

Abstract
Mendelian disorders of the epigenetic machinery are a newly delineated group of multiple congenital anomaly and intellectual disability syndromes resulting from mutations in genes encoding components of the epigenetic machinery. The gene products affected in these inherited conditions act in trans and are expected to have widespread epigenetic consequences. Many of these syndromes demonstrate phenotypic overlap with classical imprinting disorders and with one another. The various writer and eraser systems involve opposing players, which we propose must maintain a balance between open and closed chromatin states in any given cell. An imbalance might lead to disrupted expression of disease-relevant target genes. We suggest that classifying disorders based on predicted effects on this balance would be informative regarding pathogenesis. Furthermore, strategies targeted at restoring this balance might offer novel therapeutic avenues, taking advantage of available agents such as histone deacetylase inhibitors and histone acetylation antagonists.
Keywords: DNA methylation, histone tail modifications, epigenetics, epigenomics, chromatin, therapeutic development
INTRODUCTION
With the increasing use of genome-wide tools to interrogate the integrity of entire individual genomes, there has been an explosion in the identification of the class of disorders (e.g., (25, 41, 48, 54, 60, 86, 105, 106, 116, 119, 123) that we propose to call the Mendelian disorders of the epigenetic machinery. These inherited disorders result from mutations in the genes encoding various components of the epigenetic machinery (11), and the pathogenesis of these disorders is likely to be caused in part by downstream epigenetic consequences.
Herein, we review important concepts that define both classical imprinting disorders and Mendelian disorders of the epigenetic machinery. We briefly discuss classical imprinting disorders, which result mainly from disruptions occurring in cis, as well as multilocus methylation defects, which implicate trans-acting factors as playing a role in imprinting disorders. The bulk of this review, however, focuses on Mendelian disorders of the epigenetic machinery, highlighting unique characteristics and the interconnectedness of the various systems. We emphasize the epigenetic imbalance created by some of these conditions, which may lead to novel therapeutic strategies for patients with these disorders.
PRINCIPLES ILLUSTRATED BY CLASSICAL IMPRINTING DISORDERS
The term imprinting refers to the differential expression of alleles for a particular gene depending on the parental origin of the allele. Each allele contains a distinct set of epigenetic modifications or marks (see sidebar, Epigenetic Modifications), which influence chromatin structure and regulate gene expression at particular imprinted loci (46). The regulation of differential gene expression at imprinted loci is quite complex and involves DNA methylation, characteristic epigenetic signatures of associated covalent posttranslational modifications of histone tails, noncoding RNAs, and transacting factors, all of which play key roles in the process (72).
Disrupting these epigenetic marks, and subsequently tipping the balance of parent-of-origin-specific gene expression, results in classical imprinting disorders. These disorders are characterized by the universal features of abnormal growth and/or intellectual disability as well as by individual characteristic dysmorphisms, subsets of which are shared among related disorders. Classical imprinting disorders, resulting from alterations in gene dosage, illustrate many of the key concepts that have defined the field of epigenetics, including the ideas that epigenetic marks are often locus specific and that they act in cis. For example, Prader–Willi syndrome (PWS) and Angelman syndrome (AS), two classical imprinting disorders, result from opposing molecular abnormalities at an imprinted gene locus on 15q11 and have characteristic neurobehavioral phenotypes depending on whether the maternal or paternal allele is affected (22, 77). AS is caused by a lack of maternal UBE3A expression resulting from disrupted imprinting via a variety of genetic and epigenetic mechanisms occurring in cis. AS is characterized by intellectual disability, severe speech impairment, ataxia, seizures, and microcephaly (77). A lack of the paternal contribution of genes in the region leads to PWS, which is characterized by severe neonatal hypotonia and failure to thrive along with subsequent intellectual disability, hyperphagia, obesity, short stature, small hands and feet, and hypogonadism (22).
