

Abstract
Modulation of Hsp90 C-terminal function represents a promising therapeutic approach for the treatment of cancer and neurodegenerative diseases. Current drug discovery efforts toward Hsp90 C-terminal inhibition focus on novobiocin, an antibiotic that was transformed into an Hsp90 inhibitor. Based on structural information obtained during the development of novobiocin derivatives and molecular docking studies, scaffolds containing a biphenyl moiety in lieu of the coumarin ring present in novobiocin were identified as new Hsp90 C-terminal inhibitors. Structure-activity relationship studies produced new derivatives that inhibit the proliferation of breast cancer cell lines at nanomolar concentrations, which corresponded directly with Hsp90 inhibition.
Keywords: Heat shock protein 90, Hsp90 C-terminal inhibitors, Biphenyl, Structure-activity relationship, Breast cancer
Graphical abstract

1. Introduction
The 90 kDa heat shock proteins (Hsp90) are highly conserved molecular chaperones responsible for the conformational stability of more than 200 client proteins, many of which are essential to cancer cell survival [1-3]. Abnormal expression of Hsp90 has been implicated in a variety of disease states: In cancer, over-expression of Hsp90 is critical for the maturation and biological activity of numerous oncogenic proteins (eg., Her2, Raf1, Akt, CDK4, Src, c-Met, etc.) that are distributed amongst all six hallmarks of cancer [4, 5]. In neurodegenerative diseases, Hsp90 serves as the master regulator of the prosurvival heat shock response, and provides buffering capabilities for damaged proteins that accumulate beyond normal concentrations and can result in neuronal death [6]. Research has demonstrated that small molecule Hsp90 N-terminal inhibitors manifest two cellular activities, the first of which is induced degradation of proteins that are dependent upon the Hsp90 protein folding machinery. The second is concomitant induction of the pro-survival heat shock response (HSR). The HSR expands the chaperone buffering capacity to counter misfolded proteins that accumulate upon exposure to cellular stress, and thus, aids cell survival. These contradictory effects can provide unique therapeutic opportunities for the treatment of cancer and neurodegenerative diseases, if segregated [7, 8]. 17 Small molecule Hsp90 N-terminal inhibitors have entered clinical trials for the treatment of various cancers, however, the heat shock response manifested by these compounds appears detrimental, as the concentration needed for client protein degradation also induces the pro-survival response [9]. Similarly, these two effects hinder their application as neuroprotective agents, as cytotoxic client protein degradation is observed at the same concentration that induces the prosurvival HSR.
Recent studies have identified small molecules that bind the Hsp90 C-terminus and allosterically modulate Hsp90 function [10, 11]. In contrast to N-terminal inhibitors, C-terminal inhibitors can segregate the heat shock response from client protein degradation, thus providing a therapeutic opportunity for the treatment of neurodegenerative diseases or elimination of the pro-survival, heat shock response for cancer [12, 13]. Although several scaffolds are now known to bind the C-terminus (Figure 1) [13-16], medicinal chemistry efforts have been most focused on analogs of novobiocin, which was the first Hsp90 C-terminal inhibitor identified [17]. The identification of new chemical scaffolds that target the Hsp90 C-terminal domain is needed to dissect the role played by Hsp90 C-terminal inhibitors during the Hsp90 protein folding cycle as well as to improve upon inhibitory activity.
Figure 1.

Small molecules that target the Hsp90 C-terminus.
2. Result and Discuss
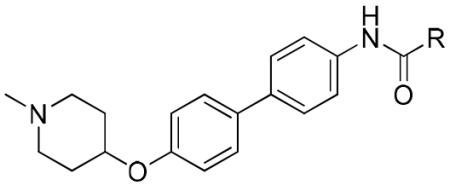
Prior modifications to novobiocin have revealed some structure-activity relationships and identified analogues that exhibit improved inhibitory activity [14, 18-23]. As summarized in Figure 2, these studies identified the benzamide side chain as critical for anti-proliferative activity, and modifications to this region can further increase inhibitory activity. The noviose sugar contributes to solubility and efficacy, however, replacement with ionizable amines results in analogues that also exhibit improved inhibitory activity, but do not induce the HSR (6, Figure 2). The amide linker not only provides important hydrogen bonding interactions, but it also serves to orient the aromatic side chain for interactions with the binding site. Recently, it was discovered that replacement of the amide with urea led to analogues that manifest greater anti-proliferative activity (7, Figure 2), presumably due to an extended hydrogen bonding network [24, 25]. In contrast to these modifications, studies on the coumarin ring system have produced only minor effects. Moreover, substitutions on the coumarin ring did not produce compounds with significantly altered activity, suggesting that the coumarin ring may serve to orient of the sugar and benzamide side chains within the binding pocket. Therefore, it was proposed that the coumarin ring could be replaced without compromising activity [19, 26].
Figure 2.

Rationale for proposed coumarin replacements.
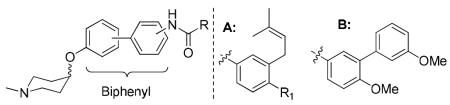
Recently, it was observed that the optimum distance between the piperidine nitrogen and the hydrogen-bonding network of the amide/urea is critical for inhibitory activity [25, 27]. Based on this observation, it was hypothesized that replacement of the coumarin core with scaffolds that maintain this distance may provide compounds upon which new inhibitors could be developed. Attempts to replace the coumarin with fused ring systems did not produce improved inhibitory activities [19, 26], suggesting that a flexible ring system may be beneficial for projection of the amino and benzamide side chain. The biphenyl ring system is relatively flexible and could therefore adopt different conformations within the binding pocket, which may present additional interactions with the protein. As a privileged-structure, compounds derived from this scaffold are known to manifest diverse activities, including anti-tumor activity [28]. In addition, the substitution pattern on this moiety can be modified and the distance between the ionizable amine and amide tuned. Therefore, molecules enlisting biphenyl as a coumarin replacement were pursued. Since no co-crystal structure of a ligand bound to the Hsp90 C-terminus exists, three substitution patterns on the biphenyl core (para-meta, meta-meta and para-para) were pursued to identify structural requirements for this scaffold. As shown in Figure 2, a piperidine was used in lieu of the noviose sugar and a prenylated benzylamide side chain was chosen for attachment to the biphenyl core.
Based on existing models for Hsp90 C-terminal inhibition [29, 30], computational docking studies utilizing the C-terminal binding site were conducted and identified compounds 8d [31] and 8e, which contain the para-meta or meta-meta substitution pattern, to overlay well with the novobiocin lead compound, 6 (Figure 3A). In contrast, compound 8f, which contains a para-para substitution, overlaid with the more active, urea-based analogue, 7 (Figure 3B). Interestingly, molecular studies suggested that compound 8f, which contains para-para substitution, may project the N-methylpiperidine deeper into the binding pocket and increase interactions with the protein (Figure 3C).
Figure 3.

Molecular docking in the putative Hsp90 C-terminal binding site: A. overlay of compounds 6 (red) and 8e (green); B. overlay of compounds 7 (red) and 8f (green); C. molecular overlay of novobiocin (green) and 8f (magenta) docked into the Hsp90 C-terminal binding site (line representation).
Encouraged by these computational studies, compounds 8 and analogs thereof were pursued along with investigation of the aryl substitution pattern. As shown in Scheme 1, these analogs were envisioned for assembly via an amide coupling reaction between amine 9 and acid chloride 10. The key intermediate, 9, could then be obtained through a Suzuki coupling reaction between piperidine-containing iodide 11 and phenylboronic acid, 12.
Scheme 1.

Retrosynthesis of biphenyl inhibitors.
Preparation of the biphenylamides that serve as novobiocin mimics is described in Scheme 2. Mitsunobo etherification of 1-methyl-4-hydroxypiperidine (13) and iodophenols, 14a or 14b, afforded iodides 11a–b, which underwent subsequent Suzuki coupling with 3- or 4-aminophenylboronic acid to produce anilines 9a-c (these compounds contain all three patterns of substitution; 9a: para-meta; 9b: meta-meta; 9c: para-para). Amide coupling of anilines 9a–c with prenylated acid chloride (10a) gave amides 8a–c, while the same conditions gave compounds 8g-i when treated with acid chloride 10b. Solvolysis of the acetate present in 8a-c in a solution of 10% triethylamine in methanol gave phenols 8d-f in good yields.
Scheme 2.

Synthesis of novobiocin mimics that contain a biphenyl moiety.
Reagents and conditions: a Ph3P, DIAD, THF, r. t., 12 h, 46%~77%; b Pd(dppf)2Cl2, 3- or 4-amino phenylboronic acid, 2M K2C03, Dioxane, 110 °C, 12 h, 52%~67%; c Pd/C, MeOH, r. t., 2 h, 100%; d pyridine, DCM, r. t., 4h, 52%~78%; e 10% Et3N/MeOH, r. t., 24 h, 72~86%.
Upon construction of this biphenyl-containing novobiocin library, the compounds were evaluated for anti-proliferative activity against SKBr3 (estrogen receptor negative, HER2 over-expressing breast cancer cells) and MCF-7 (estrogen receptor positive breast cancer cells) cell lines. Her2 and the ER are driving factors for these two cancers and are both Hsp90-dependent substrates. As shown in Table 1, the biphenyl-containing mimics exhibited low micromolar anti-proliferative activity, which is similar to that manifested by their coumarin counterparts. For analogues that contain a prenylated benzamide side chain (8a-f), the acetylated phenols (8a-c) exhibited comparable activity to the corresponding phenols (8d-f). Compounds containing the meta-meta (8b) and para-para (8c) biphenyl substitution patterns produced similar inhibitory activity and were more active than those containing the para-meta linkage (8a). Analogues containing the biaryl side chain (8g-i) showed improved anti-proliferative activity, and a para-para substituted biphenyl derivative 8i exhibited submicromolar activity against both breast cancer cell lines, approximately 2~3-fold better than its para-meta and meta-meta counterparts.
Table 1.
Anti-proliferative activity of novobiocin mimics.
| Entry | Biphenyl | R | SKBr3 (IC50, μM) |
MCF-7 (IC50, μM) |
|---|---|---|---|---|
| 5 | -- | -- | ~700 | -- |
| 6 | -- | -- | 0.76±0.14 a | 1.09±0.08 |
| 7 | -- | -- | 0.39±0.06 | 0.37±0.05 |
| 8a | para-meta | A (R1 = OAc) | 3.47±0.47a | 2.71±0.40 |
| 8b | meta-meta | A (R1 = OAc) | 1.76±0.16 | 1.70±0.21 |
| 8c | para-para | A (R1 = OAc) | 1.82±0.21 | 1.37±0.18 |
| 8d | para-meta | A (R1 = OH) | 2.94±0.11 | 2.21±0.06 |
| 8e | meta-meta | A (R1 = OH) | 2.79±0.40 | 1.17±0.05 |
| 8f | para-para | A (R1 = OH) | 2.27±0.08 | 1.85±0.32 |
| 8g | para-meta | B | 3.65±0.14 | 1.25±0.02 |
| 8h | meta-meta | B | 1.62±0.07 | 2.00±0.07 |
| 8i | para-para | B | 0.47±0.06 | 0.71±0.02 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
To confirm the observed anti-proliferative activities manifested by these biphenyl analogues resulted from Hsp90 inhibition, Western blot analyses of cell lysates following incubation with these compounds were performed. Compounds 8e, 8f, 8h and 8i induced the degradation of Hsp90-dependent client proteins, including Her2, Raf and Akt, at concentrations near their anti-proliferative IC50 value. Since Hsp90-dependent client protein degradation occurs at concentrations that mirror those needed for cellular efficacy, it is clear that Hsp90 inhibition is directly linked to cell viability. In addition, Hsp90 levels remained constant at both low and high concentrations, which is a hallmark of C-terminal inhibition.
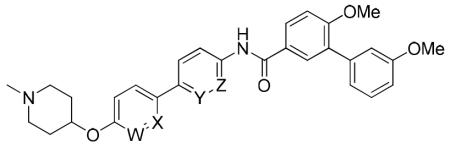
These biological assays suggest the biphenyl moiety can serve as a replacement for the coumarin ring and as a platform for the development of new Hsp90 C-terminal inhibitors. Considering the increased flexibility associated with this moiety in comparison to the coumarin ring, it was expected that the introduction of substituents onto the biphenyl system would provide additional interactions with the binding pocket. Since molecules containing a para-para substituted biphenyl moiety manifested superior Hsp90 inhibitory activity, modifications to this system were pursued. Prior SAR studies on the coumarin scaffold demonstrated that replacement of the lactone with quinoline resulted in slightly increased inhibitory activity [32]. Therefore, structural modifications were initiated by the inclusion of nitrogen at various positions throughout the biphenyl system. As illustrated in Scheme 2, the synthesis of derivatives containing nitrogen in the A ring commenced by Mitsunobo etherification of 1-methyl-4-hydroxypiperidine (13) and pyridinol 15a to give bromide 16, followed by a Suzuki coupling reaction to afford the nitro aromatic, 18a. Alternatively, direct Suzuki coupling of 15b gave phenol 17, which then underwent Mitsunobu etherification to give 18b. Subsequent reduction of the nitro group (18a-b) and coupling with 10b produced amides 19a and 19b. For construction of B-ring pyridines, the amide coupling reaction was performed first, between anilines 20a- b and biaryl acid chloride 10b, which enabled construction of bromides 21a-b. These bromides were then converted to phenols 22a-b via a Suzuki coupling reaction with 4-hydroxyphenylboronic acid. Finally, etherification of 22a-b with 1-methyl-4-hydroxypiperidine (13) afforded compounds 19c-d in good yields.
Upon construction of these nitrogen containing biphenyl derivatives, their anti-proliferative activity against SKBr3 and MCF-7 was investigated. As shown in Table 2, insertion of a nitrogen atom into the biphenyl ring system was detrimental, as all four derivatives manifested a 2-3 fold reduction in anti-proliferative activity. It appears that inclusion of a nitrogen atom into the B ring (19c-d) results in compounds that exhibit slightly better activity than inclusion of nitrogen into the A-ring, which correlates with the location of the lactone present in the coumarin ring system of novobiocin.
Table 2.
Anti-proliferative activity manifested by pyridine biphenyl derivatives.
| Entry | W | X | Y | Z | SKBr3 (IC50, μM) |
MCF-7 (IC50, μM) |
|---|---|---|---|---|---|---|
| 8i | C | C | C | C | 0.47±0.06 | 0.71±0.02 |
| 19a | N | C | C | C | 1.67±0.09 | 1.56±0.19 |
| 19b | C | N | C | C | 1.91±0.21 | 1.30±0.15 |
| 19c | C | C | N | C | 1.21±0.13 | 1.02±0.01 |
| 19d | C | C | C | N | 1.07±0.01 | 1.15±0.16 |
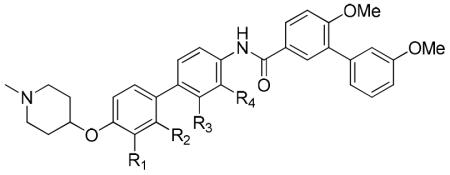
Although it was disappointing that nitrogen containing compounds did not manifest improved activity, the data suggested the binding site may be apolar. Therefore, to probe the surrounding binding pocket, additional functionalities were incorporated into the biphenyl ring system, which included a methyl, chloro, methoxy, nitro, amino or an acetamide, at all four positions. As outlined in Scheme 3, the synthesis of derivatives containing a methyl, chloride or methoxy substituent were pursued via a Suzuki coupling between bromides 23a-f and 4-nitrophenylboronic acid, or 4-hydroxyphenylboronic acid and bromides 26a-f, to afford phenols 24a-f or 27a-f, respectively. Mitsunobu etherification of the free phenols gave nitro derivatives, 25a-f or 28a-f, which underwent reduction and subsequent amide coupling with 10b to afford biphenyl derivatives, 29a-i.
Scheme 3.

Synthesis of pyridine biphenyl derivatives.
Reagents and conditions: a Ph3P, DIAD, THF, r. t., 12 h, 58%~68%; b Pd(dppf)2Cl2, 2M K2CO3, Dioxane, 110 °C, 12 h, 75%%~92%; c Pd/C, MeOH, r. t., 2 h, 100%; d pyridine, DCM, r. t., 4 h, 45%~87%.
The synthetic route used for the preparation of derivatives containing the nitro substituent was slightly altered. The Boc-protected 4-aminophenylboronic ester (31) was coupled with ortho or meta substituted nitro phenylbromides (30a or 30b) to give phenols 32a-b, which underwent Mitsunobu etherification to afford 33a-b. Deprotection to afford the corresponding aniline in the presence of trifluoroacetic acid, followed by an amide coupling with acid chloride 10b, produced the nitro-substituted derivatives, 34a-b. The synthetic route used to produce analogs that contain a nitro substituent on the B-ring were pursued via nitro substituted 4-bromoanilines, 35a-b, which were then reacted with acid chloride 10b to afford amides 36a-b, followed by Suzuki coupling with 4-hydroxyphenylboronic acid. Mitsunobo etherification of the resulting phenols with N-methyl piperidine gave nitro-derivatives, 34c-d. Subsequent reduction gave anilines 38a- d, and acylation afforded the acetamides 39a-d, respectively.
Anti-proliferative activity manifested by the substituted biphenyl derivatives was determined against SKBr3 and MCF-7 breast cancer cells. As shown Table 3, such modifications to the biphenyl ring system did not significantly affect inhibitory activity for most derivatives. It appears that substitution ortho to the amide (29d, 29h, 29l, 34d, 38d and 39d) is not tolerable, potentially due to disruption of the hydrogen bonding network and orientation of the amide side chain. Methyl and methoxy substituents at the C-2′ and C-3′ positions of the A ring and at C-3 of the B ring generated compounds (29a-c and 29e-g) that manifested similar anti-proliferative activities. For electron-withdrawing groups (Cl and NO2), it appears that substitution at C-2′ is more tolerable than at C-3′ (29j vs 29i, 34b vs 34a). Decreased activity manifested by 38 and 39, when compared with 29a-h, indicates that a hydrogen bond donor is less favorable.
Table 3.
Anti-proliferative activity manifested by substituted biphenyl derivatives.
| Entry | R1 | R2 | R3 | R4 | SKBr3 (IC50, μM) |
MCF-7 (IC50, μM) |
|---|---|---|---|---|---|---|
| 8i | H | H | H | H | 0.47±0.06 | 0.71±0.02 |
| 29a | Me | H | H | H | 0.83±0.03 | 1.69±0.08 |
| 29b | H | Me | H | H | 1.18±0.11 | 1.21±0.03 |
| 29c | H | H | Me | H | 0.97±0.01 | 1.57±0.56 |
| 29d | H | H | H | Me | 2.47±0.39 | 1.43±0.35 |
| 29e | OMe | H | H | H | 0.68±0.13 | 1.32±0.08 |
| 29f | H | OMe | H | H | 1.41±0.35 | 1.35±0.16 |
| 29g | H | H | OMe | H | 0.90±0.08 | 1.50±0.08 |
| 29h | H | H | H | OMe | 3.92±0.21 | 1.22±0.04 |
| 29i | Cl | H | H | H | 1.84±0.57 | 1.48±0.12 |
| 29j | H | Cl | H | H | 1.28±0.14 | 1.48±0.33 |
| 29k | H | H | Cl | H | 2.21±0.18 | 3.44±0.21 |
| 29l | H | H | H | Cl | 4.29±0.65 | 1.80±0.19 |
| 34a | NO2 | H | H | H | 2.07±0.17 | 1.23±0.25 |
| 34b | H | NO2 | H | H | 1.18±0.15 | 1.30±0.12 |
| 34c | H | H | NO2 | H | 2.48±0.77 | 3.32±0.25 |
| 34d | H | H | H | NO2 | 3.40±0.14 | 1.15±0.01 |
| 38a | NH2 | H | H | H | 2.23±0.49 | 5.95±1.22 |
| 38b | H | NH2 | H | H | 2.13±0.06 | 1.76±0.37 |
| 38c | H | H | NH2 | H | 3.90±0.18 | 2.07±0.23 |
| 38d | H | H | H | NH2 | 3.21±0.45 | 2.25±0.49 |
| 39a | NHAc | H | H | H | 2.66±0.76 | 1.84±0.43 |
| 39b | H | NHAc | H | H | 3.39±0.66 | 1.36±0.23 |
| 39c | H | H | NHAc | H | 2.52±0.26 | 4.66±0.49 |
| 39d | H | H | H | NHAc | 3.51±0.56 | 1.66±0.59 |
Although structural modification to the biphenyl moiety did not produce improved activities against these breast cancer cell lines, it did reflect a similar trend observed for the coumarin core of novobiocin, suggesting the biphenyl moiety is playing a similar role for orientation of the sugar and benzamide side chains. Since SAR studies on novobiocin demonstrated modification to the benzamide side chain produced analogues that exhibit improved anti-proliferative activity, SAR studies on the amide side chain were sought [32]. Electron-donating, electron-withdrawing and sterically bulky substituents were installed onto the benzylamide side chain by a straightforward coupling reaction between aniline 9c and substituted benzoyl chlorides (40a–40s), in the presence of pyridine to give 41a–41s (Scheme 6).
Scheme 6.

