

Abstract
Purpose/Aim
Acute Respiratory Distress Syndrome (ARDS) is an important clinical and public health problem. Why some at-risk individuals develop ARDS and others do not is unclear but may be related to differences in inflammatory and cell signaling systems. The Receptor for Advanced Glycation Endproducts (RAGE) and Granulocyte-Monocyte Stimulating Factor (GM-CSF) pathways have recently been implicated in pulmonary pathophysiology; whether genetic variation within these pathways contributes to ARDS risk or outcome is unknown.
Materials and Methods
We studied 842 patients from three centers in Utah and 14 non-Utah ARDS Network centers. We studied patients at risk for ARDS and patients with ARDS to determine whether Single Nucleotide Polymorphisms (SNPs) in the RAGE and GM-CSF pathways were associated with development of ARDS. We studied 29 SNPs in 5 genes within the two pathways and controlled for age, sepsis as ARDS risk factor, and severity of illness, while targeting a false discovery rate of ≤5%. In a secondary analysis we evaluated associations with mortality.
Results
Of 842 patients, 690 had ARDS, and 152 were at-risk. Sepsis was the risk factor for ARDS in 250 (30%) patients. When controlling for age, APACHE III score, sepsis as risk factor, and multiple comparisons, no SNPs were significantly associated with ARDS. In a secondary analysis, only rs743564 in CSF2 approached significance with regard to mortality (OR 2.17, unadjusted p = 0.005, adjusted p = 0.15).
Conclusions
Candidate SNPs within 5 genes in the RAGE and GM-CSF pathways were not significantly associated with development of ARDS in this multi-centric cohort.
Keywords: acute respiratory distress syndrome, GM-CSF, genetics, RAGE
INTRODUCTION
Acute Respiratory Distress Syndrome (ARDS), [1] is a serious clinical and public health burden [2]. Short-term mortality associated with ARDS is 20–40%, with more recent studies suggesting mortality nearer 20% [3–6]. ARDS arises from a complex, dysfunctional interplay among biological systems, involving immune/repair response, cytotoxicity and tissue injury, microvascular coagulopathy, and endothelial dysfunction. They are complicated by iatrogenic factors such as ventilator-associated lung injury and iatrogenic volume overload, among others [7]. Treatments that focused exclusively on the inflammatory response have failed to produce clinical benefits in ARDS, [2] or in sepsis [8], the most common risk factor for ARDS. Recent laboratory and clinical work have suggested that the multiligand pattern recognition Receptor for Advanced Glycation End-products (RAGE) and the Granulocyte-Monocyte Colony Stimulating Factor (GM-CSF, also known as CSF-2) pathways may be useful therapeutic targets in ARDS. These novel pathways both influence inflammation and epithelial cell signaling and function.
Based on considerations suggesting significant potential impact of these pathways on the pathobiology of ARDS, we evaluated whether single nucleotide polymorphisms (SNPs) within key genes identified in prior work as possible targets within the GM-CSF [9–11] or RAGE [12] pathways were associated with development of ARDS among at-risk patients.
Materials and Methods
Setting
At-risk patients were drawn from the ICUs at Intermountain Medical Center, a 450-bed tertiary-care, academic hospital in Murray, Utah; LDS Hospital, a 300-bed secondary-care, academic hospital in Salt Lake City, Utah; and the University of Utah Health Sciences Center, a 470-bed, tertiary-care, academic hospital in Salt Lake City, Utah. Patients with ARDS were drawn from the same Utah hospital ICUs and from patients at 14 non-Utah centers who participated in the NHLBI ARDS Network ALVEOLI [13], FACTT [14, 15], and ALTA [16] clinical trials.
Patients
We studied two basic patient populations: (1) Individuals at-risk for ARDS who never met criteria for ARDS and (2) Patients with ARDS. The at-risk patients came exclusively from Utah ICUs at Intermountain Medical Center, LDS Hospital, and the University of Utah Health Sciences Center. The ARDS population came from Utah ICUs and NHLBI ARDS Network patients.
Risk factors for ARDS included sepsis, pneumonia, multiple transfusions, significant trauma, or gastric acid aspiration, as described by Gong et al. [17, 18]. Specific criteria are displayed in eTable 1 of the online supplement. We enrolled at-risk patients within 48 hours of meeting at-risk criteria. The presence of a single risk factor classified a patient as at-risk. If at-risk patients subsequently met criteria for ARDS within 7 days of enrollment, they were removed from the at-risk group and reclassified as ARDS patients.