Beckwith–Wiedemann syndrome (BWS) and Russell–Silver syndrome (RSS) can result from opposing molecular abnormalities at another imprinted region, 11p15 (24, 29). These disorders are characterized by overgrowth and growth failure, respectively. The 11p15 region contains two separate imprinting domains: the IGF2/H19 locus [imprinting control region 1 (ICR1)] and the CDKN1C/KCNQOT1 locus (ICR2). When either locus is disrupted, tipping the balance toward increased expression of paternal genes, the overgrowth disorder BWS results. This dosage imbalance occurs primarily via epigenetic mechanisms acting in cis, most commonly loss of methylation at ICR2, gain of methylation at ICR1, or paternal uniparental disomy of the entire 11p15 region (24). Individuals with BWS can exhibit pre- and postnatal somatic overgrowth, hemihyperplasia, macroglossia, neonatal hypoglycemia, craniofacial dysmorphisms, and abdominal wall defects, and they are at increased risk of certain embryonal tumors. RSS, in contrast, is characterized by poor pre- and postnatal growth, failure to thrive, hemihypoplasia, and characteristic dysmorphisms (including a prominent forehead) as well as occasional hypoglycemia. The majority of RSS results from paternal hypomethylation at ICR1, a gene dosage imbalance that leads to increased maternal gene expression with no paternal contribution (29).
Genomic imprinting and other epigenetic phenomena demonstrate environmental sensitivity (59). For instance, BWS and AS occur with an increased incidence in the offspring of infertile couples conceived using artificial reproductive technologies (4), which involve a multitude of environmental disruptions that could potentially impact malleable epigenetic marks. There is also an increased incidence of BWS in monozygotic twins, and, notably, many of these twin pairs are discordant for both phenotype and epigenotype, suggesting that epigenetic alterations contribute to phenotypic variability even among genetically identical individuals. Whether somatic mutations may also play a role is unknown, however. Most examples of discordant monozygotic twins with BWS involve the same molecular epigenetic abnormality, namely DNA hypomethylation at ICR2 (90). These observations suggest an environmental or stochastic contribution to the molecular epigenotype, which subsequently leads to a disease phenotype.
The classical imprinting disorders have traditionally been thought to result exclusively from individual locus-specific effects occurring in cis (9). However, this idea has been called into question by recent studies reporting that a subset of patients with classical imprinting disorders have DNA methylation defects at multiple imprinted sites throughout the genome, including at both maternally and paternally inherited alleles (96). This suggests that an upstream trans-acting genetic factor may be causative. In fact, pathological homozygous or compound heterozygous mutations have been identified in a gene encoding a zinc finger–containing transcription factor thought to be involved in maintaining imprints (ZFP57) in some individuals with multilocus methylation defects and transient neonatal diabetes mellitus, making this the first genetic factor identified as causing a classical imprinting disorder in trans (78). Importantly, multilocus methylation defects have been reported in BWS, RSS, transient neonatal diabetes mellitus, and other disorders, but they appear to be much less common in PWS and have not yet been described for AS (96). Thus, genetic factors causing classical imprinting disorders in trans probably account for only a small fraction of imprinting disorders. More common are the newly appreciated Mendelian disorders of the epigenetic machinery, in which genetic alterations cause global epigenetic effects in trans (11).
MENDELIAN DISORDERS OF THE EPIGENETIC MACHINERY
The DNA methylation machinery and the histone machinery affect the expression of many genes in trans (11, 130). Within this group, genetic mutations may occur in writers, erasers, or readers of epigenetic marks as well as in chromatin remodelers (Table 1). The writers of epigenetic marks, which can be conceptualized as a set of highlighters, place the appropriate modifications on particular regions of the genome based on the cell type, developmental stage, and metabolic state of the cell. These marks “highlight” individual regions for use or disuse depending on whether the mark favors a more open or more closed chromatin state (Figure 1). The erasers of epigenetic marks remove these same marks, favoring the opposite chromatin states (Figure 1). The readers of epigenetic marks recognize and interpret particular marks locally and theoretically give cells a mechanism for keeping track of the overall chromatin state (Figure 1).
Table 1.