Synthesis of biphenyl derivatives containing a modified benzylamide side chain.
To compensate for the entropic penalty paid by replacement of the rigid coumarin ring with a more flexible biaryl moiety, fused ring systems (such as naphthalene, quinolone, indole, and benzo[b]thiophenyl) were introduced into the side chain. Similar to those reported earlier, these compounds were synthesized through an amide coupling between aniline 9c and the corresponding acid chlorides (42a-b, 43a-b and 45a-b), in the presence of pyridine to give 42a-b, 44a-b and 46a-b.
Biphenyl derivatives containing the modified benzamide side chain were evaluated in anti-proliferative assays against both breast cancer cell lines. As shown in Table 4, a large number of the substituted derivatives were found to exhibit increased inhibitory activity compared to the unsubstituted compound 41a, (except p-phenyl and o-phenyl substituted derivatives 41s and 41q). Compounds containing a para-halogen (41b-d, Cl, Br, I) or methoxy (41f) on the benzamide side chain manifested the most potent anti-proliferative activities, which were comparable to biaryl derivative 8i. However, shifting the substitution from para to meta (41b vs 41h, 41f vs 41i) resulted in decreased anti-proliferative activity. Consistent with this observation, installation of a meta substituent onto 41b or 41e manifested decreased inhibitory activity (41b vs 41j, 41e vs 41n and 41p). Interestingly, introduction of a meta-iodo substituent maintained activity (41e vs 41m), suggesting that a bulky substituent at the meta position may provide beneficial interactions with the binding site. In fact, this phenomena was observed for the phenyl substituted derivatives as well, although para- and ortho- substitutions (41q and 41s) did not produce compounds with enhanced anti-proliferative activity. However, meta-substitution (41r) produced inhibitors with comparable activity to lead compound 8i. Introduction of a fused ring system onto the side chain resulted in interesting activity. Compounds containing a 1- or 2-naphthoxyl amide side chain (43a and 43b) manifested good anti-proliferative activity. However, insertion of a nitrogen atom into the fused ring system (45a and 45b) decreased activity. 2-Indonyl (47a), not 2-benzo[b]thiophenyl (47b), exhibited comparable activity to 43a and 43b, suggesting that inclusion of a hydrogen bond donor is favored over a hydrogen bond acceptor.
Table 4.
Anti-proliferative activity manifested by biphenyl derivatives that contain a modified benzylamide side chain.
| Entry | R | SKBr3 (IC50, μM) | MCF-7 (IC50, μM) |
|---|---|---|---|
| 41a | phenyl | 4.13±0.22 | 3.95±0.13 |
| 41b | 4-chlorophenyl | 0.57±0.01 | 0.52±0.03 |
| 41c | 4-bromophenyl | 0.52±0.21 | 0.52±0.15 |
| 41d | 4-iodophenyl | 0.31±0.09 | 0.58±0.01 |
| 41e | 4-methylphenyl | 0.98±0.19 | 1.27±0.13 |
| 41f | 4-methoxyphenyl | 0.49±0.01 | 0.65±0.04 |
| 41g | 4-t-butylphenyl | 1.26±0.37 | 1.08±0.08 |
| 41h | 3-chlorophenyl | 1.94±0.37 | 2.83±0.69 |
| 41i | 3 -methoxyphenyl | 2.87±0.51 | 5.31±0.70 |
| 41j | 3 -methyl -4 -chl orophenyl | 1.11±0.42 | 1.03±0.16 |
| 41k | 3-chloro-4-methylphenyl | 1.96±0.24 | 2.28±0.49 |
| 41l | 3 -bromo-4-methylphenyl | 2.80±0.18 | 3.35±0.36 |
| 41m | 3-iodo-4-methylphenyl | 0.93±0.20 | 1.17±0.20 |
| 41n | 3,4-dichl orophenyl | 1.20±0.08 | 1.60±0.16 |
| 41o | 3,5-dichl orophenyl | 0.81±0.28 | 1.68±0.13 |
| 41p | 2,4-dichlorophenyl | 0.80±0.22 | 1.37±0.33 |
| 41q | 2-biphenyl | 6.26±1.54 | 6.67±0.83 |
| 41r | 3-biphenyl | 0.73±0.07 | 1.15±0.18 |
| 41s | 4-biphenyl | 4.59±0.06 | 4.44±0.60 |
| 43a | 1-naphthoyl | 0.22±0.13 | 0.58±0.02 |
| 43b | 2-naphthoyl | 0.35±0.02 | 0.49±0.11 |
| 45a | 2-quinolinyl | 2.42±0.62 | 2.76±0.76 |
| 45b | 6-quinolinyl | 1.31±0.18 | 2.07±0.16 |
| 47a | 2-indolyl | 0.64±0.08 | 0.58±0.02 |
| 47b | 2-benzo[b]thiophenyl | 1.32±0.23 | 2.01±0.58 |
| 8i | -- | 0.47±0.06 | 0.71±0.02 |
Confirmation that these molecules manifest their anti-proliferative activity through Hsp90 inhibition was determined by western blot analyses following incubation of these compounds with Mcf-7 cells for 24h. Analogs containing a halogen atom (41b, 41c, 41d) did not induce client protein degradation, while compounds 41f, 41r, 43a, 43b and 47a induced the degradation of Hsp90-dependent client proteins Her2, Raf, and Akt. Hsp90 levels remained constant, indicating these compounds manifest their inhibitory activity through C-terminal inhibition.
Structure-activity relationships obtained from these biphenylamide inhibitors largely reflect the trends observed with novobiocin, suggesting that successful modifications to novobiocin may also be applied to this scaffold. Compounds 41q-s suggest the biphenyl side chain is well accommodated, and comparison between compounds 41r and 8i indicate that additional substitutions may lead to even better inhibitory activity. To further verify the individual function of the two methoxy groups on compound 8i and potential locations for further modification, 49a, which lacks the 3′-methoxy on 8i, and compound 49b, which lacks the 4-methoxy substitution, additional compounds were synthesized. Biological evaluation of these compounds indicated the 4-methoxy is more beneficial (49a vs 49b, vide infra). Additional modifications were sought to install substitutions onto the second phenyl ring, with the aim of exploiting interactions at this location (Scheme 8). These compounds were synthesized through an amide coupling reaction between aniline 9c and acid chlorides 48c-h, which were synthesized according to reported procedures [23, 25]. Ester hydrolysis of 49d and 49e gave phenols 50a and 50b, while nitro reduction converted 49h to aniline 50c. 50c was then transformed to acetamide 50d upon acylation.
Scheme 8.

Synthesis of biphenyl derivatives containing a substituted biaryl side chain.
Reagents and conditions: a pyridine, DCM, r. t., 4 h, 56%~81%; b Et3N, MeOH, r. t., 24 h, 85%~90%; c Pd/C, MeOH, r. t., 12 h, 76%, d Ac2O, pyridine, r. t., 12 h, 76%.
As shown in Table 5, the anti-proliferative activities manifested by compounds containing a modified biaryl side chain suggest the 4-methoxy, not the 3′-methoxy is beneficial, since compound 49a exhibited similar activity, while 49b manifested decreased inhibitory activity compared to 8i. Installation of a phenolic ester onto the second phenyl ring also appears beneficial (49d and 49e vs 8i). Although hydrolysis of the 4′-ester did not alter the anti-proliferative activity (50b), hydrolysis of the 3′-ester led to decreased activity (50a). The introduction of chlorine at the 3′-position resulted in increased activity, whereas installation of chlorine at the 4′-position did not (49f and 49g vs 49a). Replacement of the 3′-methoxy with a nitro group retained anti-proliferative activity against both breast cancer cell lines, however, the corresponding aniline and acetamide failed to exhibit improved potency.
Table 5.
Anti-proliferative activity of biphenyl derivatives with a modified biaryl side chain.
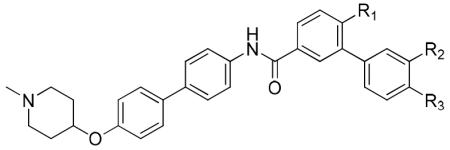
| Entry | R1 | R2 | R3 | SKBr3 | MCF-7 |
|---|---|---|---|---|---|
| 41r | H | H | H | 0.73±0.07 | 1.15±0.18 |
| 8i | OMe | OMe | H | 0.47±0.06 | 0.71±0.02 |
| 49a | OMe | H | H | 0.51±0.11 | 0.84±0.01 |
| 49b | H | OMe | H | 0.81±0.14 | 1.02±0.08 |
| 49c | OMe | H | OMe | 0.63±0.04 | 0.79±0.13 |
| 49d | OMe | OAc | H | 0.27±0.05 | 0.62±0.07 |
| 50a | OMe | OH | H | 1.56±0.35 | 1.08±0.34 |
| 49e | OMe | H | OAc | 0.14±0.01 | 0.64±0.08 |
| 50b | OMe | H | OH | 0.13±0.02 | 0.50±0.01 |
| 49f | OMe | Cl | H | 0.33±0.03 | 0.32±0.09 |
| 49g | OMe | H | Cl | 1.06±0.05 | 0.82±0.13 |
| 49h | OMe | NO2 | H | 0.40±0.07 | 1.09±0.28 |
| 50c | OMe | NH2 | H | 1.52±0.55 | 1.67±0.68 |
| 50d | OMe | NHAc | H | 3.37±0.74 | 1.43±0.28 |
Western blot analyses were performed to determine whether these compounds manifested their anti-proliferative activity through Hsp90 inhibition. Compounds, 49a, 49e and 49f induced client protein degradation (Her2, Raf and Akt) at concentrations that mirrored their anti-proliferative IC50 values. Hsp90 levels remained constant or slightly decreased, suggesting these compounds modulate Hsp90 through C-terminal inhibition. Subsequent dose-dependent analysis of Hsp90-dependent client proteins in MCF-7 cells upon administration of compound 49e (Figure 6B) demonstrated that Her2, Raf-1 and Akt underwent degradation in a concentration-dependent manner when exposed to 49e, reflecting that anti-proliferative activity is directly linked to Hsp90 inhibition.
Figure 6.

