

Abstract
Background
In recent years, much evidence suggested that vitamin D plays an important role in decreasing the risk of type 2 diabetes. The purpose of this study was to investigate whether 1, 25 (OH) 2D3 can modulate inflammation and lipid metabolism in type 2 diabetic rat liver.
Methods
Type 2 diabetes was induced in SD rat with high-fat and high-sugar diets and multiple low-dose streptozotocin. The levels of serum calcium, phosphorus, glucose, TC, TG, AST, ALT and hepatic TG were determined. H & E staining were performed to assess the effects of vitamin D treatment on pathological changes in the liver tissues. Immunohistology, real-time PCR and Western blot were used to evaluate the expressions of NF-κ B, MCP-1, ICAM-1, TGF-β1, PPAR-α and CPT-1.
Results
The administration of 1, 25 (OH) 2D3 reduced liver weight. Compared to DM rats, 1, 25 (OH) 2D3-treated DM rats had lower liver weight. Moreover, compared to healthy or 1, 25 (OH) 2D3-treated DM rats, DM rats had increased hepatic transcription factors (NF-κ B), monocyte chemoattractant protein −1 (MCP-1), intercellular adhesion molecule −1 (ICAM-1), transforming growth factor-β1 (TGF-β1) expressions, but had fewer hepatic PPAR- α and CPT-1 expressions.
Conclusions
1, 25 (OH) 2D3 significantly modulated the liver inflammation and lipid metabolism in diabetic rat models, which may be caused by its regulations on hepatic signaling NF-κ B pathway and PPAR- α.
Keywords: Diabetes-induced liver injury, Vitamin D, NF-κ B, PPAR-α
Introduction
Type 2 diabetes mellitus (T2DM) is one of the main noncommunicable chronic diseases with a growing prevalence; it affected 382 million people worldwide in the year of 2013 and is expected to affect 592 million by 2035. Chronic complications of diabetes, which involve coronary artery disease, renal, liver and ophthalmologic diseases, is the primary cause of disability and mortality in diabetes mellitus (DM) patients. It is reported that the standardized mortality rate from end-stage liver disease (i.e. cirrhosis) is higher than that for cardiovascular disease among patients with diabetes [1,2]. Liver disease is one of diabetic complications, and should be well addressed [3].
Vitamin D is a fat-soluble vitamin, which is an essential micronutrient with major implications for human health [4]. The biologically active form of vitamin D is 1, 25 (OH) 2D3 (also known as active vitamin D3 [5]. Vitamin D receptors are widely distributed in more than 38 tissues [6]. Macrophages and dendrite cells constitutively express Vitamin D receptors, which indicates vitamin D plays an important role in regulating the inflammatory response [7,8]. Several studies have confirmed the involvement of vitamin D in modulating the inflammatory response [9,10]. In recent years, much evidence suggested that vitamin D plays an important role in decreasing the risk of many chronic diseases, including type 2 diabetes [11], the metabolic syndrome [12] and cardiovascular disease [13].
Diabetes-induced liver injury includes inflammatory response, lipid accumulation and liver fibrosis [14]. T2DM produces a state of chronic hyperglycemia and insulin resistance, which leads to the increasing of intracellular reactive oxygen species (ROS) levels. The accumulated ROS can activate the NF-κ B pathway, leading to the occurrence of liver inflammation [15]. Previous study demonstrated that vitamin D reduces the nuclear translocation of NF-κ B by up-regulating the inhibitor of NF-κ B (I κ B-α) in LPS-stimulated murine macrophages [16]. However, the regulatory effect of 1, 25 (OH) 2D3 on NF-κ B and its downstream inflammatory cytokines expressions in DM hepatocyte remains unclear. In addition, the accumulation of triglycerides within hepatic cell is the mainly characteristic of fatty liver with type 2 diabetes [17]. Peroxisome proliferator-activated receptor α (PPAR-α), which is mainly expressed in the liver, plays a pivotal role in the regulation hepatic lipid metabolism [18]. Motiwala [19] and Zittermann [20] have shown that vitamin D modulates lipid metabolism by decreasing the level of serum triglyceride (TG) in overweight subjects. A recent study have found that 1, 25 (OH) 2D3 modulates cardiac lipid metabolism by affecting the expression of PPAR-α in a DM rat model [21]. It has not yet been studied whether vitamin D can modulate hepatic lipid metabolism by regulating PPAR-α in type 2 diabetic rat liver.
Increasing evidence suggests that the circulating concentration of 25-OH vitamin D was negatively associated with the risk of liver disease [22,23]. However, given that the regulatory mechanisms of 1, 25 (OH) 2D3 on liver have not been completely elucidated, the purpose of this study was to investigate whether 1, 25 (OH) 2D3 can modulate inflammation and lipid metabolism in type 2 diabetic rat liver.
Materials and methods
Animal modeling and grouping
The experimental designs and protocols for animal studies were reviewed and approved by Xinjiang management committee for medical laboratory animal sciences. A total of 50 male SD rats were provided by the Xinjiang Disease Control and Prevention Center. Rats were housed in standard cages and maintained on rat chow and tap water ad libitum. All rats were housed for 1 week prior to diet intervention. We randomly selected 15 rats as normal control group (NC group), which were fed with normal diet. The remaining rats were used to establish DM models. They were fed with high-fat and high-sugar diets, containing 10% refiing lard, 20% sucrose, 2% cholesterol, 8% custard powder, and 60% of normal diet, for indicated duration (eight weeks). Then these rats were subjected to the intraperitoneal injection of 35 mg/kg streptozotocin (STZ; Sigma, St. Louis, MO, USA). One week later, the fasting plasma glucose (FPG) and 2-h plasma glucose (2hPG) were measured with a complete blood glucose monitor (ACCU-CHEK, Germany), rats with FPG ≥ 7.0 mmol/L and/or 2hPG ≥ 11.1 mmol/L were considered as DM models.