We used the NIH/NHLBI ARDS Network inclusion and exclusion criteria for ARDS patients at all centers. We defined ARDS by consensus criteria [19, 20], as displayed in eTable 2 of the online supplement. ARDS patients were enrolled within 48 hours of meeting consensus criteria.
Clinical Data
We determined sex and race for all patients. We calculated enrollment APACHE III [21], organ dysfunction using the Brussels score [22] and risk factors for ARDS for both at-risk and ARDS patients. We determined the number of days off mechanical ventilation, days out of the ICU, and days without organ dysfunction, through day 28, using standard methodology [23].
Study Endpoints
For the primary candidate gene SNP analysis, the association of interest was between ARDS and at-risk status for each SNP. Exploratory, secondary candidate gene SNP analysis was restricted to the cohort of patients with ARDS, and the outcome of interest was 28-day mortality (the primary mortality outcome of the ARDS Network trials) with additional exploratory analyses of ventilator-free days and organ-failure-free days to post-enrollment day 28.
Gene Candidates
We evaluated five [5] candidate genes within our candidate pathways (Table 2): 11 SNPs within the GM-CSF pathway and 18 SNPs within the RAGE pathway. We used a TagSNP approach to select candidate SNPs. We selected TagSNP according to the following parameters: linkage disequilibrium (LD) blocks defined using a Caucasian LD map and an r2 = 0.8; Caucasian minor allele frequency (MAF) >0.1; range = −1,500 bps from the initiation codon to +1,500 bps from the termination codon; and 1 SNP/LD bin.
TABLE 2.
Candidate Genes and SNPs
| Pathway | Gene | SNP | MAF* | Genotype | ARDS (N) | At risk (N) | OR | p value** |
|---|---|---|---|---|---|---|---|---|
| GM-CSF | CSF2 | rs2069616 | 0.43 | AA | 241 | 48 | 1 | NA |
| AG | 316 | 69 | 0.99 | 0.66 | ||||
| GG | 130 | 35 | 0.96 | 0.22 | ||||
| rs25881 | 0.17 | GG | 473 | 101 | 1 | NA | ||
| AG | 190 | 43 | 0.99 | 0.77 | ||||
| AA | 23 | 5 | 1 | 0.97 | ||||
| rs25882 | 0.2 | AA | 437 | 97 | 1 | NA | ||
| AG | 208 | 45 | 1 | 0.9 | ||||
| GG | 32 | 8 | 0.98 | 0.77 | ||||
| rs27438 | 0.21 | GG | 436 | 94 | 1 | NA | ||
| AG | 219 | 50 | 0.99 | 0.77 | ||||
| AA | 33 | 8 | 0.98 | 0.78 | ||||
| rs743564 | 0.4 | AA | 262 | 55 | 1 | NA | ||
| AG | 311 | 66 | 1 | 0.96 | ||||
| GG | 117 | 31 | 0.96 | 0.35 | ||||
| CSF2RB | rs10222232 | 0.48 | GG | 188 | 43 | 1 | NA | |
| AG | 338 | 75 | 1.00 | 0.89 | ||||
| AA | 158 | 32 | 1.02 | 0.64 | ||||
| rs11705394 | 0.25 | AA | 368 | 82 | 1 | NA | ||
| AG | 263 | 59 | 1 | 0.97 | ||||
| GG | 30 | 9 | 0.95 | 0.45 | ||||
| rs131842 | 0.17 | AA | 468 | 104 | 1 | NA | ||
| AG | 205 | 42 | 1.01 | 0.69 | ||||
| GG | 17 | 6 | 0.92 | 0.33 | ||||
| rs1534880 | 0.44 | GG | 226 | 40 | 1 | NA | ||
