

Abstract
The Na+/Ca2+ exchanger (NCX) is the main Ca2+ extrusion mechanism of the cardiac myocyte and thus is crucial for maintaining Ca2+ homeostasis. It is involved in the regulation of several parameters of cardiac excitation contraction coupling, such as cytosolic Ca2+ concentration, repolarization and contractility. Increased NCX activity has been identified as a mechanism promoting heart failure, cardiac ischemia and arrhythmia. Transgenic mice as well as pharmacological interventions have been used to support the idea of using NCX inhibition as a future pharmacological strategy to treat cardiovascular disease.
1. INTRODUCTION
The great majority of cardiac disorders are associated with at least one of three main pathophysiologies: arrhythmia, ischemia and heart failure. In clinical settings these entities often occur in combination sometimes mutually reinforcing and/or depending upon one another. As a clinical example, human heart failure renders the heart more susceptible to arrhythmia [1]. Conversely, ischemia can result in heart failure and increased arrhythmia burden [2]. Both extracellular and cellular mechanisms have been identified in the causal chains leading to either heart failure, arrhythmia or ischemia and some of these have been successfully identified as therapeutic targets [3].
In this issue of Current Drug Targets, cardiac Ca2+ regulating proteins such as the ryanodine receptor [4], TRP channels [5] and mitochondrial Ca2+ handling proteins [6] are reviewed regarding their role in cardiac pathophysiology and their potential as future pharmacological targets.
Another Ca2+ handling protein, the Na+/Ca2+ exchanger (NCX), also operates in cardiac myocytes. In the physiological setting, NCX serves as the main Ca2+ extrusion mechanism of the cardiac myocyte [7]. However, increased activity of NCX, induced experimentally, can promote arrhythmia; exacerbate ischemic necrosis and cause heart failure. Since increased exchanger activity has also been observed in several models of these diseases, increased NCX activity could be interpreted as a common denominator in the mediation of these three pathophysiologies. Several groups [8–11], including ours [12–14] have been able to define experimental conditions under which genetic or pharmacological inhibition of NCX activity can indeed protect from arrhythmia and ischemia. Although the above studies have demonstrated a promising therapeutic potential of NCX inhibition, pharmacological NCX inhibition has not, to our knowledge, been tested on a chronic condition. Some information on the chronic effects of altered NCX activity stems from transgenic murine models. We have investigated adaptive mechanisms and pathophysiological aspects of chronically altered NCX activity in transgenic overexpressors of NCX and in cardiac specific NCX knockout (KO) mice (for review see [15–18]). Mice are viable with both ventricular-specific KO and 3-fold overexpression of NCX. Although homozygous cardiac specific KO of NCX eventually leads to hypertrophy and a heart failure phenotype in later development, this does not seem to be the case in heterozygotes when NCX expression is modestly reduced to about 50% [19]. However, these observations do not speak to the safety of chronic pharmacological NCX inhibition since murine cardiac physiology is drastically different than humans. Furthermore, transgenic manipulation in early development may offer unrecognized but beneficial adaptive mechanisms that may not operate during pharmacological suppression started during later development.
Thus, before pharmacological or genetic NCX antagonism can be tested as treatment for human cardiac disease, a considerable amount of data defining bioavailability, safety, and reliability has yet to be collected. Nevertheless, the animal data currently available is encouraging. The conduct of future studies may in the end make available a new therapeutic concept by which we can defend against the “triple threat” of cardiac arrhythmia, ischemia and heart failure by countering one single molecular target.
It is noteworthy that NCX has also been characterized in extracardiac tissue such as arterial smooth muscle [20] and neuronal tissue [21]. Though not directly involved in cardiac disease, arterial and neuronal NCX may play a significant role in arterial hypertension and brain ischemia. These diseases are also part of the burden of cardiovascular disease, however, the role of NCX in these pathophysiologies has been reviewed elsewhere [22, 23].
Here we will give an overview on the role of NCX in cardiac physiology and pathophysiology. We will furthermore review the current data on experimental therapeutic approaches targeting NCX to treat ischemia, arrhythmia and heart failure.
2. NCX IN CARDIAC EXCITATION CONTRACTION COUPLING
In cardiac myocytes, the contractile cycle is initiated by Ca2+ entry via voltage activated L-type Ca2+ channels (dihydropyridine receptor (DHPR)). This “trigger Ca2+” binds to the ryanodine receptor (RyR) of the sarcoplasmic reticulum (SR) and thus induces further release of Ca2+ into the cytosol, which allows contraction of the myofilaments. During diastole, Ca2+ is shifted back into the sarcoplasmic reticulum by the Ca2+ ATPase of the sarcoplasmic reticulum (SERCA) (Fig. 1, for review, see [24]).
Fig. 1.

Role of NCX in cellular Ca2+ cycling during excitation-contraction coupling in cardiac myocytes. During the action potential Ca2+ enters the myocyte via the voltage dependent sarcolemmal L-type Ca2+ channel, and thus triggers further Ca2+ release from the sarcoplasmic reticulum via sarcoplasmic Ca2+ release channels (ryanodine receptors). The released Ca2+ diffuses towards the myofilaments and induces the contraction. During diastole Ca2+ is shifted back into sarcoplasmic reticulum by the Ca2+ ATPase of the sarcoplasmic reticulum (SERCA). The Na+/Ca2+ exchanger extrudes Ca2+ from the cytosol into the extracellular space.
To maintain Ca2+ homeostasis and to ensure diastolic relaxation of the myocyte, the same amount of Ca2+ that enters the cell with each contractile cycle has to be extruded. As the main cardiac Ca2+ removal mechanism, the Na+/Ca2+ exchanger extrudes 1 Ca2+ ion in exchange for 3 Na+ ions [25–28]. As an additional Ca2+ removal mechanism, the plasma membrane Ca2+ ATPase (PMCA) also extrudes Ca2+ from the cell, however in far smaller quantities than NCX [29].
The RyR and the DHPR are colocalized to the cleft at the t-tubular sarcoplasmic-sarcolemmal junctions [30]. The proximity of the RyR and the DHPR is believed to ensure a high fidelity of excitation-contraction coupling. There is also mounting evidence that a sizeable proportion of NCX is also expressed within the dyadic cleft [31–33] and that NCX could thus modulate cleft Ca2+ concentration which again may have important physiological implications [34, 35].
Under certain conditions, NCX transport mode can reverse to mediate influx of Ca2+ into the myocyte instead of extruding Ca2+ [36–38]. Thus, in the presence of increased intracellular Na+, the driving force for Ca2+ extrusion is reduced or even reversed, so that Na+ leaves the cell and is exchanged for Ca2+ entering the cell. Since NCX is electrogenic, membrane depolarization during early systole also favours NCX reverse mode. It has been a matter of debate whether NCX reverse mode may contribute to systolic Ca2+ influx and thus add to force development in cardiac myocytes. Indeed, it was demonstrated that NCX reverse mode induced by long depolarizing voltage clamps could evoke contractions [39, 40]. We have recently produced new evidence from our genetic models that this reverse NCX mechanism is a manifest contributor to trigger Ca2+ [41]. As will be discussed below, NCX reverse mode is believed to play a significant role in the pathophysiology of ischemia-reperfusion injury.
In 1990, Nicoll et al. [42] first sequenced and published their initial analysis of the transsarcolemmal structure of NCX. It was found that the protein consists of 9 transmembrane segments and a long cytoplasmic loop, which separates the first 5 from the following 4 α helical transmembrane segments. Currently, three isoforms of the Na+/Ca2+ exchanger have been characterized, that have about 70% amino acid identity. While NCX1 is the predominant isoform of the heart [43, 44], NCX2 and NCX3 are detected in the central nervous system and in skeletal muscle [45, 46]. An as yet not cloned form of NCX is also thought to be present in mitochondria. Interestingly, this form of NCX does not seem to be electrogenic [47].