Mendelian disorders of the epigenetic machinery
| Epigenetic function | Gene | Gene function | Disease | Inheritance pattern | Reference(s) |
|---|---|---|---|---|---|
| Disorders of the DNA methylation machinery | |||||
| Writer | DNMT1 | DNA methyltransferase | Hereditary sensory and autonomic neuropathy with dementia and hearing loss (HSAN1E) | AD | 67 |
| Autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCADN) | AD | 129 | |||
| DNMT3b | DNA methyltransferase | Immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome | AR | 47 | |
| Reader | MeCP2 | Methyl-CpG-binding protein | Rett syndrome | XL | 3 |
| MBD5 | Methyl-CpG-binding protein | 2q23.1 microdeletion/microduplication syndrome | AD | 52, 85, 115 | |
| Disorders of the histone machinery | |||||
| Writer | CREBBP | Histone acetyltransferase | Rubinstein–Taybi syndrome (RTS) | AD | 93 |
| EP300 | Histone acetyltransferase | Rubinstein–Taybi syndrome (RTS) | AD | 101 | |
| KAT6B | Histone acetyltransferase | Genitopatellar syndrome (GPS) | AR | 21 | |
| Say–Barber–Biesecker–Young– Simpson (SBBYS) syndrome | AD | 25 | |||
| MLL2 | Histone methyltransferase (H3K4) | Kabuki syndrome (KS) | AD | 86 | |
| MLL | Histone methyltransferase (H3K4) | Wiedemann–Steiner syndrome (WSS) | AD | 61 | |
| EHMT1 | Histone methyltransferase (H3K9) | Kleefstra syndrome (KLFS) | AD | 66 | |
| EZH2 | Histone methyltransferase (H3K27) | Weaver syndrome (WS) | AD | 41, 116 | |
| NSD1 | Histone methyltransferase (H3K36, H4K20) | Sotos syndrome (SS) | AD | 69 | |
| Eraser | HDAC4 | Histone deacetylase | Brachydactyly–mental retardation (BDMR) syndrome | AD | 128 |
| HDAC8 | Histone deacetylase | Cornelia de Lange syndrome 5 (CDLS5) | XL | 27 | |
| Wilson–Turner syndrome (WTS) | XL | 48 | |||
| KDMSCa | Histone demethylase (H3K4) | Claes–Jensen syndrome (CJS) | XL | 60 | |
| KDM6Aa | Histone demethylase (H3K27) | Kabuki syndrome (KS) | XL | 71 | |
| PHF8b | Plant homeodomain finger protein | Siderius X-linked mental retardation syndrome (MRXSSD) | XL | 70 | |
| Reader | PHF6 | Plant homeodomain finger protein | Börjeson-Forssman-Lehmann syndrome (BFLS) | XL | 75 |
| BRWD3 | Bromodomain-containing protein | X-linked mental retardation and macrocephaly | XL | 36 | |
| Disorders of chromatin remodelers | |||||
| Remodeler | ATRX | SWI/SNF ATP-dependent chromatin remodeler | Alpha-thalassemia/mental retardation X-linked (ATRX) syndrome | XL | 39 |
| ARID1A | SWI/SNF ATP-dependent chromatin remodeler | Mental retardation autosomal dominant 14 (MRD 14; Coffin–Siris syndrome) | AD | 119 | |
| ARID1B | SWI/SNF ATP-dependent chromatin remodeler | Mental retardation autosomal dominant 12 (MRD 12; Coffin–Siris syndrome) | AD | 54, 105, 119 | |
| SMARCA4 | SWI/SNF ATP-dependent chromatin remodeler | Mental retardation autosomal dominant 16 (MRD 16; Coffin–Siris syndrome) | AD | 119 | |
| Rhabdoid tumor predisposition syndrome 2 | AD | 107 | |||
| SMARCB1 | SWI/SNF ATP-dependent chromatin remodeler | Mental retardation autosomal dominant 15 (MRD 15; Coffin–Siris syndrome) | AD | 119 | |
| Schwannomatosis | AD | 113 | |||
| Rhabdoid tumor predisposition syndrome 1 | AD | 110 | |||
| SMARCA2 | SWI/SNF ATP-dependent chromatin remodeler | Nicolaides–Baraitser syndrome | AD | 123 | |
| SRCAP | INO80/SWR1 ATP-dependent chromatin remodeler | Floating harbor syndrome | AD | 53 | |
| CHD7 | CHD ATP-dependent chromatin remodeler | CHARGE syndrome | AD | 125 | |
| Disorders of other chromatin-associated proteins | |||||
| Insulator | CTCF | Chromatin-organizing zinc finger protein | Mental retardation autosomal dominant 21 (MRD 21) | AD | 43 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; XL, X linked.
Known to escape X inactivation.
Encodes a component of the epigenetic machinery that functions as both an eraser and a reader.
Figure 1.