| CG | 335 | 82 | 0.95 | 0.13 | ||||
| CC | 129 | 30 | 0.96 | 0.32 | ||||
| rs738149 | 0.29 | GG | 336 | 86 | 1 | NA | ||
| AG | 292 | 52 | 1.05 | 0.06 | ||||
| AA | 59 | 13 | 1.02 | 0.64 | ||||
| rs909486 | 0.45 | GG | 212 | 48 | 1 | NA | ||
| AG | 335 | 70 | 1.01 | 0.7 | ||||
| AA | 142 | 34 | 0.99 | 0.82 | ||||
| RAGE | AGER | rs3131300 | 0.17 | AA | 475 | 103 | 1 | NA |
| AG | 192 | 41 | 1 | 0.94 | ||||
| GG | 17 | 8 | 0.87 | 0.07 | ||||
| rs3134943 | 0.15 | GG | 503 | 107 | 1 | NA | ||
| AG | 165 | 42 | 0.97 | 0.37 | ||||
| AA | 22 | 3 | 1.06 | 0.48 | ||||
| HMGB1 | rs3742305 | 0.23 | GG | 409 | 86 | 1 | NA | |
| CG | 241 | 56 | 0.99 | 0.6 | ||||
| CC | 39 | 8 | 1 | 0.95 | ||||
| NOX4 | rs10830277 | 0.12 | AA | 528 | 126 | 1 | NA | |
| AG | 148 | 23 | 1.06 | 0.08 | ||||
| GG | 14 | 3 | 1.02 | 0.86 | ||||
| rs11821838 | 0.05 | GG | 619 | 142 | 1 | NA | ||
| AG | 67 | 9 | 1.07 | 0.14 | ||||
| AA | 3 | 1 | 0.94 | 0.74 | ||||
| rs12799930 | 0.35 | AA | 277 | 69 | 1 | NA | ||
| AC | 319 | 66 | 1.03 | 0.33 | ||||
| CC | 81 | 14 | 1.05 | 0.24 | ||||
| rs1827428 | 0.08 | GG | 567 | 116 | 1 | NA | ||
| AG | 96 | 20 | 1 | 0.95 | ||||
| AA | 4 | 1 | 0.97 | 0.86 | ||||
| rs1847137 | 0.32 | CC | 319 | 77 | 1 | NA | ||
| CG | 291 | 57 | 1.03 | 0.28 | ||||
| GG | 75 | 15 | 1.03 | 0.54 | ||||
| rs2164521 | 0.12 | GG | 535 | 121 | 1 | NA | ||
| AG | 141 | 31 | 1 | 0.9 | ||||
| AA | 13 | 0 | 1.2 | 0.09 | ||||
| rs2202150 | 0.39 | AA | 245 | 65 | 1 | NA | ||
| AG | 338 | 66 | 1.05 | 0.11 | ||||
| GG | 107 | 21 | 1.05 | 0.26 | ||||
| rs317150 | 0.09 | GG | 560 | 131 | 1 | NA | ||
| AG | 120 | 20 | 1.05 | 0.19 | ||||
| AA | 7 | 1 | 1.07 | 0.64 | ||||
| rs317155 | 0.45 | AA | 204 | 53 | 1 | NA | ||
| AG | 329 | 74 | 1.02 | 0.46 | ||||
| GG | 156 | 25 | 1.07 | 0.07 | ||||
| rs317187 | 0.4 | AA | 247 | 63 | 1 | NA | ||
| AC | 322 | 70 | 1.02 | 0.4 | ||||
| CC | 120 | 19 | 1.07 | 0.09 | ||||
| rs319029 | 0.41 | GG | 209 | 36 | 1 | NA | ||
| CG | 222 | 45 | 0.98 | 0.5 | ||||
| CC | 107 | 19 | 1 | 0.92 | ||||
| rs319030 | 0.45 | GG | 198 | 53 | 1 | NA | ||
| AG | 340 | 76 | 1.03 | 0.35 | ||||
| AA | 150 | 23 | 1.08 | 0.04 | ||||
| rs521765 | 0.15 | GG | 492 | 114 | 1 | NA | ||
| CG | 178 | 37 | 1.02 | 0.6 | ||||
| CC | 19 | 1 | 1.15 | 0.11 | ||||
| rs595518 | 0.17 | CC | 463 | 106 | 1 | NA | ||
| AC | 197 | 43 | 1.01 | 0.81 | ||||
| AA | 18 | 2 | 1.09 | 0.33 | ||||
| rs957140 | 0.46 | GG | 189 | 51 | 1 | NA | ||
| AG | 361 | 73 | 1.05 | 0.15 | ||||
| AA | 138 | 28 | 1.04 | 0.26 |
SNP = Single Nucleotide Polymorphism; MAF = Minor Allele Frequency; ARDS = Acute Respiratory Distress Syndrome; OR = Odd; Ratio
MAF determined from the study sample, both cases and controls.
P value and Odds Ratio determined from univariate logistic regression.