NCX activity is regulated by a variety of mechanisms among them Ca2+ and Na+, that besides being substrates for NCX also exert separate regulatory influences (for review, see [7]). At least one study has found that Na+/Ca2+ exchange current (INCX) may respond to adrenergic stimulation [48] but most studies investigating this effect have been negative [49–51].
3. PRINCIPLES OF NCX INHIBITION
Experimental NCX inhibition has been an important tool in defining the role of NCX in cardiac physiology and pathophysiology. Both pharmacological and genetic inhibition of NCX have been used experimentally to evaluate the therapeutic potential of NCX suppression. The potential of a future therapy of cardiovascular disease by inhibition of NCX will depend directly on the reliability, specificity and safety of the means available to suppress NCX activity in vivo. We will therefore give a brief review on the tools that are currently available to suppress NCX activity.
Pharmacological Inhibition
Synthetic NCX inhibitors have been available since the mid-1990s and have since been used in numerous studies investigating the physiology and pathophysiology of Na+/Ca2+ exchange and Ca2+ cycling. A common reservation about the use of pharmacologic NCX inhibition vs. genetic ablation is the potential lack of specificity. Indeed, KB-R 7943, the first synthetic NCX inhibitor widely used experimentally shows interactions with several extracardiac [52, 53] and cardiac ion channels and functional proteins. Among the latter are L-type-Ca2+, K+ and Na+ channels [54], the RyR [55] and mitochondrial uniporters [56]. SEA0400, a synthetic inhibitor which became available in 2001 [57] appears to offer a higher specificity [54] though there is still evidence that it may also modify cardiac function via a mechanism independent of NCX [58].
Further synthetics with NCX inhibitory potential are under development [59, 60].
Genetic Knockout (KO) of NCX
Global KO of NCX is embryonically lethal in mice [61, 62], while mice with inducible cardiac specific knockout survive into adulthood. Mice with modest (=heterozygous KO) [19, 63] and complete (=homozygous KO) [64] genetic ablation of NCX have been investigated. Cardiac myocytes from NCX KO mice with complete ablation of NCX do not exhibit significant alterations of resting or systolic Ca2+ concentration, or sarcoplasmic reticular (SR) Ca2+ load when compared to WT littermates. NCX inward current is absent and the decrease of the Ca2+ transient is drastically slowed during caffeine exposure, indicating that no alternative Ca2+ extrusion mechanism is upregulated to compensate for the absence of NCX. Instead, peak L-type Ca2+ current (ICa) is decreased [32] and AP duration is reduced [65] resulting in a further reduction of net Ca2+ entry into the myocyte to 20% [35]. Thus, in the absence of NCX, transsarcolemmal Ca2+ traffic is drastically reduced. The plasma membrane Ca2+ ATPase (PMCA) has been estimated to provide 10–25% of myocyte Ca2+ efflux from the cytosol into the extracellular space depending on species variation [29, 66, 67]. Thus, a Ca2+ influx reduced to 20% as observed in NCX KO myocytes is well within the range of capacity of the PMCA (Fig. 2a). Despite these alterations, there is no difference in the cellular Ca2+ transients implying an increase in the gain of excitation-contraction coupling in KO cells. The converse has been observed in NCX overexpressor mice [68] (Fig. 2b). Though there are considerable differences between murine cardiac physiology and that of higher mammals, these studies provide information on the cellular adaptations that may be active during future therapeutic NCX manipulation.
Fig. 2.

Concept of cellular mechanisms of adaptation to reduced and increased NCX activity as modelled from studies in NCX knockout (a, [32, 35]) and overexpressor mice (b, [68]).
Gene-transfection methods to acutely inhibit NCX in isolated cardiac myocytes are currently available [69] and may be investigated as therapeutic tools in future studies.
4. NCX AND CARDIAC ARRHYTHMIA
NCX activity and Action Potential (AP) Kinetics
To understand the involvement of NCX in the generation of cardiac arrhythmia, it is necessary to define the role that NCX plays in the repolarization of the cardiac AP. During systole, when cytosolic Ca2+ concentration is high, NCX extrudes Ca2+ from the cytosol into the extracellular space. Due to the electrogenic nature of this process, NCX generates an inwardly directed membrane current (INCX) during SR Ca2+ release. It has been assumed that via this mechanism NCX would contribute to maintain the plateau of the cardiac action potential. Indeed, computer modelling revealed that increased INCX activity significantly delays repolarization of the cardiac AP [70]. Sham and Spencer [71] also demonstrated in guinea pig cardiomyocytes that an inhibition of NCX caused an abbreviation of the AP while a prolongation was observed when NCX activity was maximal. Many groups including ours have used transgenic models with cardiac specific alteration of NCX expression to further define the role of NCX in AP repolarization. A prolonged AP plateau was observed in NCX overexpressor mice, and – conversely – a shortened AP in NCX knockout mice [35] (Fig. 3), although in knockout mice, additional mechanisms contribute to shortening AP duration [65].
Fig. 3.

NCX activity regulates AP repolarization in adult cardiac myocytes. Middle column: typical “pyramid”-like mouse AP recorded in a myocyte isolated from a WT animal. Left column: A lack of the plateau phase due to a faster repolarization of the AP is observed in myocytes isolated from NCX KO mice. Right column: In myocytes isolated from mice that homozygously overexpress NCX (HOM), a slowed repolarization with a pronounced plateau phase can be observed (modified from [35, 68]).
In addition to a direct influence of NCX inward current on AP repolarization, an alternative mechanism may explain NCX-mediated repolarization changes: we have proposed a functional interplay between NCX and L-type Ca2+ channels. One of the strongest regulators of ICa is cytosolic Ca2+ itself. Thus, at high intracellular Ca2+ concentrations, Ca2+ binds to calmodulin, thereby inactivating ICa. At low cytosolic Ca2+ concentrations, when no Ca2+ is bound, L-type Ca2+ current is considered to be maximal [72, 73]. The DHPR is primarily located at sarcolemmal SR junctions in close vicinity to the RyR and NCX [30]. It has been accepted that Ca2+ release from the SR is the primary mechanism to influence and terminate L-type Ca2+ current [74, 75]. However, due to its close vicinity, NCX activity may also influence ICa. We have reported that blockade of NCX may increase cleft Ca2+ concentration [32–34]. This may have important implications for ICa regulation via Ca2+ induced inactivation: In the absence of NCX activity elevated cleft Ca2+ decreases ICa open probability and accelerates ICa inactivation. Conversely, when NCX activity is high, this may increase ICa open probability and delay ICa inactivation. Indeed, we could demonstrate that in NCX knockout mice, ICa is reduced via a Ca2+ dependent mechanism [32], while – conversely - ICa in NCX overexpressor mice is increased, a mechanism that also appears Ca2+ dependent [68]. Via this functional interplay with ICa, NCX may – indirectly - influence AP repolarization in the same direction as it does directly via reduced or increased NCX inward current as explained above. Thus, increased INCX would also increase net ICa thereby further slowing repolarization. Conversely, reduced INCX would also reduce ICa thus reducing AP duration.
NCX and Afterdepolarizations: Pathophysiological Concept
It has been hypothesized that increased NCX activity could induce afterdepolarizations of the ventricular AP that could then trigger tachyarrhythmias [76–78]. Afterdepolarizations are defined as oscillations of the electric membrane potential during (early afterdepolarization [EAD]) or after (delayed afterdepolarization [DAD]; Fig. 4) the cardiac action potential. Afterdepolarizations can induce reentry and are commonly regarded as a major trigger of cardiac tachyarrhythmias.
Fig. 4.