Western blot analyses of Hsp90-dependent client proteins from MCF-7 breast cancer cell lysate upon treatment with biphenyl derivatives. Concentrations (in μM) were indicated above each lane. H represents a concentration equal to 5-fold of the anti-proliferative activity. L represents a concentration equal to 0.5-fold of the anti-proliferative activity. Geldanamycin (G, 0.5 μM) and dimethylsulfoxide (D, 100%) were employed as positive and negative controls.
3. Conclusions
In conclusion, a small library of Hsp90 C-terminal inhibitors containing a biphenyl scaffold was designed and synthesized. These biphenyl derivatives were shown to serve as a suitable replacement for the coumarin ring system found in novobiocin. Western blot analyses demonstrated these compounds to manifest anti-proliferative activity through Hsp90 inhibition. Structural modifications to this scaffold led to structure-activity relationships and ultimately, small molecules that exhibit improved activities against these two breast cancer cell lines. Many of these molecules were shown to exhibit lead-like properties for the development of new Hsp90 C-terminal inhibitors. Identification of the biphenyl ring system provides rapid access to modifications that should enable succinct discovery of both structure-activity relationships and more potent Hsp90 C-terminal inhibitors.
4. Experimental section
4.1. Docking calculations
Initial receptor preparation before docking runs was performed using Schrödinger’s ‘Protein Preparation Wizard’ program (see www.Schrodinger.com), starting from the most representative protein conformation of previous MD simulations of Hsp90 in complex with novobiocin in the ATP bound state [25]. Bond orders and atomic charges were assigned and hydrogen atoms were added. The assignments of protonation states for basic and acidic residues were based on the optimization of hydrogen bonding patterns. The final minimization of the protein was performed with the Preparation Wizard default.
The shape and properties of the resulting binding site were mapped onto a grid with dimensions of 36 Å (enclosing box) and 14 Å (ligand diameter midpoint box), centered on the centroid of novobiocin.
Rigid receptor and flexible ligand docking calculations were performed using the program Glide (version 5.8 Schrödinger, LLC, New York, NY, 2012) [33, 34]. Docking calculations were performed in Standard Precision mode (SP) with standard OPLS-AA (2001) force field [35], non-planar conformations of amide bonds were penalized, Van der Waals radii were scaled by 0.80 and the partial charge cut off was fixed to 0.15. No further modifications were applied to the default settings.
4.2. Chemistry
1HNMR were recorded at 400 or 500 MHz (Bruker DRX-400 Bruker with a H/C/P/F QNP gradient probe) spectrometer and 13C NMR spectra were recorded at 100 or 125 MHz (Bruker DRX 500 with broadband, inverse triple resonance, and high resolution magic angle spinning HR-MA probe spectrometer); chemical shifts are reported in δ (ppm) relative to the internal reference chloroform-d (CDCl3, 7.27 ppm) or dimethyl sulfoxide-d6 (DMSO-d6, 2.50 ppm). High resolution mass spectra (FAB) were recorded with a LCT Premier (Waters Corp., Milford, MA) spectrometer. The purity of all compounds was determined to be >95% as determined by 1HNMR and 13CNMR spectra, unless otherwise noted. The most active 10 compounds were verified for >95% purity by HPLC analyses. TLC was performed on glassbacked silica gel plates (Uniplate) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure.
4.2.1. 4-(4-iodophenoxy)-1-methylpiperidine (11a): General procedure for the synthesis of compound 11a-b through Mitsunobu etherification
Diisopropylazodicarboxylate (1.89 g, 9.36 mmol) was added to an ice-cooled solution of iodophenol (0.92 g, 4.18 mmol), N-methyl-4-hydroxy-piperidine (480 mg, 4.18 mmol) and triphenylphosphine (2.46 g, 9.36 mmol) in anhydrous THF (10 mL). The reaction mixture was then allowed to stir at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated under reduced pressure and the residue was purified via column chromatography (SiO2, CH2Cl2: methanol, 10:1) to afford a thick oil (1.02 g, 77 %). 1H NMR (500 MHz, Chloroform-d) δ 7.54 (d, J = 8.9 Hz, 2H), 6.69 (d, J = 2.0 Hz, 2H), 4.27 (m, 1H), 2.73 – 2.59 (m, 2H), 2.31 (s, 3H), 2.30 (m, 2H), 1.98 (m, 2H), 1.82 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 157.34, 138.34, 118.50, 82.91, 72.18, 52.61, 46.28, 30.71. HRMS (ESI+) m/z: [M + H+] calcd for C12H17INO 318.0355; found 318.0357.
4.2.2. 4-(3-iodophenoxy)-1-methylpiperidine (11b)
Compound 11b was obtained as a yellow amorphous solid (611.1 mg, 46%). 1H NMR (500 MHz, Chloroform-d) δ 7.26 (m, 2H), 6.98 (t, J = 9.3 Hz, 1H), 6.87 (dd, J = 7.8, 2.3 Hz, 1H), 4.39 – 4.20 (m, 1H), 2.75 – 2.58 (m, 2H), 2.35 – 2.30 (m, 2H), 2.32 (s, 3H), 1.99 (m, 2H), 1.84 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 158.22, 131.03, 130.11, 125.39, 115.72, 94.61, 72.37, 52.67, 46.36, 30.81. HRMS (ESI+) m/z: [M + H+] calcd for C12H17INO 318.0355; found 318.0356.
4.2.1.1. 4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-amine (9a): General procedure for synthesis of 9a-c through Suzuki coupling
A mixture of iodide 11a (250 mg, 0.79 mmol) aminophenylboronic acid (216 mg, 1.58 mmol), potassium carbonate solution (2M, 100 μL) and [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (57 mg, 0.08 mmol) was suspended in dry dioxane (15 mL) and purged with argon for 15 min. After 15 min, the mixture was heated in a sealed tube at 110 °C for 12 hours before concentrated to dryness. The residue so obtained was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford a brownish amorphous solid (149 mg, 67 %). 1H NMR (500 MHz, Chloroform-d) δ 7.50 (d, J = 8.7 Hz, 2H), 7.22 (t, J = 7.8 Hz, 1H), 6.99 – 6.92 (m, 3H), 6.88 (s, 1H), 6.66 (dd, J = 7.9, 2.3 Hz, 1H), 4.45 – 4.34 (m, 1H), 3.74 (s, 2H), 2.79 (ddd, J = 11.8, 7.8, 3.8 Hz, 2H), 2.48 – 2.42 (m, 2H), 2.39 (s, 3H), 2.11 (ddt, J = 11.5, 7.3, 3.6 Hz, 2H), 1.94 (ddt, J = 14.0, 7.9, 3.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 156.77, 146.69, 141.94, 134.13, 129.65, 128.16, 117.28, 116.15, 113.60, 113.49, 71.45, 52.37, 45.96, 30.42. HRMS (ESI+) m/z: [M + H+] calcd for C18H23N2O 283.1810; found, 283.1808.
4.2.2.1. 3′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-amine (9b)
Compound 9b was obtained as a brownish amorphous (116 mg, 52%). 1H NMR (500 MHz, Chloroform-d) δ 7.19 (t, J = 7.9 Hz, 1H), 7.09 (t, J = 7.8 Hz, 1H), 7.03 (m, 2H, NH2), 6.98 – 6.96 (m, 1H), 6.86 – 6.83 (m, 1H), 6.80 (t, J = 2.0 Hz, 1H), 6.77 – 6.73 (m, 1H), 6.68 – 6.64 (m, 1H), 6.59 (dd, J = 8.1, 2.3 Hz, 1H), 4.36 (m, 1H), 2.66 (m, 2H), 2.37 (m, 2H), 2.25 (s, 3H), 1.92 (s, 2H), 1.80 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 157.29, 146.62, 143.06, 142.03, 129.70, 129.59, 119.98, 117.78, 115.03, 114.74, 114.64, 114.14, 72.15, 54.63, 45.46, 29.85. HRMS (ESI+) m/z: [M + H+] calcd for C18H23N2O 283.1810; found, 283.18108.
4.2.1.2. 4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-amine (9c)
Compound 9c was obtained as a yellowish amorphous solid (855.4 mg, 65%). 1H NMR (500 MHz, Chloroform-d) δ 7.89 (d, J = 4.3 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 4.3 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 4.42 (m, 1H), 2.79 – 2.61 (m, 2H), 2.39 (m, 2H), 2.36 (s, 3H), 2.08 (m, 2H), 1.91 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.13, 152.41, 149.27, 129.99, 127.82, 124.74, 121.22, 116.52, 72.02, 52.41, 46.08, 30.53. HRMS (ESI+) m/z: [M + H+] calcd for C18H23N2O 283.1810; found, 283.1811.
4.2.3. 2-(3-methylbut-2-en-1-yl)-4-((4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)carbamoyl)phenyl acetate (8a): General procedure for the synthesis of compounds 8a-c and 8g-i through amide coupling
A solution of acid chloride (75 mg, 0.27 mmol) in anhydrous dichloromethane (1 mL) was added to a solution of the aniline (50 mg, 0.18 mmol) and triethylamine (0.13 mL, 0.94 mmol) in anhydrous dichloromethane (1 mL). The resulting solution was allowed to stir at room temperature for 4 h. After 4 h, the solvent was removed and the residue was purified by column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford product as a white amorphous solid (48 mg, 59%). 1H NMR (500 MHz, Chloroform-d) δ 8.39 (d, J = 2.9 Hz, 1H, NH), 7.94 (t, J = 2.0 Hz, 1H), 7.79 (d, J = 2.3 Hz, 1H), 7.72 (dd, J = 8.4, 2.3 Hz, 1H), 7.59 – 7.57 (m, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.37 (t, J = 7.9 Hz, 1H), 7.33 – 7.29 (m, 1H), 7.07 (d, J = 8.3 Hz, 1H), 6.92 (d, J = 8.7 Hz, 2H), 5.30 – 5.06 (m, 1H), 4.54 – 4.25 (m, 1H), 3.26 (d, J = 7.3 Hz, 2H), 2.84 (ddd, J = 12.3, 8.9, 3.5 Hz, 2H), 2.62 (d, J = 8.1 Hz, 2H), 2.44 (s, 3H), 2.32 (s, 3H), 2.13 (m, 2H), 1.94 (m, 2H), 1.71 (s, 3H), 1.68 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.29, 165.64, 156.77, 151.67, 141.58, 138.73, 134.50, 134.01, 133.76, 133.00, 129.75, 129.49, 128.47, 125.97, 122.89, 122.72, 120.97, 118.85, 118.79, 116.33, 70.11, 51.69, 45.36, 29.51, 29.02, 25.88, 21.04, 18.05. HRMS (ESI+) m/z: [M + H+] calcd for C32H37N2O4 513.2753; found, 513.2752.
4.2.4. 2-(3-methylbut-2-en-1-yl)-4-((3′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)carbamoyl)phenyl acetate (8b)
Compound 8b was obtained as a white amorphous solid (46 mg, 72%). 1H NMR (500 MHz, Chloroform-d) δ 8.33 (s, 1H, NH), 7.85 (s, 1H), 7.71 (s, 1H), 7.66 – 7.61 (m, 1H), 7.61 – 7.56 (m, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.25 – 7.15 (m, 2H), 7.12 – 7.09 (m, 1H), 7.06 (d, J = 2.2 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.82 – 6.75 (m, 1H), 5.10 (m, 1H), 4.41 – 4.32 (m, 1H), 3.17 (d, J = 7.3 Hz, 2H), 2.74 (m, 2H), 2.46 (m, 2H), 2.32 (s, 3H), 2.24 (s, 3H), 2.01 (m, 2H), 1.90 – 1.77 (m, 2H), 1.63 (s, 3H), 1.60 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.13, 165.52, 157.39, 151.55, 149.71, 142.36, 141.71, 138.60, 134.37, 133.87, 132.81, 129.89, 129.37, 125.82, 123.20, 122.59, 120.82, 120.05, 119.41, 119.08, 115.13, 114.78, 70.49, 51.85, 45.45, 29.71, 28.88, 25.85, 20.93, 17.94. HRMS (ESI+) m/z: [M + H+] calcd for C32H37N2O4 513.2753; found 513.2758.
4.2.5. 2-(3-methylbut-2-en-1-yl)-4-((4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)carbamoyl)phenyl acetate (8c)
Compound 8c was obtained as a white amorphous solid (38 mg, 65%). 1H NMR (500 MHz, Chloroform-d) δ 7.81 (d, J = 2.3 Hz, 1H), 7.76 (dd, J = 8.3, 2.4 Hz, 1H), 7.72 (d, J = 8.6 Hz, 2H), 7.54 – 7.49 (m, 4H), 7.10 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 8.7 Hz, 2H), 5.20 (m, 1H), 4.56 (m, 1H), 3.28 (d, J = 7.2 Hz, 2H), 3.03 (m, 2H), 2.95 – 2.84 (m, 2H), 2.60 (s, 3H), 2.32 (s, 3H), 2.19 (m, 2H), 2.09 – 2.00 (m, 2H), 1.72 (s, 3H), 1.70 (s, 3H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 169.75, 166.51, 155.99, 151.39, 137.20, 136.62, 134.11, 133.92, 133.68, 132.94, 129.75, 127.98, 126.91, 126.14, 122.38, 121.09, 120.89, 116.27, 68.57, 50.89, 44.31, 28.86, 28.40, 25.48, 20.64, 17.63. HRMS (ESI+) m/z: [M + H+] calcd for C32H37N2O4 513.2753; found 513.2756.
4.2.3.1. 4-hydroxy-3-(3-methylbut-2-en-1-yl)-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)benzamide (8d): General procedure for the synthesis of 8d-f through ester hydrolysis
Compound 8a (24 mg, 0.047 mmol) was dissolved in a solution of 10% Et3N in methanol (1 mL) and stirred at room temperature for 24 hours before concentrated to dryness. The light brown residue so obtained was purified by flash chromatography using dichloromethane and methanol (v/v, 10:1) as eluent to afford a light brown amorphous solid (19 mg, 86%). 1H NMR (500 MHz, Chloroform-d) δ 7.89 – 7.82 (m, 2H), 7.70 (d, J = 2.4 Hz, 1H), 7.63 (dd, J = 8.3, 2.4 Hz, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.40 (t, J = 7.8 Hz, 1H), 7.35 – 7.30 (m, 1H), 6.96 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.3 Hz, 1H), 5.37 – 5.28 (m, 1H), 4.40 (m, 1H), 3.41 (d, J = 7.3 Hz, 2H), 2.77 (m, 2H), 2.45 (m, 2H), 2.37 (s, 3H), 2.10 – 2.02 (m, 2H), 1.96 – 1.89 (m, 2H), 1.78 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 165.93, 158.42, 157.18, 141.89, 138.74, 135.18, 133.63, 129.59, 129.52, 128.50, 128.15, 126.80, 126.73, 122.87, 121.46, 118.75, 118.68, 116.46, 115.78, 71.58, 52.43, 46.10, 30.47, 29.63, 26.07, 18.17. HRMS (ESI+) m/z: [M + H+] calcd for C30H35N2O3 471.2648; found 471.2644.
4.2.4.1. 4-hydroxy-3-(3-methylbut-2-en-1-yl)-N-(3′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)benzamide (8e)
Compound 8e was obtained as a light brown amorphous solid (26 mg, 72%). 1H NMR (400 MHz, Chloroform-d) δ 7.77 (d, J = 2.1 Hz, 1H), 7.54 (d, J = 2.3 Hz, 1H), 7.48 (dd, J = 8.3, 2.3 Hz, 1H), 7.45 – 7.41 (m, 1H), 7.22 (td, J = 7.8, 1.5 Hz, 1H), 7.19 – 7.12 (m, 2H), 7.07 – 7.02 (m, 1H), 7.00 (t, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.1, 2.4 Hz, 1H), 6.68 (dd, J = 8.3, 1.5 Hz, 1H), 5.17 (m, 1H), 4.40 (m, 1H), 3.23 – 3.09 (m, 2H), 2.80 (td, J = 10.8, 9.4, 3.5 Hz, 2H), 2.63 (d, J = 11.5 Hz, 2H), 2.36 (s, 3H), 1.99 – 1.90 (m, 2H), 1.89 – 1.77 (m, 2H), 1.56 (s, 3H), 1.54 (s, 3H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 167.21, 158.50, 156.92, 142.61, 141.33, 138.86, 132.96, 129.92, 129.17, 129.14, 128.44, 126.57, 125.47, 122.84, 121.85, 120.32, 119.83, 119.50, 115.01, 114.73, 114.55, 68.65, 51.02, 46.34, 28.50, 28.28, 25.59, 17.59. HRMS (ESI+) m/z: [M + H+] calcd for C30H35N2O3 471.2648; found 471.2648.
4.2.5.1. 4-hydroxy-3-(3-methylbut-2-en-1-yl)-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (8f)
Compound 8f was obtained as a white amorphous solid (16 mg, 78%). 1H NMR (400 MHz, Chloroform-d) δ 7.73 (m, 3H), 7.68 (dd, J = 8.4, 2.4 Hz, 1H), 7.58 – 7.47 (m, 4H), 7.00 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.4 Hz, 1H), 5.37 (m, 1H), 4.63 (m, 1H), 3.38 – 3.34 (m, 2H), 3.26 – 3.13 (m, 2H), 3.11 (m, 2H), 2.73 (s, 3H), 2.25 (m, 2H), 2.12 (m, 2H), 1.75 (s, 3H), 1.74 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.21, 158.39, 155.60, 140.93, 137.36, 136.09, 134.00, 132.66, 129.13, 128.29, 127.84, 126.46, 125.37, 121.81, 121.05, 116.11, 114.35, 69.04, 50.58, 46.24, 28.14, 27.73, 25.39, 17.38. HRMS (ESI+) m/z: [M + H+] calcd for C30H35N2O3 471.2648; found 471.2651.
4.2.6. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)-[1,1′-biphenyl]-3-carboxamide (8g)
Compound 8g was obtained as a white amorphous solid (48 mg, 52%). 1H NMR (500 MHz, Chloroform-d) δ 7.97 (s, 1H), 7.90 – 7.82 (m, 2H), 7.78 (s, 1H), 7.52 – 7.44 (m, 3H), 7.36 – 7.21 (m, 3H), 7.09 – 6.92 (m, 3H), 6.90 – 6.79 (m, 3H), 4.47 (s, 1H), 2.93 (ddd, J = 13.3, 10.4, 3.4 Hz, 2H), 2.84 – 2.73 (m, 2H), 2.26 (td, J = 10.5, 4.9 Hz, 2H), 2.04 – 1.91 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.26, 159.37, 159.32, 156.30, 141.41, 138.81, 138.61, 133.98, 130.67, 129.62, 129.43, 129.16, 128.47, 127.02, 122.66, 121.99, 118.63, 118.58, 116.15, 115.34, 112.93, 111.08, 68.68, 55.87, 55.35, 51.03, 44.81, 28.52. HRMS (ESI+) m/z: [M + H+] calcd for C33H35N2O4 523.2597; found 523.2599.
4.2.7. 3′,6-dimethoxy-N-(3′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-3-yl)-[1,1′-biphenyl]-3-carboxamide (8h)
Compound 8h was obtained as a white amorphous solid (114.7 mg, 62%). 1H NMR (500 MHz, Chloroform-d) δ 8.54 (s, 1H, NH), 7.98 (s, 1H), 7.89 – 7.81 (m, 2H), 7.77 (s, 1H), 7.65 – 7.53 (m, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.30 – 7.22 (m, 2H), 7.13 (dd, J = 7.7, 1.7 Hz, 1H), 7.08 (d, J = 2.5 Hz, 1H), 7.08 – 6.92 (m, 3H), 6.83 (m, 2H), 4.41 (m, 1H), 3.81 (s, 3H), 3.77 (s, 3H), 2.77 (m, 2H), 2.57 – 2.47 (m, 2H), 2.37 (s, 3H), 2.08 (m, 2H), 1.90 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.47, 159.57, 159.51, 157.58, 150.01, 142.61, 142.01, 138.99, 130.87, 130.08, 129.79, 129.63, 129.36, 128.65, 127.21, 123.32, 122.16, 120.32, 119.43, 119.14, 115.52, 115.42, 114.94, 113.13, 111.27, 70.53, 56.05, 55.53, 51.96, 45.72, 29.91. HRMS (ESI+) m/z: [M + H+] calcd for C33H35N2O4 523.2597; found 523.2593.
4.2.8. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (8i)
Compound 8i was obtained as a white amorphous solid (1.10 g, 78%). 1H NMR (500 MHz, Chloroform-d) δ 7.93 (dd, J = 8.6, 2.4 Hz, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 7.2 Hz, 2H), 7.49 (d, J = 7.2 Hz, 2H), 7.32 (t, J = 7.9 Hz, 1H), 7.12 (dt, J = 7.6, 1.3 Hz, 1H), 7.09 (dd, J = 2.6, 1.6 Hz, 1H), 7.04 (d, J = 8.7 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.89 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 4.57 (m, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.05 (m, 2H), 2.99 (m, 2H), 2.63 (s, 3H), 2.20 (ddt, J = 14.3, 10.4, 3.4 Hz, 2H), 2.12 – 1.98 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.41, 159.27, 159.17, 155.86, 148.91, 138.93, 137.31, 136.39, 134.01, 130.38, 130.07, 129.04, 128.58, 128.00, 126.91, 122.01, 121.08, 116.23, 115.26, 112.75, 110.88, 68.10, 55.69, 55.21, 50.60, 44.10, 28.06. HRMS (ESI+) m/z: [M + H+] calcd for C33H35N2O4 523.2597; found 523.2561.
4.2.9. 5-bromo-2-((1-methylpiperidin-4-yl)oxy)pyridine (16)
Diisopropylazodicarboxylate (809 mg, 4.0 mmol) was added to a solution of 5-bromopyridin-2-ol (348 mg, 2.0 mmol), N-methyl-4-hydroxy-piperidine (230 mg, 2.0 mmol) and triphenylphosphine (1.08 g, 4.0 mmol) in anhydrous THF (40 mL), and the resulting mixture was stirred at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness. The residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a thick oil (368 mg, 68%). 1H NMR (500 MHz, Chloroform-d) δ 8.15 (s, 1H), 7.62 (dd, J = 8.8, 2.5 Hz, 1H), 6.62 (dd, J = 8.8, 0.8 Hz, 1H), 5.01 (dt, J = 8.3, 4.2 Hz, 1H), 2.72 (m, 2H), 2.40 – 2.33 (m, 2H), 2.32 (s, 3H), 2.10 – 2.00 (m, 2H), 1.83 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 162.13, 147.58, 141.33, 113.49, 111.53, 70.64, 53.11, 46.28, 30.90. HRMS (ESI+) m/z: [M + H+] calcd for C11H16BrN2O 271.0446; found 271.0442.
4.2.10. 6-(4-nitrophenyl)pyridin-3-ol (17)
[1,1′-Bis(diphenylphosphino)ferrocene] dichloropalladium(II) (42 mg, 0.05 mmol) and potassium carbonate solution (2M, 100 μL) were added to a solution of 6-bromopyridin-3-ol (174 mg, 1.0 mmol) and 4-nitrophenylboronic acid (334 mg, 2.0 mmol) in dioxane (15 mL) and purged with argon for 15 min. After 15 min, the mixture was heated at 110 °C for 12 hours before concentrated to dryness. The brown residue was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a brown amorphous solid (162 mg, 75 %).1H NMR (500 MHz, Chloroform-d) δ 8.17 (m, 3H), 7.92 (dd, J = 9.1, 2.0 Hz, 2H), 7.59 (dd, J = 8.7, 1.6 Hz, 1H), 7.22 – 7.14 (m, 1H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 154.26, 147.30, 146.13, 145.30, 138.20, 126.93, 124.01, 123.67, 122.60. HRMS (ESI+) m/z: [M + H+] calcd for C11H18N2O3 226.1317; found 226.1319.
4.2.11. 2-((1-methylpiperidin-4-yl)oxy)-5-(4-nitrophenyl)pyridine (18a)
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (43 mg, 0.05 mmol) and potassium carbonate solution (2M, 100 μL) were added to a solution of bromide 16 (250 mg, 0.92 mmol) and 4-nitrophenylboronic acid (462 mg, 2.76 mmol) in dioxane (15 mL) and purged with argon for 15 min. After 15 min, the mixture was heated at 110 °C for 12 hours before concentrated to dryness. The brown residue so obtained was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a brown amorphous solid (246 mg, 85%). 1H NMR (500 MHz, Chloroform-d) δ 8.31 (d, J = 2.6 Hz, 1H), 8.21 (d, J = 8.8 Hz, 2H), 7.78 (dd, J = 8.6, 2.6 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.6 Hz, 1H), 5.07 (m, 1H), 2.72 (m, 2H), 2.41 (m, 2H), 2.29 (s, 3H), 2.04 (m, 2H), 1.92 – 1.76 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 163.45, 147.00, 145.46, 144.39, 137.76, 127.77, 127.17, 124.37, 112.09, 69.69, 52.49, 45.60, 30.15. HRMS (ESI+) m/z: [M + H+] calcd for C17H20N3O3 314.1505; found 314.1502.
4.2.12. 5-((1-methylpiperidin-4-yl)oxy)-2-(4-nitrophenyl)pyridine (18b)
Diisopropyl azodicarboxylate (279 mg, 1.38 mmol) was added to a solution of pridinol 17 (150 mg, 0.69 mmol), N-methyl-4-hydroxy-piperidine (80 mg, 0.69 mmol) and triphenylphosphine (362 mg, 1.38 mmol) in anhydrous THF (20 mL), and the resulting mixture was stirred at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated to the dryness and the remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (126 mg, 58%). 1H NMR (500 MHz, Chloroform-d) δ 8.46 – 8.38 (m, 1H), 8.28 (d, J = 8.9 Hz, 2H), 8.09 (d, J = 8.9 Hz, 2H), 7.74 (dd, J = 8.8, 0.7 Hz, 1H), 7.30 (dd, J = 8.7, 2.9 Hz, 1H), 4.45 (m, 1H), 2.72 (m, 2H), 2.40 – 2.35 (m, 1H), 2.33 (s, 3H), 2.11 – 2.00 (m, 2H), 1.92 – 1.86 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 154.01, 147.65, 147.22, 145.14, 139.63, 127.05, 124.20, 123.12, 121.88, 72.94, 52.50, 46.27, 30.70. HRMS (ESI+) m/z: [M + H+] calcd for C17H20N3O3 314.1505; found 314.1506.
4.2.13. 3′,6-dimethoxy-N-(4-(6-((1-methylpiperidin-4-yl)oxy)pyridin-3-yl)phenyl)-[1,1′-biphenyl]-3-carboxamide (19a): General procedure for the synthesis of 19a-b through reduction/amide coupling
Palladium on carbon (10 mg) was added to a solution of nitro phenyl 18a (82 mg, 0.27 mmol) in dry methanol (5 mL). The resulting mixture was stirred under hydrogen atmosphere for 2 hours. After 2 hours, the reaction mixture was filtered through celite. The filtrate was concentrated to dryness and used as such without further purification in the next step.
The amine (from the previous step) was dissolved in dry dichloromethane (0.5 ml) and added dropwise to an ice-cooled solution of acid chloride 10b (150 mg, 0.54 mmol) and pyridine (42 mg, 0.54 mmol) in dry dichloromethane (1 ml). The resulting mixture was stirred at room temperature for additional 4 hours before concentrated to dryness. The remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (68 mg, 48%). 1H NMR (500 MHz, Chloroform-d) δ 8.21 (s, 1H), 7.85 (dd, J = 7.7, 3.1 Hz, 1H), 7.80 (d, J = 2.5 Hz, 1H), 7.72 – 7.70 (m, 1H), 7.68 – 7.63 (m, 2H), 7.40 (d, J = 8.9 Hz, 2H), 7.23 (dd, J = 9.5, 6.4 Hz, 1H), 7.00 (m, 3H), 6.82 – 6.78 (m, 1H), 6.70 (d, J = 8.7 Hz, 1H), 5.13 (s, 1H), 3.78 (s, 3H), 3.74 (s, 3H), 2.98 (m, 2H), 2.90 – 2.71 (m, 2H), 2.52 (s, 3H), 2.16 (m, 2H), 2.02 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.42, 161.69, 159.35, 159.22, 144.52, 138.95, 137.98, 137.73, 133.34, 130.45, 130.07, 129.08, 128.65, 126.98, 126.96, 122.04, 121.22, 121.18, 115.32, 112.79, 111.38, 110.93, 66.64, 55.75, 55.26, 51.48, 44.36, 28.65. HRMS (ESI+) m/z: [M + H+] calcd for C32H34N3O4 524.2549; found 524.2551.
4.2.14. 3′,6-dimethoxy-N-(4-(5-((1-methylpiperidin-4-yl)oxy)pyridin-2-yl)phenyl)-[1,1′-biphenyl]-3-carboxamide (19b)
Compound 19b was obtained as a light brown solid (19 mg, 45%). 1H NMR (400 MHz, Methanol-d4) δ 8.33 (d, J = 2.9 Hz, 1H), 7.99 (dd, J = 8.6, 2.4 Hz, 1H), 7.95 (d, J = 2.4 Hz, 1H), 7.89 – 7.79 (m, 4H), 7.72 (d, J = 8.7 Hz, 1H), 7.42 (dd, J = 8.8, 3.0 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.16 (dt, J = 7.6, 1.2 Hz, 1H), 7.13 (dd, J = 2.6, 1.5 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.93 (ddd, J = 8.3, 2.7, 1.0 Hz, 1H), 4.79 (m, 1H), 3.91, (s, 3H), 3.87 (s, 3H), 3.36 (m, 4H), 2.87 (s, 3H), 2.48 – 2.40 (sm 2H), 2.34 – 2.16 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 166.57, 159.31, 159.15, 151.72, 150.82, 139.02, 138.89, 138.36, 134.22, 130.38, 130.13, 128.98, 128.58, 126.97, 126.86, 123.72, 121.96, 121.47, 120.79, 115.19, 112.72, 110.83, 66.83, 55.63, 55.13, 49.50, 43.31, 26.93. HRMS (ESI+) m/z: [M + H+] calcd for C32H34N3O4 524.2549; found 524.2546.
4.2.15. 5′-((6-bromopyridin-3-yl)carbamoyl)-2′-methoxy-[1,1′-biphenyl]-3-yl acetate (21a)
A solution of acid chloride 10b (300 mg, 1.16 mmol) in dichloromethane (1 ml) was added to a solution of 6-bromopyridin-3-amine (200 mg, 1.16 mmol) and pyridine (162mg, 2.32 mmol) in dry dichloromethane (5 mL). The solution was then stirred at room temperature for 4 hours. After 4 hours, the reaction mixture was concentrated to dryness and the remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (416 mg, 87%). 1H NMR (500 MHz, Chloroform-d) δ 8.47 (d, J = 2.8 Hz, 1H), 8.21 (s, 1H, NH), 8.17 (dd, J = 8.7, 2.9 Hz, 1H), 7.88 (dd, J = 8.6, 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.09 – 6.98 (m, 3H), 6.90 (dd, J = 8.2, 2.7 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.77, 159.94, 159.48, 141.64, 138.72, 136.00, 134.87, 130.96, 130.41, 129.84, 129.37, 128.84, 128.23, 126.05, 122.07, 115.57, 113.06, 111.32, 56.07, 55.52. HRMS (ESI+) m/z: [M + H+] calcd for C21H18BrN2O4 441.0450; found 441.0453.
4.2.16. 5′-((5-bromopyridin-2-yl)carbamoyl)-2′-methoxy-[1,1′-biphenyl]-3-yl acetate (21b)
A solution of acid chloride 10b (300 mg, 1.16 mmol) in dichloromethane (1 ml) was added to a solution of 5-bromopyridin-2-amine (200 mg, 1.16 mmol) and pyridine (162mg, 2.32 mmol) in dry dichloromethane (5 mL). The solution was then stirred at room temperature for 4 hours. After 4 hours, the reaction mixture was concentrated to dryness and the remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (392 mg, 82%). 1H NMR (500 MHz, Chloroform-d) δ 8.47 (t, J = 2.3 Hz, 1H), 8.19 (dt, J = 8.7, 3.0 Hz, 1H), 7.89 (dd, J = 8.6, 2.4 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.10 – 7.06 (m, 1H), 7.05 (dd, J = 2.6, 1.6 Hz, 1H), 7.02 (d, J = 8.7 Hz, 1H), 6.90 (dd, J = 8.4, 2.6 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.93, 159.87, 159.43, 141.61, 138.79, 135.73, 135.03, 130.84, 130.44, 129.97, 129.33, 128.90, 128.18, 126.10, 122.09, 115.56, 112.99, 111.24, 56.03, 55.50. HRMS (ESI+) m/z: [M + H+] calcd for C21H18BrN2O4 441.0450; found 441.0452.
4.2.17. N-(6-(4-hydroxyphenyl)pyridin-3-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (22a)
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (42 mg, 0.05 mmol) and potassium carbonate solution (2M, 100 μL) were added to a solution of bromide 21a (150 mg, 0.36 mmol) and 4-hydrophenylboronic acid (99 mg, 0.72 mmol) in dioxane (10 mL). The mixture was heated at 110 °C for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness. The brown residue so obtained was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a brown amorphous solid (117 mg, 76 %). 1H NMR (500 MHz, Chloroform-d) δ 8.54 (d, J = 2.6 Hz, 1H), 8.28 (dd, J = 8.7, 2.6 Hz, 1H), 7.94 – 7.81 (m, 2H), 7.63 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 8.7 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.04 (dt, J = 7.6, 1.3 Hz, 1H), 7.01 (d, J = 2.6 Hz, 1H), 6.97 (d, J = 8.6 Hz, 1H), 6.83 – 6.77 (m, 3H), 3.78 (s, 3H), 3.74 (s, 3H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.82, 159.50, 159.21, 157.87, 153.08, 140.97, 138.90, 133.90, 130.48, 130.30, 130.28, 129.38, 129.06, 128.76, 128.10, 126.42, 122.04, 120.40, 115.64, 115.27, 112.82, 110.90, 55.73, 55.24. HRMS (ESI+) m/z: [M + H+] calcd for C26H23N2O4 427.1658; found 427.1655.
4.2.18. 5′-((5-(4-hydroxyphenyl)pyridin-2-yl)carbamoyl)-2′-methoxy-[1,1′-biphenyl]-3-yl acetate (22b)
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (40 mg, 0.05 mmol) and patassium carbonate solution (2M, 100 μL) were added to a solution of bromide 21b (116 mg, 0.28 mmol) and 4-hydrophenylboronic acid (78 mg, 0.56 mmol) in dioxane (10 mL). The mixture was heated at 110 °C for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness. The brown residue so obtained was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a brown amorphous solid (110 mg, 92 %). 1H NMR (500 MHz, Chloroform-d) δ 8.46 – 8.38 (m, 2H), 7.98 – 7.93 (m, 2H), 7.91 (dd, J = 8.6, 2.5 Hz, 1H), 7.42 (d, J = 8.6 Hz, 2H), 7.35 (t, J = 7.9 Hz, 1H), 7.12 (dt, J = 7.6, 1.2 Hz, 1H), 7.10 (dd, J = 2.6, 1.5 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.92 (dd, J = 8.9, 2.3 Hz, 3H), 3.89 (s, 3H), 3.85 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.44, 159.91, 159.44, 156.76, 150.25, 145.29, 138.87, 136.84, 133.07, 131.02, 130.26, 129.32, 129.21, 128.66, 128.15, 126.31, 122.20, 116.18, 115.38, 114.33, 113.28, 111.25, 56.03, 55.51. HRMS (ESI+) m/z: [M + H+] calcd for C26H23N2O4 427.1658; found 427.1660.