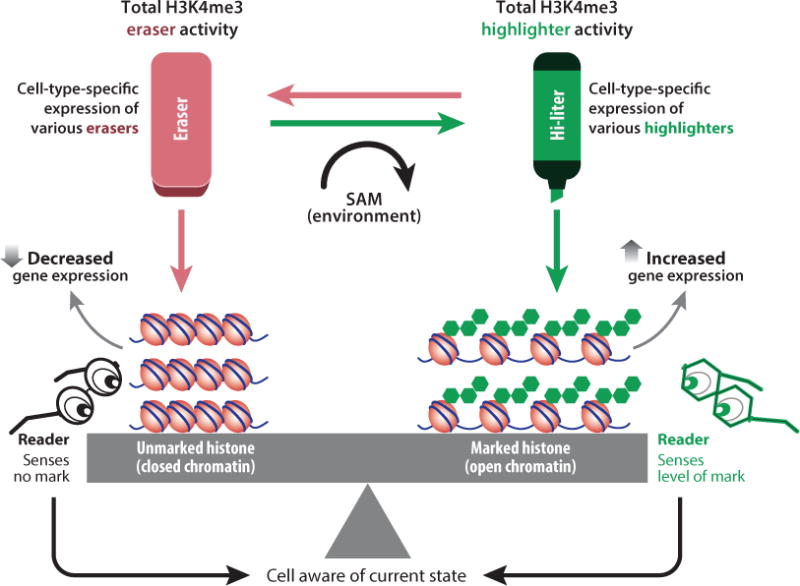
Components of the epigenetic machinery. This machinery consists of writers (highlighters) and erasers of marks [for example, trimethylation of lysine 4 on histone H3 (H3K4me3)] as well as readers of those marks. A net balance between systems that remove and add a particular mark must be achieved. In many ways, the interacting epigenetic systems have certain distinct aspects that make them powerful final integrators of cellular signals (59). For instance, many of the marks placed/removed by writers/erasers can directly affect gene expression, either in a permissive (H3K4me3, shown) or nonpermissive (H3K9me3, not shown) manner. This change in expression, presumably of multiple genes, has the potential to form feedback loops by affecting the amount and availability of the modification in question. Various internal metabolites can directly affect the prevalence of marks. For instance, S-adenosyl-methionine (SAM) is a donor for methylation reactions, including both DNA and histone methylation. Use of critical metabolic intermediates like SAM as donors for histone tail modifications or for DNA methylation allows environmental influences to impact and be integrated into the system and to potentially affect gene expression directly (76).
Another group is the chromatin remodelers, which use the energy of ATP to move nucleosomes along DNA to alter the accessibility of sites and facilitate various cellular processes involving protein–DNA interactions (49). They often function as part of large macromolecular protein complexes, and many have been implicated in Mendelian disease (11) (Table 1). They each have unique modular structures with multiple domains of differing function in addition to their ATPase activity. For example, some contain bromodomains and others contain chromodomains, both of which are readers of distinct chromatin marks (49). This modular structure of the ATP-dependent chromatin remodelers serves the important purpose of linking the various epigenetic systems.
Appropriate cell-type-specific gene expression requires achieving a balance between the activities of the two opposing systems (writers and erasers) and subsequently the placement of their respective marks (Figure 1), which ensures that the appropriate composition of chromatin is present at particular gene promoters. Although a steady-state balance of chromatin marks is likely achieved at any given time, the opposing histone systems are likely to be dynamic (35, 82), allowing the cell to rapidly respond to changes in environmental signals by altering gene expression at specific loci. The histone machinery (as well as some components of the DNA methylation machinery) is enormously redundant, perhaps reflecting the critical importance of maintaining this balance in many different cell types (Figure 1).
MOLECULAR MECHANISMS OF EPIGENETIC SYSTEMS
Understanding individual components of the epigenetic machinery and the marks that they place, remove, and interpret, as well as how they work together to affect cellular gene expression programs, is critical in appreciating the molecular pathogenesis of Mendelian disorders of the epigenetic machinery. We focus here on those marks currently known to be disrupted in the various conditions, namely histone lysine acetylation, histone lysine methylation, and DNA cytosine methylation. In general, acetylation of histones by histone acetyltransferases is associated with active gene expression, and deacetylated histones (mediated by histone deacetylases) tend to be present in transcriptionally silent regions (137). The histone acetylation mark is read by bromodomain-containing proteins. Bromodomains are found within histone acetyltransferases themselves and within chromatin remodelers, suggesting that this domain has a role in both maintaining acetylation states and recruiting chromatin-remodeling complexes to acetylated lysines, and thus to sites of active transcription (137).