Genotyping Methods
We extracted peripheral blood leukocyte DNA from 842 patients: 690 had ARDS, and 152 were classified as at-risk. We genotyped all markers using a multiplexed bead array assay run on a BeadExpress platform (Illumina, San Diego, California) as part of a larger study. We attained a genotyping call rate of 99.8%. We included 17 blinded internal replicates representing 2.0% of the sample set. The duplicate concordance rate was 99.6%. Because Caucasian patients represented the large majority of patients both locally and within ARDS Network data and there were very few non-Caucasian patients in the at risk cohort, we analyzed only samples from Caucasian patients to optimize the signal-to-noise ratio and avoid severe confounding by race.
Statistical Methods
Our primary analysis tested the association between established ARDS vs. at risk status and candidate SNPs (evaluating additive, dominant, and recessive models of inheritance) in a multivariate regression model controlling for clinical covariates. We used multivariable logistic regression models to estimate odds ratios and 95% confidence intervals for associations between genotypes (coded as homozygous wild, heterozygous, homozygous mutant) and ARDS status. We managed the risk of Type 1 statistical error for the primary outcome using a false discovery rate ≤5% that employed the technique of Benjamini and Hochberg [24, 25] adjusted for the number of SNPs evaluated. In exploratory secondary analyses to detect gene:gene interactions, we employed Multivariate Dimension Reduction (MDR) of up to 3 SNPs, with 5-fold cross validation in an additive model of inheritance [26] and Multivariate Adaptive Regression Splines (MARS)[27], which performs objective feature selection, including interaction terms, a technique that addresses usual feature selection problems in simple multivariate logistic regression. We performed statistical analysis and hypothesis testing with the R Statistical Package 3.0.2 (Vienna, Austria).[28]
We estimated power using Quanto 1.2.4.[29] Assuming a baseline ARDS risk of 6.8%,[30] and a ratio of ARDS:at-risk of 4.54, a sample size of 673 cases would have 80% power (with a two-tailed alpha of 0.05) to detect an effect with an Odds Ratio ≥1.6 for a SNP with a Minor Allele Frequency (MAF)≥ 0.16.
Ethical Considerations
This study was approved by the Institutional Review Boards (IRBs) of Intermountain Healthcare, the University of Utah Health Sciences Center, and participating NIH/NHLBI ARDS Network site IRBs. All patients, or their legal surrogates, provided written informed consent.
Results
We identified 842 patients, 152 at-risk individuals and 690 individuals with ARDS, through a process depicted in Figure 1. We obtained non-Utah ARDS Network DNA samples from 548 patients, while 294 samples came from Utah patients. Characteristics of the different populations (at-risk, ARDS cases from Utah centers, ARDS cases from the ARDS Network) are depicted in Table 1. Patients with ARDS were significantly more ill compared to the at-risk group, based on a number of physiologic measures, including APACHE III at enrollment, duration of ICU care and extent of organ failure. Those at-risk for ARDS were more likely to have sepsis and less likely to have pneumonia or aspiration as their identifying risk factor. Mortality among all ARDS patients was 17% (Table 1).
FIGURE 1.
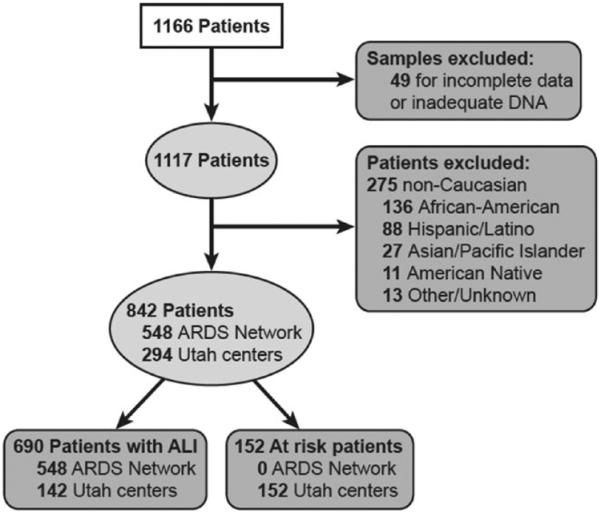
Process by which study patients were defined.
TABLE 1.
