

Abstract
Repeatedly activated T helper 1 (Th1) cells present during chronic inflammation can efficiently adapt to the inflammatory milieu, for example, by expressing the transcription factor Twist1, which limits the immunopathology caused by Th1 cells. Here, we show that in repeatedly activated murine Th1 cells, Twist1 and T-bet induce expression of microRNA-148a (miR-148a). miR-148a regulates expression of the proapoptotic gene Bim, resulting in a decreased Bim/Bcl2 ratio. Inhibition of miR-148a by antagomirs in repeatedly activated Th1 cells increases the expression of Bim, leading to enhanced apoptosis. Knockdown of Bim expression by siRNA in miR-148a antagomir-treated cells restores viability of the Th1 cells, demonstrating that miR-148a controls survival by regulating Bim expression. Thus, Twist1 and T-bet not only control the differentiation and function of Th1 cells, but also their persistence in chronic inflammation.
Keywords: Bim, miR-148a, T-bet, Th1, Twist1
Introduction
Immune responses against intracellular microorganisms are mediated by T helper type 1 (Th1) cells, which produce interferon (IFN)-γ as their hallmark cytokine [1]. The differentiation of naive CD4+ cells into Th1 cells is dynamically regulated and includes signals from (i) the T-cell receptor (TCR), (ii) the IFN-γ receptor/ signal transducer and activator of transcription (STAT)-1 and (iii) the interleukin (IL)-12 receptor/STAT4 which collectively lead to the induction and further amplification of the Th1 master transcription factor T-bet [2]. Besides their protective role in the clearance of infection, Th1 cells are involved in the initiation and maintenance of inflammatory diseases and disease models such as inflammatory bowel disease [3], OVA-induced arthritis [4], and experimental autoimmune encephalomyelitis (EAE) [5]. Repeatedly activated Th1 cells accumulate in inflamed tissues [6–8] and persist, despite therapeutic immunosuppression, in patients suffering from chronic inflammatory diseases [9]. These cells express the transcription factor Twist1, whose expression increases upon repeated restimulation of Th1 cells and limits immunopathology in chronic inflammation [9]. However, it has remained elusive how the persistence of Th1 cells in inflamed tissue is mediated.
The B-cell lymphoma 2 (Bcl-2) family proteins control the contraction of activated T cell populations after an adaptive immune response and are composed of proapoptotic and antiapoptotic proteins [10–12]. The antiapoptotic protein Bcl-2 stabilizes the mitochondrial outer membrane and inhibits its permeabilization. In the contraction phase of an adaptive immune response, withdrawal of IL-2 or IL-7 leads to the induction and activation of Bim, which interacts with and antagonizes the function of Bcl-2 and promotes the intrinsic apoptosis pathway in activated T cells [10],[13]. Excess of Bim destabilizes the mitochondrial outer membrane, leading to activation of caspase 9 and in consequence to induction of apoptosis [14]. Thus, the regulation of Bcl-2 and Bim expression is critical for the survival of T cells [15,16].
Here we show that in repeatedly activated Th1 cells, Bim expression is downregulated posttranscriptionally by miR-148a. Expression of miR-148a is induced by the transcription factors T-bet and Twist1, thus is selective for repeatedly activated Th1 cells. This mechanism promotes persistence of antigen-specific Th1 cells in long-lasting, chronic immune reactions, and identifies Twist1 as a critical regulator of chronicity in inflammation.
Results
miR-148a is upregulated in repeatedly activated Th1 cells
To address miR-148a expression in Th cells, murine naive (CD4+CD62Lhi) Th cells were activated in vitro under polarizing conditions for the generation of effector T helper type I (Th1 once), type II (Th2 once), and IL-17-producing (Th17 once) cells. Mimicking Th cells involved in chronic inflammation with a history of repeated restimulation by persistent (auto-) antigens, we repeatedly (4x) activated cells of the three different Th cell subsets. Quality of differentiation was controlled by intracellular cytokine staining for IFN-γ, IL-4, and IL-17 (Supporting Information Fig.1A–C). miR-148a was expressed in naive and once activated Th1, Th2, and Th17 and significantly upregulated up to 30-fold in repeatedly activated Th1 (Th1 rep.), but not in Th2 (Th2 rep.) or Th17 cells (Th17 rep.; 1A). Already after one round of activation, all Th1 cells uniformly expressed T-bet, qualifying them as Th1 cells [17,18] (Supporting Information Fig. 2A). Of those Th1 cells, those actually secreting IFN-γ in the restimulation and those that did not, showed the same level of miR-148a expression, and significantly less than Th1 rep. (Supporting Information Fig. 2B). miR-148a is a member of a miRNA family with miR-148b and miR-152 sharing the same seed region and potentially targeting the same genes (http://www.mirbase.org; [19]). Expression of miR-148b was sevenfold and expression of miR-152 30-fold lower than miR-148a expression in repeatedly activated Th1 cells (Fig. 1B). Both miR-148b and miR-152 were not differentially regulated between once and repeatedly activated Th1 cells (Fig. 1B). To address miR-148a expression in in vivo-differentiated Th1 cells, SMARTA1-TCR transgenic (SM TCRtg) Thy1.1+ naive CD4+ Th cells were transferred into nonlymphopenic C57BL/6 mice and infected with 200 pfu of lymphocytic choriomeningitis virus (LCMV) strain WE on day two after transfer. Seven days after infection, SM TCRtg Thy1.1+ CD4+ Th cells uniformly expressed T-bet (Fig. 1C and D) and consisted of 87% of IFN-γ producers (Supporting Information Fig. 2C). Notably, these Th1 cells expressed ninefold and 48-fold higher miR-148a levels compared to naive host CD4+ T cells on day 5 and day 7 after viral infection, respectively, (Fig. 1E). These results show that miR-148a is exclusively expressed in repeatedly activated Th1 cells in vitro and Th1 cells differentiated after LCMV infection in vivo.
Figure 1.

miR-148a is upregulated in repeatedly activated Th1 cells (A) Quantitative RT-PCR (qRT-PCR) of miR-148a expression in once (on day 6) and repeatedly (6 day intervals; three rounds of restimulation) activated Th1 cells (independent experiments, n = 7), Th2 cells (n = 7), Th17 cells (n = 3), ex vivo-isolated naive Th cells (n = 6), normalized to small nuclear RNA U6 (snU6). Each data point represents an independent experiment. Data are shown as mean + SEM pooled from three to seven independent experiments. Wilcoxon-Test for paired data, *p ≤ 0.05. (B) Expression of miRNA family members miR-148a (n = 7, same cohort as in (A)) miR-148b (independent experiments, n = 4) and miR-152 (independent experiments, n = 4) in once and repeatedly activate d Th1 cells normalized to snU6, assessed by qRT-PCR. Each data point represents an independent experiment. Data are shown as mean ± SEM pooled from four to seven independent experiments. Mann–Whitney test for unpaired data, *p ≤ 0.05, **p ≤ 0.005. (C) T-bet protein expression in SM TCRtg CD4+ T cells on day 7 after LCMV infection was assessed by intracellular staining and flow cytometry. A single histogram from an experiment performed with five samples is shown, representative of three independent experiments performed. For gating strategies see Supporting Information Fig. 7A. (D) Statistical evaluation of MFIs of T-bet as determined in (C). Data are shown as mean + SEM of n = 5 samples from a single experiment representative of three experiments. Mann–Whitney test for unpaired data, *p ≤ 0.05. (E) qRT-PCR evaluation of miR-148a expression on day 5 and 7 of LCMV infection. Data are shown as mean ± SEM, depicted is one experiment with n = 5, representative of two independent experiments. Mann–Whitney test for unpaired data, *p ≤ 0.05, **p ≤ 0.005.