These DM model rats were randomly divided into diabetic group (DM group) and vitamin D treatment group (VD group, n = 15 per group). Rats of vitamin D treatment group were administrated vitamin D (0.03 μg/kg body weight; Shanghai Roche Pharmaceuticals, Shanghai, China) in 0.05 mL peanut oil once daily via gavage, and equivalent peanut oil was administrated in diabetic group and normal control group for 8 weeks. At the end of the vitamin D intervention period, blood samples were drawn from the abdominal aorta and the serum samples were separated by centrifuge. The entire liver was dissected out, weighed, the tissues were cut into small pieces, immersed in RNA later solution at room temperature, and then stored at −80°C. Remaining liver tissues were fixed in 4% paraformaldehyde.
Biochemical analysis
Serum calcium (Ca), phosphorus (P), glucose total cholesterol (TC), TG, Alanine aminotransferase (ALT), aspartate aminotransferase (AST), were determined with an automatic biochemistry analyzer (Hitachi 7600, Tokyo, Japan). Hepatic TG contents were determined enzymatically by using commercial kits (Bio Sino Bio-technology and Science Inc, Beijing, China).
Histopathological staining
The fresh liver tissue was washed with saline, and then fixed in 10% buffered formalin. After dehydrated, these tissues were embedded with paraffin, and then cut into 3 μm sections on a microtome (Leica, Nussloch, Germany) and then stained with H & E.
Immunohistochemistry
Paraffin sections were de-waxed and re-hydrated through a graded alcohol series. The endogenous peroxidase was removed, and the sections were exposed to antigen retrieval. Then, slides were incubated, respectively, with Rabbit anti-rat MCP-1 (Boster Biological Technology, Wuhan, Hubei, China), rabbit anti-rat ICAM-1 (Boster Biological Technology, Wuhan, Hubei, China), rabbit anti-rat TGF-β1 (Boster Biological Technology, Wuhan, Hubei, China), and rabbit anti-rat CPT-1a (Bioss Biotechnology, Beijing, China) (final concentrations 1:100, 1:100, 1:200, 1:100, respectively) for over-night at 4°C. Then the sections were then incubated with secondary antibodies. After stained with DAB chromogenic reagent (ZSGB-BIO, Beijing, China) and counterstained with hematoxylin, these sections were sealed and then visualized under microscopy with the CM-2000B biomedicine image analysis system (Beihang, Beijing, China). Brown staining was considered as positive. Five fields were randomly selected under high magnification (×400), and the averaged number of positive cells were counted and calculated.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from liver tissue (100 mg) with RNeasy Mini kits (Qiagen, Hilden, Germany). QuantiTect Rev Transcription Kits (Qiagen) were used to reverse-transcribed to cDNA, according to manufactures’ instructions. The real-time quantitative PCR assays were performed with QuantiFast SYBR green PCR Master Mix containing ROX as a passive reference (Qiagen), on Bio-Rad iQ5 system (Bio-Rad, Hercules, CA, USA). Primer sequences used were listed in Table 1. Quantitative PCR amplification conditions were as follows: melt at 95°C for 5 min; and at 95°C for 50 s, 60°C for 30 s, for 40 cycles. The housekeeping gene b-actin was used as a reference gene for normalization. Primers used for rat genes were as following: NF-κ B p65: F: 5′-TCA CGG GAC CTG GCT GGG AG-3′,R: 5′-CCG CCG AAG CTG CAT GGA CA-3′;MCP-1:F: 5′-CAG CCA GAT GCA GTT AAT GCC-3′,R: 5′-AGC CGA CTC ATT GGG ATC AT-3′;ICAM-1:F: 5′-CGT GGC GTC CAT TTA CAC CT-3′,R: 5′-TTA GGG CCT CCT CCT GAG C-3′; TGF-β1:F: 5′-AGA AGT CAC CCG CGT GCT AAT-3′,R: 5′-CAC TGC TTC CCG AAT GTC TGA-3′; PPAR-α: F: 5′-CCTGCCTTCCCTGTGAACT-3′,R:5′-ATCTGCTTCAAGTGGGGAGA-3′;CPT-1:F:5′-GCCAGACGAAGAAC ATTG-3′,R: 5′-CCTTGACCATAGCCA TCC-3′; β- actin: F: 5′-AGT ACC CCA TTG AAC ACG GC-3′,R: 5′-TTT TCA CGG TTG GCC TTA GG-3′. For each gene, relative change in steady state Mrna in samples was determined using the ΔΔCt method, corrected for the housekeeper.
Table 1.
Effects of 1,25(OH)2D3 on body and liver weight
| Group | Weight(g) | Liver weight (g) | Liver⁄Body |
|---|---|---|---|
| NC | 461.71 ± 35 | 13.20 ± 1.2 | 2.86 ± 0.22 |
| DM | 346.14 ± 14* | 24.37 ± 1.5* | 7.05 ± 0.44* |
| VD | 362.87 ± 16* | 21.57 ± 1.6*# | 5.96 ± 0.53*# |
NC, normal control group; DM, diabetic group; VD, vitamin D treatment group.