Pathophysiological concept on the role of NCX in the mediation of cardiac arrhythmia (modified from [76]).
EADs have been shown to occur in animal models with slowed AP repolarization [79]. The prolonged AP duration prolongs the period of time the myocytes spend in a depolarized state which again will broaden the “time window” for voltage dependent INa or ICa to reactivate and induce an EAD (Fig. 4). This mechanism has been evoked to explain proarrhythmia in animal models of hypertrophy and heart failure where a reduction of K+ carried outward currents and an increased AP duration have been observed (for review see [80]). Likewise, specific mutations of K+ channel subunits resulting in loss of function, or Na+ channel gain-of-function mutations [81], can both slow AP repolarization and cause EADs resulting in ventricular tachyarrhythmia [81–83]. Since NCX also slows repolarization it may be a further candidate for the promotion of EADs.
NCX is also essential for the development of DADs. The initiating event of a DAD consists of a spontaneous release of Ca2+ from the SR, as occurs in the setting of SR Ca2+ overload. This sudden increase of cytosolic Ca2+ then leads to an activation of NCX inward current which depolarizes the sarcolemmal membrane. If this reaches the threshold for Na+ channel activation, a new and premature AP is triggered [76, 84] (Fig. 4).
Initial studies in isolated cardiac myocytes from several different species have indeed demonstrated that DADs can be triggered via Ca2+ overload of the SR [85–88]. An increased occurrence of afterdepolarizations of the cardiac AP has also been observed in animal models of hypertrophy and heart failure [89–94], where NCX is known to be overexpressed [95, 96].
Studies testing the proarrhythmic potential of increased NCX expression have mostly been limited to hypertrophied and failing myocardium. Therefore, their informations on the role of NCX in the generation of cardiac arrhythmia are somewhat limited because a multitude of structural, expressional and functional changes other than the upregulation of NCX have been observed in human and animal failing and hypertrophic myocardium (for review, see [97]) and any one or a combination of these could be potential promoters of cardiac arrhythmia. The NCX overexpressing mouse on the other hand has increased NCX activity in the absence of cellular hypertrophy or reduced K+ channel activity. Before 14 weeks, these animals have normal whole heart and cellular function and structure and preserved K+ outward currents. We have found that these animals are prone to whole heart ventricular tachyarrhythmia. In isolated cardiac myocytes, APs are prololonged and there is an increased occurrence of EADs as well as DADs [14]. These observations indicate that NCX may be a promoter for arrhythmia independent of structural or ionic remodelling associated with heart failure.
Experimental Inhibition of NCX to Counter Proarrhythmia
Early studies investigating the antiarrhythmic potential of the NCX inhibitor SEA0400 yielded inconsistent results. While Nagy et al. [8] observed that the NCX inhibitor SEA0400 inhibited EADs induced by dofelitide and barium in guinea pig cardiac muscle strips, Amran et al. did not observe a protective effect of SEA0400 in either in vivo studies or isolated cardiac myocytes when monitoring aconitine-induced arrhythmia in the same species [98]. Two independent studies investigated the effects of SEA0400 on arrhythmias induced by ouabain. Tanaka et al. [9] showed an antiarrhythmic effect in guinea pig right ventricular muscle strips. In an in vivo dog model, Nagasawa and coworkers [99] observed that pharmacological inhibition of NCX protected against ouabain-induced arrhythmia but not against arrhythmia induced by acute ischemia. A recent study has specifically investigated the effect of SEA0400 on cardiac myocytes isolated from the pulmonary vein regions of guinea pig left atria [100]. Spontaneous electrical activity generated by this cell population is regarded to be the trigger for atrial fibrillation, one of the most frequent tachyarrhythmias.
We have also attempted to characterize the antiarrhythmic effect of pharmacological NCX inhibition. To produce proarrhythmic experimental conditions in which afterdepolarizations occur, we utilized a recently developed intact heart model of the rabbit heart mimicking the electrophysiological alterations of long QT 2 and 3 syndromes (LQT 2 and 3) [101–103]. Under these experimental conditions, SEA0400 significantly reduced AP duration and suppressed EADs and the Torsade-de-Pointes form of ventricular tachycardia [13].
The discrepancies evident in studies investigating the antiarrhythmic potential of acute NCX inhibition may be due to the following factors: 1. Proarrhythmic interventions of a different nature have been applied (i.e. aconitine-, vs. ouabain-, vs. dofetilide- vs. ischemia-induced arrhythmia), 2. Different species (guinea pig vs. dog vs. rabbit) have been investigated and species differences in the composition of repolarizing ion channels are well known [104]. 3. Finally, different preparations of cardiac tissue ranging from in vivo models over trabecular preparations to patch clamp experiments in isolated cardiac myocytes have been used.
5. NCX AND ISCHEMIA
Role of NCX in the Pathophysiology of Ischemia Reperfusion Injury
During cardiac ischemia, a rise in intracellular Na+ occurs due to a reduction of Na+/K+ ATPase activity. This increase in Na+ concentration is followed by a rise in intracellular Ca2+. This accumulation of cytosolic Ca2+ concentration is dependent on the rise in cytosolic Na+ concentration and it has therefore been suggested that the process of Ca2+ accumulation during ischemia is mediated via NCX working in reverse mode [105–110]. As mentioned above, depending on the transsarcolemmal Na+ and Ca2+ gradients and the membrane potential, NCX can switch into reverse mode and transport Ca2+ into the cell instead of extruding Ca2+ [7]. In reverse mode, NCX extrudes 3 Na+ in exchange for 1 Ca2+ ion entering the cell. NCX reverse mode is thus favored by the high cytosolic Na+ concentration characteristic for ischemic conditions. Consistent with this hypothesis, mice with heterozygous overexpression of NCX exhibit increased susceptibility to ischemia-reperfusion injury [10, 111].
However, during ischemia, there is also a rise in cytosolic H+ concentration that contributes to the rise in cytosolic Na+ concentration via activation of the Na+/H+ exchanger (NHE). This increased Na+ favors NCX reverse mode and cytosolic Ca2+ accumulation. Inhibition of NHE blocks the rise in Na+ and Ca2+ and is cardioprotective during ischemia [110, 112]. Some studies have suggested that preventing the rise in cytosolic Na+ concentration via NHE inhibition may be cardioprotective, independent of the effect on Ca2+ accumulation [113, 114]. Both cytosolic accumulation of Ca2+ and Na+ are inhibited by NHE inhibitors, and it is therefore difficult to distinguish between the relative protective effects of preventing the increase in Na+ vs. Ca2+ concentration.
The development of transgenic mice and genetic transfection techniques has provided a unique tool to further investigate this question. We have demonstrated that NCX KO mice are significantly less susceptible to ischemic injury [12]. Cellular studies have demonstrated that there is no detectible Ca2+ entry during experimental lowering of the transsarcolemmal Na+ gradient in NCX KO myocytes while WT myocytes exhibited significant cytosolic Ca2+ accumulation under these conditions (Fig. 5). During ischemia-reperfusion, hearts from NCX KO mice exhibited less necrosis, better post-ischemic recovery of cardiac performance, higher levels of high-energy phosphates, and improved recovery of Na+ homeostasis when compared to WT animals [12]. Maddaford et al. [115] have demonstrated that acute targeted genetic suppression of NCX from isolated cardiac myocytes can also protect from ischemic injury. Similarly, it was shown that introduction of specific splice variants of NCX into neonatal cardiac myocytes via transfection prevented ischemia induced cytosolic Ca2+ accumulation [116]. These observations add further support to the hypothesis that NCX is a direct mediator of ischemic injury. Moreover, these data indicate that inhibition of NCX seems to be protective independent of a direct inhibition of H+ accumulation via NHE.
Fig. 5.