Demographics and Outcomes Among Three Patient Groups
| Variable | At Risk | ARDS Utah | ARDS ARDSNet | p value* |
|---|---|---|---|---|
| APACHE III at enrollment (points) | 70.56 | 83.14 | 92.28 | <0.001 |
| PF Ratio at enrollment | 207.1 | 130.71 | 135.28 | <0.001 |
| Age (years) | 52.85 | 52.39 | 50.88 | <0.001 |
| Female sex (percent) | 51% | 56% | 47% | 0.59 |
| 28-day mortality (percent) | 4% | 14% | 18% | <0.001 |
| Risk factor | ||||
| Trauma | 14% | 6% | 8% | 0.03 |
| Sepsis | 58% | 21% | 24% | <0.001 |
| Transfusion | 3% | 4% | 2% | 0.38 |
| Aspiration | 2% | 14% | 18% | <0.001 |
| Pneumonia | 19% | 49% | 41% | <0.001 |
| Ventilator-free days | 25.57 | 17.44 | 15.21 | <0.001 |
| ICU-free days | 17.5 | 11.92 | 14.21 | <0.001 |
| Organ-failure free days | 27.29 | 26.25 | 10.93 | <0.001 |
ARDS = Acute Respiratory Distress Syndrome; ARDSNet = National Institutes of Health ARDS Clinical Trials Network.
For comparison of all ARDS vs. all at risk patients.
For our primary analysis, we controlled for age (OR 0.98 [0.97–0.995] for each additional year, p 0.007), APACHE III score (OR 1.04 [1.03–1.05] for each additional point, p < 0.001), and presence of sepsis as the risk factor for ARDS (OR 0.13 [0.08–0.20], p < 0.001) in multivariate regression. We did not control for PaO2/FiO2 ratio at enrollment due to extensive missing data for this parameter. A heatmap displaying correlations among the candidate SNPs is displayed in the eFigure of the online data supplement. No candidate SNPs were significantly associated with the primary outcome. Our exploratory MDR and MARS analyses did not detect any significant interactions among candidate SNPs.
For our secondary, exploratory analyses, sepsis as risk factor was not associated with mortality and was excluded from the base regression model. After controlling for age (OR 1.03 [1.02–1.05], p < 0.001) and APACHE III score (OR 1.04 [1.03–1.04], p < 0.001), there was no significant association of candidate SNPs with mortality. One SNP, rs743564 in CSF2, approached significance on a recessive model of inheritance (OR 2.17 [1.25–3.74], unadjusted p = 0.005, adjusted p The = 0.15). minor allele frequency (MAF) of rs743564 is 0.224 in a general population of 1000 genomes.[31] In the ARDS cohort, the MAF was 0.39 (0.39 among survivors, 0.43 among non-survivors).
In further exploratory analysis using linear regression of ventilator, ICU-, and organ-free days, there was no significant association with any candidate SNPs, after controlling for age and APACHE III score.
DISCUSSION
In this multicenter cohort of patients with or at risk for ARDS, no candidate SNP within two novel inflammatory or epithelial signaling pathways achieved the pre-specified threshold for significance; only one SNP within CSF2 approached significance on a secondary analysis. While the GM-CSF and RAGE pathways have been associated with important observations about the nature of inflammation and its interaction with epithelial cells, our study did not identify a significant role for genetic variation among the tested SNPs in the development of or outcome after ARDS.
The Receptor for Advanced Glycation Endproducts (RAGE) is a multi-ligand pattern-recognition receptor implicated in inflammation. In mammals, RAGE is predominantly expressed by alveolar epithelial cell [32–34] and transduces the biological impact of discrete families of endogenous danger signals present at sites of cellular injury or death, including advanced glycation end products (AGE), members of the S100/calgranulin family, high mobility group box-1 (HMGB1), membrane-activated complex-1 and amyloid-peptides. By activating RAGE, these ligands up-regulate a program of inflammatory and tissue injury-provoking genes. Although the lung has the capacity to heal fully following ARDS, in many patients there is aberrant repair leading to ongoing inflammation and ultimately to fibrosis. RAGE may promote this fibrotic response through activation of a distinct NADPH oxidase in the lung parenchyma, NOX-4, resulting in production of reactive oxygen species that propagate ongoing inflammation, further injury, and ultimately, fibrosis [12, 34–39].