Values are mean ± SD, n = 15 per group.
*P < 0 .05 vs. NC; #P < 0 .05 vs. DM.
Western blotting
Liver tissue was homogenized in RIPA Lysis Buffer containing PMSF (a protease inhibitor), and Proteins were collected by centrifuging at 12,000 turn at 4°C in centrifuge for 10 min. The protein concentration was measured by Bradford assay. About 50 micrograms of protein were loaded on 5–10% gradient gels. Proteins were transferred to a PVDF membrane (Thermo Scientific) for 120 min at 80 V. The membranes were incubated in blocking buffer for 2 hours. Then they were incubated with different primary antibodies overnight at 4°C, washed with TBST and incubated with secondary antibody for 2 h at room temperature. Antigen antibody complexes were then visualized using Western Breeze (invitrogen, USA).The primary antibodies used here include NF- k B p65(1:1000, Cell Signaling Technology) and PPAR-α(1:1000, Cell Signaling Technology). The intensity of protein bands were quantified using the Quantity One software (Bio-Rad, USA).
Statistical analysis
All quantitative data are expressed as the mean ± the standard error of the mean (SEM). SPSS 17.0 software was used for statistical analysis. Comparisons between two groups were analyzed via t-test, and comparisons between more than two groups were analyzed via one-way ANOVA to identify differences among means. Pearson correlations were adopted to note the correlation. A value of P < 0.05 was considered statistically significant.
Result
Effect of 1,25(OH)2D3 and diabetes on body and liver weight
About the final weight, rats of DM and VD group showed a lower body weight compared to rats of NC group (P < 0.05), whereas the body weight was similar between the DM and VD group (P > 0.05, Table 1).
The liver weight was measured and showed a significant increase in DM rats than those in NC rats (P < 0.05) , the ratio of liver to body weight was still significantly higher. The administration of 1,25 (OH) 2D3 led to a significant reduction in both liver weight and the ratio of liver to body weigh 1,25 (OH) 2D3-treated DM rats compared to DM rats (P < 0.05, Table 1).
Effect of 1,25 (OH) 2D3 on serum metabolic parameters and hepatic TG
There was no significant difference in serum calcium and phosphorus levels of all groups (P > 0.05). Serum glucose level in DM and VD groups was significantly higher than that in NC group (P < 0.05). Rats in the DM group exhibited impaired metabolic function as shown by increased serum triglyceride, TC. In the VD group, 1, 25 (OH) 2D3 reduced the level of serum TG, but not serum TC. Our data showed that serum ALT and AST concentrations were extremely elevated in rats of DM group. In the VD group, 1, 25 (OH) 2D3 treatment significantly lowered serum ALT and AST concentrations compared with the DM group. The level of hepatic TG in the DM group was significantly higher than that in NC group (P < 0.05), In the VD group, 1, 25 (OH) 2D3 reduced the level of hepatic TG (Table 2).
Table 2.
Effects of 1,25(OH)2D3 on serum metabolic parameters and hepatic TG
| Group | Ca | P | Glucose | TC | TG | ALT | AST | Hepatic TG |
|---|---|---|---|---|---|---|---|---|
| (mmol/L) | (mmol/L) | (mmol/L) | (mmol/L) | (mmol/L) | (U/L) | (U/L) | (mg/g) | |
| NC | 2.57 ± 0.10 | 1.52 ± 0.11 | 5.61 ± 0.62 | 1.11 ± 0.09 | 0.86 ± 0.11 | 43.09 ± 7.6 | 69.86 ± 9.9 | 8.35 ± 1.34 |
| DM | 2.61 ± 0.13 | 1.55 ± 0.08 | 26.52 ± 3.62* | 1.25 ± 0.14* | 1.30 ± 0.09* | 121.87 ± 14* | 150.16 ± 12* | 67.74 ± 8.89* |
| VD | 2.63 ± 0.17 | 1.53 ± 0.08 | 26.09 ± 1.92* | 1.29 ± 0.11* | 1.11 ± 0.13*# | 95.99 ± 16. *# | 105.18 ± 15*# | 31.72 ± 7.49*# |
Ca, serum calcium; P, serum phosphorus; ALT, alanine aminotransferase; AST, aspartate aminotransferase; NC, normal control group; DM, diabetic group; VD, vitamin D treatment group.
Values are mean ± SD, n = 15 per group.
*P < 0 .05 vs. NC; # P < 0 .05 vs. DM.
1, 25 (OH) 2D3 attenuated diabetes-induced hepatic damage
Liver pathology with H & E staining is presented in Figure 1. The hepatic cell structure in normal control group was normal and clear without inflammation and necrosis. In the liver of DM group, diabetes increased hepatic damage, and the morphological change was obviously necrotic and/or inflammatory foci. However, in the liver of 1, 25 (OH) 2D3-treated DM group, the morphological change was less severe with less inflammatory and/or necrotic foci as compared to the liver of Diabetes group.
Figure 1.
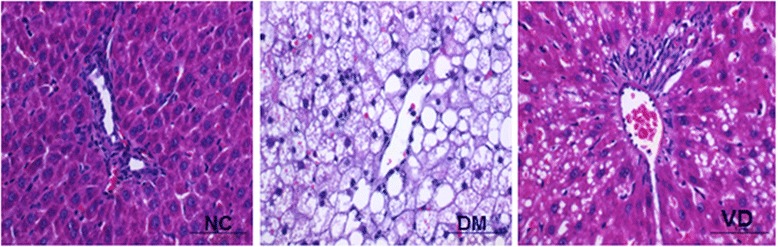
Effects of 1,25 (OH) 2D3 on liver histology. Histological analysis of steatosis in liver sections stained with H & E (magnification 400 ×). NC, normal control group; DM, diabetic group; VD, vitamin D treatment group.