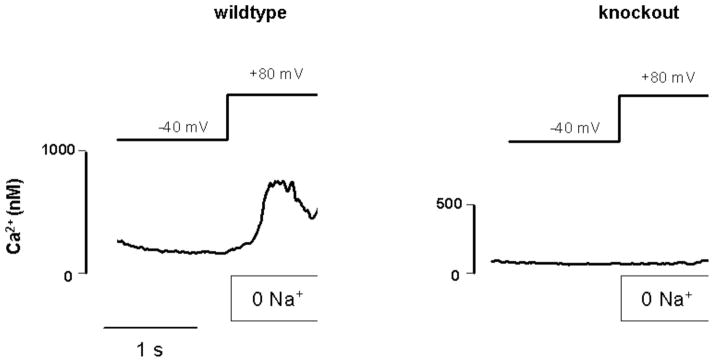
Ca2+ entry induced by Na+ depletion is absent in NCX KO myocytes: Recording of cytosolic Ca2+ concentration in patch clamped cardiac myocytes isolated from NCX KO and WT mice. Ca2+ entry via NCX reverse mode was induced in WT myocytes by hyperpolarization and simultaneous rapid removal of external Na+. Under these conditions, no increase in cytosolic Ca2+ concentration was observed in NCX KO cells. (Modified from [12]).
NCX Inhibition in Models of Ischemia-Reperfusion Injury
Pharmacological studies in animal models have also been conducted to investigate the therapeutic potential of NCX inhibitors as a means to protect against ischemia-reperfusion injury.
The most frequently used agents to test the effect of NCX inhibition on ischemia reperfusion injury are KB-R7943 and more recently SEA0400. KB-R7943 has been demonstrated to reduce ischemic injury under a variety of experimental conditions ranging form cellular to in vivo models. Thus, it has been shown to inhibit ischemia-reperfusion induced cytolsolic Ca2+ accumulation in isolated cardiac myocytes [117, 118] and to improve post-ischemic contractility in trabecular preparations [119, 120] and whole heart models [121–123].
In a similar range of experimental settings, SEA0400 has as well been demonstrated to reduce cellular Ca2+ entry upon ischemia-reperfusion [124], limit myocardial infarction size [125] and improve post-ischemic contractility [124, 125] even several days after the ischemia-reperfusion event [126]. A new NCX inhibitor, NCC-135, has also been shown to inhibit Ca2+ overload and reduce ischemia-reperfusion injury [127]. One comparative study using a whole heart rabbit model to investigate the protective effect of SEA0400 and KB-R7943 under identical experimental conditions has observed a similar efficiency of both drugs in reducing infarct size. However in this model only SEA0400 improved post-ischemic cardiac contractility while KB-R7943 significantly reduced heart rate and cardiac output [125]. This may be a consequence of the previously reported lack of KB-R7943’s specificity.
6. NCX AND CHRONIC HEART FAILURE
NCX and Chronic Heart Failure: Pathophysiological Concept
In the failing human heart, contractile force is reduced and relaxation is retarded. These alterations are caused in part by defective intracellular Ca2+-handling (for review see [128]). In failing myocytes, there is an increase in diastolic and a decrease of systolic Ca2+ [129] resulting in a reduced Ca2+ transient and contractile force, especially at increased cardiac frequencies [130]. These alterations have been attributed to decreased sarcoplasmic Ca2+-load, which could be caused by insufficient Ca2+-reuptake in diastole due to decreased activity of the SERCA [131, 132] or to excessive Ca2+ leak through hyperphosphorylated ryanodine receptors [133]. NCX is upregulated in the failing and hypertrophic heart in both activity and expression [93, 96]. It is unknown whether increased NCX activity is a cause or a consequence of chronic heart failure. Evidence suggests that increased NCX activity may provide an adaptive mechanism which protects against diastolic Ca2+ overload [95]. Yet, it may also be possible that the increased NCX current in the failing heart competes with the reduced SERCA of failing myocardium for cytosolic Ca2+, a maladaptive mechanism contributing to sarcoplasmic reticular Ca2+ loss and reduced contractility.
Cardiac glycosides such as digoxin have been used for over a 100 years to manipulate this relationship between SERCA and NCX in order to restore contractility in failing myocardium. Digoxin inhibits the Na+/K+ ATPase, resulting in increased cytoplasmic Na+. This increased Na+ concentration alters the transmembrane gradient for Na+, which reduces Ca2+ efflux via NCX and allows SERCA to accumulate Ca2+. In heart failure, the restored SR Ca2+ load provided by this indirect manipulation of NCX improves contractility. As noted above, this can unfortunately result in SR Ca2+ overload, DADs and triggered arrhythmias, which likely explains why digoxin does not provide a mortality benefit [128]. If intracellular Na+ could be more carefully regulated, it might reduce the arrhythmias associated with digoxin. The approach of reducing digoxin target levels may help in this regard.
Therapeutic Approaches and Lessons from Transgenic Mice
As mentioned above, there is evidence, that in acute heart failure induced by ischemia NCX inhibition can be beneficial (for review see [134]). However, experimental testing of the therapeutic potential of NCX inhibition in chronic heart failure is difficult. In an ideal experimental setup, the development of chronic heart failure would be monitored in the absence and presence of pharmacological NCX inhibition. However, depending on the model, the development from a non-failing phenotype to overt heart failure may take up to several weeks. Alternatively, one could choose an approach in which NCX inhibitors are administered in a phenotype that already exhibits heart failure to test whether a reduction of NCX activity may cause reverse remodelling. However, to detect macro- or microstructural remodelling in chronic heart failure, one would require an observation period of at least weeks. To our knowledge there is no study that has administered pharmacological NCX inhibitors over such a long period of time. This may in part be due to the fact that there is no published information available on the bioavailability or plasma removal rates of pharmacological NCX inhibitors.
Some information on the effect of altered NCX activity on the development of heart failure has been gathered in transgenic mice. In accordance with the hypothesis that increased NCX activity can promote chronic heart failure, transgenic mice with 3-fold overexpression develop clinically overt heart failure under experimental conditions under which their WT littermates do not [135].
In mice with complete ablation of NCX using cre/lox technology, cellular excitation-contraction coupling and Ca2+ handling can adapt so that cellular contractility is preserved [16, 35, 64]. Nevertheless, with progressing age, these animals also exhibit overt clinical heart failure and premature death [19]. This does not necessarily contradict the hypothesis that reduction of NCX activity may protect from heart failure since a beneficial effect may only be evident with modest NCX inhibition. Accordingly, heterozygous NCX KO mice show normal cardiac function and structure with normal lifespan [19]. Thus the heterozygous model may be better suited to further investigate the potential of reduced NCX activity in the therapy of chronic heart failure.
7. FUTURE DIRECTIONS: NCX INHIBITION AS A NEW THERAPEUTIC TOOL?
A wide variety of therapeutic tools exist to treat cardiovascular disease. These consist of catheter-based, surgical and pharmacological approaches. Nevertheless, cardiovascular disease remains the leading cause of death in western industrial nations. Since the 1980s, several innovations have been made in the field of interventional therapies such as catheter based coronary revascularization and arrhythmia ablation, implantable cardioverter/defibrillators and cardiac resynchronization therapy to name only a few. Innovations have also been made in the pharmacological field. Since the advent of angiotensin converting enzyme blockers (ACE inhibitors), novel promising drugs such as ranolazine or ivabradine have emerged. The overwhelming experimental data reviewed here point towards a benefit of using NCX inhibition as a novel therapy as well. However, research on NCX inhibition – may it be pharmacological or genetical – has to be taken to the next level. Long term animal feeding studies will have to be conducted to give some information on the safety of pharmacological NCX inhibitors. This will be a precondition of eventually testing NCX inhibitors on their safety in humans.