miR-148a targets the proapoptotic gene Bim
Candidate miR-148a targets in repeatedly activated Th1 cells were identified by target screens with PicTar (http://pictar.mdc-berlin.de/; [20]) and TargetScan (http://www.targetscan.org/; [21]), in combination with global transcriptome data of once versus repeatedly activated Th1 cells (Niesner et al. [9] and Supporting Information Table 1). Genome wide, 581 genes were identified as potential targets of miR-148a. Of these, 361 were expressed in Th1 cells and 130 genes were differentially expressed with a foldchange ≥ 1.4 in once versus repeatedly activated Th1 cells with 61 genes being down- and 69 genes being up-regulated in repeatedly activated Th1 cells. Among the predicted miR-148a targets was the proapoptotic gene Bim. Bim suited the hypothesis of a miR-148a mediated Th1 specific survival mechanism, which was raised from the observation that Th cells from inflamed tissues of autoimmune patients persist despite immunosuppressive therapy. Expression levels of Bim mRNA and protein were reduced twofold in repeatedly activated Th1 cells as compared to once activated Th1 cells (Supporting Information Table 1 and Fig. 2A and B). Ectopic overexpression of miR-148a in activated Th1 cells decreased levels of Bim mRNA and Bim protein by 50% (Fig. 2C–E). The two predicted miR-148a binding sites (bs) in the Bim 3′-UTR were validated in reporter assays. The Bim 3′-UTR was cloned downstream of a human CD4 reporter gene (hCD4) [22] (Supporting Information Fig. 3). Reporter gene expression (MFI of hCD4) was reduced by 30% in activated Th1 cells (d5) in the presence of a miR-148a overexpression vector (Fig. 2F). When both bs were destroyed by mutation (Supporting Information Fig. 3), this suppression was abrogated (Fig. 2F). By treating repeatedly activated Th1 cells with specific antagomirs [23], the inhibition of Bim expression by endogenous miR-148a was demonstrated. Antagomir-148a reduced miR-148a expression levels up to 98%, as compared to cells treated with a scrambled control antagomir, and increased Bim mRNA 1.8-fold while Bcl2 expression remained unchanged (data not shown). On the protein level, Bim expression, as measured by intracellular immunofluorescence, increased 1.6-fold in repeatedly activated Th1 cells treated with antagomir-148a compared to cells treated with the scrambled antagomir (Fig. 2G and H). Bcl-2 protein expression remained similar in antagomir-148a treated cells (Fig. 2G and H and Supporting Information Fig.4A) leading to a significant shift in the ratio of Bim to Bcl2 expression in favor of Bim (Fig. 2H).
Figure 2.

miR-148a targets Bim in repeatedly activated Th1 cells. (A) Bim mRNA expression in once and repeatedly activated Th1 cells was assessed by qRT-PCR, normalized to HPRT and presented relative to values obtained with once-activated Th1 cells. Data are shown as mean ± SEM, n = 1, each pooled from four independent experiments. (B) Bim protein expression in once and repeatedly activated Th1 cells assessed by intracellular protein staining and flow cytometry, presented as MFI of Bim, relative to once-activated Th1 cells. Data are shown as mean ± SEM, n = 1, each pooled from three independent experiments. For gating strategies see Supporting Information Fig. 7B. (C and D) Overexpression of miR-148a and a scrambled control (miR-scr) in CD4+ T cells assessed by qRT-PCR, analyzed on day 5 postactivation. miR-148a and Bim expression was normalized to snU6 or HPRT and presented relative to values obtained with miR-scr. Data are shown as mean ± SEM, n = 1, each pooled from six (miR148a) or three (Bim) independent experiments. (E) Bim expression was validated by immunoblotting and represented relative to values obtained with miR-scr. Data are shown as mean ± SEM, n = 1, each pooled from two (miR-scr) or three (miR-148a) independent experiments. (F) Reporter gene expression in activated Th1 cells cotransduced with Bim3′-utr reporter vector containing miR-148a bs (Bim bs) or mutated bs for the miR-148a (BimMUTbs) and an overexpression vector for miR-148a (miR-148a) or a scrambled overexpression vector (miR-scr), assessed by flow cytometry of the MFI of human CD4 on day 5 after activation and presented relative to values obtained for BimMUTbs/miR-148a. Data are shown as mean ± SEM, n = 1, each pooled from four independent experiments. For gating strategies see Supporting Information Fig. 7C. (G) Representative intracellular protein staining of Bim and Bcl2 in repeatedly activated Th1 cells after treatment with antagomir-148a or scrambled control (antagomir-scr) on day 3 postrestimulation with αCD3/αCD28, assessed by flow cytometry. Data shown are from one experiment representative of two independent experiments. For gating strategies see Supporting Information Fig. 7B. (H) Statistical evaluation of MFIs of Bim, Bcl2, and the ratio Bim/Bcl2 after treatment with antagomir-148a presented relative to values obtained with antagomir-scr. Data are shown as mean ± SEM, n = 5 pooled from two independent experiments. (A–F and H) Mann–Whitney test for unpaired data, *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001.
miR-148a has been reported to target other apoptosis regulators, for example, phosphatase and tensin homolog (Pten). The 3′-UTR of Pten contains conserved bs for miR-148a and a downregulation of Pten by miR-148a has been shown in hepatocytes [24]. In once activated Th1 cells, ectopic miR-148a overexpression downregulated the expression of a reporter construct containing the Pten 3′ -UTR depending on the presence of the miR-148a bs (p < 0.05; Fig. 3A) and Pten mRNA, the latter however, with low significance (p = 0.125; Fig. 3B). In contrast, inhibition of endogenous miR-148a expression levels in repeatedly activated Th1 cells, by antagomir-148a, did not change expression of endogenous Pten mRNA or protein (Fig. 3C and D). Another potential target is Broad-complex-Tramtrack-Bric-a-Brac and Cap'n'collar homology 1 bZip transcription factor 2 (Bach2) which is differentially expressed between once and repeatedly activated Th1 cells (Supporting Information Table 1). However, inhibition of endogenous miR-148a expression in repeatedly activated Th1 cells by antagomirs did not enhance expression of Bach2 (Fig. 3E).
Figure 3.