Increased NF-κ B and its downstream inflammatory cytokines expression in liver tissues from the DM group and suppressed by 1, 25 (OH) 2D3
To test whether hepatic NF-κ B signaling pathway involves in the mechanism of diabetes-induced hepatic damage, we examined their expression by different methods. IHC staining for MCP-1, ICAM-1, and TGF-β1 in livers from the DM group were both strongly positive, whereas they were almost negative in the normal control group , the amount of brown staining granules and the covered area were significantly decreased in the 1, 25 (OH) 2D3-treated DM group, compared with the DM group (Figure 2). Western blot analysis showed that hepatic NF-κ B protein levels in the DM group were significantly increased compared with those of the normal control group; 1, 25 (OH) 2D3 treatment significantly reduced NF-κ B protein levels compared with DM group (Figure 3A), as was hepatic NF-κ B, MCP-1, ICAM-1, and TGF-β1 mRNA (Figure 4). 1, 25 (OH) 2D3 administration suppressed the expression of NF-κ B, MCP-1, ICAM-1, and TGF-β1. Hepatic NF-κ B, MCP-1, ICAM-1, and TGF-β1 mRNA levels of the 1,25 (OH) 2D3treated DM group were all strongly reduced than those from the DM group (Figure 4), as was hepatic NF-κ B protein levels (Figure 3A).
Figure 2.
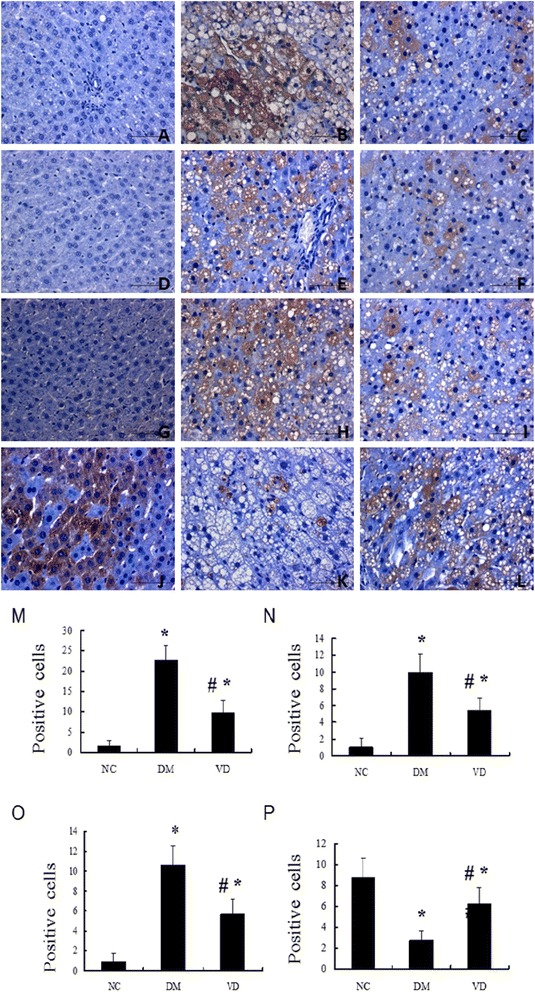
Immunohistochemistry (IHC) staining for MCP-1, ICAM-1, TGF-β1, CPT-1. IHC staining for MCP-1 (A-C; ×400); IHC staining for ICAM-1 (D-F; ×400); IHC staining for TGF-β1 (G-I; ×400), IHC staining for CPT-1 (J-L; ×400). The averaged number of positive cells of MCP-1 (M), ICAM-1 (N), TGF-β1 (O), CPT-1 (P). NC, normal control group; DM, diabetic group; VD, vitamin D treatment group. *P < 0 .05 vs. NC; #P < 0 .05 vs. DM.
Figure 3.
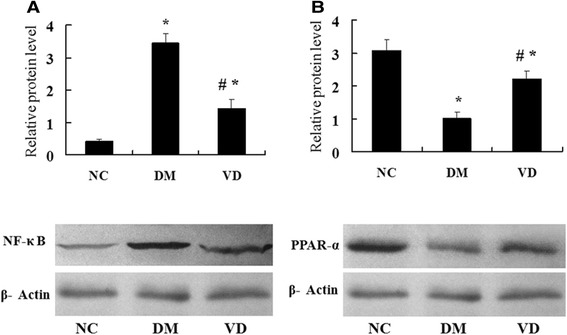
Western blot analysis of protein expression NF-κ B and PPAR-α. Representative western blot images and quantitative analysis of NF-κ B (A), PPAR-α (B). β-actin was used as a loading control. NC, normal control group; DM, diabetic group; VD, vitamin D treatment group. *P < 0 .05 vs. NC; #P < 0 .05 vs. DM.
Figure 4.