Contemplating the broad spectrum of pathophysiologies that depend on NCX activity (arrhythmia, ischemia, heart failure, arterial hypertension) the authors imagine, that once established in terms of safety and benefit, inhibition of NCX for the treatment of cardiovascular disease may have a similar impact on the field as the advent of ACE inhibitors in the 1980s.
Acknowledgments
This work was supported by grants Interdisziplinäre Medizinische Forschung (IMF Po 12 06 07), a stipend by the Deutsche Forschungsgemeinschaft (DFG Po 1004-1/2) (both to C.P.), NIH (R01 HL070828) to J.I.G. L.E. holds the Peter-Osypka Professorship for Clinical and Experimental electrophysiology.
References
- 1.Eckardt L, Haverkamp W, Breithardt G. Antiarrhythmic therapy in heart failure. Heart Fail Monit. 2002;2:110–9. [PubMed] [Google Scholar]
- 2.Barth AS, Tomaselli GF. Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol. 2009;2:327–35. doi: 10.1161/CIRCEP.108.817320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott J. Pathophysiology and biochemistry of cardiovascular disease. Curr Opin Genet Dev. 2004;14:271–9. doi: 10.1016/j.gde.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Dulhunty AF, Casarotto MG, Beard NA. The ryanodine receptor: a pivotal Ca2+ regulatory protein and potential therapeutic drug target. Curr Drug Targets. :XXX. doi: 10.2174/138945011795378595. [DOI] [PubMed] [Google Scholar]
- 5.Jiang LH, Gamper N, Beech DJ. Properties and therapeutic potential of TRP channels with putative roles in adversity. Curr Drug Targets. :XXX. doi: 10.2174/138945011795378568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viola HM, Hool LC. Targeting calcium and the mitochondria in prevention of pathology in the heart. Curr Drug Targets. :XXX. doi: 10.2174/138945011795378603. [DOI] [PubMed] [Google Scholar]
- 7.Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–33. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 8.Nagy ZA, Virag L, Toth A, Biliczki P, Acsai K, Banyasz T, et al. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–31. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka H, Shimada H, Namekata I, Kawanishi T, Iida-Tanaka N, Shigenobu K. Involvement of the Na+/Ca2+ exchanger in ouabain-induced inotropy and arrhythmogenesis in guinea-pig myocardium as revealed by SEA0400. J Pharmacol Sci. 2007;103:241–6. doi: 10.1254/jphs.fp0060911. [DOI] [PubMed] [Google Scholar]
- 10.Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res. 1998;83:1215–23. doi: 10.1161/01.res.83.12.1215. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi T, Takahashi K, Onishi M, Suzuki T, Tanaka Y, Ota T, et al. Effects of SEA0400, a novel inhibitor of the Na+/Ca2+ exchanger, on myocardial stunning in anesthetized dogs. Eur J Pharmacol. 2004;505:163–8. doi: 10.1016/j.ejphar.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 12.Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res. 2005;97:916–21. doi: 10.1161/01.RES.0000187456.06162.cb. [DOI] [PubMed] [Google Scholar]
- 13.Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5:1444–52. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 14.Pott C, Muszynski A, Ruhe M, Schulte JS, Milberg P, Fabritz L, Goldhaber JI, Breithardt G, Philipson KD, Kirchhof P, Müller FU. Proarrhythmia in a non-failing murine model of Na+/Ca2+ exchanger overexpression: Cellular and molecular mechanisms; abstract. Heart Rhythm Meeting; Denver. 2010. [Google Scholar]
- 15.Pott C, Goldhaber JI, Philipson KD. Genetic manipulation of cardiac Na+/Ca2+ exchange expression. Biochem Biophys Res Commun. 2004;322:1336–40. doi: 10.1016/j.bbrc.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 16.Pott C, Henderson SA, Goldhaber JI, Philipson KD. Na+/Ca2+ exchanger knockout mice: plasticity of cardiac excitation-contraction coupling. Ann N Y Acad Sci. 2007;1099:270–5. doi: 10.1196/annals.1387.015. [DOI] [PubMed] [Google Scholar]
- 17.Goldhaber JI, Henderson SA, Reuter H, Pott C, Philipson KD. Effects of Na+-Ca2+ exchange expression on excitation-contraction coupling in genetically modified mice. Ann N Y Acad Sci. 2005;1047:122–6. doi: 10.1196/annals.1341.011. [DOI] [PubMed] [Google Scholar]
- 18.Reuter H, Pott C, Goldhaber JI, Henderson SA, Philipson KD, Schwinger RH. Na+-Ca2+ exchange in the regulation of cardiac excitation-contraction coupling. Cardiovasc Res. 2005;67:198–207. doi: 10.1016/j.cardiores.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 19.Jordan MC, Henderson SA, Han T, Fishbein MC, Philipson KD, Roos KP. Myocardial Function With Reduced Expression of the Sodium-Calcium Exchanger. J Card Fail. 2010;16:786–796. doi: 10.1016/j.cardfail.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci. 1973;265:87–94. doi: 10.1098/rstb.1973.0011. [DOI] [PubMed] [Google Scholar]
- 21.Blaustein MP, Oborn CJ. The influence of sodium on calcium fluxes in pinched-off nerve terminals in vitro. J Physiol. 1975;247:657–86. doi: 10.1113/jphysiol.1975.sp010951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blaustein MP, Zhang J, Chen L, Song H, Raina H, Kinsey SP, et al. The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension. 2009;53:291–8. doi: 10.1161/HYPERTENSIONAHA.108.119974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffs GJ, Meloni BP, Bakker AJ, Knuckey NW. The role of the Na+/Ca2+ exchanger (NCX) in neurons following ischaemia. J Clin Neurosci. 2007;14:507–14. doi: 10.1016/j.jocn.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 25.Bers DM, Bridge JH, Spitzer KW. Intracellular Ca2+ transients during rapid cooling contractures in guinea-pig ventricular myocytes. J Physiol. 1989;417:537–53. doi: 10.1113/jphysiol.1989.sp017817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science. 1990;248:376–8. doi: 10.1126/science.2158147. [DOI] [PubMed] [Google Scholar]
- 27.Crespo LM, Grantham CJ, Cannell MB. Kinetics, stoichiometry and role of the Na-Ca exchange mechanism in isolated cardiac myocytes. Nature. 1990;345:618–21. doi: 10.1038/345618a0. [DOI] [PubMed] [Google Scholar]
- 28.Reuter H, Seitz N. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J Physiol. 1968;195:451–70. doi: 10.1113/jphysiol.1968.sp008467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–93. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–91. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, et al. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–37. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Pott C, Yip M, Goldhaber JI, Philipson KD. Regulation of cardiac L-type Ca2+ current in Na+-Ca2+ exchanger knockout mice: functional coupling of the Ca2+ channel and the Na+-Ca2+ exchanger. Biophys J. 2007;92:1431–7. doi: 10.1529/biophysj.106.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, Goldhaber JI. Sodium-Calcium Exchange is Essential for Effective Triggering of Calcium Release in Mouse Heart. Biophys J. 2010;99:755–64. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH, Weiss JN. Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium-calcium exchange. J Physiol. 1999;520(Pt 2):431–8. doi: 10.1111/j.1469-7793.1999.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pott C, Philipson KD, Goldhaber JI. Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circ Res. 2005;97:1288–95. doi: 10.1161/01.RES.0000196563.84231.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–6. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- 37.Levesque PC, Leblanc N, Hume JR. Role of reverse-mode Na+-Ca2+ exchange in excitation-contraction coupling in the heart. Ann N Y Acad Sci. 1991;639:386–97. doi: 10.1111/j.1749-6632.1991.tb17327.x. [DOI] [PubMed] [Google Scholar]
