure 5. The elimination of infected CD4+ T cells is mediated by direct killing by autologous CD8+ T cells.
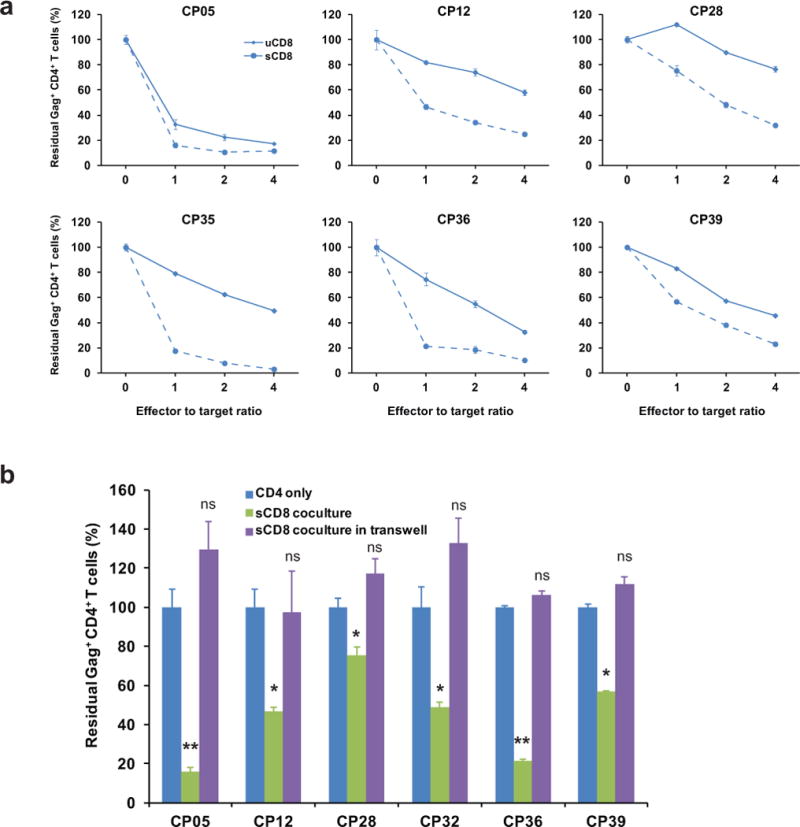
a, Killing of infected CD4+ T cells is enhanced by increased E:T ratios for both pre-stimulated and non-stimulated CD8+ T cells. b, Killing of the infected CD4+ T cells depends on direct cell-cell contact between CD4+ T cells and CTLs. All results were normalized to the CD4 only control group. Error bars represent s.e.m., n = 3. *: p<0.05; **: p<0.01; ***: p<0.001; ns: p>0.05, paired t test.
Extended Data Figure 6. Viral dynamics and depletion of CD4+ T cells in humanized mice.
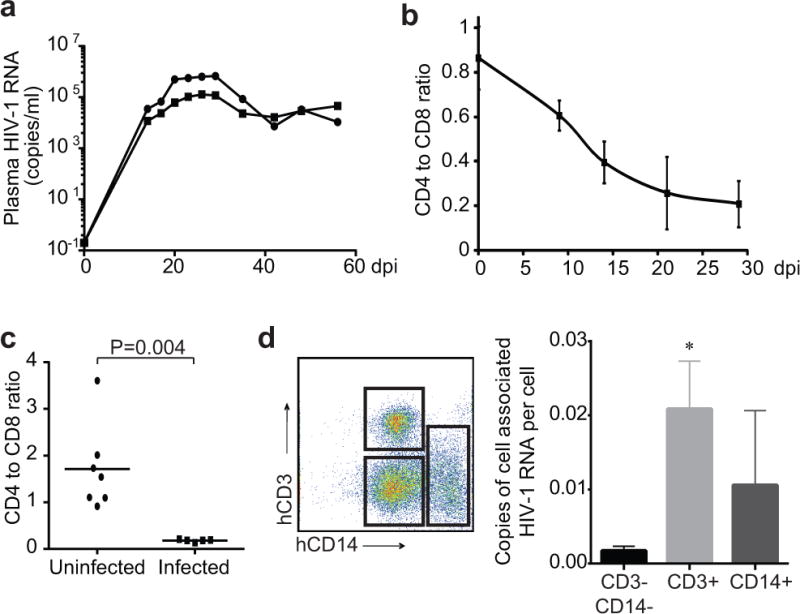
a, Viral dynamics in CP18-infected MIS(KI)TRG mice. CP18-derived MIS(KI)TRG mice were infected with autologous HIV-1. Plasma HIV-1 RNA levels were measured from day 0 to day 56. b, Depletion of CD4+ T cells in peripheral blood of HIV-1 BaL-infected mice. MIS(KI)TRG mice engrafted with fetal liver CD34+ cells were infected with HIV-1 BaL. CD4 to CD8 ratio in peripheral blood was measured by FACS from day 0 to day 29 after infection. c, Depletion of CD4+ T cells in spleen of HIV-1 BaL-infected mice. MIS(KI)TRG mice engrafted with fetal liver CD34+ cells were infected