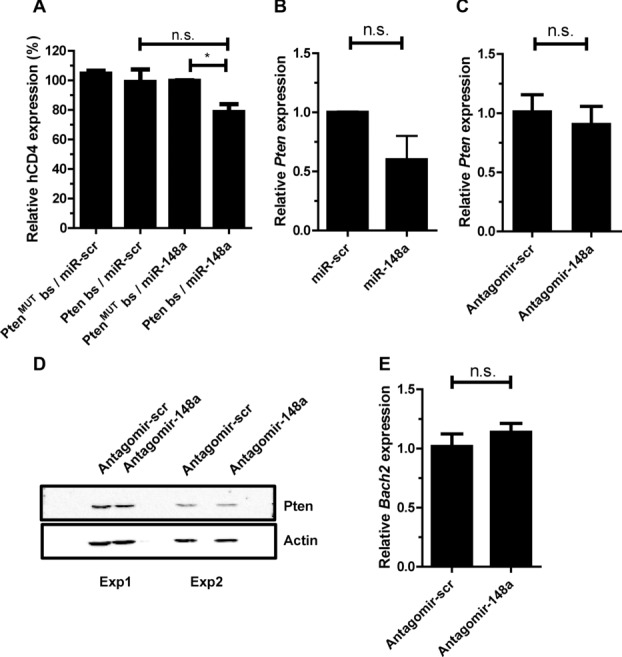
Mir-148a does no target Pten in repeatedly activated Th1 cells. (A) Reporter gene expression in activated Th1 cells cotransduced with Pten3′-uTR reporter vector containing miR-148a bs (Pten bs) or mutated bs for the miR-148a (PtenMUTbs) and an overexpression vector for miR-148a (miR-148a) or a scrambled overexpression vector (miR-scr), assessed by flow cytometry. MFI of human CD4 on day 5 after activation are presented relative to values obtained for PtenMUTbs/miR-148a. Data are shown as mean ± SEM, n = 4, pooled from two independent experiments. Mann–Whitney test for unpaired data, one-tailed, *p ≤ 0.05, n.s. p = 0.125. For gating strategies see Supporting Information Fig. 7C. (B) Overexpression of miR-148a and miR-scr in CD4+ T cells, analyzed on day 5 postactivation. Pten expression was normalized to HPRT and are presented relative to values obtained with miR-scr. Data are shown as mean ± SEM, n = 1 each pooled from three independent experiments. Mann–Whitney test for unpaired data, n.s. p = 0.125. (C) Pten expression of repeatedly activated Th1 cells after antagomir treatment with antagomir-148a or antagomir-scr on day 3 post restimulation with αCD3/αCD28, normalized to HPRT and presented relative to values obtained with antagomir-scr. Data are shown as mean ± SEM, n = 1, each pooled from three independent experiments. (D) Pten expression in repeatedly activated Th1 cells after treatment with antagomir-148a or antagomir-scr on day 3 postrestimulation with αCD3/αCD28 validated by immunoblotting data from two independent experiments are shown. (E) Bach2 expression in repeatedly activated Th1 cells after antagomir treatment, assessed by qRT-PCR, normalized to HPRT and presented relative to values obtained for antagomir-scr. Data are shown as mean ± SEM, n = 3, from one experiment representative of two independent experiments. Mann–Whitney test for unpaired data, one-tailed.
Inhibition of miR-148a increases apoptosis of repeatedly activated Th1 cells after reactivation
It needed to be investigated whether the increase of Bim after miR-148a inhibition impacted on the survival of repeatedly activated Th1 cells. Treatment with antagomir-148a significantly decreased the numbers of repeatedly activated Th1 cells to 50 and 70% compared to control-treated cells on day 3 and 4 after restimulation, respectively (Fig. 4A). This was due to an increased apoptosis rate and not due to reduced proliferation (Supporting Information Fig. 4B). On day 3, antagomir-148a treated cells showed 30% more apoptotic cells after reactivation than controls (Fig. 4B and C). In repeatedly activated Th2 and Th17 cells antagomir mediated inhibition of miR-148a did not result in reduced numbers of viable cells (Supporting Information Fig. 5A–D), probably because of unchanged apoptosis rates (Supporting Information Fig. 5–E) and Bim/Bcl2 ratio (Supporting Information Fig. 5–F). With respect to regulation of apoptosis in repeatedly activated Th1 cells, Bim is a main target of miR-148a in repeatedly activated Th1 cells, since knocking down Bim expression with a specific siRNA (siBim) in antagomir-148a treated cells restored their viability. In such cells, Bim expression was restored to levels observed in cells treated with a scrambled antagomir and a control siRNA (siScr) not targeting Bim (Fig. 4D–E). The numbers of viable repeatedly activated Th1 cells were reconstituted by 50% (Fig. 4F). Together, these results demonstrate that the survival mediated by miR-148a targeting Bim is unique in repeatedly activated Th1 cells.
Figure 4.

Inhibition of miR-148a results in increased apoptosis of repeatedly activated Th1 cells after reactivation. (A) The numbers of viable repeatedly activated Th1 cells after antagomir treatment and restimulation with αCD3/αCD28, was assessed by flow cytometry with n = 5 for day 1, 2, and 3, n = 3 for day 4. Data are shown as mean ± SEM, n = 5 (day 1–3) or n = 3 (day 4), pooled from two independent experiments. Two-way ANOVA with Bonferroni correction. For gating strategies see Supporting Information Fig. 7D. (B) Representative annexinV/PI staining of repeatedly activated Th1 cells and (C) frequencies of live cells (annexinV−PI−), apoptotic cells (annexinV+PI−), and dead cells (annexinV+PI+) after antagomir treatment assessed by annexinV/PI staining followed by flow cytometry on day 3 postrestimulation with αCD3/αCD28. Data are shown as mean + SEM, n = 6/group, pooled from four independent experiments. For gating strategies see Supporting Information Fig. 7E. (D–F) Repeatedly activated Th1 cells were treated with antagomir-148a / antagomir-scr and siRNA against Bim (siBim) or a nontargeting control (siScr) prior to restimulation with αCD3/αCD28. (D and E) qRT-PCR was used to assess (D) miR-148a and (E) Bim expression, which was normalized to snU6 or HPRT and presented relative to values obtained with antagomir-scr and siScr. (F) The number of viable Th1 cells was determined by flow cytometry. Data are shown as mean ± SEM, n = 7/group, pooled from three independent experiments. (C–F) Mann–Whitney test for unpaired data, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Expression of miR-148a in Th1 cells is induced by T-bet and Twist1
As expression of miR-148a is upregulated selectively in repeatedly activated Th1 cells (Fig. 1A) it is likely that the expression of the miRNA is regulated by the Th1 specific transcription factors T-bet and Twist1. In repeatedly activated Th1 cells we observed a twofold induction of expression of the Th1 master transcription factor Tbx21 (gene encoding for T-bet), as compared to once activated T cells (Fig. 5A). When activated under Th1-polarizing conditions, Tbx21-deficient CD4+ T cells showed a four- to six-fold reduced induction of miR-148a expression (Fig. 5B). It is unlikely that T-bet directly induces miR-148a expression, since it has been shown that T-bet does not bind upstream of the miR-148a locus [25]. Ectopic overexpression of T-bet induced miR-148a expression 1.4-fold (p = 0.091) compared to an empty retroviral control vector. In accordance, overexpression of the dominant-negative T-bet mutant Y182S [26] significantly reduced miR-148a expression (Fig. 5C). In repeatedly activated Th1 cells we observed a 12-fold induction of expression of the transcription factor Twist1 (Fig. 5D) [9]. Twist1-deficient Th cells isolated ex vivo showed a significant reduction in miR-148a expression (data not shown), while Bim levels were significantly increased by 60%, as compared to Twist1-sufficient cells (Fig. 5E). In vitro, repeatedly activated Twist1-deficient Th1 cells showed a threefold decreased miR-148a expression compared to Twist1-sufficient cells (Fig. 5F). Chromatin immunoprecipitation (ChIP) of Twist1 revealed a functional Twist1 bs 2.1 kb upstream of the miR-148a gene (Fig. 5G and Supporting Information Fig. 6). We used the IFN-γ promoter (IFN-γ -0.4) and CNS-34 (IFN-γ -34)[27], both containing E-box binding sites, as positive controls. These results imply an indirect role of T-bet and a direct involvement of Twist1 in regulating miR-148a expression in Th1 cells.
Figure 5.