- 38.Lipp P, Niggli E. Sodium current-induced calcium signals in isolated guinea-pig ventricular myocytes. J Physiol. 1994;474:439–46. doi: 10.1113/jphysiol.1994.sp020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barcenas-Ruiz L, Beuckelmann DJ, Wier WG. Sodium-calcium exchange in heart: membrane currents and changes in [Ca2+]i. Science. 1987;238:1720–2. doi: 10.1126/science.3686010. [DOI] [PubMed] [Google Scholar]
- 40.Hume JR, Uehara A. Properties of “creep currents” in single frog atrial cells. J Gen Physiol. 1986;87:833–55. doi: 10.1085/jgp.87.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larbig R, Torres N, Bridge JH, Goldhaber JI, Philipson KD. Activation of Reverse Na+-Ca2+ Exchange by the Na+ Current during an Action Potential Augments the Calcium Transient: Evidence from NCX Knockout Mice. J Physiol. 2010 Jul 19; doi: 10.1113/jphysiol.2010.187708. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicoll DA, Longoni S, Philipson KD. Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science. 1990;250:562–5. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- 43.Kofuji P, Hadley RW, Kieval RS, Lederer WJ, Schulze DH. Expression of the Na-Ca exchanger in diverse tissues: a study using the cloned human cardiac Na-Ca exchanger. Am J Physiol. 1992;263:C1241–9. doi: 10.1152/ajpcell.1992.263.6.C1241. [DOI] [PubMed] [Google Scholar]
- 44.Komuro I, Wenninger KE, Philipson KD, Izumo S. Molecular cloning and characterization of the human cardiac Na+/Ca2+ exchanger cDNA. Proc Natl Acad Sci U S A. 1992;89:4769–73. doi: 10.1073/pnas.89.10.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Matsuoka S, Hryshko LV, Nicoll DA, Bersohn MM, Burke EP, et al. Cloning of the NCX2 isoform of the plasma membrane Na+-Ca2+ exchanger. J Biol Chem. 1994;269:17434–9. [PubMed] [Google Scholar]
- 46.Nicoll DA, Quednau BD, Qui Z, Xia YR, Lusis AJ, Philipson KD. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J Biol Chem. 1996;271:24914–21. doi: 10.1074/jbc.271.40.24914. [DOI] [PubMed] [Google Scholar]
- 47.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2009;107:436–41. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang YH, Hinde AK, Hancox JC. Anti-adrenergic effect of adenosine on Na+-Ca2+ exchange current recorded from guinea-pig ventricular myocytes. Cell Calcium. 2001;29:347–58. doi: 10.1054/ceca.2001.0197. [DOI] [PubMed] [Google Scholar]
- 49.Ballard C, Schaffer S. Stimulation of the Na+/Ca2+ exchanger by phenylephrine, angiotensin II and endothelin 1. J Mol Cell Cardiol. 1996;28:11–7. doi: 10.1006/jmcc.1996.0002. [DOI] [PubMed] [Google Scholar]
- 50.Main MJ, Grantham CJ, Cannell MB. Changes in subsarcolemmal sodium concentration measured by Na-Ca exchanger activity during Na-pump inhibition and beta-adrenergic stimulation in guinea-pig ventricular myocytes. Pflugers Arch. 1997;435:112–8. doi: 10.1007/s004240050490. [DOI] [PubMed] [Google Scholar]
- 51.Ginsburg KS, Bers DM. Isoproterenol does not enhance Ca-dependent Na/Ca exchange current in intact rabbit ventricular myocytes. J Mol Cell Cardiol. 2005;39:972–81. doi: 10.1016/j.yjmcc.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 52.Liang GH, Kim JA, Seol GH, Choi S, Suh SH. The Na+/Ca2+ exchanger inhibitor KB-R7943 activates large-conductance Ca2+-activated K+ channels in endothelial and vascular smooth muscle cells. Eur J Pharmacol. 2008;582:35–41. doi: 10.1016/j.ejphar.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 53.Pezier A, Bobkov YV, Ache BW. The Na+/Ca2+ exchanger inhibitor, KB-R7943, blocks a nonselective cation channel implicated in chemosensory transduction. J Neurophysiol. 2009;101:1151–9. doi: 10.1152/jn.90903.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol. 2002;135:1096–100. doi: 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrientos G, Bose DD, Feng W, Padilla I, Pessah IN. The Na+/Ca2+ exchange inhibitor 2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)isothiourea methanesulfonate (KB-R7943) also blocks ryanodine receptors type 1 (RyR1) and type 2 (RyR2) channels. Mol Pharmacol. 2009;76:560–8. doi: 10.1124/mol.109.057265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santo-Domingo J, Vay L, Hernandez-Sanmiguel E, Lobaton CD, Moreno A, Montero M, et al. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br J Pharmacol. 2007;151:647–54. doi: 10.1038/sj.bjp.0707260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, et al. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–56. [PubMed] [Google Scholar]
- 58.Reuter H, Henderson SA, Han T, Matsuda T, Baba A, Ross RS, et al. Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ Res. 2002;91:90–2. doi: 10.1161/01.res.0000027529.37429.38. [DOI] [PubMed] [Google Scholar]
- 59.Niu CF, Watanabe Y, Ono K, Iwamoto T, Yamashita K, Satoh H, et al. Characterization of SN-6, a novel Na+/Ca2+ exchange inhibitor in guinea pig cardiac ventricular myocytes. Eur J Pharmacol. 2007;573:161–9. doi: 10.1016/j.ejphar.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 60.Secondo A, Pannaccione A, Molinaro P, Ambrosino P, Lippiello P, Esposito A, et al. Molecular pharmacology of the amiloride analog 3-amino-6-chloro-5-[(4-chloro-benzyl)amino]-n-[[(2,4-dimethylbenzyl)-amino ]iminomethyl]-pyrazinecarboxamide (CB-DMB) as a pan inhibitor of the Na+-Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in stably transfected cells. J Pharmacol Exp Ther. 2009;331:212–21. doi: 10.1124/jpet.109.152132. [DOI] [PubMed] [Google Scholar]
- 61.Koushik SV, Wang J, Rogers R, Moskophidis D, Lambert NA, Creazzo TL, et al. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. Faseb J. 2001;15:1209–11. doi: 10.1096/fj.00-0696fje. [DOI] [PubMed] [Google Scholar]
- 62.Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, et al. Cardiac excitation-contraction coupling in the absence of Na+ - Ca2+ exchange. Cell Calcium. 2003;34:19–26. doi: 10.1016/s0143-4160(03)00018-6. [DOI] [PubMed] [Google Scholar]
- 63.Komuro I, Ohtsuka M. Forefront of Na+/Ca2+ exchanger studies: role of Na+/Ca2+ exchanger-lessons from knockout mice. J Pharmacol Sci. 2004;96:23–6. doi: 10.1254/jphs.fmj04002x5. [DOI] [PubMed] [Google Scholar]
- 64.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, et al. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–11. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 65.Pott C, Ren X, Tran DX, Yang MJ, Henderson S, Jordan MC, et al. Mechanism of shortened action potential duration in Na+-Ca2+ exchanger knockout mice. Am J Physiol Cell Physiol. 2007;292:C968–73. doi: 10.1152/ajpcell.00177.2006. [DOI] [PubMed] [Google Scholar]
- 66.Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–9. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 67.Lamont C, Eisner DA. The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflugers Arch. 1996;432:961–9. doi: 10.1007/s004240050223. [DOI] [PubMed] [Google Scholar]