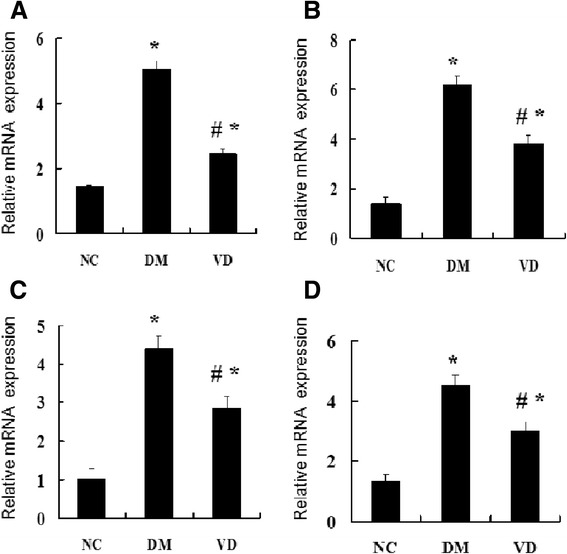
Effects of 1,25 (OH) 2D3 on the expression of hepatic NF-κ B and the related genes. The mRNA expression of hepatic NF-κ B and related genes were assessed. The relative mRNA levels were determined by quantitative RT-PCR and normalized to β-actin. Each gene expression was determined by the 2-ΔΔCt method. (A) The mRNA level of hepatic NF-κ B. (B) The mRNA level of MCP-1, (C) The mRNA level of hepatic ICAM-1, (D) The mRNA level of hepatic TGF-β1. Data are expressed as mean ± SD of each group (n = 15 per group). NC, normal control group; DM, diabetic group; VD, vitamin D treatment group. *P < 0 .05 vs. NC; #P < 0 .05 vs. DM.
1,25 (OH) 2D3 up-regulated the PPAR-α and CPT-1 expression, which involved in lipid oxidation
The cytokines involved in the regulation of hepatic lipid b-oxidation were assessed by different methods. IHC staining for PPAR-α and CPT-1 in livers from the normal control group were both strongly positive, whereas the amount of brown staining granules and the covered area were significantly decreased in the DM group. 1, 25 (OH) 2D3 treatment significantly increased the amount of brown staining granules and the covered area compared with DM group (Figure 2). Western blot analysis showed that a drastic decrease in hepatic protein level of PPAR-α was found in the DM group when compared to that of normal control group, 1, 25 (OH) 2D3 treatment reversed the expression of PPAR-α (Figure 3B), as was hepatic PPAR-α and CPT-1 mRNA, 1, 25 (OH) 2D3 administration up-regulated the expression of PPAR-α and CPT-1 in liver (Figure 5).
Figure 5.
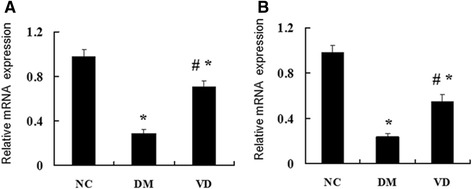
Effects of 1,25 (OH) 2D3 on the expression of hepatic PPAR-α and the related genes CPT-1. The mRNA expression of hepatic PPAR-α and related genes were assessed. The relative mRNA levels were determined by quantitative RT-PCR and normalized to β-actin. Each gene expression was determined by the 2-ΔΔCt method. (A) The mRNA level of hepatic PPAR-α. (B) The mRNA level of CPT-1. Data are expressed as mean ± SD of each group (n = 15 per group). NC, normal control group; DM, diabetic group; VD, vitamin D treatment group. *P < 0 .05 vs. NC; #P < 0 .05 vs. DM.
Discussion
In the present study, we have demonstrated that 1, 25 (OH) 2D3 has protective effects on livers of diabetic rat by modulating inflammation and lipid metabolism. Moreover, the data reported here suggest that 1, 25 (OH) 2D3 down-regulates the expression of the NF-κ B signaling pathway and up-regulates the expression of PPAR-α to attenuate the diabetes-induced liver injury.
Chronic inflammation is involved in the development of hepatic disease [24,25].With the increasing severity of liver disease, the expression of hepatic pro-inflammatory cytokines also increased [26]. The transcription factor NF-κ B and related mechanisms have gradually become a new hot spot in related research field. Multiple studies have demonstrated that the transcription factor NF-κ B play a crucial role in the decision between life and death of a hepatocyte [27]. It is the “genetic switch” of inflammation chain and regulates the gene expression including cellular proliferation, the inflammatory response, chemokines and cell adhesion molecules [28]. Cai D [29] reported that the activity of NF-κ B in the diabetic liver was significantly higher. MCP-1, which is produced mainly by macrophages and endothelial cells, is a potent chemotactic factor for monocytes [30]. It is considered that MCP-1 was specifically activated by NF-κ B in the presence of high glucose [31]. Several studies have reported a correlation between blood and hepatic levels of MCP-1 and the extent of inflammation both in human and animals [32,33]. Moreover, it is suggested that MCP-1 contributes to liver fibrosis, independent of effects on steatosis and inflammation [34]. ICAM-1 is a cytokine responsive integral membrane receptor expressing on the vascular endothelium, and it has been known to play an important role in the development of diabetic pathology [35]. HG-induced up-regulation of NF-κ B promotes ICAM-1 expression through PKC and mitogen-activated protein kinase (MAPK) pathways [36]. TGF-β1 is a multifunctional cytokine, it participates in inflammatory processes, induces fibrosis, causes the suppression of immune response [37,38]. Liver cells can produce TGF-β1, and it plays an important role in inflammation in the liver [39]. It was also suggested that TGF-β1 is a potent profibrogenic factor secreted by activated HSCs, which contributes to the extra cellular matrix (ECM) expression and liver fibrosis [40]. Our results showed that the expression of intra-hepatic NF-κ B and its downstream inflammatory cytokines MCP-1, ICAM-1, and TGF-β1 in DM group are significantly higher than the normal control group, indicating that the liver injury in diabetic rats may be partly associated with the NF-κ B signaling pathway. Emerging data have demonstrated that vitamin D has anti-inflammatory and antioxidative effects in streptozotocin-induced diabetes [41]. In this study, the effectiveness of 1, 25 (OH) 2D3 in attenuating the increasing of NF-κ B, MCP-1, ICAM-1, and TGF-β1 in liver of DM rats suggests that 1,25 (OH) 2D3 might serve as an anti-inflammatory agent for DM liver disease.