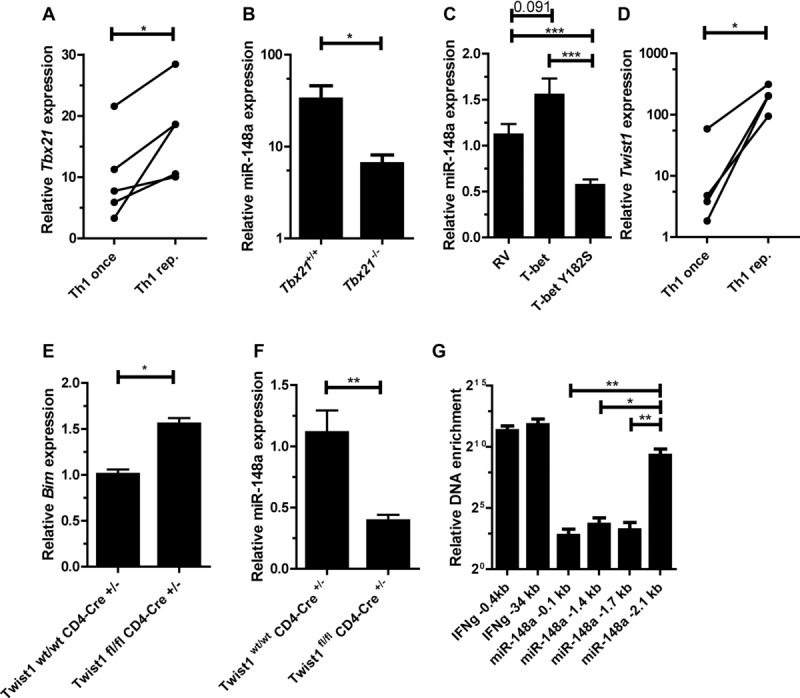
Expression of miR-148a in Th1 cells is induced by T-bet and Twist1. (A) Tbx21 expression in once and repeatedly activated Th1 cells was assessed by qRT-PCR, normalized to HPRT and presented relative to values obtained for naive Th cells. Each data point represents an individual experiment performed with n = 1. Wilcoxon-test for paired data, *p ≤ 0.05. (B) miR-148a expression in Tbx21−/− Th1 cells and wt control (Tbx21+/+) 48 h after activation, assessed by qRT-PCR, normalized to snU6 and presented relative to values obtained in naive Tbx21+/+ or Tbx21−/− Th cells. Data are shown as mean ± SEM, n = 11 (Tbx21+/+) and n = 12 (Tbx21−/−), pooled from three independent experiments. (C) Overexpression of T-bet and a T-bet mutant (T-bet Y182S) in activated Th1 cells, assessed by qRT-PCR, analyzed on day 5 postactivation. miR-148a was normalized to snU6 and presented relative to values obtained with an empty retroviral control vector (RV). Data are shown as mean ± SEM, n = 8 (RV), n = 7 (T-bet), and n = 6 (T-bet Y182S), pooled from three independent experiments. (D) Twist1 expressionin once and repeatedly activated Th1 cells, assessed by qRT-PCR, normalized to HPRT, and presented relative to values obtained for naive Th cells. Each data point represents an individual experiment, performed with n = 1 (Wilcoxon-test for paired data, *p ≤ 0.05). (E) Bim expression in ex vivo isolated Twist1fl/fl CD4-Cre+/− cells and Twist1wt/wt CD4-Cre+/− control, assessed by qRT-PCR, normalized to HPRT and presented relative to values obtained for Twist1wt/wt CD4-Cre+/−. Data are shown as mean ± SEM, n = 1, each pooled from two independent experiments. (F) miR-148a expression in repeatedly activated Twist1fl/fl CD4-Cre+/− Th1 cells and Twist1wt/wt CD4-Cre+/− control, assessed by qRT-PCR, normalized to snU6 and presented relative to values obtained for Twist1wt/wt CD4-Cre+/−. Data are shown as mean ± SEM, n = 1, each pooled from three independent experiments. (G) Twist1 binding to miR-148a locus determined by chromatin immunoprecipitation (ChIP), normalized to total DNA and presented relative to samples obtained from Twist1fl/fl CD4-Cre+/− Th1cells. Data are shown as mean ± SEM, n = 1, each pooled from four independent experiments. (B, C, and E–G) Mann–Whitney test for unpaired data, *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001.
miR-148a is expressed in T cells from rheumatoid arthritis patients
CD3+CD4+CD14−CD45RO+ T cells, isolated from inflamed tissue of patients suffering from rheumatic arthritis (RA) exhibit significantly higher levels of miR-148a, both directly ex vivo as well as after 3 h of mitogenic restimulation with PMA/Ionomycin, compared to cells isolated from peripheral blood of healthy donors (Fig. 6A). To demonstrate the selectivity of this upregulation, we also quantified miR-363 and miR-150 expression in these cells [28]. We did not detect significant differential expression between cells from peripheral blood and cells from synovial fluid, isolated ex vivo (Fig. 6B and C). Memory/effector cells isolated from the synovial fluid of arthritic patients are enriched for Th1 cells, as assessed by chemokine receptor expression, including CXCR3, CCR5, CCR4, and CCR6 [29] (data not shown). A positive correlation of miR-148a expression and Twist1 expression in these cells was observed (Supporting Information Fig. 6B), further supporting the role of Twist1 as a key inducer of miR-148a expression.
Figure 6.
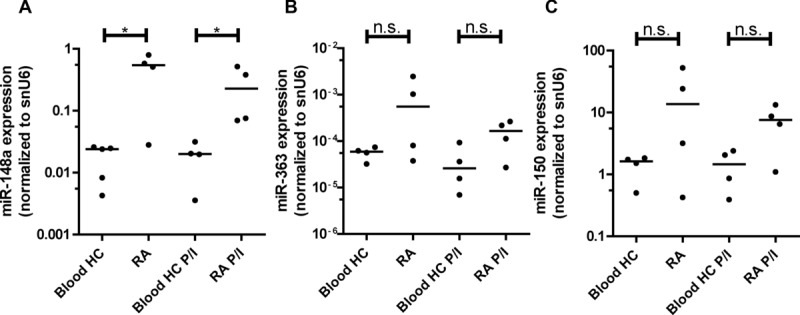
miR-148a is expressed in patients suffering from rheumatoid arthritis. (A) miR-148a, (B) miR-363, and (C) miR-150 expression, assessed by qRT-PCR, in CD3+CD4+CD14−CD45RO+ T cells isolated from synovial fluids of patients suffering from rheumatoid arthritis (RA) or blood from healthy control (HC) donors either ex vivo (n = 4 RA / n = 5 Blood HC) or after 3 h of mitogenic restimulation with PMA/Ionomycin (P/I) (n = 4 RA P/I / n = 4 Blood HC P/I), normalized to snU6. Data are representative of two independent experiments; each data point represents an individual donor, horizontal bar; median. (Mann–Whitney test for unpaired data, *p ≤ 0.05).
Discussion
We have previously shown that the transcription factor Twist1 (i) is upregulated in repeatedly activated Th1 cells, (ii) is highly upregulated in restimulated CD3+CD4+ cells isolated from the inflamed tissue of patients with chronic inflammation of the joint, and (iii) limits immunopathology in a murine model of arthritis [9]. Here we show that Twist1, together with T-bet, also controls the persistence of repeatedly activated Th1 cells, by upregulating expression of miR-148a, which in turn targets the proapoptotic gene Bim.