- 68.Pott C, Goldhaber JI, Philipson KD. Homozygous overexpression of the Na+-Ca2+ exchanger in mice: evidence for increased transsarcolemmal Ca2+ fluxes. Ann N Y Acad Sci. 2007;1099:310–4. doi: 10.1196/annals.1387.019. [DOI] [PubMed] [Google Scholar]
- 69.Hurtado C, Wigle JT, Dibrov E, Maddaford TG, Pierce GN. A comparison of adenovirally delivered molecular methods to inhibit Na+/Ca2+ exchange. J Mol Cell Cardiol. 2007;43:49–53. doi: 10.1016/j.yjmcc.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 70.Noble D. Influence of Na/Ca exchange stoichiometry on model cardiac action potentials. Ann N Y Acad Sci. 2002;976:133–6. doi: 10.1111/j.1749-6632.2002.tb04731.x. [DOI] [PubMed] [Google Scholar]
- 71.Spencer CI, Sham JS. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2552–62. doi: 10.1152/ajpheart.00274.2003. [DOI] [PubMed] [Google Scholar]
- 72.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–58. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 73.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 74.Sham JS, Cleemann L, Morad M. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U S A. 1995;92:121–5. doi: 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, et al. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U S A. 1998;95:15096–101. doi: 10.1073/pnas.95.25.15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–6. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Sipido KR, Varro A, Eisner D. Sodium calcium exchange as a target for antiarrhythmic therapy. Handb Exp Pharmacol. 2006:159–99. doi: 10.1007/3-540-29715-4_6. [DOI] [PubMed] [Google Scholar]
- 78.Goldhaber JI. Sodium-calcium exchange: the phantom menace. Circ Res. 1999;85:982–4. doi: 10.1161/01.res.85.11.982. [DOI] [PubMed] [Google Scholar]
- 79.Eckardt L, Haverkamp W, Borggrefe M, Breithardt G. Experimental models of torsade de pointes. Cardiovasc Res. 1998;39:178–93. doi: 10.1016/s0008-6363(98)00043-1. [DOI] [PubMed] [Google Scholar]
- 80.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–29. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 81.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–7. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 82.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–80. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 83.Salama G, London B. Mouse models of long QT syndrome. J Physiol. 2007;578:43–53. doi: 10.1113/jphysiol.2006.118745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–18. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- 85.De Ferrari GM, Viola MC, D’Amato E, Antolini R, Forti S. Distinct patterns of calcium transients during early and delayed afterdepolarizations induced by isoproterenol in ventricular myocytes. Circulation. 1995;91:2510–5. doi: 10.1161/01.cir.91.10.2510. [DOI] [PubMed] [Google Scholar]
- 86.Kimura S, Cameron JS, Kozlovskis PL, Bassett AL, Myerburg RJ. Delayed afterdepolarizations and triggered activity induced in feline Purkinje fibers by alpha-adrenergic stimulation in the presence of elevated calcium levels. Circulation. 1984;70:1074–82. doi: 10.1161/01.cir.70.6.1074. [DOI] [PubMed] [Google Scholar]
- 87.Sicouri S, Antzelevitch C. Afterdepolarizations and triggered activity develop in a select population of cells (M cells) in canine ventricular myocardium: the effects of acetylstrophanthidin and Bay K 8644. Pacing Clin Electrophysiol. 1991;14:1714–20. doi: 10.1111/j.1540-8159.1991.tb02753.x. [DOI] [PubMed] [Google Scholar]
- 88.Tseng GN, Wit AL. Effects of reducing [Na+]o on catecholamine-induced delayed afterdepolarizations in atrial cells. Am J Physiol. 1987;253:H115–25. doi: 10.1152/ajpheart.1987.253.1.H115. [DOI] [PubMed] [Google Scholar]
- 89.de Groot SH, Schoenmakers M, Molenschot MM, Leunissen JD, Wellens HJ, Vos MA. Contractile adaptations preserving cardiac output predispose the hypertrophied canine heart to delayed afterdepolarization-dependent ventricular arrhythmias. Circulation. 2000;102:2145–51. doi: 10.1161/01.cir.102.17.2145. [DOI] [PubMed] [Google Scholar]
- 90.Meszaros J, Khananshvili D, Hart G. Mechanisms underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. Am J Physiol Heart Circ Physiol. 2001;281:H903–14. doi: 10.1152/ajpheart.2001.281.2.H903. [DOI] [PubMed] [Google Scholar]
- 91.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–19. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 92.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 93.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, et al. Enhanced Ca2+ elease and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–44. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 94.Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34:348–59. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 95.Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, et al. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–8. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 96.Reinecke H, Studer R, Vetter R, Holtz J, Drexler H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc Res. 1996;31:48–54. [PubMed] [Google Scholar]
- 97.Liew CC, Dzau VJ. Molecular genetics and genomics of heart failure. Nat Rev Genet. 2004;5:811–25. doi: 10.1038/nrg1470. [DOI] [PubMed] [Google Scholar]
- 98.Amran MS, Hashimoto K, Homma N. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in guinea pigs in vivo, in vitro, and in computer simulation studies. J Pharmacol Exp Ther. 2004;310:83–9. doi: 10.1124/jpet.104.066951. [DOI] [PubMed] [Google Scholar]
- 99.Nagasawa Y, Zhu BM, Chen J, Kamiya K, Miyamoto S, Hashimoto K. Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur J Pharmacol. 2005;506:249–55. doi: 10.1016/j.ejphar.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 100.Namekata I, Tsuneoka Y, Takahara A, Shimada H, Sugimoto T, Takeda K, et al. Involvement of the Na+/Ca2+ exchanger in the automaticity of guinea-pig pulmonary vein myocardium as revealed by SEA0400. J Pharmacol Sci. 2009;110:111–6. doi: 10.1254/jphs.08159sc. [DOI] [PubMed] [Google Scholar]
- 101.Milberg P, Ramtin S, Monnig G, Osada N, Wasmer K, Breithardt G, et al. Comparison of the in vitro electrophysiologic and proarrhythmic effects of amiodarone and sotalol in a rabbit model of acute atrioventricular block. J Cardiovasc Pharmacol. 2004;44:278–86. doi: 10.1097/01.fjc.0000129581.81508.78. [DOI] [PubMed] [Google Scholar]
- 102.Milberg P, Reinsch N, Wasmer K, Monnig G, Stypmann J, Osada N, et al. Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res. 2005;65:397–404. doi: 10.1016/j.cardiores.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 103.Eckardt L, Haverkamp W, Mertens H, Johna R, Clague JR, Borggrefe M, et al. Drug-related torsades de pointes in the isolated rabbit heart: comparison of clofilium, d,l-sotalol, and erythromycin. J Cardiovasc Pharmacol. 1998;32:425–34. doi: 10.1097/00005344-199809000-00013. [DOI] [PubMed] [Google Scholar]
- 104.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 105.Imahashi K, Kusuoka H, Hashimoto K, Yoshioka J, Yamaguchi H, Nishimura T. Intracellular sodium accumulation during ischemia as the substrate for reperfusion injury. Circ Res. 1999;84:1401–6. doi: 10.1161/01.res.84.12.1401. [DOI] [PubMed] [Google Scholar]