On the other hand, hepatic steatosis is one of the mechanisms of liver injury in diabetes. Previous investigation showed that an impaired fatty acid oxidation can promote the development of hepatic steatosis [18]. Hepatic lipid metabolism is mainly regulated by several nuclear receptors and transcription factors. Peroxisome proliferator-activated receptor a (PPAR-α) is one of the major regulators in fatty acid oxidation, prominently expressed in liver, heart and skeletal muscle, which plays an important role in regulation of genes that are involved in lipids utilization and storage [42]. CPT-1 is a rate-limiting enzyme of mitochondria mediated fatty acids β-oxidation, which can facilitate the first step of long-chain fatty acids entering into mitochondria. Further, CPT-1 was declined in NAFLD state and its gene expression is under the regulation of PPAR-α [43,44]. It is reported that PPAR-α-defective mice exhibit macrovesicular steatosis because of failing to induce fatty acid oxidation in liver [45]. The expression of hepatic PPAR-a was significantly decreased in the state of NAFLD [43]. PPAR-α agonists was shown to prevent the development of steatosis by promoting fatty acid oxidation [46]. To explore whether the protective effects of 1, 25 (OH) 2D3 was related to PPAR a activation, the expression of PPAR-α and its target gene was assessed. Our results showed that the expression of PPAR- α and CPT-1 was significantly declined in DM group, whereas 1, 25 (OH) 2D3 increased the PPAR-α and CPT-1 expression. Up-regulated PPAR-α by 1, 25 (OH) 2D3 in liver may promote β-oxidation and results in decreased TG levels. CPT-1 was benefit for FFA transport into mitochondrial for further oxidation and thus decreased lipid deposition. Our study only examined lipid profiles in serum but not in liver tissues, because elevated serum cholesterol and triglyceride concentrations are closely associated with hyperglycemia and obesity. Our data suggested that the level of serum TG decreased with the increasing of PPAR-α and CPT-1 expression. These results indicated that PPAR-a pathway contributed to the therapeutic role of 1, 25 (OH) 2D3 on diabetes-induced hepatic steatosis. Moreover, we suggest that 1, 25 (OH) 2D3 may play as PPAR-α agonists that could decrease plasma TG levels to modulate lipid metabolism. Nevertheless, the detailed mechanism behind PPAR-α up-regulation by 1, 25 (OH) 2D3 considered as complex and remains unclear.
Recent studies have suggested that PPAR-α down-regulation may facilitate the activity of hepatic pro-inflammatory cytokines [47]. Devchand et al. [48] have demonstrated that PPAR-α plays a role in acute inflammation control by using the PPAR-α-deficient mice. Thus, the PPAR-α signaling pathway may become a potentially interesting target for anti-inflammatory drug development. Furthermore, several studies have shown that inflammation regulates expression of PPAR isoforms, transcriptional interference between PPAR-α and NF-κ B occurs reciprocally, since NF-κ B can inhibit PPAR-α-mediated activation of a PPAR response element-driven promoter [49]. In this study, we have demonstrated that 1, 25 (OH) 2D3 can down-regulates the expression of the NF-κ B up-regulates the expression of PPAR-α in liver, and the expression of NF-κ B and PPAR-α were negatively correlated. Concordantly with a previous report [47-49], we speculate that further investigation of the molecular interaction between NF-κ B and PPAR-α in liver may help to develop therapeutic approaches for the treatment of hepatic diseases associated with diabetes mellitus.
In summary, our data provided some evidences that 1, 25 (OH) 2D3 can inhibit NF-κ B and its downstream inflammatory cytokines expression and promote PPAR-α and CPT-1 expression in liver of diabetic rat, which may partly contribute to its protective effects for diabetic liver injury. Further studies are required to identify how 1, 25 (OH) 2D3 can regulate the gene expression involved in hepatic proinflammatory cytokines and lipid metabolism. The relationship between vitamin D response element and the direct impact of 1, 25 (OH) 2D3 on NF-κ B and PPAR-α gene expression in liver may need further investigation.
Conclusion
This study links the effects of the 1, 25 (OH) 2D3 in regulating proinflammatory cytokines and lipid metabolism in a DM liver model. 1, 25 (OH) 2D3 significantly changed liver functional characteristics and fatty acid regulation, which may have been caused by its effects on hepatic PPAR-α and proinflammatory cytokines.
Acknowledgments
This work was supported by Natural science foundation of China grant (No. 8160116). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: JZ, CN, LL, GL; Performed the experiments: CN, LL, GL; YY, YZ, RY, YW. Contributed reagents/materials/analysis tools: GL, YW. Wrote the paper: JZ, CN. All authors read and approved the final manuscript.
Contributor Information
Conghua Ning, Email: xch623@126.com.
Lina Liu, Email: 2559435673@qq.com.
Guodong Lv, Email: xx_1112@sina.cn.