We have previously demonstrated that Twist1 is only expressed in Th1 cells, as its induction was directly dependent on IL-12/STAT4 signaling in combination with nuclear factor of activated T cells (NFAT) and nuclear factor κB (NF-κB) [9]. In Th1 cells, the expression of Twist1 gradually increased upon repeated rounds of restimulation, which is in line with the induction of miR-148a in repeatedly activated Th1 cells. In such cells, Twist1 binds to a functional E-box motif 2.1 kb upstream of the miR-148a gene and presumably directly regulates miR-148a expression. We also observed a contribution of T-bet on the expression of miR-148a by analyzing Tbx21-deficient Th cells as well as by ectopic overexpression of T-bet and a dominant-negative T-bet mutant (T-bet Y182S) in vitro. Since we could not identify potential T-bet binding sites in the regulatory elements of the miR-148a locus, we suggest that T-bet enhances expression of miR-148a indirectly. This is in line with the results of Nakayamada and colleagues, who have shown that T-bet does not bind within a 200 kb region upstream of the miR-148a locus [25]. T-bet is not required for the induction of Twist1 [9], but it may induce expression of an as yet unknown transcription factor required for miR-148a transcription.
Expression of miR-148a is not only induced in Th cells activated and polarized in vitro to develop into Th1 cells, but also in murine LCMV-specific Th1 cells generated in vivo, upon infection of mice with LCMV. In humans, we had described earlier, that TWIST1 expression is significantly upregulated in Th cells isolated from inflamed tissues of patients with chronic inflammatory diseases, as compared to Th cells from blood or healthy colon [9]. Here we show that Th cells isolated from synovial fluid of patients with rheumatoid arthritis have upregulated expression of miR-148a. Interestingly, upregulation of miR-148a correlates with the expression levels of TWIST1. miR-148a-mediated suppression of Bim, as discussed below, does promote the persistence and viability of the cells and may contribute to the resistance of the cells toward currently available therapies. Thus, Twist1 has a dual role as (i) an attenuator of Th1 effector function, to minimizing immunopathology, and (ii) a promotor of persistence of these Th1 cells in situations of repeated restimulation. These functions identify Twist1 as a master switch of chronicity in inflammation.
The proapoptotic protein Bim regulates the survival of memory Th cells by antagonizing the antiapoptotic protein Bcl2 [15]. The molecular mechanisms controlling the critical balance between Bcl2 and Bim in T cells are poorly understood. In reactivated Th1 cells as compared to Th17 cells, Bcl2 is downregulated [30]. This suppression of Bcl2 is probably mediated by T-bet. T-bet suppresses IL-2 expression [17], and IL-2 is required for the upregulation of Bcl2 [31]. Thus, in order to survive and maintain an antiapoptotic Bim/Bcl2 ratio, Th1 cells have to suppress expression of Bim, too. As we show here, in repeatedly activated Th1 cells, an efficient mechanism to suppress Bim expression is its posttranscriptional downregulation by miR-148a, which is induced by T-bet and Twist1. T-bet and Twist1 are key transcription factors of Th1 cells. T cells of other differentiation lineages seem to have developed different mechanisms and depend less on activation induced cell death. Repeatedly activated Th17 cells upregulate Bcl2 expression [30,32] and downregulate the Fas-L [33–35] when compared to reactivated Th1 cells. Likewise, Th2 cells maintain or upregulate the expression of Bcl2 via IL-4 signaling [36,37] and reduce Fas and FAS-L expression [38–40]. Neither of the T cell lineages upregulates miR-148a expression. This is in line with our observation, that antagomir-148a treatment does not influence the survival of repeatedly activated Th2 and Th17 cells.
We here demonstrate that Bim is the main target of miR-148a regarding the control of survival of Th1 cells. Complementing the antagomir-mediated inhibition of miR-148a with siRNA-mediated suppression of Bim largely restores viability of the Th1 cells. The physiological level of miR-148a in repeatedly activated Th1 cells is apparently phylogenetically optimized for the suppression of Bim with regard to its role in maintaining viability. SiRNA-mediated reduction of Bim expression in repeatedly activated Th1 cells with endogenous levels of miR-148a expression did not further increase the number of viable cells. Our results are supported by the notion that miR-148a supports the survival of glioblastoma cells by targeting BIM [41].
Theoretically, other miRNAs could possibly target Bim in repeatedly activated Th1 cells, for example, miRNAs of the miR-17-92 cluster [42]. However, among the 13 miRNAs selectively upregulated by repeatedly activated Th1 cells, as compared to Th2 and Th17 cells, none, except miR-148a, has candidate seed sequences in the 3′-UTR of Bim. miRNAs of the miR-17-92 cluster are not upregulated (data not shown). Therefore, we conclude that posttranscriptional regulation of Bim in repeatedly activated Th1 cells is mediated by miR-148a.
For miR-148a other potential targets affecting viability have been described for other cell types, namely, Pten and Bcl2 for hepatocytes and colorectal cancer cells, respectively [24,43]. However, both Pten and Bcl2 are not differentially expressed between once and repeatedly activated Th1 cells. Expression of Bcl2 protein is increased by 10% in repeatedly activated Th1 cells upon miR-148a inhibition by antagomirs. Since Bcl2 mRNA levels do not change detectably, the difference in protein expression, tiny as it is, could be due to either inhibition of translation of Bcl2 mRNA by miR-148a at physiological levels, or to enhanced stabilization of Bcl2 protein by the elevated levels of Bim protein [44] in antagomir-148a treated Th1 cells. With respect to Pten, a negative regulator of cell cycle progression and indirect inducer of Bim expression [45,46], we did not observe any effect on its endogenous expression levels when we blocked miR-148a in repeatedly activated Th1 cells. Although miR-148a overexpression in once activated Th1 cells regulated expression of a reporter gene with the Pten 3′-UTR, in dependency of the presence of the miR-148a target sequence, endogenous Pten levels were not significantly affected. Thus, the lack of regulation of Pten by miR-148a in repeatedly activated Th1 cells either reflects a context-dependent activity of miR-148a, or the quantitative difference of endogenous versus ectopic miR-148a expression levels. In any case, physiological expression of miR-148a in repeatedly activated Th1 cells does not regulate Pten expression.
Repeatedly activated Th1 cells express candidate target genes of miR-148a, which affect biological functions other than survival (Supporting Information Table 1). These genes influence functions such as migration (e.g. S1PR1 and RAB11B), differentiation (e.g. Bach2) and miRNA maturation (Dicer1). Not all of them may be true targets of miR-148a in the context of repeatedly activated Th1 cells. In the accompanying paper by Porstner et al., the authors identified Mitf and Bach2 as targets of miR-148a in B lymphocytes. miR-148a promotes the differentiation of activated B cells into plasma cells, by repressing Bach2 and Mitf. The transcription factor Mitf is neither expressed in once nor in repeatedly activated Th1 cells. Bach2 is a transcriptional repressor involved in regulation of effector functions in Th cells [47]. Bach2 expression is inversely correlated to miR-148a expression in Th1 cells. However, inhibition of miR-148a in repeatedly activated Th1 cells did not increase expression of Bach2, suggesting that Bach2 in these cells is not a physiological target. The present data confirm that it is essential to analyze target gene regulation by miRNAs in under physiological conditions. While inhibition of miR-148a in these cells sufficed to regulate Bim, no effect on the expression of Pten, Bach2, and Bcl2 was observed, all of which are described targets of miR-148a in other cells [24,43] (accompanying paper by Porstner et al.). Beyond its significant effect on viability, by regulation of Bim, it remains to be shown, to what extend miR-148a is regulating function and fate of Th1 cells in chronic or recurrent immune reactions.
Materials and methods
Mice
OTII, C57BL/6, BALB/c, C57BL/6SMARTA, Twistfl/fl CD4-Cre+/−, TwistWT/WT CD4-Cre+/−, and Tbx21−/− mice were housed and bred under specific pathogen-free conditions. Mice were handled in accordance with good animal practice as defined by German animal welfare bodies and sacrificed by cervical dislocation. All experiments were approved by the federal state institution “Landesamt für Gesundheit und Soziales” (G0325/12) in Berlin, Germany.