4.2.19. 3′,6-dimethoxy-N-(6-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyridin-3-yl)-[1,1′-biphenyl]-3-carboxamide (19c)
Diisopropylazodicarboxylate (36 mg, 0.18 mmol) was added to a solution of phenol 22a (38 mg, 0.09 mmol), N-methyl-4-hydroxy-piperidine (21 mg, 0.18 mmol) and triphenylphosphine (47 mg, 0.18 mmol) in anhydrous THF (1 mL), and the resulting mixture was stirred at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness and the remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (31 mg, 67%). 1H NMR (500 MHz, Chloroform-d) δ 8.73 (d, J = 2.6 Hz, 1H), 8.56 (s, 1H), 8.32 (dd, J = 8.7, 2.7 Hz, 1H), 7.93 (dd, J = 8.6, 2.5 Hz, 1H), 7.88 – 7.82 (m, 3H), 7.62 (d, J = 8.6 Hz, 1H), 7.31 (t, J = 7.9 Hz, 1H), 7.11 – 7.05 (m, 2H), 6.99 (d, J = 8.6 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.89 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 4.48 – 4.31 (m, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 2.76 (m, 2H), 2.47 (m, 2H), 2.37 (s, 3H), 2.08 (m, 2H), 1.91 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 165.91, 159.65, 159.43, 157.97, 152.85, 141.46, 138.87, 133.61, 131.94, 130.76, 130.03, 129.29, 128.80, 128.71, 128.11, 126.57, 122.12, 119.94, 116.25, 115.50, 113.04, 111.15, 71.14, 55.99, 55.47, 52.24, 45.89, 30.22. HRMS (ESI+) m/z: [M + H+] calcd for C32H34N3O4 524.2549; found 524.2549.
4.2.20. 3′,6-dimethoxy-N-(5-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyridin-2-yl)-[1,1′-biphenyl]-3-carboxamide (19d)
Diisopropylazodicarboxylate (40 mg, 0.2 mmol) was added to a solution of phenol 22b (43 mg, 0.1 mmol), N-methyl-4-hydroxy-piperidine (24 mg, 0.2 mmol) and triphenylphosphine (52 mg, 0.2 mmol) in anhydrous THF (5 mL), and the resulting mixture was stirred at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness and the remaining residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford desired product as a light brown solid (34 mg, 65%). 1H NMR (500 MHz, Chloroform-d) δ 8.35 (d, J = 2.5 Hz, 1H), 8.30 (d, J = 8.7 Hz, 1H), 7.90 – 7.81 (m, 3H), 7.42 (d, J = 8.6 Hz, 2H), 7.24 (t, J = 8.0 Hz, 1H), 7.04 – 6.98 (m, 3H), 6.92 (d, J = 8.7 Hz, 2H), 6.82 – 6.78 (m, 1H), 4.58 (m, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.08 (m,, 4H), 2.65 (s, 3H), 2.25 (m, 2H), 2.06 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 165.73, 159.79, 159.20, 156.29, 150.46, 145.25, 138.69, 136.70, 132.33, 130.75, 130.71, 130.14, 129.08, 128.51, 128.13, 125.97, 121.96, 116.43, 115.17, 114.38, 112.96, 111.07, 66.72, 55.76, 55.21, 50.22, 43.80, 27.47. HRMS (ESI+) m/z: [M + H+] calcd for C32H34N3O4 524.2549; found 524.2548.
4.2.21. 3-methyl-4′-nitro-[1,1′-biphenyl]-4-ol (24a): General procedure for the synthesis of 24a-f through Suzuki coupling
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (42 mg, 0.05 mmol) and patassium carbonate solution (2M, 100 μL) were added to a solution of bromide 23a (187 mg, 1.00 mmol) and 4-nitrophenylboronic acid (249 mg, 1.50 mmol) in dioxane (5 mL). The mixture was refluxed at 110 °C for 12 hours before concentrated to dryness. The resulted brown residue was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a yellow amorphous solid (134 mg, 59%). Compound 24a was prepared following the general procedure B to afford a yellow amorphous solid (134 mg, 59%). 1H NMR (400 MHz, Chloroform-d + CD3OD) δ 8.28 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 8.9 Hz, 2H), 7.44 (s, 1H), 7.42 – 7.36 (m, 1H), 6.91 (d, J = 8.3 Hz, 1H), 2.36 (s, 3H). 13C NMR (126 MHz, CDCl3) δ154.94, 146.75, 145.29, 129.30, 129.07, 125.97, 125.05, 123.15, 122.42, 114.36, 15.18. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO3: 230.0817; found 230.0815.
4.2.22. 2-methyl-4′-nitro-[1,1′-biphenyl]-4-ol (24b)
Compound 24b was obtained as a yellow amorphous solid (185 mg, 40%). 1H NMR (400 MHz, Chloroform-d) δ 8.13 (d, J = 2.4 Hz, 1H), 8.06 (dd, J = 8.4, 2.5 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.26 – 7.14 (m, 2H), 7.00 – 6.91 (m, 2H), 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.57, 148.36, 146.85, 137.40, 132.38, 130.81, 130.34, 125.35, 121.08, 115.49, 20.90. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO3: 230.0817; found 230.0822.
4.2.23. 3-chloro-4′-nitro-[1,1′-biphenyl]-4-ol (24c)
Compound 24c was obtained as a yellow amorphous solid (180 mg, 74%). 1H NMR (500 MHz, CDCl3) δ 8.34 – 8.23 (m, 2H), 7.71 – 7.64 (m, 2H), 7.62 (d, J = 2.3 Hz, 1H), 7.48 (dd, J = 8.5, 2.3 Hz, 1H), 7.15 (d, J = 8.5 Hz, 1H), 5.74 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 152.10, 146.93, 145.83, 132.28, 127.88, 127.50, 127.24 (2C), 124.24 (2C), 120.75, 116.95. HRMS (ESI−) m/z [M-H+] calcd for C12H8ClNO3 248.0114, found 248.0117.
4.2.24. 2-chloro-4′-nitro-[1,1′-biphenyl]-4-ol (24d)
Compound 24d was obtained as a yellow amorphous solid (180 mg, 74%). 1H NMR (500 MHz, Chloroform-d) δ 8.25 – 8.14 (m, 2H), 7.58 – 7.43 (m, 2H), 7.11 (dt, J = 8.4, 1.8 Hz, 1H), 6.92 (t, J = 2.4 Hz, 1H), 6.80 – 6.71 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 158.21, 146.82, 146.34, 132.64, 131.88, 130.64, 129.48, 123.32, 117.12, 114.74. HRMS (ESI−) m/z [M+K]+ calcd for C12H8ClNO3 288.0214, found 288.2896.
4.2.25. 3-methoxy-4′-nitro-[1,1′-biphenyl]-4-ol (24e)
Compound 24e was obtained as a yellow amorphous solid (200 mg, 56%). 1H NMR (400 MHz, Chloroform-d) δ 8.27 (d, J = 8.9 Hz, 2H), 7.68 (d, J = 8.9 Hz, 2H), 7.17 (dd, J = 8.2, 2.1 Hz, 1H), 7.10 (d, J = 2.1 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 5.78 (s, 1H, OH), 3.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 147.70, 147.16, 146.86, 131.21, 127.37, 124.28, 120.99, 115.22, 109.76, 56.23. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO4: 246.0766; found 246.0762.
4.2.26. 2-methoxy-4′-nitro-[1,1′-biphenyl]-4-ol (24f)
Compound 24f was obtained as a yellow amorphous solid (160 mg, 44%). 1H NMR (500 MHz, Chloroform-d) δ 8.23 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 8.2 Hz, 1H), 6.62 – 6.45 (m, 2H), 4.96 (s, 1H, OH), 3.82 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 157.18, 156.88, 145.60, 144.69, 130.92, 129.41, 122.65, 120.40, 107.07, 98.88, 55.01. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO4: 246.0766; found 246.0769.
4.2.27. 2′-methyl-4′-nitro-[1,1′-biphenyl]-4-ol (27a): General procedure for the synthesis of 27a, 27c and 27e-f through Suzuki coupling
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (82 mg, 0.10 mmol) and patassium carbonate solution (2M, 100 μL) were added to a solution of 26a (621 mg, 2.36 mmol) and 4-hydroxyphenylboronic acid (326 mg, 2.36 mmol) in dioxane (40 mL). The mixture was refluxed at 110 °C for 12 hours before concentrated to dryness. The resulted brown residue was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a yellow amorphous solid (120 mg, 46%). 1H NMR (500 MHz, Chloroform-d) δ 8.14 (s, 1H), 8.08 (dd, J = 8.4, 2.4 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.6 Hz, 2H), 5.03 (s, 1H, OH), 2.38 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.63, 148.42, 146.92, 137.46, 132.45, 130.88, 130.41, 125.42, 121.14, 115.56, 20.97. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO3: 230.0817; found 230.0819.
4.2.28. 3′-methyl-4′-nitro-[1,1′-biphenyl]-4-ol (27b)
A mixture of boronic acid (300 mg, 2.175 mmol), 4-chloro-2-methyl-1-nitrobenzene (373 mg, 2.175 mmol), Pd(OAc)2 (5 mg, 0.022 mmol), TBAB (723 mg, 2.175 mmol) and 2M Na2CO3 was irridated by microwave at 175° C for 10 min. The reaction mixture was then extracted by ethyl acetate. The organic layer was collected, dried (over Na2SO4) and concentrated under reduced pressure. The brown residue was purified by flash column chromatography (SiO2, 10:1, EtOAc:Hexane) to afford desired product as a yellowish amorphous solid (80 mg, 17 %). 1H NMR (500 MHz, Chloroform-d) δ 8.09 (d, J = 9.0 Hz, 1H), 7.62 – 7.40 (m, 4H), 7.03 – 6.85 (m, 2H), 4.89 (s, 1H), 2.69 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.97, 147.51, 145.34, 144.15, 134.24, 133.92, 131.29, 130.48, 128.50, 125.32, 124.59, 115.74, 20.87. HRMS (ESI+) m/z [M+] calcd for C13H11NO3 229.0739, found 229.0742.
4.2.29. 2′-methoxy-4′-nitro-[1,1′-biphenyl]-4-ol (27c)
Compound 27c was obtained as a yellow amorphous solid (152 mg, 27%). 1H NMR (500 MHz, Chloroform-d) δ 7.91 (dd, J = 8.4, 2.2 Hz, 1H), 7.82 (d, J = 2.2 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 7.45 (s, 1H), 6.93 (d, J = 8.6 Hz, 2H), 3.93 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 156.87, 155.93, 147.77, 137.21, 131.11, 130.84, 129.03, 116.38, 115.45, 106.37, 56.29. HRMS (ESI+) m/z: [M + H+] calcd for C13H12NO4: 246.0766; found 246.0763.
4.2.30. 3′-methoxy-4′-nitro-[1,1′-biphenyl]-4-ol (27d)
A mixture of boronic acid (300 mg, 2.18 mmol), 4-chloro-2-methoxy-1-nitrobenzene (408 mg, 2.18 mmol), Pd(OAc)2 (5 mg, 0.022 mmol), TBAB (723 mg, 2.18 mmol) and 2M Na2CO3 (3.27 ml, 6.54 mmol) was irridated by microwave at 175° C for 10 min. The reaction mixture was then extracted by ethyl acetate. The organic layer was collected, dried (over Na2SO4) and concentrated under reduced pressure. The brown residue was purified by column chromatography (SiO2, 10:1, EtOAc:Hexane) to afford desired product as a yellowish amorphous solid (95 mg, 18 %). 1H NMR (400 MHz, Chloroform-d) δ 7.93 (d, J = 8.5 Hz, 1H), 7.48 – 7.42 (m, 2H), 7.19 – 7.11 (m, 2H), 6.95 – 6.87 (m, 2H), 4.00 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 156.80, 152.76, 146.98, 136.59, 129.71, 127.71, 125.69, 117.48, 115.07, 110.41, 55.59. Exact Mass, Calculated for C13H11NO4 (M-H): 244.0546; found (M-H): 244.0542.
4.2.31. 2′-chloro-4′-nitro-[1,1′-biphenyl]-4-ol (27e)
Compound 27e obtained as a yellow amorphous solid (300 mg, 59%). 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J = 2.4 Hz, 1H), 8.16 (dd, J = 8.5, 2.3 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.44 – 7.31 (m, 2H), 6.99 – 6.91 (m, 2H), 4.91 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 156.08, 147.01, 146.55, 133.51, 131.81, 130.77 (2C), 129.87, 125.33, 121.83, 115.35 (2C). HRMS (ESI−) m/z [M-H+] calcd for C12H8ClNO3 248.0114, found 248.0118.
4.2.32. 3′-chloro-4′-nitro-[1,1′-biphenyl]-4-ol (27f)
Compound 27f was obtained as a yellow amorphous solid (259 mg, 42%). 1H NMR (500 MHz, Chloroform-d) δ 7.95 (d, J = 8.5 Hz, 1H), 7.67 (d, J = 1.9 Hz, 1H), 7.52 (dd, J = 8.5, 2.0 Hz, 1H), 7.47 – 7.41 (m, 2H), 6.95 – 6.86 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 154.32, 142.77, 141.49, 125.48, 124.80, 124.64, 123.86, 122.46, 121.13, 112.19. HRMS (ESI−) m/z [M-H+] calcd for C12H8ClNO3 248.0114, found 248.0108.
4.2.33. 1-methyl-4-((3-methyl-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)piperidine (25a): General procedure for the synthesis of 25a-f and 28a-f
Diisopropylazodicarboxylate (0.94 mL, 6.20 mmol) was added to a solution of phenol (280 mg, 1.20 mmol), PPh3 (1.28g, 6.20 mmol) and 4-hydroxy N-methyl piperidine(280 mg, 2.40 mmol) in THF (8 mL) at room temperature. The reaction mixture was stirred for 18 hours before the removal of solvent under reduced pressure. The remaining residue was purified by silica gel column chromatography (eluting with methylene chloride: methanol = 99:1 to 20:1) to yield 25a as a light brown amorphous solid (180mg, 46%). Compound 25a was prepared following the general procedure A to afford a yellow amorphous solid (180 mg, 46%). 1H NMR (500 MHz, Chloroform-d) δ 8.35-8.18 (m, 2H), 7.75-7.62 (m, 2H), 7.49-7.36 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 4.45 (s, 1H), 2.66 (s, 2H), 2.44-2.34 (m, 2H), 2.33 (s, 3H), 2.31 (s, 3H), 2.11-1.86 (m, 4H). 13C NMR (126 MHz, CDCl3) δ156.59, 147.57, 146.54, 130.70, 130.04, 128.86, 127.15, 124.23, 113.06, 52.57, 46.47, 30.92, 29.85, 16.80. Exact Mass Calculated for C19H23N2O3 (M+H+): 327.1709; found (M+H+) 327.1724
4.2.35. 1-methyl-4-((2-methyl-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)piperidine (25b)
Compound 25b was obtained as a yellow amorphous solid (300 mg, 61%). 1H NMR (500 MHz, Chloroform-d) δ 8.26 (d, J = 8.9 Hz, 2H), 7.68 (d, J = 8.9 Hz, 2H), 7.47 – 7.37 (m, 2H), 6.92 (d, J = 8.5 Hz, 1H), 4.49 (s, 1H), 2.77 – 2.68 (m, 2H), 2.50 (s, 2H), 2.39 (s, 3H), 2.31 (s, 3H), 2.15 – 2.08 (m, 2H), 2.02 – 1.92 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 156.51, 147.61, 146.68, 130.96, 130.20, 127.28, 124.35, 113.11, 52.42, 46.27, 30.57, 16.90. IR 2954, 2923, 2852, 2358, 2341, 1593, 1514, 1485, 1340, 1307, 1274, 1247, 1135, 1108, 1039 cm−1. Exact Mass: Calculated for C19H22N2O3 (M+Na+) 349.1528; found 349.1528.
4.2.36. 4-((3-methoxy-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (25c)
Compound 25c was obtained as a yellow amorphous solid (200 mg, 80%). 1H NMR (500 MHz, Chloroform-d: Acetone d6 (10:1)) δ 8.25 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 8.8 Hz, 2H), 7.18 – 7.08 (m, 2H), 7.00 (d, J = 8.4 Hz, 1H), 4.44 (dp, J = 6.9, 3.4 Hz, 1H), 3.92 (s, 3H), 2.90 (ddd, J = 11.9, 8.6, 3.4 Hz, 2H), 2.70 −2.60 (m, 1H), 2.43 (s, 3H), 2.11 (ddd, J = 12.5, 8.5, 3.8 Hz, 2H), 1.99 (s, 3H). 13C NMR (126 MHz, CDCl3: Acetone d6 (10:1)) δ 176.70, 151.31, 146.78, 132.86, 127.43, 124.20, 120.20, 117.41, 111.54, 56.26, 44.99, 22.83. Exact Mass: Calculated for C19H22N2O4 Na(M+Na) 365.1477; found 365.1473.
4.2.37. 4-((2-methoxy-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (25d)
Compound 25d was obtained as a yellow amorphous solid (191 mg, 78%). 1H NMR (500 MHz, Chloroform-d) δ 8.23 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.8 Hz, 1H), 6.59 (m, 2H), 4.41 (m, 1H), 3.82 (s, 3H), 2.75 (m, 2H), 2.38 (m, 2H), 2.36 (s, 3H), 2.06 (m, 2H), 2.00 – 1.82 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.53, 157.90, 146.38, 145.52, 131.49, 130.18, 123.45, 121.25, 106.92, 101.09, 70.09, 55.80, 52.72, 46.31, 30.88. Exact Mass: Calculated for C19H22N2O4 (M+H) 343.1658; found 365.1658.
4.2.38. 4-((3-chloro-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (25e)
Compound 25e was obtained as a yellow amorphous solid (200 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 8.8 Hz, 2H), 7.77 – 7.52 (m, 3H), 7.46 (dd, J = 8.5, 2.4 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 4.81 (m, 1H), 3.49 – 3.37 (m, 2H), 3.23 (m, 2H), 2.87 (s, 3H), 2.32 (m, 2H), 2.24 – 2.10 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 147.99, 142.90, 141.28, 129.32, 125.35, 123.24, 123.19, 122.97, 120.71, 120.08, 120.04, 111.82, 67.46, 54.11, 44.12, 26.76. HRMS (ESI+) m/z [M+H+] calcd for C18H19ClN2O3 347.1163; found 347.1159.
4.2.39. 4-((2-chloro-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (25f)
Compound 25f was obtained as a yellow amorphous solid (150 mg, 78%). 1H NMR (500 MHz, Chloroform-d) δ 8.28 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 0.7 Hz, 1H), 7.07 (d, J = 2.5 Hz, 1H), 6.92 (dd, J = 8.5, 2.5 Hz, 1H), 4.42 (m, 1H), 2.76 (m, 2H), 2.42 (m, 2H), 2.39 (s, 3H), 2.11 (m, 2H), 1.93 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 158.30, 147.19, 145.92, 133.15, 131.99, 130.72, 126.23, 123.55, 117.64, 115.13, 72.44, 52.65, 46.15, 30.40. HRMS (ESI+) m/z [M+H+] calcd for C18H19ClN2O3 347.1163; found 347.1158.
4.2.40. 1-methyl-4-((2′-methyl-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)piperidine (28a)
Compound 28a was obtained as a yellow amorphous solid (120 mg, 73%). 1H NMR (500 MHz, Methanol-d4) δ 8.14 (d, J = 2.4 Hz, 1H), 8.09 – 8.02 (m, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.31 – 7.24 (m, 2H), 7.03 (d, J = 8.7 Hz, 2H), 4.49 (q, J = 5.1, 4.6 Hz, 1H), 2.76 (s, 2H), 2.50 – 2.40 (m, 2H), 2.37 (s, 3H), 2.33 (s, 3H), 2.05 (ddd, J = 12.7, 6.5, 3.1 Hz, 2H), 1.91 – 1.81 (m, 2H). 13C NMR (126 MHz, MeOD) δ 158.59, 149.66, 148.08, 138.64, 133.53, 131.82, 131.22, 126.00, 124.48, 121.80, 116.90, 112.62, 79.50, 53.25, 46.10, 31.30, 20.88, 16.60. Exact Mass Calculated for C19H22N2O4Na (M+Na): 365.1477; found 365.1481.
4.2.41. 1-methyl-4-((3′-methyl-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)piperidine (28b)
Compound 28b was obtained as a yellow amorphous solid (80 mg, 75%). 1H NMR (500 MHz, Chloroform-d) δ 8.08 (d, J = 9.1 Hz, 1H), 7.55 (d, J = 8.7 Hz, 2H), 7.52 – 7.46 (m, 2H), 7.01 (d, J = 8.8 Hz, 2H), 4.46 (s, 1H), 2.83 – 2.79 (m, 2H), 2.69 (s, 3H), 2.56 – 2.48 (m, 2H), 2.42 (s, 3H), 2.18 – 2.14 (m, 2H), 1.98 – 1.94 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 158.10, 147.64, 145.77, 134.69, 131.60, 130.90, 128.79, 125.78, 125.02, 116.59, 71.39, 52.31, 45.99, 30.29, 21.34. Exact Mass Calculated for C19H22N2O4 (M+H): 327.1709; found: 327.1721.
4.2.42. 4-((2′-methoxy-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (28c)
Compound 28c was obtained as a yellow amorphous solid (114 mg, 65%). 1H NMR (400 MHz, Chloroform-d) δ 7.96 – 7.87 (m, 1H), 7.82 (d, J = 2.2 Hz, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 4.41 (m, 1H), 3.93 (s, 3H), 2.82 – 2.63 (m, 2H), 2.39 (m, 2H), 2.36 (s, 3H), 2.09 (m, 2H), 1.92 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 157.69, 156.87, 147.77, 137.20, 130.97, 130.83, 128.90, 116.39, 115.79, 106.37, 70.23, 56.30, 52.29, 45.98, 30.45. Exact Mass Calculated for C19H22N2O4Na (M+Na+): 365.1477; found: 327.1483.
4.2.43. 4-((3′-methoxy-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (28d)
Compound 28d was obtained as a yellow amorphous solid (85 mg, 60%). 1H NMR (400 MHz, Chloroform-d) δ 7.96 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 8.7 Hz, 2H), 7.22 – 7.14 (m, 2H), 7.01 (d, J = 8.7 Hz, 2H), 4.45 (s, 1H), 4.03 (s, 3H), 2.89 – 2.69 (m, 2H), 2.52 – 2.42 (m, 2H), 2.37 (s, 3H), 2.15 (d, J = 16.9 Hz, 2H), 1.94 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 158.23, 153.75, 147.55, 137.94, 131.76, 128.73, 126.72, 118.62, 116.53, 111.59, 56.67, 52.30, 45.99, 30.34. Exact Mass Calculated for C19H22N2O4 (M+H): 343.1658; found 343.1658
4.2.44. 4-((2′-chloro-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (28e)
Compound 28e was obtained as a yellow amorphous solid (225 mg, 81%). 1H NMR (500 MHz, Chloroform-d) δ 8.35 (d, J = 2.3 Hz, 1H), 8.15 (dt, J = 8.4, 2.2 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.43 – 7.34 (m, 2H), 7.05 – 6.94 (m, 2H), 4.41 (dt, J = 7.2, 3.7 Hz, 1H), 2.83 – 2.65 (m, 2H), 2.34 (s, 3H), 2.07 (ddd, J = 13.9, 7.1, 3.5 Hz, 2H), 1.92 (ddd, J = 13.2, 7.9, 3.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 158.01, 146.90, 146.62, 133.42, 131.78, 130.57, 129.47, 125.32, 121.80, 115.58, 71.98, 52.60, 46.17, 30.74. HRMS (ESI+) m/z [M+H+] calcd for C18H19ClN2O3 347.1163, found 347.1136.
4.2.45. 4-((3′-chloro-4′-nitro-[1,1′-biphenyl]-4-yl)oxy)-1-methylpiperidine (28f)
Compound 25c was obtained as a yellow amorphous solid (110 mg, 63%). 1H NMR (500 MHz, Chloroform-d) δ 7.99 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 1.9 Hz, 1H), 7.62 – 7.47 (m, 3H), 7.02 (d, J = 8.7 Hz, 2H), 4.44 (m, 1H), 2.76 (m, 2H), 2.42 (m, 2H), 2.38 (s, 3H), 2.10 (m, 2H), 1.93 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 158.71, 146.52, 145.96, 130.06, 129.83, 128.80, 128.10, 126.65, 125.45, 116.72, 71.93, 52.54, 46.21, 30.62. HRMS (ESI+) m/z [M+H+] calcd for C18H19ClN2O3 347.1163, found 347.1159.
4.2.46. 3′,6-dimethoxy-N-(3′-methyl-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29a): General procedure for the synthesis of 29a-l
Palladium on carbon (10% w/w, 20 mg) was added to a solution of 25a (164 mg, 0.5 mmol) in methanol. The reaction mixture was then stirred under hydrogen atmosphere overnight before filtration. The filtrate was concentrated to dryness to get aniline. The aniline was dissolved in anhydrous dichloromethane and slowly added to an ice-cooled solution of 4-(chlorocarbonyl)-2-(3-methylbut-2-en-1-yl)phenyl acetate (276 mg, 1.0 mmol) and pyridine (0.2 mL) in anhydrous dichloromethane (2 mL). The reaction mixture was allowed to stir at room temperature for 4 hours. After 4 hours, the solvent was removed and the residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford 29a as a white amorphous solid (210 mg, 78%). 1H NMR (500 MHz, Chloroform-d) δ 8.20 (s, 1H), 7.98 (dd, J = 8.6, 2.4 Hz, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H), 7.42 – 7.40 (m, 1H), 7.37 – 7.32 (m, 2H), 7.17 – 7.14 (m, 1H), 7.12 (dd, J = 2.6, 1.5 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 6.93 (dd, J = 8.3, 2.6 Hz, 1H), 6.84 (d, J = 8.5 Hz, 1H), 4.58 (m, 1H), 3.90 (s, 3H), 3.86 (s, 3H), 3.00 (m, 4H), 2.64 (s, 3H), 2.45 – 2.34 (m, 2H), 2.30 (s, 3H), 2.17 – 2.06 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.23, 159.32, 154.02, 149.81, 138.84, 137.10, 136.60, 133.49, 130.61, 129.75, 129.69, 129.15, 128.51, 127.79, 127.14, 127.08, 125.22, 122.01, 120.66, 115.33, 112.94, 112.83, 111.03, 67.89, 55.85, 55.35, 50.88, 44.64, 28.28, 16.63. HRMS (ESI+) m/z [M+H+] calcd for C34H37N2O4 537.2753; found 537.2754.
4.2.47. 3′,6-dimethoxy-N-(2′-methyl-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29b)
Compound 29b was obtained as a white amorphous solid (45 mg, 81%). 1H NMR (500 MHz, Chloroform-d) δ 7.89 (s, 1H), 7.87 (dd, J = 8.6, 2.4 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 7.34 – 7.33 (m, 1H), 7.31 – 7.26 (m, 2H), 7.06 (dt, J = 7.6, 1.3 Hz, 1H), 7.03 (dd, J = 2.7, 1.6 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 6.86 (dd, J = 8.3, 2.7 Hz, 1H), 6.79 (d, J = 8.5 Hz, 1H), 4.50 (s, 1H), 3.82 (s, 3H), 3.78 (s, 3H), 2.94 – 2.74 (m, 4H), 2.52 (s, 3H), 2.33 – 2.25 (m, 3H), 2.23 (m, 2H), 2.06 – 1.98 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.12, 159.34, 154.16, 138.83, 136.97, 136.71, 133.41, 130.69, 129.69, 129.60, 129.17, 128.45, 127.85, 127.61, 127.20, 127.10, 125.20, 121.99, 120.50, 115.33, 112.96, 112.87, 111.07, 68.06, 55.87, 55.35, 50.99, 44.88, 28.84, 16.64. HRMS (ESI+) m/z [M+H+] calcd for C34H37N2O4 537.2753; found 537.2756.
4.2.48. 3′,6-dimethoxy-N-(2-methyl-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29c)
Compound 29c was obtained as a white amorphous solid (25 mg, 76%). 1H NMR (500 MHz, Chloroform-d) δ 7.91 (s, 1H), 7.87 (dd, J = 8.6, 2.4 Hz, 1H), 7.77 (d, J = 2.3 Hz, 1H), 7.50 (d, J = 2.3 Hz, 1H), 7.44 (dd, J = 8.3, 2.3 Hz, 1H), 7.28 (t, J = 7.9 Hz, 1H), 7.16 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 8.3 Hz, 1H), 7.06 (dt, J = 7.8, 1.2 Hz, 1H), 7.03 (dd, J = 2.7, 1.5 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 6.87 – 6.83 (m, 3H), 4.46 (m, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 2.92 (m, 2H), 2.82 – 2.67 (m, 2H), 2.51 (s, 3H), 2.30 – 2.22 (m, 2H), 2.21 (s, 3H), 1.99 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.17, 159.33, 159.31, 155.62, 149.82, 138.85, 137.48, 137.01, 136.26, 134.56, 130.64, 130.56, 130.41, 129.62, 129.16, 128.44, 127.14, 122.00, 117.77, 115.46, 115.32, 112.95, 111.05, 69.02, 55.86, 55.35, 51.35, 44.89, 28.70, 20.75. HRMS (ESI+) m/z [M+H+] calcd for C34H37N2O4 537.2753; found 537.2762.
4.2.49. 3′,6-dimethoxy-N-(3-methyl-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29d)
Compound 29d was obtained as a white amorphous solid (13 mg, 58%). 1H NMR (500 MHz, Chloroform-d) δ 8.01 (s, NH), 7.90 (dd, J = 8.5, 2.4 Hz, 1H), 7.85 – 7.78 (m, 2H), 7.48 (dd, J = 8.4, 1.4 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 7.35 – 7.30 (m, 1H), 7.12 – 7.08 (m, 1H), 7.08 – 7.03 (m, 2H), 6.93 (dd, J = 8.5, 1.4 Hz, 2H), 6.91 – 6.87 (m, 1H), 4.52 – 4.38 (m, 1H), 3.86 (d, J = 1.2 Hz, 3H), 3.82 (d, J = 1.3 Hz, 3H), 2.92 – 2.54 (m, 4H), 2.44 (s, 3H), 2.34 (s, 3H), 2.11 (d, J = 10.8 Hz, 2H), 1.96 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 165.91, 159.56, 159.44, 156.55, 139.02, 138.03, 134.74, 133.89, 130.88, 130.78, 129.94, 129.33, 128.98, 128.58, 128.29 (2C), 127.10, 125.16, 124.33, 122.17, 116.44 (2C), 115.43, 113.08, 111.25, 69.95, 55.97, 55.37, 51.73, 45.43, 29.44, 18.24. HRMS (ESI+) m/z [M+H+] calcd for C34H36N2O4 537.2753, found 537.2757.
4.2.50. 3′,6-dimethoxy-N-(3′-methoxy-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29e)
Compound 29e was obtained as a white amorphous solid (33 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ 10.25 (s, 1H, NH), 8.03 (dd, J = 8.6, 2.4 Hz, 1H), 7.98 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 7.9 Hz, 1H), 7.27 (s, 1H), 7.26 (d, J = 5.7 Hz, 1H), 7.18 (d, J = 9.0 Hz, 1H), 7.14 – 7.07 (m, 3H), 6.95 (dd, J = 8.3, 2.6 Hz, 1H), 4.51 – 4.39 (m, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.80 (s, 3H), 3.09 (m, 2H), 2.83 (m, 2H), 2.57 (s, 3H), 2.05 (m, 2H), 1.83 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.74, 158.92, 158.71, 150.62, 149.53, 145.14, 138.77, 138.30, 134.92, 134.04, 129.80, 129.20, 129.14, 129.10, 126.80, 126.44, 121.73, 120.62, 118.41, 115.14, 112.54, 111.40, 110.68, 71.29, 55.84, 55.67, 55.07, 51.02, 43.45, 28.64. HRMS (ESI+) m/z [M+H+] calcd for C34H36N2O5 553.2702; found 553.2700.
4.2.51. 3′,6-dimethoxy-N-(2′-methoxy-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29f)
Compound 29f was obtained as a white amorphous solid (31 mg, 72%). 1H NMR (500 MHz, Chloroform-d) δ 7.86 (m, 2H), 7.77 (d, J = 2.4 Hz, 1H), 7.60 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 7.29 (t, J = 7.9 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H), 7.06 (dd, J = 7.6, 1.2 Hz, 1H), 7.03 (dd, J = 2.7, 1.6 Hz, 1H), 6.99 (d, J = 8.7 Hz, 1H), 6.85 (dd, J = 8.3, 2.7 Hz, 1H), 6.49 – 6.44 (m, 1H), 4.41 (m, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.71 (s, 3H), 2.90 – 2.67 (m, 2H), 2.64 (m, 2H), 2.44 (s, 3H), 2.23 – 2.10 (m, 2H), 2.01 – 1.84 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.09, 159.33, 159.30, 157.64, 138.85, 136.68, 134.22, 131.15, 130.67, 129.99, 129.61, 129.16, 128.40, 127.21, 123.46, 122.00, 119.79, 115.29, 112.99, 111.05, 106.57, 102.55, 100.79, 69.99, 55.86, 55.60, 55.35, 51.55, 45.22, 29.31. HRMS (ESI+) m/z [M+H+] calcd for C34H36N2O5 553.2702; found 553.2706.
4.2.52. 3′,6-dimethoxy-N-(2-methoxy-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29g)
Compound 29g was obtained as a white amorphous solid (42 mg, 79%). 1H NMR (500 MHz, Chloroform-d) δ 8.11 (s, 1H), 7.98 (dd, J = 8.6, 2.4 Hz, 1H), 7.88 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.48 (d, J = 8.6 Hz, 2H), 7.38 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 8.3 Hz, 1H), 7.17 – 7.13 (m, 2H), 7.12 (s, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.96 – 6.91 (m, 3H), 4.55 (m, 1H), 3.91 (s, 3H), 3.87 (s, 6H), 3.01 (m, 2H), 2.90 (m, 2H), 2.61 (s, 3H), 2.41 – 2.27 (m, 2H), 2.10 – 2.00 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.22, 159.39, 159.33, 156.73, 155.57, 149.83, 138.81, 138.52, 130.73, 130.68, 130.58, 129.61, 129.17, 128.48, 127.00, 125.88, 122.00, 115.44, 115.36, 112.93, 112.07, 111.09, 103.77, 68.43, 55.87, 55.67, 55.36, 50.95, 44.75, 28.50. HRMS (ESI+) m/z [M+H+] calcd for C34H36N2O5 553.2702; found 553.2699.
4.2.53. 3′,6-dimethoxy-N-(3-methoxy-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (29h)
Compound 29h was obtained as a white amorphous solid (50 mg, 65%). 1H NMR (500 MHz, Chloroform-d) δ 8.55 (d, J = 8.4 Hz, 1H), 8.51 (s, NH), 7.91 (dd, J = 8.5, 2.4 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.56 – 7.46 (m, 2H), 7.36 (t, J = 7.9 Hz, 1H), 7.27 (s, 1H), 7.19 (dd, J = 8.4, 1.9 Hz, 1H), 7.17 – 7.09 (m, 2H), 7.07 (dd, J = 5.3, 3.4 Hz, 2H), 6.99 – 6.95 (m, 2H), 6.93 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.59 (s, 1H), 3.97 (s, 3H), 3.87 (d, J = 15.2 Hz, 6H), 3.11 – 2.89 (m, 5H), 2.64 (s, 3H), 2.43 – 2.31 (m, 2H), 2.11 (dt, J = 14.3, 4.6 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 164.67, 159.22, 159.18, 155.89, 148.34, 138.81, 136.12, 134.27, 130.58, 129.61, 129.05, 128.10, 127.43, 126.81, 121.92, 119.95, 119.32, 116.15, 115.21, 112.87, 110.92, 108.31, 68.19, 55.82, 55.76, 55.23, 50.61, 44.45, 27.93. HRMS (ESI+) m/z [M+H+] calcd for C34H36N2O5 553.2702; found 553.2713.
4.2.54. N-(3′-chloro-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (29i)