- 106.Kusuoka H, Camilion de Hurtado MC, Marban E. Role of sodium/calcium exchange in the mechanism of myocardial stunning: protective effect of reperfusion with high sodium solution. J Am Coll Cardiol. 1993;21:240–8. doi: 10.1016/0735-1097(93)90743-k. [DOI] [PubMed] [Google Scholar]
- 107.Kusuoka H, Porterfield JK, Weisman HF, Weisfeldt ML, Marban E. Pathophysiology and pathogenesis of stunned myocardium. Depressed Ca2+ activation of contraction as a consequence of reperfusion-induced cellular calcium overload in ferret hearts. J Clin Invest. 1987;79:950–61. doi: 10.1172/JCI112906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ladilov Y, Haffner S, Balser-Schafer C, Maxeiner H, Piper HM. Cardioprotective effects of KB-R7943: a novel inhibitor of the reverse mode of Na+/Ca2+ exchanger. Am J Physiol. 1999;276:H1868–76. doi: 10.1152/ajpheart.1999.276.6.H1868. [DOI] [PubMed] [Google Scholar]
- 109.Murphy E, Cross H, Steenbergen C. Sodium regulation during ischemia versus reperfusion and its role in injury. Circ Res. 1999;84:1469–70. doi: 10.1161/01.res.84.12.1469. [DOI] [PubMed] [Google Scholar]
- 110.Murphy E, Perlman M, London RE, Steenbergen C. Amiloride delays the ischemia-induced rise in cytosolic free calcium. Circ Res. 1991;68:1250–8. doi: 10.1161/01.res.68.5.1250. [DOI] [PubMed] [Google Scholar]
- 111.Sugishita K, Su Z, Li F, Philipson KD, Barry WH. Gender influences [Ca2+](i) during metabolic inhibition in myocytes overexpressing the Na+-Ca2+ exchanger. Circulation. 2001;104:2101–6. doi: 10.1161/hc4001.097038. [DOI] [PubMed] [Google Scholar]
- 112.Hartmann M, Decking UK. Blocking Na+-H+ exchange by cariporide reduces Na+-overload in ischemia and is cardioprotective. J Mol Cell Cardiol. 1999;31:1985–95. doi: 10.1006/jmcc.1999.1029. [DOI] [PubMed] [Google Scholar]
- 113.Iwai T, Tanonaka K, Inoue R, Kasahara S, Kamo N, Takeo S. Mitochondrial damage during ischemia determines post-ischemic contractile dysfunction in perfused rat heart. J Mol Cell Cardiol. 2002;34:725–38. doi: 10.1006/jmcc.2002.2002. [DOI] [PubMed] [Google Scholar]
- 114.Sawyer DB, Suter TM, Apstein CS. The sting of salt on an old, but open, wound--is Na+ the cause of mitochondrial and myocardial injury during ischemia/reperfusion? J Mol Cell Cardiol. 2002;34:699–702. doi: 10.1006/jmcc.2002.2030. [DOI] [PubMed] [Google Scholar]
- 115.Maddaford TG, Dibrov E, Hurtado C, Pierce GN. Reduced expression of the Na+/Ca2+ exchanger in adult cardiomyocytes via adenovirally delivered shRNA results in resistance to simulated ischemic injury. Am J Physiol Heart Circ Physiol. 2010;298:H360–6. doi: 10.1152/ajpheart.00932.2009. [DOI] [PubMed] [Google Scholar]
- 116.Hurtado C, Prociuk M, Maddaford TG, Dibrov E, Mesaeli N, Hryshko LV, et al. Cells expressing unique Na+/Ca2+ exchange (NCX1) splice variants exhibit different susceptibilities to Ca2+ overload. Am J Physiol Heart Circ Physiol. 2006;290:H2155–62. doi: 10.1152/ajpheart.00958.2005. [DOI] [PubMed] [Google Scholar]
- 117.Wang J, Zhang Z, Hu Y, Hou X, Cui Q, Zang Y, et al. SEA0400, a novel Na+/Ca2+ exchanger inhibitor, reduces calcium overload induced by ischemia and reperfusion in mouse ventricular myocytes. Physiol Res. 2007;56:17–23. doi: 10.33549/physiolres.930894. [DOI] [PubMed] [Google Scholar]
- 118.Wei GZ, Zhou JJ, Wang B, Wu F, Bi H, Wang YM, et al. Diastolic Ca2+ overload caused by Na+/Ca2+ exchanger during the first minutes of reperfusion results in continued myocardial stunning. Eur J Pharmacol. 2007;572:1–11. doi: 10.1016/j.ejphar.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 119.Mukai M, Terada H, Sugiyama S, Satoh H, Hayashi H. Effects of a selective inhibitor of Na+/Ca2+ exchange, KB-R7943, on reoxygenation-induced injuries in guinea pig papillary muscles. J Cardiovasc Pharmacol. 2000;35:121–8. doi: 10.1097/00005344-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 120.Satoh H, Mukai M, Urushida T, Katoh H, Terada H, Hayashi H. Importance of Ca2+ influx by Na+/Ca2+ exchange under normal and sodium-loaded conditions in mammalian ventricles. Mol Cell Biochem. 2003;242:11–7. [PubMed] [Google Scholar]
- 121.Seki S, Taniguchi M, Takeda H, Nagai M, Taniguchi I, Mochizuki S. Inhibition by KB-r7943 of the reverse mode of the Na+/Ca2+ exchanger reduces Ca2+ overload in ischemic-reperfused rat hearts. Circ J. 2002;66:390–6. doi: 10.1253/circj.66.390. [DOI] [PubMed] [Google Scholar]
- 122.Schafer C, Ladilov Y, Inserte J, Schafer M, Haffner S, Garcia-Dorado D, et al. Role of the reverse mode of the Na+/Ca2+ exchanger in reoxygenation-induced cardiomyocyte injury. Cardiovasc Res. 2001;51:241–50. doi: 10.1016/s0008-6363(01)00282-6. [DOI] [PubMed] [Google Scholar]
- 123.Nakamura A, Harada K, Sugimoto H, Nakajima F, Nishimura N. Effects of KB-R7943, a novel Na+/Ca2+ exchange inhibitor, on myocardial ischemia/reperfusion injury. Nippon Yakurigaku Zasshi. 1998;111:105–15. doi: 10.1254/fpj.111.105. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi K, Takahashi T, Suzuki T, Onishi M, Tanaka Y, Hamano-Takahashi A, et al. Protective effects of SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injuries. Eur J Pharmacol. 2003;458:155–62. doi: 10.1016/s0014-2999(02)02732-2. [DOI] [PubMed] [Google Scholar]
- 125.Magee WP, Deshmukh G, Deninno MP, Sutt JC, Chapman JG, Tracey WR. Differing cardioprotective efficacy of the Na+/Ca2+ exchanger inhibitors SEA0400 and KB-R7943. Am J Physiol Heart Circ Physiol. 2003;284:H903–10. doi: 10.1152/ajpheart.00784.2002. [DOI] [PubMed] [Google Scholar]
- 126.Yoshiyama M, Nakamura Y, Omura T, Hayashi T, Takagi Y, Hasegawa T, et al. Cardioprotective effect of SEA0400, a selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injury in rats. J Pharmacol Sci. 2004;95:196–202. doi: 10.1254/jphs.fpj03101x. [DOI] [PubMed] [Google Scholar]
- 127.Satoh N, Kitada Y. Cardioprotective effect of MCC-135 is associated with inhibition of Ca2+ overload in ischemic/reperfused hearts. Eur J Pharmacol. 2004;499:179–87. doi: 10.1016/j.ejphar.2004.07.095. [DOI] [PubMed] [Google Scholar]
- 128.Goldhaber JI, Hamilton MA. Role of inotropic agents in the treatment of heart failure. Circulation. 2010;121:1655–60. doi: 10.1161/CIRCULATIONAHA.109.899294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–55. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 130.Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;85:1743–50. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 131.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, et al. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–8. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 132.Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, et al. Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305–9. doi: 10.1172/JCI114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wehrens LS, Reiken S, Vest JA, Wronska A, Marks AR, et al. [Google Scholar]
- 134.Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;17;103(3):511–8. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee C, Dhalla NS, Hryshko LV. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can J Cardiol. 2005;21:509–16. [PubMed] [Google Scholar]
- 136.Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, et al. Hypertrophy and heart failure in mice overexpressing the cardiac sodium-calcium exchanger. J Card Fail. 2007;13:318–29. doi: 10.1016/j.cardfail.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]