Cell culture
Naïve or CD4+ T cells were isolated from spleens of 6–10 weeks old mice and cultured under Th1, Th2, and Th17 inducing conditions as described in [48]. In brief, 3 × 106 Th cells/mL were cultured and stimulated in the presence of 0.5 mM cognate peptide OVA323–339 for OT-II cells or plate-bound αCD3/αCD28 (3 μg/mL, EBioscience) for C57BL/6, BALB/c, Twistfl/fl CD4-Cre+/−, and Tbx21−/− cells. If T cells were cultured for more than 5 days irradiated (30 Gy) CD90 depleted splenocytes from C57BL/6 or BALB/c mice were added as antigen-presenting cells to the culture. Th1 differentiation was achieved by addition of recombinant IL-12 (5 ng/mL; R&D Systems, Minneapolis, MN) and anti-IL-4 (11B11) antibody. Th2 differentiation was achieved by addition of IL-4 (100 ng/mL, culture supernatant of HEK293 T cells transfected with murine IL-4 cDNA), anti-IL-12 (C17.8), and anti-IFN-γ (AN18.17.24) antibodies. Th17 differentiation was achieved by addition of TGF-β1 (1 ng/mL), IL-6, IL-23 (20 ng/mL) (all from R&D Systems), anti-IL-4 and anti-IFN-γ. Cells were restimulated every 6 days for three times to induce a chronically activated phenotype. Recombinant IFN-γ (rIFN-γ) was added at 10 ng/mL in Tbx21 knockout experiments.
Patient material
Synovial fluids were taken from patients suffering from rheumatoid arthritis, psoriatic arthritis (PsA) or juvenile idiopathic arthritis (JA) by puncture of joints. Fluids were mixed and washed with PBS/EDTA. Cell suspension was depleted of CD15+ cells using MACS and sorted for CD3+ CD4+ CD14− CD45RO+ cells using FACS. Blood was taken from healthy donors as control. Cells were isolated by density gradient separation using LSM 1077 Lymphocyte separation medium and sorted for CD3+ CD4+ CD14− CD45RO+ cells using FACS. Half the cells were restimulated with PMA/Ionomycin for 3 h. Samples were lysed in TRIzol Reagent (Invitrogen) for RNA extraction. All human studies were approved by the Charité ethical committee and the informed consent of all participating subjects was obtained.
LCMV model
Thy1.1+ spleenocytes from C57BL/6SMARTA were naive sorted by depletion of CD11c, CD11b, CD19, Gr1, CD8, NK1.1, CD25 positive cells. A total of 5 × 106 naive CD4+ SM TCRtg T cells was transferred intravenously into C57BL/6 host mice and 2 days after transfer, mice were infected with 200 plaque-forming units of LCMV strain WE. Mice were sacrificed on day 5 and day 7, sorted for Thy1.1+, CD44high, CD62Llow cells and analyzed for miR-148a expression. T-bet expression was determined by flow cytometry in blood of animals prior to sacrifice. All animal experiments were in accordance with institutional, state and federal guidelines (Landesamt Für Gesundheit und Soziales, Berlin, Germany, accreditation number G0325/12).
Retroviral transfection
Viral supernatants were produced using 293 HEK cells transfected with pECO, pCGP, and respective transfer plasmid [9]. CD4+ T cells were isolated and activated as described above. Thirty six hours postactivation RPMI medium was removed and virus-containing medium supplemented with HEPES-buffer (20 mM) and polybrene (8 μg/mL) was added to T cells. Cells were centrifuged for 1.5 h at 32°C, 1800 rpm, supernatant was removed and Th1 medium was readded.
Plasmids
A miR-148a overexpression vector was generated by amplifying a primary form of miR-148a from murine spleen cDNA using the following primers: pri148a forward 5′-GTTAACTGTGACATTGCCACCAGA-3′ and pri148a reverse 5′-CTCGAGAAAAAAACGACGTGGCCAACA-3′; and cloned under the control of U6 promoter into pQCXIX (BD Biosciences Clontech) using HpaI and XhoI restriction enzymes. pQCXIX has a GFP marker for positive selection. As a control, a scrambled miRNA overexpression vector was constructed as described before [22]. Transfection of T cells was performed as described above. Three days posttransfection cells were harvested and sorted for GFP expression. miRNA expression levels were assessed by quantitative PCR. For cotransfection with reporter vectors, cells were analyzed by flow cytometry. For generation of Bim and Pten reporter vectors, parts of the respective 3′-UTRs were amplified from cDNA using the following primers: BIM forward 5′-CGCGGATCCCTCAAGTTCCCAGCAAAGTA-3′, BIM reverse 5′-CCCAAGCTTCACAGGTACAGTGGCAATTA-3′, PTEN forward 5′-CGCGGATCCGCTGAAAGTGGCTGACTAAA-3′, and PTEN reverse 5′-CCCAAGCTTCACCCACACAATGACAAGA-3′; and cloned downstream of the human CD4 gene within the pMSCV vector (BD Biosciences Clontech). Mutation of miR-148a binding sites (bs) in 3′-UTRs of Bim and Pten reporter vectors were induced using the site specific mutagenesis kit (Stratagene) and the following primer:
BIM1 mut forward 52-GGTATCCTTTAGTGAACAGCGGTCGTCTCTGTATAGTCCCCATCAC-32, BIM1 mut reverse 52-GTGATGGGGACTATACAGAGACGACCGCTGTTCACTAAAGGATACC-3′, BIM2 mut forward 52-CTGGCTTCCTTTACGTTTTGCGGCCATGAATTTTGACAGGGTAATTGC-32, BIM2 mut reverse 52-GCAATTACCCTGTCAAAATTCATGGCCGCAAAACGTAAAGGAAGCCAG-322, PTEN2 mut forward 52-GCAGTGGCTCTGTGTGTAAATGCTAGCCACGCAGGATACACACAAATATG-32, PTEN2 mut forward 52-CATATTTGTGTATCCTGCGTGGCTAGCATTTACACACAGAGCCACTGC-32. Transfection of T cells was performed as described above. T cells were stained with αhCD4-Cy5 (Clone TT1, purified in-house) and expression of human CD4 reporter gene was assessed by flow cytometry as described previously [22].
Inhibition of miR-148a
Inhibition of miR-148a was achieved using specific cholesterol-coupled antagomir oligonucleotides (custom synthesized by Dharmacon) [23]. Repeatedly activated Th1 cells were resuspended in serum-free medium (ACCELL, Dharmacon) supplemented with antagomir-148a or scrambled control (1 μM). After 1.5 h incubation at 37°C and 5% CO2 T cells were reactivated and cultured in Th1 polarizing medium (added in fourfold excess to T cells in ACCELL medium). Knock down efficiency was assessed by quantitative PCR.
Inhibition of Bim and Twist1
Specific ACCELL siRNAs targeting Bim (Dharmacon) were used to decrease functional Bim mRNA expression. Repeatedly activated Th1 cells were treated with 1 μM antagomir and 1 μM siRNA in serum-free ACCELL medium. After 1.5 h incubation at 37°C and 5% CO2 T cells were reactivated and cultured in Th1 polarizing medium (added 1:1 to a final concentration of 0.5 μM antagomir and siRNA). Knockdown efficiency was assessed by qRT-PCR and phenotypical effects were detected by flow cytometry. Twist1 knockdown was achieved by retroviral expression of shRNA as described before [9].