GM-CSF is a potent growth factor that was originally purified from lung. GM-CSF is a mitogen and survival factor for alveolar epithelial cells [40], and is essential for normal maturation of alveolar macrophages. It is the product of a number of different lung cell types, including alveolar epithelial cells [41, 42]. Lack of GM-CSF leads to pathological accumulation of surfactant and increased susceptibility to pulmonary infection [43–49], while over-expression of GM-CSF has been protective against hyperoxia-induced [10, 50] or bleomycin-induced [51] lung injury in mouse models. There is also some early evidence that detection of GM-CSF in BAL fluid is associated with improved clinical outcome in human ARDS [52]. One small, early study suggested a trend toward reduced mortality among ARDS patients treated with exogenous GM-CSF [53].
Several gene polymorphisms are associated with greater incidence of ARDS and/or outcome from ARDS, including myosin light chain kinase [54], surfactant protein B [18], mannose binding lectin-2 [17], FAAH [55], and POPDC3 [55].
Strengths of this study include its size, the use of data from multiple centers, and uniform inclusion and exclusion criteria applied to study patients and focus on a series of genes within two discrete pathways.
Limitations of this study include the fact that we did not exhaustively evaluate all polymorphisms in the candidate genes, that we investigated promising targets rather than all possible target genes within the pathways, the possibility that at-risk patients may differ systematically from ARDS patients in terms of non-genetic exposures that were not recorded in the study's case report forms, restriction of at-risk patients to the Utah sites, and lack of racial diversity. We controlled for measured non-genetic differences between the cohorts using multivariate logistic regression. We also acknowledge that the study was not powered to discover variants with modest effect sizes or in variants with low minor allele frequencies. Similarly, ARDS is a syndrome with heterogeneous causes and likely with heterogeneity with respect to mechanistic pathways. We cannot exclude the possibility that gene polymorphisms for the pathways of interest might have effects that may only be identified based on analysis of specific subsets of patients. Our strategy of restricting to 1,500 bps on either side of target genes may have led us to miss important SNPs in the 5′ untranslated region [56, 57] or effects mediated by the 3′ untranslated region, as enhancers may be present as much as 1 million bps from the transcription start site [58]. Our definition of at-risk status was unable to incorporate a granular measure of ARDS risk such as the Lung Injury Prevention Score, published after our present study was underway [30]. Larger studies with more complete sampling of gene candidates within the RAGE and/or GM-CSF pathways, perhaps with a more granular definition of risk, would be required to definitively rule out contributions to the pathophysiology of ARDS.
Supplementary Material
Acknowledgments
Funding: This study was funded by National Heart, Lung, and Blood Institute (NO1-HR-46062, HHSN268200536165C, and HHSN 268200536179C for the ARDS Network Studies and 1K23HL092161 to MTR) National Institute of General Medical Sciences (K23GM094465 to SMB), the National Institute of Aging (1R03AG040631 and 1R01AG04802201 to MTR), Department of Veterans Affairs (Merit Review Grant to RP), the Intermountain Research and Medical Foundation, and a Catalyst Grant from the University of Utah School of Medicine.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- [1].ARDS Definition Task Force. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- [2].Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- [3].Spragg RG, Bernard GR, Checkley W, Curtis JR, Gajic O, Guyatt G, et al. Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med. 2010;181(10):1121–1127. doi: 10.1164/rccm.201001-0024WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133(5):1120–1127. doi: 10.1378/chest.07-2134. [DOI] [PubMed] [Google Scholar]
- [5].Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, et al. Has mortality from acute respiratory distress syndrome decreased over time?: A systematic review. Am J Respir Crit Care Med. 2009;179(3):220–227. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- [6].Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD, Network NNA. Recent trends in acute lung injury mortality: 1996–2005. Crit Care Med. 2009;37(5):1574–1579. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. The J Clin Invest. 2012;122(8):2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Russell JA. Management of sepsis. N Engl J Med. 2006;355(16):1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- [9].Sturrock A, Seedahmed E, Mir-Kasimov M, Boltax J, Mc-Manus ML, Paine R., 3rd GM-CSF provides autocrine protection for murine alveolar epithelial cells from oxidant-induced mitochondrial injury. Am J Physiol Lung Cell Mol Physiol. 2012;302(3):L343–51. doi: 10.1152/ajplung.00276.2011. [DOI] [PubMed] [Google Scholar]