Ye Yang, Email: cbd_xj@sina.cn.
Yuanyuan Zhang, Email: lf_1112@sina.cn.
Rui Yu, Email: yyn_1112@sina.cn.
Yongtao Wang, Email: mx_1112@sina.cn.
Jun Zhu, Email: zhujun6677@163.com.
References
- 1.Harrison SA. Liver disease in patients with diabetes mellitus. J Clin Gastroenterol. 2006;40:68–76. doi: 10.1097/01.mcg.0000190774.91875.d2. [DOI] [PubMed] [Google Scholar]
- 2.de Marco R, Locatelli F, Zoppini G, Verlato G, Bonora E, Muggeo M. Cause-specific mortality in type 2 diabetes. The Verona Diabetes Study. Diabetes Care. 1999;22:756–61. doi: 10.2337/diacare.22.5.756. [DOI] [PubMed] [Google Scholar]
- 3.Manna P, Das J, Ghosh J, Sil PC. Contribution of type 1 diabetes to rat liver dysfunction and cellular damage via activation of NOS, PARP, IkappaBalpha/NF-kappaB, MAPKs, and mitochondria-dependent pathways: Prophylactic role of arjunolic acid. Free Radic Biol Med. 2010;48:1465–84. doi: 10.1016/j.freeradbiomed.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 4.Holick MF. Vitamin D, deficiency. N Engl J Med. 2007;357:266–81. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 5.Holick MF. Vitamin D: evolutionary, physiological and health perspectives. Curr Drug Targets. 2011;12:4–18. doi: 10.2174/138945011793591635. [DOI] [PubMed] [Google Scholar]
- 6.James WP. 22nd Marabou Symposium: the changing faces of vitamin D. Nutr Rev. 2008;66:286–90. doi: 10.1111/j.1753-4887.2008.00034.x. [DOI] [PubMed] [Google Scholar]
- 7.Palomer X, Gonzalez-Clemente JM, Blanco-Vaca F, Mauricio D. Role of vitamin D in the pathogenesis of type 2 diabetes mellitus. Diabetes Obes Metab. 2008;10:185–97. doi: 10.1111/j.1463-1326.2007.00710.x. [DOI] [PubMed] [Google Scholar]
- 8.Melamed ML, Michos ED, Post W, Astor B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med. 2008;168:1629–37. doi: 10.1001/archinte.168.15.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Etten E, Mathieu C. Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. J Steroid Biochem Mol Biol. 2005;97:93–101. doi: 10.1016/j.jsbmb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8:285–93. doi: 10.1038/ni1433. [DOI] [PubMed] [Google Scholar]
- 11.Harris SS. Does vitamin D deficiency contribute to increased rates of cardiovascular disease and type 2 diabetes in African Americans? Am J Clin Nutr. 2011;93:1175S–8. doi: 10.3945/ajcn.110.003491. [DOI] [PubMed] [Google Scholar]
- 12.Ganji V, Zhang X, Shaikh N, Tangpricha V. Serum 25-hydroxyvitamin D concentrations are associated with prevalence of metabolic syndrome and various cardiometabolic risk factors in US children and adolescents based on assay-adjusted serum 25-hydroxyvitamin D data from NHANES 2001–2006. Am J Clin Nutr. 2011;94:225–33. doi: 10.3945/ajcn.111.013516. [DOI] [PubMed] [Google Scholar]
- 13.Anagnostis P, Athyros VG, Adamidou F, Florentin M, Karagiannis A. Vitamin D and cardiovascular disease: a novel agent for reducing cardiovascular risk? Curr Vasc Pharmacol. 2010;8:720–30. doi: 10.2174/157016110792006978. [DOI] [PubMed] [Google Scholar]
- 14.Zhang C, Lu X, Tan Y, Li B, Miao X, Jin L, et al. Diabetes-induced hepatic pathogenic damage, inflammation, oxidative stress, and insulin resistance was exacerbated in zinc deficient mouse model. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes. 2005;54:3458–65. doi: 10.2337/diabetes.54.12.3458. [DOI] [PubMed] [Google Scholar]
- 16.Cohen-Lahav M, Shany S, Tobvin D, Chaimovitz C, Douvdevani A. Vitamin D decreases NFkappaB activity by increasing IkappaBalpha levels. Nephrol Dial Transplant. 2006;21:889–97. doi: 10.1093/ndt/gfi254. [DOI] [PubMed] [Google Scholar]
- 17.Allison J, Thomas H, Beck D, Brady JL, Lew AM, Elefanty A, et al. Transgenic overexpression of human Bcl-2 in islet beta cells inhibits apoptosis but does not prevent autoimmune destruction. Int Immunol. 2000;12:9–17. doi: 10.1093/intimm/12.1.9. [DOI] [PubMed] [Google Scholar]
- 18.Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141:603–10. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motiwala SR, Wang TJ. Vitamin D and cardiovascular risk. Curr Hypertens Rep. 2012;14:209–18. doi: 10.1007/s11906-012-0262-y. [DOI] [PubMed] [Google Scholar]
- 20.Zittermann A, Frisch S, Berthold HK, Gotting C, Kuhn J, Kleesiek K, et al. Vitamin D supplementation enhances the beneficial effects of weight loss on cardiovascular disease risk markers. Am J Clin Nutr. 2009;89:1321–7. doi: 10.3945/ajcn.2008.27004. [DOI] [PubMed] [Google Scholar]