Apoptosis assay
T cells were stained with αmCD4-Cy5 (GK1.5; purified in-house). Apoptosis was examined using an AnnexinV-FITC/Propidiumiodide Assay (Roche) and analyzed by flow cytometry.
Total numbers of viable T cells were determined by flow cytometry (MACSQuant Analyzer, Miltenyi Biotec).
Chromatin immunoprecipitation
ChIP was performed as described previously [48] with a polyclonal Twist1 antibody (4 μg/mL; sc-6070; Santa Cruz Biotechnology). Immunoprecipitated DNA was measured by quantitative PCR. Putative binding sites in conserved regions of the miR-148a locus (chromosome 6; 51269812-51269910 (–)) were identified with the web server of the comparative tool rVista based on the professional V10.2 library of the TRANSFAC database. The following primers were used: miR-148 -0.1kb forward 5′- CCTCTGGAAGTTTCGTCCTGC-3′, miR-148 -0.1kb reverse 5′-TATTCTTCTTTGCCTTCACTGGG-3′, miR-148 -1.4kb forward 5′-ATTTGGGTTTGGAGACGACC-3′, miR-148 -1.4kb reverse 5′-AATAGCAAGAGCAGCCGTGAC-3′, miR-148 -1.7kb forward 5′-AGCAGAGTGAGAAATGGAAACCTT-3′, miR-148 -1.7kb reverse 5′-TCTCAGTTCTTGTAACACTCAGCCC-3′, miR-148 -2.1kb forward 5′-AACTCAAGGTGCTCAGAATTGTCC-3′, miR-148 -2.1kb reverse 5′-CCTTTCTTCTACAAAGCACGCCT-3′. Regulatory regions of the IFN-γ promoter described by [27] served as positive controls.
RNA extraction and quantitative PCR
Total RNA was isolated with RNeasy kit (Qiagen) or Direct-zol RNA kit (Zymo Research). Mature miR-148a and U6 small nuclear RNA (snRNA) were detected by quantitative PCR with Taqman MicroRNA Reverse Transcription kit in combination with TaqMan MicroRNA Assays (Applied Biosystems) according to manufacturer's recommendations. For normalization the expression values were compared to values of snU6 by the change-in-threshold method (2−ΔCT).
Reverse transcription of mRNA was performed using the Reverse Transcription kit (Applied Biosystems) and cDNA was quantified by SYBR Green based real-time PCR (Roche) using the following primer pairs: hypoxanthine guanine phosphoribosyltransferase (HPRT) forward 5′-TCCTCCTCAGACCGCTTTT-3′, HPRT reverse 5′-CATAACCTGGTTCATCATCGC-3′, BIM forward 5′-CCCGGAGATACGGATTGCA-3′, BIM reverse 5′-AACACCCTCCTTGTGTAAGTTTCGT-3′, BCL2 forward 5′-TGAACCGGCATCTGCACA -3′, BCL2 reverse 5′-CAGAGGTCGCATGCTGGG-3′, PTEN forward 5′-GCGGAACTTGCAATCCTCAGT-3′, PTEN reverse 5′- AGGCAATGGCTGAGGGAACT-3′, TBET forward 5′-TCCTGCAGTCTCTCCACAAGT-3′, TBET reverse 5′-CAGCTGAGTGATCTCTGCGT-3′, TWIST1 forward 5′-CGCACGCAGTCGCTGAACG-3′, TWIST1 reverse 5′-GACGCGGACATGGACCAGG -3′. For normalization, the expression values were compared to values of HPRT by the change-in-threshold method (2−ΔCT).
Immunoblot analysis
Immunoblot analysis was performed as described previously [49]. Membranes were probed with primary antibodies to total Pten (D4.3, Cell Signaling), Bim (3C5, Enzo Life Sciences) and Bcl2 (50E3, Cell Signaling), and polyclonal antibody to actin (sc-1616; Santa Cruz) for normalization. Immunoreactive bands were detected by chemiluminescence using ECL reagent (GE Healthcare) and quantified by densitometry (Fuji LAS-4000 software).
Intracellular protein/cytokine staining
Intracellular cytokine staining was performed after every round of (re)stimulation. Cells were stimulated with PMA/Ionomycin for 3 h and fixed in 2% paraformaldehyde. Cytokine specific antibodies were administered in 0.5% Saponin in PBS, incubated for 20 min at 4°C and analyzed by flow cytometry. Intracellular protein staining was performed using primary antibodies to Bim (14A8, Merck Chemicals) and Bcl2 (3F11, BD Biosciences). Cells were fixed and stained using the Foxp3 staining buffer set (eBioscience) according to manufacturer's recommendation with extended fixation overnight. Samples were stained for 30 min at 4°C and analyzed by flow cytometry.
Isolation of IFN-γ-secreting cells
Separation of IFN-γ-producing and nonproducing Th1 cells was performed in principle as described previously [50–52]. Once activated Th1 cells were restimulated with PMA/Ionomycin for 3 h. IFN-γ-catch and -detection reagents (aIFN-γ-allophycocyanin) were obtained from Miltenyi Biotec and used according to the manufacturer's guidelines.
CFSE dilution assay
To test the proliferative capacity of antagomir-148a or antagomir-scr treated Th1 rep. cells, we performed a CFSE dilution assay according to a published protocol [53].
Statistics
All statistical analyses used the Mann–Whitney test for unpaired data, unless stated otherwise. A p-value of equal or less than 0.05 was considered significant.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (GRK1121; BSRT (DFG-GSC203), SFB 618, SFB 650, SFB 633, and SFB TR52), the International Max Planck Research School for Infectious Diseases and Immunology, the IMI JU funded project BTCure, no 115241, the FORSYS (Forschungseinheiten zur Systembiologie)and the “e:Bio – Innovationswettbewerb Systembiologie” program of the Federal Ministry of Education. J.W. and H.-M.J. were supported in part by the Interdisciplinary Center for Clinical Research (IZKF) Erlangen (project D7) and research grants GRK1660, FOR832 (JA968/4) and TRR130 from the Deutsche Forschungsgemeinschaft (DFG). We would like to thank Jenny Kirsch and Toralf Kaiser for technical support. This work has been supported by the ERC Advanced Grant (ERC-2010-AdG_20100317 Grant 268987) to A.R.