Compound 29i was obtained as a white amorphous solid (20 mg, 47%). 1H NMR (500 MHz, Chloroform-d) δ 8.08 (s, 1H), 7.94 (dd, J = 8.6, 2.4 Hz, 1H), 7.85 (d, J = 2.4 Hz, 1H), 7.76 – 7.69 (m, 2H), 7.61 (d, J = 2.3 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.41 (dd, J = 8.5, 2.3 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.13 (dt, J = 7.6, 1.2 Hz, 1H), 7.10 (dd, J = 2.6, 1.6 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.92 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.55 (s, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.05 – 2.87 (m, 2H), 2.78 (d, J = 16.2 Hz, 2H), 2.52 (s, 3H), 2.32 – 2.18 (m, 2H), 2.05 (dq, J = 14.6, 4.6 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.19, 159.35, 159.30, 151.60, 138.78, 137.59, 135.18, 134.98, 130.64, 129.63, 129.14, 128.77, 128.46, 127.18, 126.97, 125.98, 124.76, 121.96, 120.57, 116.34, 115.32, 112.91, 111.03, 55.84, 55.32, 51.08, 45.24, 29.69, 29.06. HRMS (ESI+) m/z [M+H+] calcd for C33H33ClN2O4 557.2207; found 557.2215.
4.2.55. N-(2′-chloro-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (29j)
Compound 29j was obtained as a white amorphous solid (90 mg, 87%). 1H NMR (400 MHz, Chloroform-d) δ 8.56 (s, 1H), 7.99 (dd, J = 8.6, 2.4 Hz, 1H), 7.91 (d, J = 2.3 Hz, 1H), 7.83 – 7.72 (m, 2H), 7.41 – 7.30 (m, 3H), 7.22 (d, J = 8.4 Hz, 1H), 7.17 – 7.09 (m, 2H), 7.03 (d, J = 8.6 Hz, 1H), 6.98 (d, J = 2.5 Hz, 1H), 6.94 – 6.87 (m, 1H), 6.81 (dd, J = 8.5, 2.5 Hz, 1H), 4.52 (s, 1H), 3.85 (d, J = 11.2 Hz, 6H), 3.01 (d, J = 8.7 Hz, 4H), 2.62 (s, 3H), 2.36 (d, J = 7.4 Hz, 2H), 2.12 – 1.98 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.20, 159.35, 159.31, 149.80, 138.79, 137.31, 135.06, 133.06, 132.06, 131.96, 130.69, 130.22, 130.15, 129.60, 129.15, 128.41, 127.04, 121.97, 119.69, 117.24, 115.29, 114.70, 112.97, 111.03, 67.62, 55.85, 55.33, 50.82, 45.61, 29.86. HRMS (ESI+) m/z [M+] calcd for C33H33ClN2O4 557.2207; found 557.2209.
4.2.56. N-(2-chloro-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (29k)
Compound 29k was obtained as a white amorphous solid (35 mg, 39%). 1H NMR (500 MHz, Chloroform-d) δ 7.77 (m, 3H), 7.32 – 7.20 (m, 1H), 7.11 (d, J = 8.6 Hz, 2H), 7.07 (t, J = 7.9 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 6.86 (s, 1H), 6.84 (d, J = 9.2 Hz, 1H), 6.72 (d, J = 8.2 Hz, 2H), 6.62 (dd, J = 8.3, 2.7 Hz, 1H), 4.45 (m, 1H), 3.63 (s, 3H), 3.57 (s, 3H), 3.11 – 2.90 (m, 4H), 2.54 (s, 3H), 2.02 (m, 2H), 1.89 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.39, 160.09, 159.95, 140.43, 139.92, 135.32, 132.62, 131.98, 131.70, 131.14, 130.57, 130.25, 129.89, 127.79, 127.40, 122.88, 122.30, 119.94, 117.34, 116.36, 116.31, 113.42, 111.78, 69.59, 56.68, 56.10, 50.37, 44.39, 27.88. HRMS (ESI+) m/z [M+H+] calcd for C33H33ClN2O4 557.2207; found 557.2211.
4.2.57. N-(3-chloro-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (29l)
Compound 29l was obtained as a white amorphous solid (92 mg, 86%). 1H NMR (500 MHz, Chloroform-d) δ 8.61 (d, J = 8.6 Hz, 1H), 8.43 (s, NH), 7.95 (dd, J = 8.5, 2.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.60 (d, J = 2.1 Hz, 1H), 7.55 – 7.48 (m, 3H), 7.38 (t, J = 7.9 Hz, 1H), 7.17 – 7.08 (m, 4H), 6.98 (d, J = 8.8 Hz, 2H), 4.72 (s, 1H), 3.92 (s, 3H), 3.87 (s, 3H), 3.35 – 3.25 (m, 2H), 3.24 – 3.10 (m, 2H), 2.78 (s, 3H), 2.65 – 2.55 (m, 2H), 2.26 – 2.15 (m, 2H).13C NMR (126 MHz, CDCl3) δ 164.78, 159.68, 159.34, 138.69, 136.88, 133.72, 131.00, 129.84, 129.20, 128.26, 128.20, 127.27, 126.94, 126.79, 126.02, 123.38, 121.98, 121.69, 120.48, 116.24, 115.31, 113.06, 111.12, 71.59, 55.91, 55.34, 50.01, 43.62, 29.95. HRMS (ESI+) m/z [M+] calcd for C33H33ClN2O4 557.2207; found 557.2199.
4.2.48. tert-butyl (4′-hydroxy-3′-nitro-[1,1′-biphenyl]-4-yl)carbamate (32a): General procedure for the synthesis of 32a-b
Palladium tetraphenylphosphine (115 mg, 0.10 mmol) and potassium carbonate solution (2M, 100 μL) were added to a solution of 4-bromo-2-nitrophenol (150 mg, 0.69 mmol) and boronic ester (300 mg, 0.82 mmol) in dioxane (40 mL) and the mixture was refluxed at 110 °C for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness and the residue so obtained was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a yellow amorphous solid (136 mg, 60%). 1H NMR (500 MHz, Chloroform-d) δ 10.58 (s, 1H), 8.29 (d, J = 2.3 Hz, 1H), 7.81 (dd, J = 8.7, 2.4 Hz, 1H), 7.61 – 7.37 (m, 4H), 7.25 – 7.21 (m, 1H), 6.55 (s, 1H), 1.55 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.11, 151.21, 138.75, 138.34, 136.01, 135.15, 132.82, 127.25, 122.26, 120.39, 118.90, 81.15, 28.35. HRMS (ESI−) m/z [M-H+] calcd for C17H18N2O5 329.1137, found 329.1133.
4.2.49. tert-butyl (4′-hydroxy-2′-nitro-[1,1′-biphenyl]-4-yl)carbamate (32b)
Compound 32b was obtained as a yellow amorphous solid (210 mg, 72%). 1H NMR (500 MHz, Chloroform-d) δ 7.40 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 2.6 Hz, 1H), 7.28 (d, J = 5.0 Hz, 1H), 7.23 – 7.17 (m, 2H), 7.07 (dd, J = 8.4, 2.6 Hz, 1H), 6.53 (s, 1H), 5.47 (s, 1H), 1.54 (s, 9H).13C NMR (126 MHz, CDCl3) δ 155.35, 153.00, 149.83, 138.39, 133.34, 132.05, 129.00, 128.64, 119.86, 118.99, 111.40, 81.16, 28.64. HRMS (ESI−) m/z [M-H+] calcd for C17H18N2O5 329.1137, found 329.1132.
4.2.50. tert-butyl (4′-((1-methylpiperidin-4-yl)oxy)-3′-nitro-[1,1′-biphenyl]-4-yl)carbamate (33a): General procedure for the synthesis of 33a-b
Diisopropylazodicarboxylate (83 mg, 0.41 mmol) was added to an ice-cooled solution of phenol 32a (75 mg, 0.23 mmol), N-methyl-4-hydroxy-piperidine (31.5 mg, 0.27 mmol) and triphenylphosphine (150 mg, 0.54 mmol) in anhydrous THF (2 mL). The reaction mixture was then allowed to stir at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated under reduced pressure and the residue was purified via column chromatography (SiO2, CH2Cl2: methanol, 10:1) to afford a yellow amorphous semi-solid (80 mg, 82%). 1H NMR (500 MHz, Chloroform-d) δ 8.01 (d, J = 2.4 Hz, 1H), 7.70 (dd, J = 8.7, 2.4 Hz, 1H), 7.51 – 7.41 (m, 4H), 7.13 (d, J = 8.8 Hz, 1H), 6.57 (s, 1NH), 4.70 (s, 1H), 2.85 (s, 2H), 2.70 (s, 2H), 2.48 (s, 3H), 2.24 (s, 2H), 2.06 (ddd, J = 15.0, 7.7, 3.9 Hz, 2H), 1.54 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 153.49, 150.08, 141.99, 139.23, 134.69, 133.72, 132.74, 128.15, 124.54, 119.81, 117.21, 81.78, 51.83, 46.40, 30.62, 30.18, 29.24. HRMS (ESI+) m/z [M+H+] calcd for C23H29N3O5 428.2186; found 428.2177.
4.2.51. tert-butyl (4′-((1-methylpiperidin-4-yl)oxy)-2′-nitro-[1,1′-biphenyl]-4-yl)carbamate (33b)
Compound 32b was obtained as a yellow amorphous solid (175 mg, 89%). 1H NMR (500 MHz, Chloroform-d) δ 7.41 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 2.6 Hz, 1H), 7.29 (d, J = 33.1 Hz, 1H), 7.23 – 7.19 (m, 2H), 7.13 (dd, J = 8.6, 2.6 Hz, 1H), 6.53 (s, NH), 4.42 (s, 1H), 2.73 (s, 2H), 2.36 (s, 3H), 2.15 – 2.01 (m, 2H), 1.91 (d, J = 11.4 Hz, 2H), 1.65 – 1.54 (m, 2H), 1.54 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 156.95, 152.90, 149.98, 138.48, 133.18, 131.97, 128.97, 128.51, 120.36, 118.84, 111.29, 81.09, 52.25, 46.19, 30.42, 30.03, 28.63. HRMS (ESI+) m/z [M+H+] calcd for C23H29N3O5 428.2186; found 428.2182.
4.2.52. N-(4-bromo-3-nitrophenyl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (36a): General procedure for the synthesis of 36a-b
A solution of acid chloride 10b (200 mg, 0.72 mmol) in dry dimethylformamide (0.5 ml) was added slowly to a solution of aniline 35a (150 mg, 0.69 mmol) and pyridine (160 mg, 2.30 mmol) in dimethylformamide (1 mL) and heated at 90 °C for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness; diluted with water and extracted with ethyl acetate (3 × 10 ml). The organic layers were combined, dried (over Na2SO4) and concentrated. The residue was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a light brown solid (283 mg, 90%). 1H NMR (400 MHz, Chloroform-d) δ 8.28 (d, J = 2.5 Hz, 1H), 7.97 (broad, 1H, NH), 7.90 (dd, J = 8.6, 2.4 Hz, 1H), 7.79 (d, J = 2.4 Hz, 1H), 7.76 (d, J = 2.5 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.13 – 7.05 (m, 3H), 6.94 (dd, J = 8.3, 2.7 Hz, 1H), 3.91 (s, 3H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.35, 160.13, 159.57, 150.09, 138.71, 138.57, 135.53, 131.18, 129.76, 129.45, 128.81, 125.97, 124.32, 122.10, 116.92, 115.61, 113.18, 111.45, 108.40, 56.14, 55.56. HRMS (ESI+) m/z [M+H+] calcd for C21H18BrN2O5 457.0399, found 457.0402.
4.2.53. N-(4-bromo-2-nitrophenyl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (36b)
Compound 36b was obtained as a yellow amorphous solid (80 mg, 43%). 1H NMR (500 MHz, Chloroform-d) δ 11.30 (s, NH), 8.97 (d, J = 9.1 Hz, 1H), 8.43 (d, J = 2.4 Hz, 1H), 8.05 – 7.93 (m, 2H), 7.80 (dd, J = 9.1, 2.4 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.20 – 7.06 (m, 3H), 6.94 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 3.93 (s, 3H), 3.87 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.81, 160.78, 159.94, 139.61, 139.05, 137.11, 135.41, 131.81, 130.97, 129.82, 128.99, 126.57, 124.11, 122.53, 115.78, 115.65, 113.81, 111.79, 56.54, 55.92. HRMS (ESI+) m/z [M+H+] calcd for C21H17BrN2O5 457.0399, found 457.0402.
4.2.54. N-(4′-hydroxy-2-nitro-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (37a): General procedure for the synthesis of 37a-b
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (42 mg, 0.05 mmol) and potassium carbonate solution (2M, 100 μL) were added to a solution of bromide 36a (120 mg, 0.26 mmol) and 4-hydrophenylboronic acid (72 mg, 0.52 mmol) in dioxane (10 mL) and the mixture was refluxed at 110 °C for 12 hours. After 12 hours, the reaction mixture was concentrated to dryness and the residue so obtained was purified via column chromatography (SiO2, 100:1, CH2Cl2: acetone) to afford desired product as a brown amorphous solid (52 mg, 43 %). 1H NMR (400 MHz, Chloroform-d) δ 9.83 (s, 1H, NH), 8.16 (t, J = 1.8 Hz, 1H), 7.91 – 7.83 (m, 3H), 7.29 (d, J = 8.3 Hz, 1H), 7.25 – 7.22 (m, 1H), 7.05 (m, 3H), 7.03 – 6.97 (m, 2H), 6.82 (dd, J = 8.3, 2.6 Hz, 1H), 6.79 – 6.72 (m, 2H), 3.80 (s, 3H), 3.76 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.66, 159.52, 159.17, 156.95, 149.16, 138.84, 138.32, 131.98, 131.09, 130.42, 130.24, 129.07, 129.02, 128.78, 128.18, 127.52, 126.35, 123.76, 121.98, 115.52, 115.24, 112.74, 110.87, 55.67, 55.18. HRMS (ESI+) m/z [M+Na+] calcd for C27H22N2O6Na 493.1376, found 493.1371.
4.2.55. N-(4′-hydroxy-3-nitro-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (37b)
Compound 37b was obtained as a yellow amorphous solid (110 mg, 92%). 1H NMR (500 MHz, Chloroform-d) δ 11.35 (s, OH), 9.05 (d, J = 8.8 Hz, 1H), 8.45 (d, J = 2.3 Hz, 1H), 8.09 – 7.95 (m, 2H), 7.90 (dd, J = 8.8, 2.3 Hz, 1H), 7.59 – 7.47 (m, 2H), 7.38 (t, J = 7.9 Hz, 1H), 7.21 – 7.05 (m, 3H), 6.98 – 6.89 (m, 3H), 3.93 (s, 3H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.90, 160.62, 159.94, 156.48, 139.16, 137.21, 136.56, 134.72, 134.65, 131.73, 131.41, 130.97, 129.81, 128.96, 128.75, 126.96, 123.77, 123.11, 122.58, 116.65, 115.77, 113.82, 111.76, 56.53, 55.93. HRMS (ESI+) m/z [M+Na]+ calcd for C27H22N2O6 493.1376, found 493.3180.
4.2.56. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-3′-nitro-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (34a): General procedure for the synthesis of 34a-b
A solution of trifluoroacetic acid (0.5 ml) in anhydrous dichloromethane (0.5 ml) was added to an ice-cooled solution of boc-protected aniline 33a (65mg, 0.15 mmol) in anhydrous dichloromethane (0.5 ml) and allowed to stir at room temperature for 2 hours. After 2 hours, the reaction mixture was concentrated under high vacuum to afford a brownish amorphous semi-solid (48 mg, 98%), which was used as such without further purification in the next step.
Acid chloride (50 mg, 0.36 mmol) was added to a solution of aniline (50 mg, 0.18 mmol, obtained from previous step) and triethylamine (0.13 mL, 0.94 mmol) in anhydrous dichloromethane (5 mL) and stirred at room temperature for 4 hours. After 4 hours, the reaction mixture was concentrated and the residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford 34a as a yellow amorphous solid (30 mg, 63%). 1H NMR (500 MHz, Chloroform-d) δ 8.28 (s, 1H), 8.05 (d, J = 2.4 Hz, 1H), 7.97 (dd, J = 8.6, 2.4 Hz, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.81 (d, J = 8.3 Hz, 2H), 7.75 (dd, J = 8.7, 2.4 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.9 Hz, 1H), 7.17 – 7.09 (m, 3H), 7.06 (d, J = 8.7 Hz, 1H), 6.92 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 4.84 (s, 1H), 3.87 (d, J = 19.2 Hz, 6H), 3.30 – 2.89 (m, 4H), 2.67 (s, 3H), 2.54 – 2.37 (m, 2H), 2.16 – 2.09 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.28, 159.41, 159.29, 148.60, 140.80, 138.72, 138.34, 134.09, 133.66, 132.21, 130.59, 129.71, 129.13, 128.58, 127.17, 126.81, 123.85, 121.95, 120.77, 116.04, 115.35, 112.86, 111.03, 69.03, 55.84, 55.32, 49.78, 44.39, 27.82. HRMS (ESI+) m/z [M+H+] calcd for C33H33N3O6 568.2448, found 568.2445.
4.2.57. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-2′-nitro-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (34b)
Compound 34b was obtained as a yellow amorphous solid (125 mg, 63%). 1H NMR (500 MHz, Chloroform-d) δ 7.95 (s, 1H), 7.87 (dd, J = 8.5, 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.66 – 7.61 (m, 2H), 7.32 – 7.26 (m, 3H), 7.20 (d, J = 2.0 Hz, 1H), 7.06 (ddd, J = 9.0, 5.3, 1.9 Hz, 2H), 7.04 – 6.97 (m, 2H), 6.86 (ddd, J = 8.3, 2.7, 1.0 Hz, 1H), 4.50 (s, 1H), 3.80 (d, J = 18.6 Hz, 6H), 2.86 (t, J = 10.5 Hz, 2H), 2.48 (s, 3H), 2.23 (s, 3H), 2.04 – 1.88 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.20, 159.43, 159.33, 156.29, 149.67, 138.77, 138.05, 133.07, 132.82, 130.74, 129.68, 129.17, 128.71, 128.56, 128.43, 126.92, 121.98, 120.33, 119.86, 115.29, 113.01, 111.24, 111.08, 70.27, 55.87, 55.35, 51.14, 44.96, 29.71. HRMS (ESI+) m/z [M+H+] calcd for C33H33N3O6 568.2448, found 568.2446.
4.2.58. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-2-nitro-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (34c): General procedure for the synthesis of 34c-d
Diisopropylazodicarboxylate (93 mg, 0.46 mmol) was added to an ice-cooled solution of phenol 37a (110 mg, 0.23 mmol), N-methyl-4-hydroxy-piperidine (27 mg, 0.23 mmol) and triphenylphosphine (128 mg, 0.46 mmol) in anhydrous THF (10 mL). The reaction mixture was then allowed to stir at room temperature for 12 hours. After 12 hours, the reaction mixture was concentrated under reduced pressure and the residue was purified via column chromatography (SiO2, CH2Cl2: methanol, 10:1) to afford a yellow amorphous solid (85 mg, 65%). 1H NMR (500 MHz, Chloroform-d) δ 8.15 (s, 1H), 7.93 (dd, J = 8.5, 2.5 Hz, 2H), 7.90 (d, J = 8.7 Hz, 1H), 7.85 (s, 1H), 7.31 (dd, J = 8.4, 2.5 Hz, 2H), 7.30 – 7.27 (m, 1H), 7.17 – 7.13 (m, 1H), 7.07 (d, J = 7.7 Hz, 1H), 7.03 (d, J = 2.1 Hz, 1H), 7.01 (dd, J = 8.7, 2.4 Hz, 1H), 6.85 (m, 3H), 4.35 (s, 1H), 3.82 (s, 3H), 3.78 (s, 3H), 2.70 (m, 2H), 2.52 – 2.40 (m, 2H), 2.30 (s, 3H), 1.99 (m, 2H), 1.85 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.45, 159.66, 159.30, 157.16, 149.24, 138.93, 138.60, 132.22, 130.87, 130.58, 130.21, 129.71, 129.33, 129.18, 128.93, 126.43, 123.83, 122.11, 116.10, 115.66, 115.44, 112.86, 111.06, 70.57, 55.87, 55.39, 51.93, 45.64, 29.74. HRMS (ESI+) m/z [M+K+] calcd for C33H33N3O6K 606.2006, found 606.2007.
4.2.59. 3′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-3-nitro-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (34d)
Compound 34d was obtained as a yellow amorphous solid (25 mg, 39%) 1H NMR (500 MHz, Chloroform-d) δ 11.34 (s, 1H), 9.05 (d, J = 8.8 Hz, 1H), 8.45 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 8.1 Hz, 2H), 7.90 (dd, J = 8.8, 2.3 Hz, 1H), 7.58 – 7.52 (m, 2H), 7.37 (t, J = 7.9 Hz, 1H), 7.18 – 7.10 (m, 3H), 7.04 – 6.99 (m, 2H), 6.96 – 6.92 (m, 1H), 4.43 (s, 1H), 3.92 (s, 3H), 3.87 (s, 3H), 2.83 – 2.72 (m, 2H), 2.51 – 2.41 (m, 2H), 2.39 (s, 3H), 2.11 (ddd, J = 11.3, 8.1, 3.7 Hz, 2H), 1.93 (tdd, J = 10.9, 7.2, 3.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.23, 159.99, 159.32, 157.55, 138.55, 136.58, 135.90, 134.07, 134.03, 131.09, 130.67, 130.34, 129.19, 128.33, 127.95, 126.34, 123.12, 122.47, 121.95, 116.52, 115.15, 113.19, 111.14, 71.62, 55.81, 55.36, 52.44, 45.84, 29.73. HRMS (ESI+) m/z [M+] calcd for C33H33N3O6 567.2348, found 567.2339.
4.2.60. N-(3′-amino-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (38a): General procedure for the synthesis of 38a-d
Palladium on carbon (10%, 10 mg) was added to a solution of Nitro 34a (60 mg, 0.1 mmol), followed by two drops of acetic acid. The resulted suspension was degased and stirred under hydrogen atmosphere for 12 hours before filtration. The filtrate was concentrated to dryness to afford aniline 38a as a white amorphous solid (46 mg, 85%). 1H NMR (400 MHz, Chloroform-d) δ 7.94 (d, J = 8.7 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.3 Hz, 2H), 7.37 (t, J = 7.9 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 2.1 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 7.00 (s, 1H), 6.97 – 6.91 (m, 2H), 6.85 – 6.81 (m, 1H), 4.63 (m, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.16 (m, 4H), 2.72 (s, 3H), 2.42 (m, 2H), 2.20 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.31, 159.57, 159.54, 143.58, 139.02, 137.56, 137.24, 137.04, 135.04, 130.92, 129.77, 129.38, 128.63, 127.48, 127.28, 122.19, 120.62, 117.47, 115.54, 114.63, 114.31, 113.16, 111.29, 69.10, 56.08, 55.56, 50.61, 44.37, 28.10. HRMS (ESI+) m/z [M+H+] calcd for C33H36N3O4 538.2706, found 538.2707.
4.2.61. N-(2′-amino-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (38b)
Compound 38b was obtained as a white amorphous solid (28 mg, 39%). 1H NMR (500 MHz, Chloroform-d) δ 8.03 (d, J = 2.9 Hz, 1H), 7.94 (dd, J = 8.5, 2.4 Hz, 1H), 7.86 (d, J = 2.5 Hz, 1H), 7.71 (dd, J = 8.5, 2.7 Hz, 2H), 7.42 (dd, J = 8.7, 2.6 Hz, 2H), 7.36 (td, J = 7.9, 2.8 Hz, 1H), 7.17 – 7.09 (m, 2H), 7.05 (ddd, J = 18.4, 8.5, 2.7 Hz, 2H), 6.93 (dd, J = 8.2, 2.6 Hz, 1H), 6.38 (dt, J = 8.5, 2.6 Hz, 1H), 6.33 (d, J = 2.5 Hz, 1H), 4.42 (s, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.80 (s, NH2), 2.95 – 2.81 (m, 2H), 2.67 (s, 2H), 2.49 (s, 3H), 2.29 – 2.14 (m, 2H), 1.99 (d, J = 14.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.25, 159.34, 159.30, 157.35, 144.89, 138.79, 136.90, 135.13, 131.38, 130.65, 129.70, 129.63, 129.14, 128.45, 127.00, 121.96, 120.69, 120.60, 115.32, 112.91, 111.04, 106.02, 103.05, 69.81, 55.84, 55.32, 51.86, 45.30, 29.69. HRMS (ESI+) m/z [M+H+] calcd for C33H35N3O4 538.2706, found 538.2704.
4.2.62. N-(2-amino-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (38c)
Compound 38c was obtained as a white amorphous solid (21 mg, 85%). 1H NMR (400 MHz, Chloroform-d) δ 7.92 (dd, J = 8.5, 2.3 Hz, 1H), 7.82 (d, J = 2.4 Hz, 1H), 7.77 (s, 1H), 7.39 (d, J = 8.7 Hz, 3H), 7.14 (d, J = 7.9 Hz, 1H), 7.10 (d, J = 2.3 Hz, 1H), 7.07 (dd, J = 8.5, 2.2 Hz, 2H), 6.97 (d, J = 8.5 Hz, 2H), 6.94 (dd, J = 8.3, 2.7 Hz, 1H), 6.86 (dd, J = 8.2, 2.1 Hz, 1H), 4.58 (m, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.02 (m, 4H), 2.63 (s, 3H), 2.37 (m, 2H), 2.10 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.05, 159.33, 159.30, 155.73, 144.29, 138.82, 138.19, 132.21, 130.79, 130.69, 130.45, 129.51, 129.16, 128.34, 127.24, 123.26, 121.97, 116.17, 115.30, 112.96, 111.05, 110.21, 107.01, 69.03, 55.86, 55.34, 50.67, 44.57, 28.34. HRMS (ESI+) m/z [M+H+] calcd for C33H36N3O4 538.2706, found 538.2709.
4.2.63. N-(3-amino-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (38d)
Compound 38d was obtained as a white amorphous solid (25 mg, 90%). 1H NMR (400 MHz, Chloroform-d) δ 7.91 (dd, J = 9.0, 2.4 Hz, 1H), 7.87 (s, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.29 (t, J = 7.9 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.09 – 6.94 (m, 5H), 6.89 (d, J = 8.8 Hz, 2H), 6.85 (dd, J = 8.3, 2.6 Hz, 1H), 4.51 (m, 1H), 3.83 (s, 3H), 3.79 (s, 3H), 2.99 – 2.74 (m, 4H), 2.54 (s, 3H), 2.15 (m, 2H), 2.06 – 1.94 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.52, 159.32, 156.19, 141.22, 139.75, 138.96, 134.13, 130.65, 130.31, 129.19, 128.84, 128.42, 128.27, 126.33, 126.25, 123.71, 122.11, 118.33, 116.42, 116.23, 115.37, 112.96, 111.03, 68.80, 55.88, 55.39, 50.92, 44.54, 28.39. HRMS (ESI+) m/z [M+K+] calcd for C33H35N3O4K 576.2265, found 576.2264.
4.2.64. N-(3′-acetamido-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (39a): General procedure for the synthesis of 39a-d
Aniline 38a (23 mg, 0.04 mmol) was added to a solution of acetic anhydride in pyridine (1:3, v/v) and the resulting mixture was stirred at room temperature for 4 hours before being concentrated to dryness. The remaining residue was further dried under vacuum overnight to afford acetamide 39a as a light brown amorphous solid (25 mg, 100%). 1H NMR (500 MHz, Chloroform-d) δ 8.01 (s, 1H), 7.87 (dd, J = 8.6, 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.73 (s, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.29 (t, J = 7.9 Hz, 1H), 7.18 – 7.15 (m, 1H), 7.06 (dt, J = 7.6, 1.2 Hz, 1H), 7.03 (s, 1H), 6.99 (d, J = 8.7 Hz, 1H), 6.85 (ddd, J = 8.3, 2.7, 1.0 Hz, 1H), 6.80 (d, J = 8.6 Hz, 1H), 4.50 (m, 1H), 3.82 (s, 3H), 3.78 (s, 3H), 3.15 (m, 2H), 2.94 – 2.82 (m, 2H), 2.61 (s, 3H), 2.35 – 2.26 (m, 2H), 2.18 (s, 3H), 2.17 – 2.07 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 174.51, 168.78, 165.41, 159.58, 159.53, 139.02, 137.46, 136.40, 134.43, 130.89, 129.89, 129.37, 128.65, 127.52, 127.20, 125.01, 123.84, 123.04, 122.20, 121.88, 120.81, 115.54, 113.15, 111.26, 69.90, 56.07, 55.55, 51.00, 44.13, 29.92, 28.34. HRMS (ESI+) m/z [M+K+] calcd for C35H37N3O5K 618.2370, found 618.2373.
4.2.65. N-(2′-acetamido-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (39b)
Compound 39b was obtained as a white amorphous solid (11 mg, 100%). 1H NMR (400 MHz, Chloroform-d) δ 7.82 (dd, J = 8.6, 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.58 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 2.5 Hz, 1H), 7.23 – 7.15 (m, 3H), 7.04 (d, J = 8.4 Hz, 1H), 6.99 (dt, J = 7.7, 1.3 Hz, 1H), 6.97 – 6.93 (m, 2H), 6.78 – 6.74 (m, 1H), 6.63 (dd, J = 8.5, 2.6 Hz, 1H), 4.42 (m, 1H), 3.75 (s, 3H), 3.70 (s, 3H), 2.89 (m, 2H), 2.73 (m, 2H), 2.44 (s, 3H), 2.12 – 2.00 (m, 2H), 1.99 – 1.92 (m, 2H), 1.89 (s, 3H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 168.45, 165.21, 158.75, 158.51, 138.57, 138.17, 135.32, 132.82, 130.44, 129.80, 129.30, 128.91, 128.75, 128.52, 126.81, 126.77, 121.55, 120.50, 120.40, 114.95, 112.09, 110.31, 110.24, 70.60, 55.30, 54.76, 51.21, 44.76, 29.06, 23.74. HRMS (ESI+) m/z [M+K+] calcd for C35H37N3O5K 618.2370, found 618.2372.
4.2.66. N-(2-acetamido-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (39c)
Compound 39c was obtained as a white amorphous solid (15 mg, 100%). 1H NMR (500 MHz, Chloroform-d) δ 7.76 (dd, J = 8.5, 2.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 2.2 Hz, 1H), 7.56 (dd, J = 8.4, 2.2 Hz, 1H), 7.14 (t, J = 7.9 Hz, 1H), 7.10 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.4 Hz, 1H), 6.96 – 6.93 (m, 1H), 6.92 (s, 1H), 6.90 (d, J = 8.6 Hz, 1H), 6.80 (d, J = 8.6 Hz, 2H), 6.71 (dd, J = 8.2, 2.6 Hz, 1H), 4.42 (m, 1H), 3.70 (s, 3H), 3.65 (s, 3H), 2.91 (m, 2H), 2.80 (m, 2H), 2.47 (s, 3H), 2.02 (m, 2H), 1.92 – 1.87 (m, 2H), 1.82 (s, 3H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 170.20, 166.68, 159.26, 159.09, 156.03, 138.85, 137.89, 134.07, 131.30, 130.75, 130.48, 130.34, 130.26, 130.12, 128.87, 128.49, 126.70, 121.88, 118.69, 117.00, 115.93, 115.06, 112.68, 110.74, 69.73, 55.51, 50.71, 43.95, 29.51, 28.22. HRMS (ESI+) m/z [M+K+] calcd for C35H37N3O5K 618.2370, found 618.2367.
4.2.67. N-(3-acetamido-4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′,6-dimethoxy-[1,1′-biphenyl]-3-carboxamide (39d)
Compound 39b was obtained as a white amorphous solid (10 mg, 100%). 1H NMR (400 MHz, Chloroform-d) δ 7.81 (m, 2H), 7.61 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 2.3 Hz, 1H), 7.32 (dd, J = 8.4, 2.1 Hz, 1H), 7.21 (t, J = 7.9 Hz, 1H), 7.02 (d, J = 7.9 Hz, 1H), 7.00 – 6.98 (m, 1H), 6.98 – 6.94 (m, 1H), 6.84 (d, J = 8.9 Hz, 2H), 6.77 (dd, J = 8.2, 2.5 Hz, 1H), 4.46 (m, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 2.91 – 2.75 (m, 4H), 2.50 (s, 3H), 2.09 (m, 2H), 2.06 (s, 3H), 1.95 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 170.95, 166.15, 159.53, 159.19, 156.19, 138.82, 138.31, 133.28, 130.63, 130.56, 130.37, 129.94, 129.03, 128.35, 128.14, 126.19, 126.10, 124.58, 122.95, 121.96, 116.21, 115.17, 112.87, 110.98, 68.17, 55.71, 55.18, 50.64, 44.20, 28.12, 23.18. HRMS (ESI+) m/z [M+K+] calcd for C35H37N3O5K 618.2370, found 618.2368.
4.2.68.1. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41a): General procedure for the synthesis of 41a-41s, 43a-b, 45a-b, 47a-b and 49a-h
Acid chloride 40a (50 mg, 0.36 mmol) was added to a solution of aniline 9c (50 mg, 0.18 mmol) and triethylamine (0.13 mL, 0.94 mmol) in anhydrous dichloromethane (5 mL). After 12 h, the solvent was removed and the residue was purified via column chromatography (SiO2, 10:1, CH2Cl2: methanol) to afford 41a as a white amorphous solid (51 mg, 74%). 1H NMR (500 MHz, Chloroform-d) δ 7.95 (d, J = 6.9 Hz, 2H), 7.77 (d, J = 8.6 Hz, 2H), 7.61 – 7.53 (m, 5H), 7.53 – 7.49 (m, 2H), 7.02 (d, J = 8.7 Hz, 2H), 4.71 (s, 1H), 3.27 (m, 4H), 2.82 (s, 3H), 2.43 – 2.29 (m, 2H), 2.25 – 2.11 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.69, 156.00, 137.67, 136.89, 135.17, 134.64, 132.09, 128.83, 128.43, 127.71, 127.27, 121.56, 116.58, 67.03, 50.55, 46.71, 27.76. HRMS (ESI+) m/z: [M + H+] calcd for C25H27N2O2 387.2073; found 387.2071.
4.2.68.2. 4-chloro-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41b)
Compound 41b was obtained as a white amorphous solid (20 mg, 65%). 1H NMR (500 MHz, DMSO-d6) δ 8.00 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.7 Hz, 2H), 7.62 (m, 4H), 7.05 (d, J = 8.8 Hz, 2H), 4.53 (s, 1H), 2.93 (m, 1H), 2.68 – 2.55 (m, 1H), 2.45 (s, 3H), 2.03 (m, 2H), 1.79 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.32, 156.16, 137.82, 136.36, 135.08, 133.56, 132.34, 129.61, 128.44, 127.44, 126.23, 120.67, 116.29, 69.80, 51.33, 44.20, 29.09. HRMS (ESI+) m/z: [M + H+] calcd for C25H26ClN2O2 421.1683; found 421.1681.
4.2.68.3. 4-bromo-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41c)
Compound 41c was obtained as a white amorphous solid (25 mg, 70%).Compound 41c was prepared from 19c using general procedure C and acid chloride 7 to afford a white amorphous solid (28 mg, 88%). 1H NMR (400 MHz, Chloroform-d) δ 7.66 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 4.31 (m, 1H), 2.65 (d, J = 9.7 Hz, 2H), 2.40 (s, 2H), 2.25 (s, 3H), 1.93 (m, 2H), 1.79 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.49, 156.37, 137.82, 135.18, 133.98, 132.21, 131.43, 129.83, 127.46, 126.27, 125.36, 120.71, 116.27, 71.02, 52.10, 45.12, 29.96. HRMS (ESI+) m/z: [M + H+] calcd for C25H26BrN2O2 465.1178; found 465.1181.
4.2.68.4. 4-iodo-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41d)
Compound 41d was obtained as a white amorphous solid (32 mg, 80%).1H NMR (400 MHz, Chloroform-d) δ 7.86 – 7.73 (m, 2H), 7.64 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.7 Hz, 2H), 6.94 – 6.76 (m, 2H), 4.39 (m, 1H), 2.74 (m, 2H), 2.48 (m, 2H), 2.34 (s, 3H), 2.13 – 1.97 (m, 2H), 1.88 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 166.30, 156.37, 137.69, 136.95, 136.87, 134.34, 133.60, 129.01, 127.90, 126.91, 121.11, 116.30, 98.61, 69.97, 51.89, 45.12, 29.41. HRMS (ESI+) m/z: [M + H+] calcd for C25H26IN2O2 513.1039; found 513.1042.
4.2.68.5. 4-methyl-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41e)
Compound 41e was obtained as a white amorphous solid (35 mg, 69%).1H NMR (500 MHz, Chloroform-d) δ 7.82 (d, J = 8.2 Hz, 2H), 7.74 – 7.68 (m, 2H), 7.52 (ddd, J = 8.6, 4.5, 2.2 Hz, 4H), 7.36 – 7.18 (m, 2H), 7.06 – 6.76 (m, 2H), 4.60 (d, J = 10.7 Hz, 1H), 3.10 (m, 2H), 2.95 (m, 2H), 2.65 (s, 3H), 2.41 (s, 3H), 2.17 (m, 2H), 2.07 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.24, 155.74, 142.02, 137.08, 136.34, 133.76, 131.64, 128.78, 127.63, 127.13, 126.49, 121.01, 116.00, 69.53, 50.91, 43.73, 28.31, 20.66. HRMS (ESI+) m/z: [M + H+] calcd for C26H29N2O2 401.2229; found 401.2231.