- 21.Lee TI, Kao YH, Chen YC, Tsai WC, Chung CC, Chen YJ. Cardiac metabolism, inflammation, and peroxisome proliferator-activated receptors modulated by 1,25-dihydroxyvitamin D3 in diabetic rats. Int J Cardiol. 2014;176:151–7. doi: 10.1016/j.ijcard.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Jablonski KL, Jovanovich A, Holmen J, Targher G, McFann K, Kendrick J, et al. Low 25-hydroxyvitamin D level is independently associated with non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2013;23:792–8. doi: 10.1016/j.numecd.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhee EJ, Kim MK, Park SE, Park CY, Baek KH, Lee WY, et al. High serum vitamin D levels reduce the risk for nonalcoholic fatty liver disease in healthy men independent of metabolic syndrome. Endocr J. 2013;60:743–52. doi: 10.1507/endocrj.EJ12-0387. [DOI] [PubMed] [Google Scholar]
- 24.Targher G, Bertolini L, Padovani R, Zenari L, Zoppini G, Falezza G. Relation of nonalcoholic hepatic steatosis to early carotid atherosclerosis in healthy men: role of visceral fat accumulation. Diabetes Care. 2004;27:2498–500. doi: 10.2337/diacare.27.10.2498. [DOI] [PubMed] [Google Scholar]
- 25.Targher G, Bertolini L, Poli F, Rodella S, Scala L, Tessari R, et al. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005;54:3541–6. doi: 10.2337/diabetes.54.12.3541. [DOI] [PubMed] [Google Scholar]
- 26.Targher G, Bertolini L, Padovani R, Poli F, Scala L, Zenari L, et al. Non-alcoholic fatty liver disease is associated with carotid artery wall thickness in diet-controlled type 2 diabetic patients. J Endocrinol Invest. 2006;29:55–60. doi: 10.1007/BF03349177. [DOI] [PubMed] [Google Scholar]
- 27.Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: effects on liver homeostasis and beyond. Endocr Rev. 2007;28:365–86. doi: 10.1210/er.2006-0031. [DOI] [PubMed] [Google Scholar]
- 28.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 29.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-βand NF-κB. Nat Med. 2005;11:183–90. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol. 2002;13:894–902. doi: 10.1681/ASN.V134894. [DOI] [PubMed] [Google Scholar]
- 32.Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol. 2008;28:2195–201. doi: 10.1161/ATVBAHA.108.168633. [DOI] [PubMed] [Google Scholar]
- 33.Tous M, Ferre N, Rull A, Marsillach J, Coll B, Alonso-Villaverde C, et al. Dietary cholesterol and differential monocyte chemoattractant protein-1 gene expression in aorta and liver of apo E-deficient mice. Biochem Biophys Res Commun. 2006;340:1078–84. doi: 10.1016/j.bbrc.2005.12.109. [DOI] [PubMed] [Google Scholar]
- 34.Kassel KM, Guo GL, Tawfik O, Luyendyk JP. Monocyte chemoattractant protein-1 deficiency does not affect steatosis or inflammation in livers of mice fed a methionine-choline-deficient diet. Lab Invest. 2010;90:1794–804. doi: 10.1038/labinvest.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregoire FM, Zhang Q, Smith SJ, Tong C, Ross D, Lopez H, et al. Diet-induced obesity and hepatic gene expression alterations in C57BL/6 J and ICAM-1-deficient mice. Am J Physiol Endocrinol Metab. 2002;282:E703–13. doi: 10.1152/ajpendo.00072.2001. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe N, Shikata K, Shikata Y, Sarai K, Omori K, Kodera R, et al. Involvement of MAPKs in ICAM-1 expression in glomerular endothelial cells in diabetic nephropathy. Acta Med Okayama. 2011;65:247–57. doi: 10.18926/AMO/46850. [DOI] [PubMed] [Google Scholar]
- 37.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–807. doi: 10.2741/gressner. [DOI] [PubMed] [Google Scholar]
- 38.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 39.Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316:701–5. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- 40.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salum E, Kampus P, Zilmer M, Eha J, Butlin M, Avolio AP, et al. Effect of vitamin D on aortic remodeling in streptozotocin-induced diabetes. Cardiovasc Diabetol. 2012;11:58. doi: 10.1186/1475-2840-11-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–80. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi Y, Yu Z e, Min X, Luo X q, Lu X f, Wenhua L. Vitamin D attenuates high fat diet–induced hepatic steatosis in rats by modulating lipid metabolism. Eur J Clin Invest. 2012;42(11):1189–96. doi: 10.1111/j.1365-2362.2012.02706.x. [DOI] [PubMed] [Google Scholar]
- 45.Yi Y, Yu Z e, Min X, Luo X q, Lu X f, Wenhua LingRao MS, et al. PPARalpha in the pathogenesis of fatty liver disease. Hepatology. 2004;40:783–6. doi: 10.1002/hep.20453. [DOI] [PubMed] [Google Scholar]
- 46.Chen WL, Chen YL, Chiang YM, Wang SG, Lee HM. Fenofibrate lowers lipid accumulation in myotubes by modulating the PPARalpha/AMPK/FoxO1/ATGL pathway. Biochem Pharmacol. 2012;84:522–31. doi: 10.1016/j.bcp.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 47.Videla LA, Pettinelli P. Misregulation of PPAR functioning and its pathogenic consequences associated with nonalcoholic fatty liver disease in human obesity. PPAR Res. 2012;2012:107434. doi: 10.1155/2012/107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 49.Delerive P, De Bosscher K, Besnard S, Vanden BW, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–54. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]