Glossary
- Bcl2
B-cell lymphoma 2
- Bim
Bcl2-interacting mediator of cell death
- HPRT
hypoxanthine guanine phosphoribosyltransferase
- LCMV
lymphocytic choriomeningitis virus
- miR/miRNA
microRNA
- RAB11B
Ras-related protein Rab-11B
- S1Pr1
Sphingosine-1-phosphate receptor 1
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- 1.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA. Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 2.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janke M, Peine M, Nass A, Morawietz L, Hamann A, Scheffold A. In-vitro-induced Th17 cells fail to induce inflammation in vivo and show an impaired migration into inflamed sites. Eur. J. Immunol. 2010;40:1089–1098. doi: 10.1002/eji.200939487. [DOI] [PubMed] [Google Scholar]
- 5.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 7.Morita Y, Yamamura M, Kawashima M, Harada S, Tsuji K, Shibuya K, Maruyama K, et al. Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:1669–1676. doi: 10.1002/1529-0131(199809)41:9<1669::AID-ART19>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 8.Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008;67:1299–1304. doi: 10.1136/ard.2007.080341. [DOI] [PubMed] [Google Scholar]
- 9.Niesner U, Albrecht I, Janke M, Doebis C, Loddenkemper C, Lexberg MH, Eulenburg K, et al. Autoregulation of Th1-mediated inflammation by twist1. J. Exp. Med. 2008;205:1889–1901. doi: 10.1084/jem.20072468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snow AL, Pandiyan P, Zheng L, Krummey SM, Lenardo MJ. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol. Rev. 2010;236:68–82. doi: 10.1111/j.1600-065X.2010.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurtulus S, Tripathi P, Opferman JT, Hildeman DA. Contracting the ‘mus cells’–does down-sizing suit us for diving into the memory pool? Immunol. Rev. 2010;236:54–67. doi: 10.1111/j.1600-065X.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shenoy AR, Kirschnek S, Hacker G. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur. J. Immunol. 2014;44:2500–2507. doi: 10.1002/eji.201344238. [DOI] [PubMed] [Google Scholar]
- 13.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc. Natl. Acad. Sci. U S A. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr. Opin. Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, et al. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J. Exp. Med. 2007;204:1665–1675. doi: 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai YG, Hou MS, Lo A, Huang ST, Huang YW, Yang-Yen HF, Liao NS. IL-15 modulates the balance between Bcl-2 and Bim via a Jak3/1-PI3K-Akt-ERK pathway to promote CD8alphaalpha+ intestinal intraepithelial lymphocyte survival. Eur. J. Immunol. 2013;43:2305–2316. doi: 10.1002/eji.201243026. [DOI] [PubMed] [Google Scholar]
- 17.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 18.Rebhahn JA, Deng N, Sharma G, Livingstone AM, Huang S, Mosmann TR. An animated landscape representation of CD4(+) T-cell differentiation, variability, and plasticity: Insights into the behavior of populations versus cells. Eur. J. Immunol. 2014;44:2216–2229. doi: 10.1002/eji.201444645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 21.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 22.Stittrich AB, Haftmann C, Sgouroudis E, Kuhl AA, Hegazy AN, Panse I, Riedel R, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat. Immunol. 2010;11:1057–1062. doi: 10.1038/ni.1945. [DOI] [PubMed] [Google Scholar]
- 23.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 24.Yuan K, Lian Z, Sun B, Clayton MM, Ng IO, Feitelson MA. Role of miR-148a in hepatitis B associated hepatocellular carcinoma. PLoS One. 2012;7:e35331. doi: 10.1371/journal.pone.0035331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, Sun HW, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. 2011;35:919–931. doi: 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008;22:2980–2993. doi: 10.1101/gad.1689708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang S, Aune TM. Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat. Immunol. 2007;8:723–731. doi: 10.1038/ni1473. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Wan Y, Guo Q, Zou L, Zhang J, Fang Y, Zhang J, et al. Altered microRNA expression profile with miR-146a upregulation in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res. Ther. 2010;12:R81. doi: 10.1186/ar3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, Frosali F, et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:2504–2515. doi: 10.1002/art.30332. [DOI] [PubMed] [Google Scholar]
- 30.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. 2010;130:418–426. doi: 10.1111/j.1365-2567.2009.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. Eur. J.Immunol. 2011;41:1862–1871. doi: 10.1002/eji.201041195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi G, Ramaswamy M, Vistica BP, Cox CA, Tan C, Wawrousek EF, Siegel RM, et al. Unlike Th1, Th17 cells mediate sustained autoimmune inflammation and are highly resistant to restimulation-induced cell death. J. Immunol. 2009;183:7547–7556. doi: 10.4049/jimmunol.0900519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat. Rev. Rheumatol. 2009;5:325–331. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 35.Tan C, Ramaswamy M, Shi G, Vistica BP, Siegel RM, Gery I. Inflammation-inducing Th1 and Th17 cells differ in their expression patterns of apoptosis-related molecules. Cell. Immunol. 2011;271:210–213. doi: 10.1016/j.cellimm.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunkle A, Dzhagalov I, He YW. Cytokine-dependent and cytokine-independent roles for Mcl-1: genetic evidence for multiple mechanisms by which Mcl-1 promotes survival in primary T lymphocytes. Cell. Death Dis. 2011;2:e214. doi: 10.1038/cddis.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rautajoki KJ, Marttila EM, Nyman TA, Lahesmaa R. Interleukin-4 inhibits caspase-3 by regulating several proteins in the Fas pathway during initial stages of human T helper 2 cell differentiation. Mol. Cell. Proteomics. 2007;6:238–251. doi: 10.1074/mcp.M600290-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Pandiyan P, Gartner D, Soezeri O, Radbruch A, Schulze-Osthoff K, Brunner-Weinzierl MC. CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J. Exp. Med. 2004;199:831–842. doi: 10.1084/jem.20031058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Brunner T, Carter L, Dutton RW, Rogers P, Bradley L, Sato T, et al. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 1997;185:1837–1849. doi: 10.1084/jem.185.10.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varadhachary AS, Perdow SN, Hu C, Ramanarayanan M, Salgame P. Differential ability of T cell subsets to undergo activation-induced cell death. Proc. Natl. Acad. Sci. U S A. 1997;94:5778–5783. doi: 10.1073/pnas.94.11.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 2001;7:913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 42.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng H, Lai M. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell. Death Differ. 2011;18:1702–1710. doi: 10.1038/cdd.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jorgensen TN, McKee A, Wang M, Kushnir E, White J, Refaeli Y, Kappler JW, et al. Bim and Bcl-2 mutually affect the expression of the other in T cells. J. Immunol. 2007;179:3417–3424. doi: 10.4049/jimmunol.179.6.3417. [DOI] [PubMed] [Google Scholar]
- 45.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haftmann C, Stittrich AB, Sgouroudis E, Matz M, Chang HD, Radbruch A, Mashreghi MF. Lymphocyte signaling: regulation of FoxO transcription factors by microRNAs. Ann. N Y Acad. Sci. 2012;1247:46–55. doi: 10.1111/j.1749-6632.2011.06264.x. [DOI] [PubMed] [Google Scholar]
- 47.Tsukumo SI, Unno M, Muto A, Takeuchi A, Kometani K, Kurosaki T, Igarashi K, et al. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. U S A. 2013;110:10735–10740. doi: 10.1073/pnas.1306691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang HD, Helbig C, Tykocinski L, Kreher S, Koeck J, Niesner U, Radbruch A. Expression of IL-10 in Th memory lymphocytes is conditional on IL-12 or IL-4, unless the IL-10 gene is imprinted by GATA-3. Eur. J. Immunol. 2007;37:807–817. doi: 10.1002/eji.200636385. [DOI] [PubMed] [Google Scholar]
- 49.Mashreghi MF, Klemz R, Knosalla IS, Gerstmayer B, Janssen U, Buelow R, Jozkowicz A, et al. Inhibition of dendritic cell maturation and function is independent of heme oxygenase 1 but requires the activation of STAT3. J. Immunol. 2008;180:7919–7930. doi: 10.4049/jimmunol.180.12.7919. [DOI] [PubMed] [Google Scholar]
- 50.Manz R, Assenmacher M, Pfluger E, Miltenyi S, Radbruch A. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc. Natl. Acad. Sci. U S A. 1995;92:1921–1925. doi: 10.1073/pnas.92.6.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brett SJ, Baxter G, Cooper H, Rowan W, Regan T, Tite J, Rapson N. Emergence of CD52-, glycosylphosphatidylinositol-anchor-deficient lymphocytes in rheumatoid arthritis patients following Campath-1H treatment. Int. Immunol. 1996;8:325–334. doi: 10.1093/intimm/8.3.325. [DOI] [PubMed] [Google Scholar]
- 52.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007;2:2049–2056. doi: 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.