with HIV-1 BaL. CD4 to CD8 ratio in spleen was measured by FACS 20 days after infection. Median and P value from Mann-Whitney test are shown. d, Detection of cell-associated HIV-1 RNA in T cells and macrophages/monocytes. CD3+ and CD14+ human cells from HIV-1-infected MIS(KI)TRG mice from spleen and lung were purified by FACS. CD3−CD14− cells were also collected as controls. Cell associated HIV-1 RNA was quantified by gag-specific qPCR. *: p<0.05, unpaired t test.
Extended Data Figure 7. HIV-1 infection occurs in peripheral blood and tissues in humanized mice.
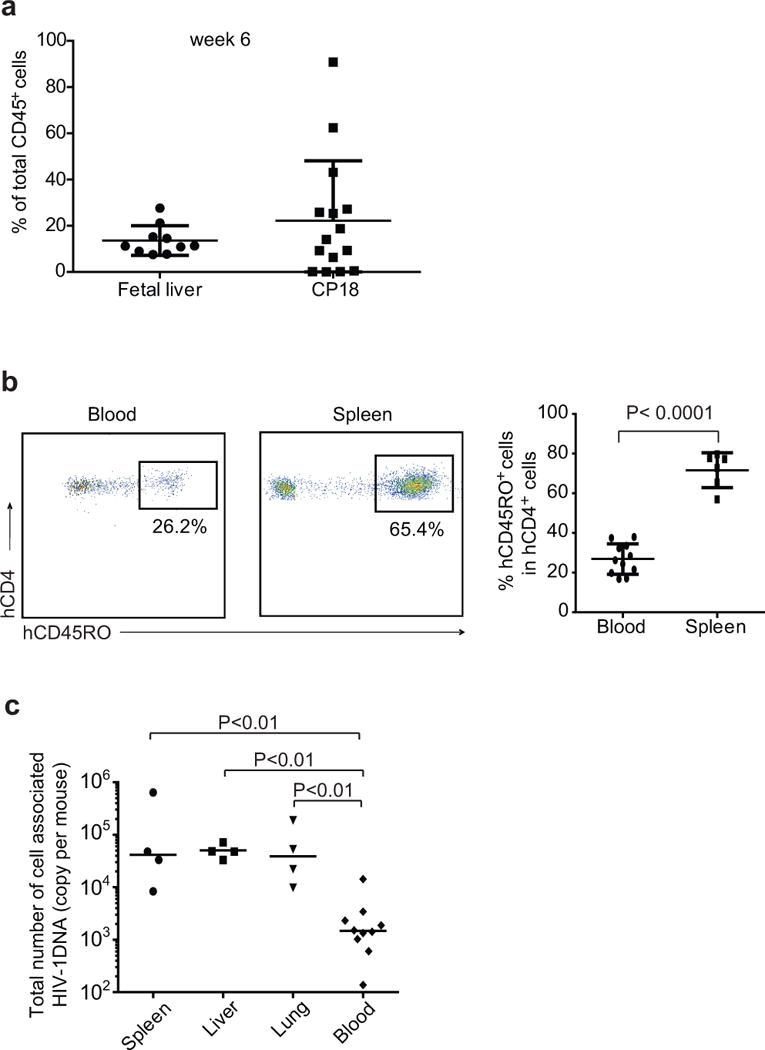
a, Engraftment levels of MIS(KI)TRG mice with fetal liver or patient CD34+ cells. b, Memory CD4+ T cells are detected in MIS(KI)TRG mice after infection. MIS(KI)TRG mice were infected with HIV-1 BaL. Peripheral blood and indicated tissues from infected mice were collected at 20 dpi. Memory CD4+ T cells were determined by CD45RO staining. c, Total number of cell associated HIV-1 DNA in blood and tissues. DNA Leukocytes from peripheral blood or indicated tissues were isolated for the measurement of total amount of cell-associated HIV-1 DNA by real-time PCR. For b and c, median and P value from Mann-Whitney test are shown.