4.2.68.6. 4-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41f)
Compound 41f was obtained as a white amorphous solid (40 mg, 75%).1H NMR (500 MHz, Chloroform-d) δ 7.76 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 6.6 Hz, 2H), 7.37 (d, J = 6.5 Hz, 2H), 6.89 – 6.75 (m, 4H), 4.43 (m, 1H), 3.72 (s, 3H), 2.94 – 2.83 (m, 2H), 2.77 (m, 2H), 2.46 (s, 3H), 2.15 – 2.01 (m, 2H), 1.94 – 1.87 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.65, 162.42, 155.92, 137.29, 136.42, 133.95, 129.23, 127.94, 126.85, 121.07, 116.22, 113.67, 68.59, 55.25, 50.89, 44.34, 28.40. HRMS (ESI+) m/z: [M + H+] calcd for C26H29N2O3 417.2178; found 417.2176.
4.2.68.7. 4-(tert-butyl)-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41g)
Compound 41g was obtained as a white amorphous solid (38 mg, 70%). 1H NMR (500 MHz, DMSO-d6) δ 10.25 (s, 1H, NH), 7.90 (d, J = 8.5 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 4.2 Hz, 2H), 7.59 (d, J = 4.3 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 4.56 (m, 1H), 3.34 (s, 3H), 2.99 (m, 2H), 2.74 (m, 2H), 2.06 (m, 2H), 1.84 (m, 2H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO) δ 165.41, 156.04, 154.37, 138.15, 134.77, 132.48, 132.21, 129.14, 127.50, 126.19, 125.12, 120.51, 116.32, 69.87, 51.08, 43.71, 34.66, 30.84, 28.67. HRMS (ESI+) m/z: [M + H+] calcd for C29H35N2O2: 443.2699; found 443.2696.
4.2.68.8. 3-chloro-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41h)
Compound 41h was obtained as a white amorphous solid (30 mg, 73%).1H NMR (400 MHz, Chloroform-d) δ 7.82 (s, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.64 – 7.58 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 7.36 – 7.28 (m, 2H), 6.85 (d, J = 7.5 Hz, 2H), 4.37 (m, 1H), 2.89 – 2.68 (m, 2H), 2.52 (m, 2H), 2.34 (s, 3H), 2.05 – 1.99 (m, 2H), 1.94 – 1.78 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.62, 156.37, 136.94, 136.90, 136.71, 134.53, 133.58, 131.61, 129.83, 127.91, 127.62, 126.92, 125.61, 121.14, 116.31, 70.25, 51.75, 45.20, 29.52. HRMS (ESI+) m/z: [M + H+] calcd for C25H26ClN2O2 421.1683; found 421.1685.
4.2.68.9. 3-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41i)
Compound 41i was obtained as a white amorphous solid (38 mg, 74%).1H NMR (500 MHz, Chloroform-d) δ 7.53 (d, J = 8.6 Hz, 2H), 7.39 – 7.22 (m, 6H), 7.18 (t, J = 7.9 Hz, 1H), 6.87 (dd, J = 8.4, 2.6 Hz, 1H), 6.77 (d, J = 8.7 Hz, 2H), 4.36 (m, 1H), 3.65 (s, 3H), 2.77 (m, 2H), 2.57 (m, 2H), 2.33 (s, 3H), 1.93 (m, 2H), 1.80 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 166.97, 159.49, 155.97, 136.97, 136.54, 136.01, 133.59, 129.28, 127.68, 126.59, 121.08, 119.30, 117.47, 116.09, 112.44, 69.15, 54.93, 51.18, 44.33, 28.72. HRMS (ESI+) m/z: [M + H+] calcd for C26H29N2O3 417.2178; found 417.2175.
4.2.68.10. 4-chloro-3-methyl-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41j)
Compound 41j was obtained as a white amorphous solid (20 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H, NH), 7.97 (d, J = 2.2 Hz, 1H), 7.89 – 7.77 (m, 3H), 7.67 – 7.53 (m, 5H), 7.05 (d, J = 8.8 Hz, 2H), 4.51 (m, 1H), 2.88 (m, 2H), 2.63 – 2.52 (m, 2H), 2.43 (s, 3H), 2.40 (s, 3H), 2.02 (m, 2H), 1.77 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.48, 156.20, 137.85, 136.51, 135.61, 135.04, 133.63, 132.29, 130.40, 128.86, 127.42, 126.88, 126.21, 120.63, 116.26, 70.60, 51.57, 44.44, 29.42, 19.62. HRMS (ESI+) m/z: [M + H+] calcd for C26H28ClN2O2 435.1839; found 435.1838.
4.2.68.11. 3-chloro-4-methyl-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41k)
Compound 41k was obtained as a white amorphous solid (40 mg, 80%). 1H NMR (500 MHz, Chloroform-d) δ 7.88 (d, J = 1.9 Hz, 1H), 7.70 – 7.65 (m, 3H), 7.52 (d, J = 8.6 Hz, 2H), 7.48 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.7 Hz, 2H), 4.46 (m, 1H), 2.93 – 2.66 (m, 2H), 2.54 (m, 2H), 2.44 (s, 3H), 2.42 (s, 3H), 2.16 – 2.07 (m, 2H), 1.98 – 1.90 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 165.03, 156.52, 140.36, 137.05, 136.88, 134.89, 134.17, 133.69, 131.29, 128.14, 128.05, 127.25, 125.60, 120.91, 116.45, 70.23, 51.78, 45.40, 29.35, 20.30. HRMS (ESI+) m/z: [M + H+] calcd for C26H28ClN2O2 435.1839; found 435.1841.
4.2.68.12. 3-bromo-4-methyl-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41l)
Compound 41l was obtained as a white amorphous solid (29 mg, 69%). 1H NMR (500 MHz, Chloroform-d) δ 7.95 (d, J = 1.9 Hz, 1H), 7.61 (dd, J = 7.9, 1.9 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 9.0 Hz, 2H), 7.33 (d,, J = 8.0 Hz, 2H), 7.18 (d, J = 7.9 Hz, 1H), 6.80 (d, J = 8.4 Hz, 2H), 4.44 – 4.27 (m, 1H), 2.77 (m, 2H), 2.60 (m, 2H), 2.36 (s, 3H), 2.28 (s, 3H), 1.98 (m, 2H), 1.84 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 165.52, 156.08, 141.83, 136.97, 136.71, 134.04, 133.72, 131.30, 130.68, 127.86, 126.79, 126.25, 124.74, 121.13, 116.21, 69.55, 51.13, 44.60, 28.73, 22.65. HRMS (ESI+) m/z: [M + H+] calcd for C26H28BrN2O2 479.1334; found 479.1333.
4.2.68.13. 3-iodo-4-methyl-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41m)
Compound 41m was obtained as a white amorphous solid (36 mg, 78%). 1H NMR (500 MHz, DMSO-d6) δ 10.33 (s, 1H, NH), 8.42 (d, J = 1.8 Hz, 1H), 7.93 (dd, J = 7.9, 1.9 Hz, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.66 – 7.53 (m, 4H), 7.50 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 4.48 (m, 1H), 2.80 (m, 2H), 2.45 (s, 3H), 2.34 (m, 2H), 2.07 – 1.93 (m, 2H), 1.80 – 1.66 (m, 2H). 13C NMR (126 MHz, DMSO) δ 163.54, 156.27, 144.65, 137.79, 137.40, 135.07, 133.99, 132.21, 129.72, 127.70, 127.41, 126.19, 120.68, 116.23, 101.09, 71.05, 52.03, 45.16, 29.83, 27.51. HRMS (ESI+) m/z: [M + H+] calcd for C26H28IN2O2 527.1195; found 527.1192.
4.2.68.14. 3,4-dichloro-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41n)
Compound 41n was obtained as a white amorphous solid (40 mg, 81%). 1H NMR (400 MHz, Chloroform-d) δ 7.77 (d, J = 2.1 Hz, 1H), 7.50 (dd, J = 8.5, 2.1 Hz, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.29 – 7.16 (m, 4H), 7.13 (t, J = 4.1 Hz, 1H), 6.67 (d, J = 8.7 Hz, 2H), 4.26 (m, 1H), 2.72 (m, 2H), 2.60 (m, 2H), 2.29 (s, 3H), 1.88 (m, 2H), 1.75 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 164.64, 156.20, 136.95, 136.88, 135.92, 134.79, 133.72, 132.80, 130.51, 129.65, 127.98, 126.94, 126.82, 121.18, 116.29, 69.59, 51.17, 44.80, 28.69. HRMS (ESI+) m/z: [M + H+] calcd for C25H25Cl2N2O2 455.1293; found 455.1291.
4.2.68.15. 3,5-dichloro-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41o)
Compound 41o was obtained as a white amorphous solid (33 mg, 70%). 1H NMR (400 MHz, Chloroform-d) δ 7.70 (s, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.37 (m, 5H), 6.82 (d, J = 8.6 Hz, 2H), 4.33 (m, 1H), 2.80 – 2.64 (m, 2H), 2.49 (m, 2H), 2.30 (s, 3H), 1.94 (m, 2H), 1.83 (m, 2H). 13C NMR (126 MHz, DMSO) δ 162.61, 156.44, 138.09, 137.46, 135.50, 134.33, 132.09, 130.95, 127.49, 126.53, 126.31, 120.78, 116.27, 71.35, 52.01, 45.26, 30.08. HRMS (ESI+) m/z: [M + H+] calcd for C25H25Cl2N2O2 455.1293; found 455.1296.
4.2.68.16. 2,4-dichloro-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzamide (41p)
Compound 41p was obtained as a white amorphous solid (22 mg, 63%). 1H NMR (500 MHz, Chloroform-d) δ 7.58 (d, J = 8.3 Hz, 2H), 7.47 – 7.33 (m, 4H), 7.27 – 7.18 (m, 2H), 7.09 (dd, J = 8.4, 2.1 Hz, 1H), 6.85 (d, J = 8.3 Hz, 2H), 4.49 (m, 1H), 3.07 – 2.90 (m, 4H), 2.58 (s, 3H), 2.13 (m, 2H), 2.00 – 1.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 164.97, 156.32, 137.13, 136.69, 136.57, 134.57, 133.67, 131.98, 130.25, 129.90, 128.03, 127.34, 127.10, 120.61, 116.34, 69.83, 51.49, 45.06, 29.19. HRMS (ESI+) m/z: [M + H+] calcd for C25H25Cl2N2O2 455.1293; found 455.1292.
4.2.68.17. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-2-carboxamide (41q)
Compound 41q was obtained as a white amorphous solid (18 mg, 66%). 1H NMR (400 MHz, Methanol-d4) δ 8.24 (d, J = 4.5 Hz, 2H), 7.55 – 7.40 (m, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.19 (m, 6H), 7.06 (m, 4H), 6.69 (dd, J = 8.4, 3.0 Hz, 2H), 4.39 – 4.25 (m, 1H), 2.89 – 2.64 (m, 4H), 2.40 (s, 3H), 2.06 – 1.91 (m, 2H), 1.83 (m, 2H). 13C NMR (126 MHz, DMSO) δ 167.65, 155.87, 139.92, 139.09, 137.87, 136.96, 134.72, 132.51, 129.88, 129.67, 128.17, 128.13, 127.69, 127.35, 127.16, 127.11, 126.17, 119.79, 116.26, 69.25, 50.77, 43.29, 28.11. HRMS (ESI+) m/z: [M + H+] calcd for C31H31N2O2 463.2386; found 463.2389.
4.2.68.18. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (41r)
Compound 41r was obtained as a white amorphous solid (30 mg, 70%). 1H NMR (500 MHz, DMSO-d6) δ 10.24 (s, 1H, NH), 8.03 (dd, J = 8.6, 2.4 Hz, 1H), 7.97 (d, J = 2.3 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 4H), 7.56 (d, J = 7.4 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.4 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.08 (d, J = 8.5 Hz, 2H), 4.63 (m, 1H), 3.15 (m, 2H), 2.96 (m, 2H), 2.64 (s, 3H), 2.11 (m, 2H), 1.96 – 1.78 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.73, 158.69, 155.89, 138.17, 137.46, 134.70, 132.63, 129.87, 129.37, 129.34, 129.08, 128.08, 127.44, 127.19, 126.88, 126.18, 120.69, 116.38, 111.38, 68.81, 50.55, 43.04, 28.02. HRMS (ESI+) m/z: [M + H+] calcd for C31H31N2O2 463.2386; found 463.2387.
4.2.68.19. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-4-carboxamide (41s)
Compound 41s was obtained as a white amorphous solid (24 mg, 75%). 1H NMR (400 MHz, Chloroform-d) δ 7.90 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 7.8 Hz, 2H), 7.43 (dd, J = 13.4, 8.2 Hz, 4H), 7.36 (t, J = 7.5 Hz, 2H), 7.29 (d, J = 6.8 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 4.37 (m, 1H), 2.75 (m, 2H), 2.54 (m, 2H), 2.36 (s, 3H), 2.08 – 1.95 (m, 2H), 1.87 (m, 2H). 13C NMR (126 MHz, DMSO) δ 165.05, 156.23, 143.05, 139.05, 138.03, 134.95, 133.63, 132.28, 129.04, 128.35, 128.13, 127.41, 126.89, 126.55, 126.20, 120.62, 116.25, 70.83, 51.67, 44.67, 29.68. HRMS (ESI+) m/z: [M + H+] calcd for C31H31N2O2 463.2386; found 463.2383.
4.2.68.20. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-2-naphthamide (43a)
Compound 43a was obtained as a white amorphous solid (36 mg, 60%). 1H NMR (500 MHz, Methanol-d4) δ 8.35 (d, J = 1.8 Hz, 1H), 7.92 – 7.84 (m, 3H), 7.81 – 7.74 (m, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.55 – 7.35 (m, 6H), 6.87 (d, J = 8.7 Hz, 2H), 4.40 (m, 1H), 2.78 (m, 2H), 2.59 (m, 2H), 2.38 (s, 3H), 2.04 (m, 2H), 1.90 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 167.04, 156.35, 137.18, 136.80, 134.88, 133.69, 132.61, 132.09, 128.98, 128.44, 127.97, 127.93, 127.84, 127.73, 127.01, 126.80, 123.86, 121.08, 116.35, 70.03, 51.61, 45.21, 29.65. HRMS (ESI+) m/z: [M + H+] calcd for C29H29N2O2 437.2229; found 437.2227.
4.2.68.21.N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-1-naphthamide (43b)
Compound 43b was obtained as a white amorphous solid (40 mg, 80%). 1H NMR (500 MHz, Chloroform-d) δ 8.17 (d, J = 8.1 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.79 – 7.73 (m, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 7.0 Hz, 1H), 7.44 – 7.37 (m, 7H), 6.85 (d, J = 8.3 Hz, 2H), 4.41 (s, 1H), 2.85 (d, J = 12.0 Hz, 2H), 2.67 (s, 2H), 2.41 (s, 3H), 2.04 (t, J = 11.1 Hz, 2H), 1.90 (d, J = 14.2 Hz, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 168.82, 156.12, 137.24, 136.77, 134.38, 133.81, 133.61, 130.68, 130.02, 128.27, 127.96, 127.06, 127.00, 126.37, 125.19, 125.06, 124.65, 120.63, 116.28, 69.38, 51.34, 44.66, 28.95. HRMS (ESI+) m/z: [M + H+] calcd for C29H29N2O2 437.2229; found 437.2231.
4.2.68.22. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)quinoline-3-carboxamide (45a)
Compound 45a was obtained as a white amorphous solid (35 mg, 78%). 1H NMR (500 MHz, Chloroform-d) δ 9.29 (d, J = 2.3 Hz, 1H), 8.79 (d, J = 2.3 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.93 (dd, J = 8.3, 1.4 Hz, 1H), 7.82 – 7.78 (m, 1H), 7.75 (d, J = 8.6 Hz, 2H), 7.60 (t, J = 8.2 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 4.51 – 4.40 (m, 1H), 2.93 – 2.79 (m, 2H), 2.69 (m, 2H), 2.45 (s, 3H), 2.11 – 2.07 (m, 2H), 2.00 – 1.88 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 164.54, 151.05, 148.52, 148.28, 138.45, 137.28, 137.08, 131.84, 131.14, 129.12, 128.46, 128.11, 127.97, 127.84, 127.25, 127.13, 121.24, 116.38, 68.04, 51.55, 45.14, 28.95. HRMS (ESI+) m/z: [M + H+] calcd for C28H28N3O2 438.2182; found 438.2181.
4.2.68.23. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)quinoline-6-carboxamide (45b)
Compound 45b was obtained as a white amorphous solid (40 mg, 74%). 1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H, NH), 9.02 (dd, J = 4.2, 1.7 Hz, 1H), 8.67 (d, J = 2.0 Hz, 1H), 8.55 (dd, J = 8.3, 1.7 Hz, 1H), 8.28 (dd, J = 8.8, 2.0 Hz, 1H), 8.15 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 8.7 Hz, 2H), 7.68 – 7.63 (m, 3H), 7.61 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 4.50 (m, 1H), 3.33 (m, 2H), 2.84 (m, 2H), 2.38 – 2.34 (m, 3H), 2.02 (m, 2H), 1.77 (m, 2H). 13C NMR (126 MHz, DMSO) δ 165.12, 156.36, 152.34, 148.73, 138.08, 137.18, 135.11, 132.80, 132.36, 129.09, 128.55, 128.14, 127.56, 127.13, 126.38, 122.37, 120.77, 116.25, 70.88, 51.89, 44.82, 29.71. HRMS (ESI+) m/z: [M + H+] calcd for C28H28N3O2 438.2182; found 438.2185.
4.2.68.24. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-1H-indole-2-carboxamide (47a)
Compound 47a was obtained as a white amorphous solid (38 mg, 80%). 1H NMR (500 MHz, DMSO-d6) δ 11.63 (d, J = 2.2 Hz, 1H, NH), 10.15 (s, 1H, NH), 7.71 (d, J = 8.7 Hz, 2H), 7.50 (dd, J = 7.9, 1.1 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 7.29 (dd, J = 8.0, 1.0 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H), 7.04 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 6.92 – 6.83 (m, 3H), 4.36 (m, 1H), 2.79 – 2.68 (m, 2H), 2.57 – 2.42 (m, 2H), 2.28 (s, 3H), 1.96 – 1.78 (m, 2H), 1.63 – 1.55 (m, 2H). 13C NMR (126 MHz, DMSO) δ 159.62, 156.10, 137.84, 136.78, 134.78, 132.42, 131.45, 127.42, 126.97, 126.30, 123.75, 121.70, 120.38, 119.88, 116.30, 112.34, 103.97, 69.99, 51.29, 44.04, 28.99. HRMS (ESI+) m/z: [M + H+] calcd for C27H28N3O2 426.2182; found 426.2179.
4.2.68.25. N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)benzo[b]thiophene-2-carboxamide (47b)
Compound 47b was obtained as a white amorphous solid (50 mg, 81%). 1H NMR (500 MHz, DMSO-d6) δ 10.68 (s, 1H, NH), 8.45 (s, 1H), 8.07 (dd, J = 7.4, 1.5 Hz, 1H), 8.05 – 7.99 (m, 1H), 7.87 (d, J = 8.7 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.53 – 7.46 (m, 2H), 7.07 (d, J = 8.8 Hz, 2H), 4.68 – 4.46 (m, 1H), 3.06 – 2.96 (m, 2H), 2.85 – 2.74 (m, 2H), 2.54 (s, 3H), 2.17 – 2.03 (m, 2H), 1.91 – 1.78 (m, 2H). 13C NMR (126 MHz, DMSO) δ 160.23, 156.09, 140.44, 140.04, 139.12, 137.50, 135.17, 132.39, 127.47, 126.34, 125.89, 125.39, 125.04, 122.83, 122.71, 120.58, 116.33, 69.70, 50.70, 43.47, 28.36. HRMS (ESI+) m/z: [M + H+] calcd for C27H27N2O2S 443.1793; found 443.1791.
4.2.68.26. 6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (49a)
Compound 49a was obtained as a white amorphous solid (33 mg, 78%). 1H NMR (500 MHz, DMSO-d6) δ 10.25 (s, 1H, NH), 8.04 (dd, J = 8.6, 2.4 Hz, 1H), 7.98 (d, J = 2.3 Hz, 1H), 7.86 (d, J = 8.6 Hz, 2H), 7.60 (dd, J = 8.6, 3.5 Hz, 4H), 7.56 (d, J = 7.3 Hz, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.37 (t, J = 7.4 Hz, 1H), 7.26 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 8.5 Hz, 2H), 4.58 (m, 1H), 3.86 (s, 3H), 3.11 – 2.93 (m, 2H), 2.78 (m, 2H), 2.52 (s, 3H), 2.08 (m, 2H), 1.85 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.68, 158.68, 156.01, 138.20, 137.49, 134.70, 132.52, 129.90, 129.36, 129.09, 128.06, 127.41, 127.17, 126.93, 126.16, 120.66, 116.33, 111.36, 69.61, 55.85, 50.89, 43.61, 28.55. HRMS (ESI+) m/z: [M + H+] calcd for C32H33N2O3 493.2491; found 493.2495.
4.2.68.27. 3′-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (49b)
Compound 49b was obtained as a white amorphous solid (52 mg, 72%). 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H, NH), 8.24 (s, 1H), 7.96 (dt, J = 7.7, 1.4 Hz, 1H), 7.93 – 7.84 (m, 3H), 7.68 – 7.57 (m, 4H), 7.44 (dd, J = 8.6, 7.1 Hz, 1H), 7.35 (dt, J = 7.8, 1.2 Hz, 1H), 7.33 (t, J = 2.1 Hz, 1H), 7.10 – 7.05 (m, 2H), 7.03 – 6.98 (m, 1H), 4.61 (m, 1H), 3.86 (s, 3H), 3.11 – 3.02 (m, 2H), 2.88 (s, 2H), 2.60 (s, 3H), 2.20 – 2.01 (m, 2H), 1.93 – 1.76 (m, 2H). 13C NMR (126 MHz, DMSO) δ 165.34, 159.78, 156.00, 140.99, 140.15, 138.02, 135.52, 134.95, 132.54, 130.08, 129.88, 129.06, 127.46, 126.97, 126.24, 125.86, 120.72, 119.23, 116.36, 113.34, 112.49, 69.66, 55.20, 50.77, 43.33, 28.33. HRMS (ESI+) m/z: [M + H+] calcd for C32H33N2O3 493.2491; found 493.2494.
4.2.68.28. 4′,6-dimethoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (49c)
Compound 49c was obtained as a white amorphous solid (23 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ 10.22 (s, 1H, NH), 7.99 (dd, J = 8.6, 2.4 Hz, 1H), 7.95 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.64 – 7.60 (m, 3H), 7.51 (d, J = 8.7 Hz, 1H), 7.24 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 8.6 Hz, 2H), 7.02 (d, J = 8.8 Hz, 1H), 4.65 (m, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.25 – 2.96 (m, 4H), 2.68 (s, 3H), 2.11 (m, 2H), 1.91 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.78, 158.67, 158.50, 155.86, 138.25, 134.63, 132.70, 130.48, 129.63, 129.04, 128.52, 127.44, 126.91, 126.18, 120.63, 116.40, 113.56, 113.52, 111.29, 66.97, 55.82, 55.12, 51.23, 42.74, 28.33. HRMS (ESI+) m/z: [M + H+] calcd for C33H35N2O4 523.2597; found 523.2602.
4.2.68.29. 2′-methoxy-5′-((4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)carbamoyl)-[1,1′-biphenyl]-3-yl acetate (49d)
Compound 49d was obtained as a white amorphous solid (32 mg, 76%). 1H NMR (500 MHz, Chloroform-d) δ 7.93 (s, 1H), 7.86 (dd, J = 8.7, 2.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 8.8 Hz, 2H), 7.35 – 7.30 (m, 1H), 7.01 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.7 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 4.43 (m, 1H), 3.81 (s, 3H), 2.90 – 2.79 (m, 2H), 2.66 (m, 2H), 2.45 (s, 3H), 2.25 (s, 3H), 2.19 (m, 2H), 1.95 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 169.64, 165.09, 159.23, 156.27, 150.37, 138.92, 137.04, 136.58, 133.73, 129.62, 129.55, 129.12, 128.82, 128.05, 127.22, 127.18, 127.03, 122.77, 120.64, 120.51, 116.27, 111.11, 69.73, 55.86, 51.43, 45.19, 29.15, 21.21. HRMS (ESI+) m/z: [M + H+] calcd for C34H35N2O5 551.2546; found 551.2543.
4.2.68.30. 2′-methoxy-5′-((4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)carbamoyl)-[1,1′-biphenyl]-4-yl acetate (49e)
Compound 49e was obtained as a white amorphous solid (55 mg, 66%). 1H NMR (500 MHz, Chloroform-d) δ 7.98 (s, 1H), 7.86 (dd, J = 8.6, 2.4 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 8.6 Hz, 1H), 7.46 (m, 5H), 7.06 (d, J = 8.6 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 4.43 (m, 1H), 3.81 (s, 3H), 2.93 – 2.78 (m, 2H), 2.66 (s, 2H), 2.45 (s, 3H), 2.26 (s, 3H), 2.26 – 2.10 (m, 2H), 1.95 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 169.68, 165.08, 159.28, 156.27, 149.97, 137.10, 136.56, 135.14, 133.71, 130.58, 129.83, 129.63, 128.59, 128.04, 127.18, 127.15, 121.29, 120.51, 116.27, 111.06, 69.72, 55.81, 51.41, 45.17, 29.16, 21.21. HRMS (ESI+) m/z: [M + H+] calcd for C34H35N2O5 551.2546; found 551.2545.
4.2.68.31. 3′-chloro-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (49f)
Compound 49f was obtained as a white amorphous solid (32 mg, 80%). 1H NMR (500 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.06 (dd, J = 8.6, 2.4 Hz, 1H), 8.00 (d, J = 2.4 Hz, 1H), 7.84 (d, J = 8.6 Hz, 2H), 7.63 – 7.57 (m, 54H), 7.55 – 7.44 (m, 3H), 7.30 (d, J = 8.7 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 4.41 (m, 1H), 3.88 (s, 3H), 2.71 – 2.57 (m, 2H), 2.20 (m, 5H), 2.12 – 1.90 (m, 2H), 1.66 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.50, 158.58, 156.38, 139.55, 138.04, 134.86, 132.73, 132.09, 129.95, 129.81, 129.71, 128.97, 128.13, 127.70, 127.35, 127.12, 127.01, 126.13, 120.67, 116.17, 111.55, 71.85, 55.98, 52.32, 45.74, 30.51. HRMS (ESI+) m/z: [M + H+] calcd for C32H32ClN2O3 527.2101; found 527.2100.
4.2.68.32. 4′-chloro-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (49g)
Compound 49g was obtained as a white amorphous solid (23 mg, 70%). 1H NMR (500 MHz, DMSO-d6) δ 10.23 (s, 1H, NH), 8.04 (dd, J = 8.6, 2.3 Hz, 1H), 7.97 (d, J = 2.3 Hz, 1H), 7.84 (d, J = 8.7 Hz, 2H), 7.60 (m, 5H), 7.52 (d, J = 8.5 Hz, 2H), 7.28 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 8.7 Hz, 2H), 4.55 (m, 1H), 3.87 (s, 3H), 3.33 (s, 3H), 2.98 (m, 2H), 2.70 (m, 2H), 2.06 (m, 2H), 1.80 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.60, 158.61, 156.07, 138.16, 136.26, 134.76, 132.48, 132.02, 131.18, 129.78, 129.44, 128.10, 127.98, 127.43, 127.04, 126.19, 120.66, 116.33, 111.51, 69.88, 55.96, 51.10, 43.86, 28.74. HRMS (ESI+) m/z: [M + H+] calcd for C32H32ClN2O3 527.2101; found 527.2105.
4.2.68.31. 6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-3′-nitro-[1,1′-biphenyl]-3-carboxamide (49h)
Compound 49h was obtained as a yellow amorphous solid (32 mg, 72%). 1H NMR (500 MHz, DMSO-d6) δ 10.27 (s, 1H, NH), 8.42 (t, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.2, 2.4 Hz, 1H), 8.11 (dd, J = 8.6, 2.3 Hz, 1H), 8.08 – 8.03 (m, 2H), 7.86 – 7.83 (m, 2H), 7.78 (t, J = 8.0 Hz, 1H), 7.65 – 7.57 (m, 4H), 7.34 (d, J = 8.8 Hz, 1H), 7.05 (d, J = 8.7 Hz, 2H), 4.52 (m, 1H), 3.91 (s, 3H), 2.91 (m, 2H), 2.66 – 2.56 (m, 2H), 2.44 (s, 3H), 2.09 – 1.96 (m, 2H), 1.85 – 1.66 (m, 2H). 13C NMR (126 MHz, DMSO) δ 164.46, 158.58, 156.14, 147.68, 138.93, 138.06, 136.08, 134.82, 132.37, 130.15, 129.96, 129.71, 127.40, 127.21, 126.80, 126.18, 123.79, 122.14, 120.68, 116.27, 111.69, 69.71, 56.10, 51.67, 44.44, 29.14. HRMS (ESI+) m/z: [M + H+] calcd for C32H32N3O5 538.2342; found 538.2346.
4.2.68.29.1. 3′-hydroxy-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (50a): General procedure for the synthesis of 50a-b
Compounds 49d (20 mg, 0.036 mmol) were dissolved in a solution of 10% Et3N in methanol (1 mL) and stirred at room temperature for 24 hours before concentrated to dryness. The light brown residue so obtained was purified by flash chromatography using dichloromethane and methanol (v/v, 10:1) as eluent to give 50a as a white amorphous solid (16 mg, 85%). 1H NMR (500 MHz, Methanol-d4) δ 7.95 (dd, J = 8.6, 2.5 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 7.27 (t, J = 7.9 Hz, 1H), 7.08 (d, J = 8.6 Hz, 2H), 7.05 – 7.03 (m, 1H), 6.99 (d, J = 8.7 Hz, 2H), 6.84 (dd, J = 8.0, 2.5 Hz, 1H), 4.49 (m, 1H), 3.90 (s, 3H), 2.86 (m, 2H), 2.63 (m, 2H), 2.46 (s, 3H), 2.12 (m, 2H), 2.02 – 1.93 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.57, 159.32, 156.47, 156.29, 138.87, 137.19, 136.60, 133.67, 130.54, 130.01, 129.01, 128.41, 127.90, 126.91, 126.88, 121.04, 120.98, 116.35, 116.31, 114.33, 110.87, 69.62, 55.62, 51.63, 45.18, 29.42. HRMS (ESI+) m/z: [M + H+] calcd for C32H33N2O4 509.2440, found 509.2442.
4.2.68.30.1. 4′-hydroxy-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (50b)
Compound 50b was obtained as a white amorphous solid (16 mg, 90%). 1H NMR (500 MHz, Methanol-d4) δ 7.90 (dd, J = 8.5, 2.4 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 8.6 Hz, 2H), 7.56 – 7.50 (m, 4H), 7.43 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.6 Hz, 1H), 4.55 (m, 1H), 3.89 (s, 3H), 3.00 (m, 2H), 2.84 (m, 2H), 2.57 (s, 3H), 2.20 (m, 2H), 2.06 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 166.53, 159.30, 156.25, 156.03, 137.23, 136.47, 133.88, 130.59, 130.56, 129.76, 128.81, 127.96, 127.80, 126.93, 126.91, 121.00, 116.26, 114.96, 110.79, 68.82, 55.63, 51.12, 44.67, 28.69. HRMS (ESI+) m/z [M+H+] calcd for C32H33N2O4 509.2440, found 509.2441.
4.2.68.31.1. 3′-amino-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (50c)
Palladium on carbon (10%, 5 mg) was added to a solution of 49h (25 mg, 0.047 mmol) in methanol (1 ml), followed by two drops of acetic acid. The resulted suspension was degased and stirred under hydrogen atmosphere for 12 hours before filtration. The filtrate was concentrated to dryness to afford aniline 50c as a white amorphous solid (18 mg, 76%). 1H NMR (400 MHz, Chloroform-d) δ 7.85 – 7.81 (m, 1H), 7.78 (s, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.43 (m, 4H), 7.11 (t, J = 7.8 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 6.87 (m, 3H), 6.81 (s, 1H), 6.64 – 6.62 (m, 1H), 4.54 (m, 1H), 3.77 (s, 3H), 3.13 – 2.99 (m, 4H), 2.63 (s, 3H), 2.25 – 2.15 (m, 2H), 2.08 – 2.00 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 159.41, 156.00, 146.03, 138.68, 137.32, 136.51, 134.07, 130.83, 129.93, 128.98, 128.53, 128.09, 127.04, 126.93, 121.02, 120.98, 120.31, 116.69, 116.31, 114.71, 110.97, 68.71, 55.76, 50.83, 44.44, 28.31. HRMS (ESI+) m/z: [M + H+] calcd for C32H34N3O3 508.2600; found 508.2598.
4.2.68.31.2. 3′-acetamido-6-methoxy-N-(4′-((1-methylpiperidin-4-yl)oxy)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-3-carboxamide (50d)
Aniline 50c (10 mg, 0.02 mmol) was added to a solution of acetic anhydride in pyridine (1:3, v/v) and the resulting mixture was stirred at room temperature for 4 hours before being concentrated to dryness. The remaining residue was further dried under vacuum overnight to afford acetamide 50d as a light brown amorphous solid (10 mg, 100%). 1H NMR (500 MHz, Methanol-d4) δ 7.76 (dd, J = 8.8, 2.7 Hz, 1H), 7.73 (s, 1H), 7.56 – 7.51 (m, 3H), 7.37 – 7.29 (m, 5H), 7.17 – 7.08 (m, 2H), 6.88 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 8.6 Hz, 2H), 4.34 (m, 1H), 3.67 (s, 3H), 2.81 (m, 2H), 2.62 (s, 1H), 2.36 (m, 2H), 1.96 (s, 3H), 1.94 (m, 2H), 1.80 (m, 2H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 170.17, 166.64, 159.19, 155.91, 138.13, 137.98, 137.25, 136.38, 133.83, 130.00, 129.93, 128.69, 128.18, 127.79, 126.78, 126.67, 125.34, 121.11, 121.07, 118.95, 116.14, 110.77, 68.98, 55.39, 51.01, 44.30, 28.59, 23.26. HRMS (ESI+) m/z [M+K+] calcd for C34H35N3O4K 588.2265, found 588.2270.
Anti-proliferation assays
Cells were maintained in a 1:1 mixture of Advanced DMEM/F12 (Gibco) supplemented with non-essential amino acids, L-glutamine (2 mM), streptomycin (500 g/mL), penicillin (100 units/mL), and 10% FBS. Cells were grown to confluence in a humidified atmosphere (37° C, 5% CO2), seeded (2000/well, 100 μL) in 96-well plates, and allowed to attach overnight. Compound or GDA at varying concentrations in DMSO (1% DMSO final concentration) was added, and cells were returned to the incubator for 72 h. At 72 h, the number of viable cells was determined using an MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells incubated in 1% DMSO were used at 100% proliferation, and values were adjusted accordingly. IC50 values were calculated from separate experiments performed in triplicate using GraphPad Prism.
Western blot analyses
MCF-7 cells were cultured as described above and treated with various concentrations of drug, GDA in DMSO (1% DMSO final concentration), or vehicle (DMSO) for 24 h. Cells were harvested in cold PBS and lysed in RIPA lysis buffer containing 1 mM PMSF, 2 mM sodium orthovanadate, and protease inhibitors on ice for 1 h. Lysates were clarified at 14000g for 10 min at 4° C. Protein concentrations were determined using the Pierce BCA protein assay kit per the manufacturer’s instructions. Equal amounts of protein (20 g) were electrophoresed under reducing conditions, transferred to a nitrocellulose membrane, and immunoblotted with the corresponding specific antibodies. Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized.
Supplementary Material
Highlights.
A small library of biphenylamide derivatives was designed and analogues synthesized.
Compounds were evaluated for anti-proliferative activity against cancer cell lines.
Hsp90 inhibition was confirmed by Western blot analysis.
Several analogues showed low nanomolar inhibitory activity.
Figure 4.