- [10].Paine R, 3rd, Wilcoxen SE, Morris SB, Sartori C, Baleeiro CE, Matthay MA, et al. Transgenic overexpression of granulocyte macrophage-colony stimulating factor in the lung prevents hyperoxic lung injury. Am J Pathol. 2003;163(6):2397–2406. doi: 10.1016/S0002-9440(10)63594-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Christensen PJ, Bailie MB, Goodman RE, O'Brien AD, Toews GB, Paine R., 3rd Role of diminished epithelial GM-CSF in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L487–L95. doi: 10.1152/ajplung.2000.279.3.L487. [DOI] [PubMed] [Google Scholar]
- [12].Reynolds PR, Schmitt RE, Kasteler SD, Sturrock A, Sanders K, Bierhaus A, et al. Receptors for advanced glycation end-products targeting protect against hyperoxia-induced lung injury in mice. Am J Respir Cell Mol Biol. 2010;42(5):545–551. doi: 10.1165/rcmb.2008-0265OC. [DOI] [PubMed] [Google Scholar]
- [13].Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, et al. Higher versus lower positive endexpiratory pressures in patients with the acute respiratory distress syndrome. New England J Med. 2004;351(4):327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- [14].National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Wheeler AP, Bernard GR, Thompson BT, Schoenfeld D, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. New England J Med. 2006;354(21):2213–2224. doi: 10.1056/NEJMoa061895. [DOI] [PubMed] [Google Scholar]
- [15].National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, et al. Comparison of two fluid- management strategies in acute lung injury. The New England J Med. 2006;354(24):2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- [16].National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Matthay MA, Brower RG, Carson S, Douglas IS, et al. Randomized, placebo-controlled clinical trial of an aerosolized beta(2)-agonist for treatment of acute lung injury. Am J Resp Crit Care Med. 2011;184(5):561–568. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gong MN, Zhou W, Williams PL, Thompson BT, Pothier L, Christiani DC. Polymorphisms in the mannose binding lectin-2 gene and acute respiratory distress syndrome. Crit Care Med. 2007;35(1):48–56. doi: 10.1097/01.CCM.0000251132.10689.F3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gong MN, Wei Z, Xu LL, Miller DP, Thompson BT, Christiani DC. Polymorphism in the surfactant protein-B gene, gender, and the risk of direct pulmonary injury and ARDS. Chest. 2004;125(1):203–211. doi: 10.1378/chest.125.1.203. [DOI] [PubMed] [Google Scholar]
- [19].Bernard GR. Acute respiratory distress syndrome: a historical perspective. Am J Respir Crit Care Med. 2005;172(7):798–806. doi: 10.1164/rccm.200504-663OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1):818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- [21].Knaus WA, Wagner DP, Draper EA, Zimmerman JE, Bergner M, Bastos PG, et al. The APACHE III prognostic system. Risk prediction of hospital mortality for critically ill hospitalized adults. Chest. 1991;100(6):1619–1636. doi: 10.1378/chest.100.6.1619. [DOI] [PubMed] [Google Scholar]
- [22].Russell JA, Singer J, Bernard GR, Wheeler A, Fulkerson W, Hudson L, et al. Changing pattern of organ dysfunction in early human sepsis is related to mortality. Crit Care Med. 2000;28(10):3405–3411. doi: 10.1097/00003246-200010000-00005. [DOI] [PubMed] [Google Scholar]
- [23].Schoenfeld DA, Bernard GR. Statistical evaluation of ventilator-free days as an efficacy measure in clinical trials of treatments for acute respiratory distress syndrome. Crit Care Med. 2002;30(8):1772–1777. doi: 10.1097/00003246-200208000-00016. [DOI] [PubMed] [Google Scholar]
- [24].Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc Ser B (Methodological) 1995;57(1):289–300. [Google Scholar]
- [25].Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. 2001. pp. 1165–1188. [Google Scholar]
- [26].Winham SJ, Motsinger-Reif AA. An R package implementation of multifactor dimensionality reduction. BioData Min. 2011;4(1):24. doi: 10.1186/1756-0381-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Friedman JH, Roosen CB. An introduction to multivariate adaptive regression splines. Stat Methods Med Res. 1995;4(3):197–217. doi: 10.1177/096228029500400303. [DOI] [PubMed] [Google Scholar]
- [28].R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- [29].Gauderman WJ, Morrison JM. QUANTO 1.2.4: A computer program for power and sample size calculations for genetic-epidemiology studies. 2009. [Google Scholar]
- [30].Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, et al. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med. 2011;183(4):462–470. doi: 10.1164/rccm.201004-0549OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M. The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2006;19(11):1437–1445. doi: 10.1038/modpathol.3800661. [DOI] [PubMed] [Google Scholar]
- [33].Mukherjee TK, Mukhopadhyay S, Hoidal JR. Implication of receptor for advanced glycation end product (RAGE) in pulmonary health and pathophysiology. Respir Physiol Neurobiol. 2008;162(3):210–215. doi: 10.1016/j.resp.2008.07.001. [DOI] [PubMed] [Google Scholar]