Extended Data Figure 8. Broad-spectrum cytotoxic T lymphocytes suppress in vivo infection of patient-derived humanized mice with autologous latent HIV-1.
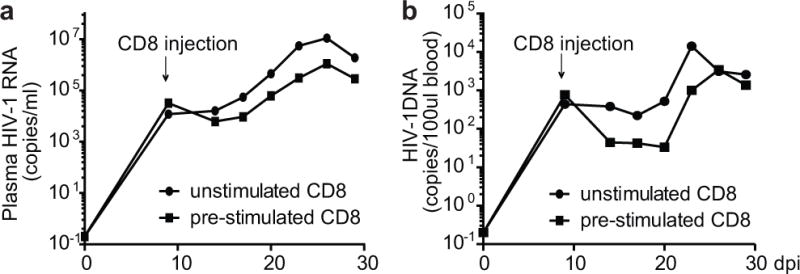
Generation of patient CP36-derived humanized mice was described in Fig. 4. Mice were infected with autologous viruses at 6 weeks old. CD8+ T cells from CP36 were pre-stimulated with the mixture of Gag peptides or left untreated for 6 days in vitro and were injected into mice by i.v. 9 days after infection. Plasma HIV-1 RNA and HIV-1 DNA in peripheral blood were measured by real-time PCR.
Extended Data Table 1.
Characteristics of study subjects.
| Patient | Year of Diagnosis | CD4 count* (cells/μl) |
Plasma HIV-1 RNA† (copies/ml) |
Time on ART (years) |
Treatment start time after infection (days) |
|---|---|---|---|---|---|
| AP01 | 2006 | 1251 | <50 | 7 | 64 |
| AP03 | 2004 | 595 | <50 | 9 | 34 |
| AP04 | 1998 | 953 | <50 | 15 | 77 |
| AP05 | 2002 | 618 | <50 | 11 | 39 |
| AP07 | 2012 | 592 | <50 | 1.5 | 67 |
| AP08 | 2008 | 780 | <50 | 6 | 28 |
| AP09 | 2012 | 1069 | <50 | 2 | 39 |
| AP10 | 2006 | 513 | <50 | 7 | 50 |
| AP11 | 2007 | 874 | <50 | 6 | 10 |
| AP12 | 2007 | 629 | <50 | 6 | 15 |
| CP05 | 2001 | 500 | <50 | 10 | >180 |
| CP12 | 1997 | 1074 | <50 | 15 | >180 |
| CP18 | 1998 | 773 | <50 | >4 | >180 |
| CP19 | 2006 | 620 | <50 | 6 | >180 |
| CP26 | 1994 | 640 | <50 | 16 | >180 |
| CP27 | 1987 | 784 | <50 | 4 | >180 |
| CP28 | 1998 | 614 | <50 | 6 | >180 |
| CP31 | 2000 | 619 | <50 | 10 | >180 |
| CP32 | 1999 | 780 | <50 | 2 | >180 |
| CP35 | 2002 | 738 | <50 | 10 | >180 |
| CP36 | 2003 | 1119 | <50 | 4 | >180 |
| CP37 | 1999 | 730 | <50 | 12 | >180 |
| CP38 | 1986 | 870 | <50 | 3 | >180 |
| CP39 | 1996 | 1152 | <50 | 10 | >180 |
| CP40 | 2002 | 544 | <50 | 7 | >180 |
| CP42 | 1987 | 684 | <50 | 16 | >180 |
| CP47 | 1986 | 792 | <50 | 14 | >180 |
| CP48 | 1998 | 641 | <50 | 8 | >180 |
| CP49 | 1992 | 864 | <50 | 5 | >180 |
| CP50 | 2001 | 964 | <50 | 4 | >180 |
Patient CD4 count was measured during this study.