Western blot analyses of Hsp90-dependent client proteins from MCF-7 breast cancer cell lysate upon treatment with biphenyl derivatives. Concentrations (in μM) were indicated above each lane. H represents a concentration equal to 5-fold of the anti-proliferative activity. L represents a concentration equal to 0.5-fold of the anti-proliferative activity. Geldanamycin (G, 0.5 μM) and dimethylsulfoxide (D, 100%) were employed as positive and negative controls.
Figure 5.

Western blot analyses of Hsp90-dependent client proteins from MCF-7 breast cancer cell lysate upon treatment with biphenyl derivatives. Concentrations (in μM) were indicated above each lane. H represents a concentration equal to 5-fold of the anti-proliferative activity. L represents a concentration equal to 0.5-fold of the anti-proliferative activity. Geldanamycin (G, 0.5 μM) and dimethylsulfoxide (D, 100%) were employed as positive and negative controls.
Scheme 4.

Synthesis of novobiocin mimics that contain a substituted biphenyl moiety.
Reagents and conditions: a Pd(dppf)2Cl2, 2M K2CO3, Dioxane, 110 °C, 12 h, 17%%~74%; b Ph3P, DIAD, THF, r. t., 12 h, 46%~80%; c Pd/C, MeOH, r. t., 2 h, 100%; d pyridine, DCM, r. t., 4 h, 39%~87%.
Scheme 5.