- [34].Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, et al. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Gene Cell : Dev Mol Cell Mech. 2004;9(2):165–174. doi: 10.1111/j.1356-9597.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- [35].Briot R, Frank JA, Uchida T, Lee JW, Calfee CS, Matthay MA. Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type I cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest. 2009;135(2):269–275. doi: 10.1378/chest.08-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, et al. Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax. 2008;63(12):1083–1089. doi: 10.1136/thx.2008.095588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wittkowski H, Sturrock A, van Zoelen MA, Viemann D, van der Poll T, Hoidal JR, et al. Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit Care Med. 2007;35(5):1369–1375. doi: 10.1097/01.CCM.0000262386.32287.29. [DOI] [PubMed] [Google Scholar]
- [38].Su X, Looney MR, Gupta N, Matthay MA. Receptor for advanced glycation end-products (RAGE) is an indicator of direct lung injury in models of experimental lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L1–5. doi: 10.1152/ajplung.90546.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L661–L73. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- [40].Huffman Reed JA, Rice WR, Zsengeller ZK, Wert SE, Dranoff G, Whitsett JA. GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol. 1997;273(4 Pt 1):L715–L725. doi: 10.1152/ajplung.1997.273.4.L715. [DOI] [PubMed] [Google Scholar]
- [41].Paine R, 3rd, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, et al. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1210–L1228. doi: 10.1152/ajplung.2001.281.5.L1210. [DOI] [PubMed] [Google Scholar]
- [42].Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15(4):557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- [43].Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. The New Engl J Med. 2003;349(26):2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- [44].LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest. 1999;103(4):563–569. doi: 10.1172/JCI5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Paine R, 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, et al. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol. 2000;164(5):2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- [46].LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. The J Clin Invest. 1999;103(4):563–569. doi: 10.1172/JCI5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Paine R, 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, et al. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol. 2000;164(5):2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- [48].Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice [see comments] J Clin Invest. 1996;97(3):649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. The J Clin Invest. 1996;97(3):649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Baleeiro CE, Christensen PJ, Morris SB, Mendez MP, Wilcoxen SE, Paine R., 3rd GM-CSF and the impaired pulmonary innate immune response following hyperoxic stress. American journal of physiology Lung cellular and molecular physiology. 2006;291(6):L1246–55. doi: 10.1152/ajplung.00016.2006. [DOI] [PubMed] [Google Scholar]
- [51].Moore BB, Coffey MJ, Christensen P, Sitterding S, Ngan R, Wilke CA, et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J Immunol. 2000;165(7):4032–4039. doi: 10.4049/jimmunol.165.7.4032. [DOI] [PubMed] [Google Scholar]
- [52].Matute-Bello G, Liles WC, Radella F, 2nd, Steinberg KP, Ruzinski JT, Hudson LD, et al. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med. 2000;28(1):1–7. doi: 10.1097/00003246-200001000-00001. [DOI] [PubMed] [Google Scholar]
- [53].Paine R, 3rd, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med. 2012;40(1):90–97. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, et al. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell aMol Biol. 2006;34(4):487–495. doi: 10.1165/rcmb.2005-0404OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tejera P, Meyer NJ, Chen F, Feng R, Zhao Y, O'Mahony DS, et al. Distinct and replicable genetic risk factors for acute respiratory distress syndrome of pulmonary or extrapulmonary origin. J Med Gen. 2012;49(11):671–680. doi: 10.1136/jmedgenet-2012-100972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dvir S, Velten L, Sharon E, Zeevi D, Carey LB, Weinberger A, et al. Deciphering the rules by which 5'-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci U S A. 2013;110(30):E2792–E2801. doi: 10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, et al. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science. 2013;340(6128):82–85. doi: 10.1126/science.1231197. [DOI] [PubMed] [Google Scholar]
- [58].Sturrock A, Mir-Kasimov M, Baker J, Rowley J, Paine R., 3rd Key role of microRNA in the regulation of granulocyte macrophage colony-stimulating factor expression in murine alveolar epithelial cells during oxidative stress. J Biol Chem. 2014;289(7):4095–4105. doi: 10.1074/jbc.M113.535922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.