Plasma HIV-1 RNA levels for all patients were <50 copies/ml for at least one year prior to this study.
Acknowledgments
We thank all study participants. We thank Dr. Joel Blankson for critical advice to the project; Linda Alston and Rebecca Hoh for coordinating patient recruitment; Jonathan Alderman, Carla Weibel and Elizabeth Henchey for technical assistance in animal study. We thank NIH AIDS Reagent Program for providing HIV-1 consensus B peptides. R.F.S. is supported by the Howard Hughes Medical Institute, by NIH grant AI43222 and AI096109, and by an ARCHE grant from amfAR (The Foundation for AIDS Research). L.S. is supported by NIH grant T32 AI07019. R.A.F. is supported by the Bill and Melinda Gates Foundation and the Howard Hughes Medical Institute.
Footnotes
Author Contributions
K.D., L.S. R.A.F and R.F.S. conceived and designed the research studies; K.D., J.L., H.Z., J.B.M. and L.S. performed the in vitro experiments; K.D. A.R., L.W., C.G., A.J.M., D.M.V., G.D.Y., T.S., P. K. and L.S. performed animal experiments; C.M.D., G.G., H.L.M. and S.G.D. provided patient samples; K.D., M.P., L.W., H.H, J.D.S., S.L.S., L.S. and R.F.S. analyzed data; K.D., L.S. and R.F.S. wrote the manuscript.
References
- 1.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–8. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 2.Strain MC, et al. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc Natl Acad Sci U S A. 2003;100:4819–24. doi: 10.1073/pnas.0736332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 4.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Richman DD, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–7. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 6.Archin NM, et al. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25:207–12. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contreras X, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem. 2009;284:6782–9. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–5. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shan L, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker BD, et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328:345–8. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- 11.Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–5. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–11. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 13.Goonetilleke N, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–72. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10:11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips RE, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–9. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 16.Koup RA. Virus escape from CTL recognition. J Exp Med. 1994;180:779–82. doi: 10.1084/jem.180.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goulder PJ, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–7. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 18.Goulder PJ, Watkins DI. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol. 2004;4:630–40. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 19.Henn MR, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu MK, et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest. 2013;123:380–93. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rolland M, Nickle DC, Mullins JI. HIV-1 group M conserved elements vaccine. PLoS Pathog. 2007;3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Letourneau S, et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS One. 2007;2:e984. doi: 10.1371/journal.pone.0000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu XG, et al. Consistent patterns in the development and immunodominance of human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T-cell responses following acute HIV-1 infection. J Virol. 2002;76:8690–701. doi: 10.1128/JVI.76.17.8690-8701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho YC, et al. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell. 2013;155:540–51. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rongvaux A, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32:364–72. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitney JB, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014:74–77. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ananworanich J, et al. Impact of multi-targeted antiretroviral treatment on gut T cell depletion and HIV reservoir seeding during acute HIV infection. PLoS One. 2012;7:e33948. doi: 10.1371/journal.pone.0033948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goulder PJ, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–8. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- 29.Le Gall S, Stamegna P, Walker BD. Portable flanking sequences modulate CTL epitope processing. J Clin Invest. 2007;117:3563–75. doi: 10.1172/JCI32047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen SG, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science. 2013;340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Streeck H, Frahm N, Walker BD. The role of IFN-gamma Elispot assay in HIV vaccine research. Nat Protoc. 2009;4:461–9. doi: 10.1038/nprot.2009.7. [DOI] [PubMed] [Google Scholar]
- 34.Siliciano JD, Siliciano RF. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol Biol. 2005;304:3–15. doi: 10.1385/1-59259-907-9:003. [DOI] [PubMed] [Google Scholar]