Synthesis of novobiocin mimics that contain nitro substitution.
Reagents and conditions: a Pd(PPh3)4, 2M K2CO3, Dioxane, 110 °C, 12 h, 60%~72%; b Ph3P, DIAD, THF, r. t., 12 h, 39%~89%; c 10% TFA/DCM, r. t., 2 h, 98%; d pyridine, DCM, r. t., 4 h, 63%; d pyridine, DMF, 90 °C, 12 h, 43%~90%; f Pd(dppf)2Cl2, 2M K2CO3, Dioxane, 120 °C, 12 h, 41%~65%; g pd/C MeOH, AcOH(cat.), r. t., 12 h, 100%; h Ac2O, pyridine, r. t., 12 h, 85~90%.
Scheme 7.

Synthesis of biphenyl derivatives containing a fused-aromatic side chain.
Acknowledgement
The authors gratefully acknowledge support of this project by the NIH/NCI (CA120458). The NMR support for this project was provided by NSF (9512331). The authors are greatly thankful to Frontier Scientific, Inc. for providing high quality boronic acids for this project. GC gratefully acknowledges AIRC (Associazione Italiana Ricerca sul Cancro) for support through the grant IG.11775 and the Flagship “INTEROMICS” project (PB.P05) funded by MIUR and CNR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- [1].Blagg BSJ, Kerr TA. Hsp90 inhibitors: Small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Medicinal Research Reviews. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- [2].Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14:e358–e369. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- [3].Picard D. http://www.picard.ch/downloads/Hsp90interactors.pdf
- [4].Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [5].Barrott JJ, Haystead TAJ. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013;280:1381–1396. doi: 10.1111/febs.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhao H, Michaelis Mary L, Blagg Brian SJ. Hsp90 modulation for the treatment of Alzheimer’s disease. Adv Pharmacol. 2012;64:1–25. doi: 10.1016/B978-0-12-394816-8.00001-5. [DOI] [PubMed] [Google Scholar]
- [7].Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Luo WJ, Rodina A, Chiosis G. Heat shock protein 90: translation from cancer to Alzheimer’s disease treatment? Bmc Neuroscience. 2008;9 doi: 10.1186/1471-2202-9-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Whitesell L, Santagata S, Lin NU. Inhibiting HSP90 to treat cancer: a strategy in evolution. Curr. Mol. Med. 2012;12:1108–1124. doi: 10.2174/156652412803306657. [DOI] [PubMed] [Google Scholar]
- [10].Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. Journal of the National Cancer Institute. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- [11].Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. Journal of Biological Chemistry. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- [12].Shelton SN, Shawgo ME, Matthews SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BSJ, Robertson JD. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 2009;76:1314–1322. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang XY, Hembruff S, Donnelly A, Rajewski RA, Blagg BSJ, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. Bmc Cancer. 2011;11 doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. Hsp90 Inhibitors Identified from a Library of Novobiocin Analogues. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- [15].Kimura H, Yukitake H, Tajima Y, Suzuki H, Chikatsu T, Morimoto S, Funabashi Y, Omae H, Ito T, Yoneda Y, Takizawa M. ITZ-1, a Client-Selective Hsp90 Inhibitor, Efficiently Induces Heat Shock Factor 1 Activation. Chemistry & Biology. 2010;17:18–27. doi: 10.1016/j.chembiol.2009.12.012. [DOI] [PubMed] [Google Scholar]
- [16].Yin ZY, Henry EC, Gasiewicz TA. (−)-Epigallocatechin-3-gallate Is a Novel Hsp90 Inhibitor. Biochemistry. 2009;48:336–345. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huiping Zhao BSJB. Hsp90 C-terminal inhibitors. In: Timothy GZ, Machajewski D, editors. Inhibitors of Molecular Chaperones As Therapeutic Agents. RSC Publishing; London: 2014. pp. 259–411. [Google Scholar]
- [18].Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- [19].Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. The Design, Synthesis, and Evaluation of Coumarin Ring Derivatives of the Novobiocin Scaffold that Exhibit Antiproliferative Activity. J. Org. Chem. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alison HZ, Donnelly C. Bhaskar Reddy Kusuma and Brian S. J. Blagg, Cytotoxic sugar analogues of an optimized novobiocin scaffold. Med. Chem. Commun. 2010;1:165–170. [Google Scholar]
- [21].Huiping Zhao BRK, Blagg Brian S. J. Synthesis and Evaluation of Noviose Replacements on Novobiocin That Manifest Antiproliferative Activity. ACS Med. Chem. Lett. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. Engineering an Antibiotic to Fight Cancer: Optimization of the Novobiocin Scaffold to Produce Anti-proliferative Agents. J. Med. Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. Development of Novobiocin Analogues That Manifest Anti-proliferative Activity against Several Cancer Cell Lines. J. Org. Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- [24].Zhao H, Moroni E, Yan B, Colombo G, Blagg BSJ. 3D-QSAR-Assisted Design, Synthesis, and Evaluation of Novobiocin Analogues. ACS Med. Chem. Lett. 2013;4:57–62. doi: 10.1021/ml300275g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moroni E, Zhao H, Blagg BSJ, Colombo G. Exploiting Conformational Dynamics in Drug Discovery: Design of C-Terminal Inhibitors of Hsp90 with Improved Activities. J. Chem. Inf. Model. 2014;54:195–208. doi: 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kusuma BR, Khandelwal A, Gu W, Brown D, Liu W, Vielhauer G, Holzbeierlein J, Blagg BSJ. Synthesis and biological evaluation of coumarin replacements of novobiocin as Hsp90 inhibitors. Bio.Med. Chem. 2014;22:1441–1449. doi: 10.1016/j.bmc.2013.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhao H, Moroni E, Yan B, Colombo G, Blagg BS. 3D-QSAR-Assisted Design, Synthesis, and Evaluation of Novobiocin Analogues. ACS medicinal chemistry letters. 2012;4:57–62. doi: 10.1021/ml300275g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Costantino L, Barlocco D. Privileged structures as leads in medicinal chemistry. Curr Med Chem. 2006;13:65–85. [PubMed] [Google Scholar]
- [29].Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. Elucidation of the Hsp90 C-Terminal Inhibitor Binding Site. Acs Chemical Biology. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morra G, Neves MAC, Plescia CJ, Tsustsumi S, Neckers L, Verkhivker G, Altieri DC, Colombo G. Dynamics-Based Discovery of Allosteric Inhibitors: Selection of New Ligands for the C-terminal Domain of Hsp90. J. Chem. Theory Comput. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhao H, Moroni E, Colombo G, Blagg BSJ. Identification of a New Scaffold for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2014;5:84–88. doi: 10.1021/ml400404s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. The Design, Synthesis, and Evaluation of Coumarin Ring Derivatives of the Novobiocin Scaffold that Exhibit Antiproliferative Activity. The Journal of Organic Chemistry. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. Journal of Medicinal Chemistry. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- [34].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of Medicinal Chemistry. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- [35].Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. Journal of the American Chemical Society. 1996;118:11225–11236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.