Histone lysine methylation by histone methyltransferases is more complex than histone lysine acetylation because lysine methylation can be associated with either open or closed chromatin and can exist in four distinct states (Figure 2). For instance, trimethylation of lysine 4 on histone H3 (H3K4me3) and H3K36me2/3 are generally associated with open chromatin. The former is preferentially associated with promoter regions, and the latter occurs mainly within gene bodies (137). Interestingly, loss of the H3K4me3 mark in some experimental systems does not correlate with significant changes in gene expression (137), although this has not been tested universally. In contrast, H3K9me2/3, H3K27me3, and H4K20me3 (and, occasionally, H3K36me2/3) are marks associated with transcriptionally silent regions (137). H3K9me2/3 is often found at inactive promoters, as is H3K27me3 (137). The latter may be accompanied by H3K4me3 at minimally transcribed bivalent “poised” promoters (14) and may target (either alone or in combination with H3K4me3) de novo DNA methylation of gene promoters in cancer (10). All three of the silencing marks have been localized to particular heterochromatic genomic regions. For example, H3K9me2/3 and H4K20me3 mark pericentromeric heterochromatin and repetitive elements, and H3K27me3 marks facultative heterochromatin, which is present along the inactive X chromosome (18). Removal of lysine methylation is catalyzed by lysine-specific demethylases, which have been identified recently (42).
Figure 2.
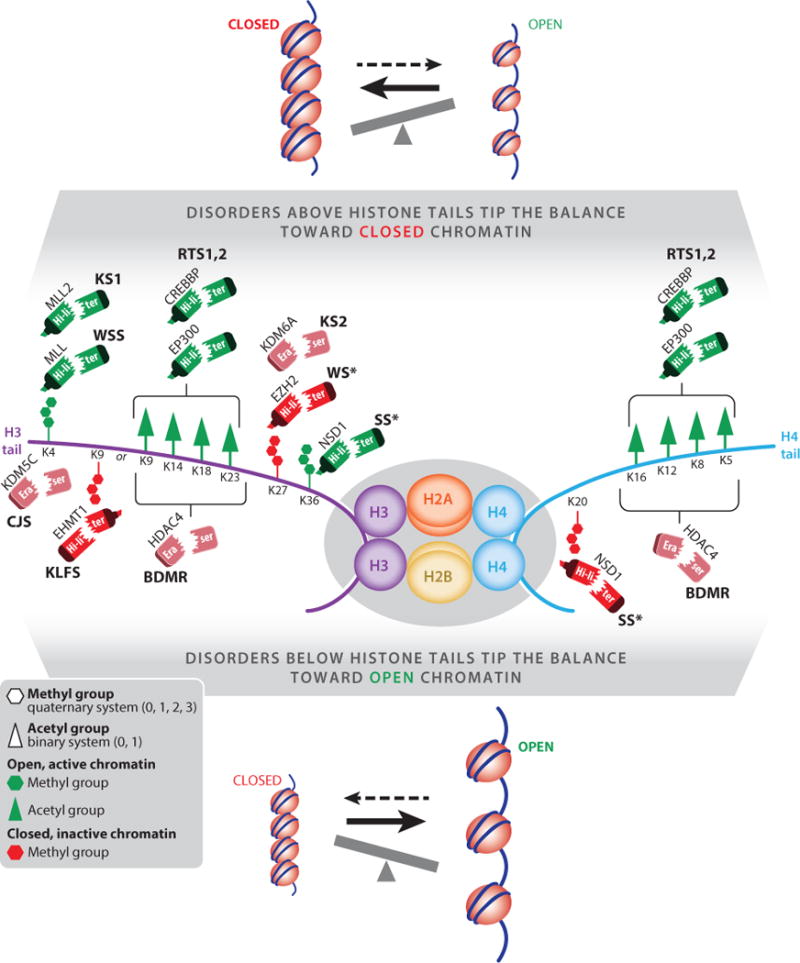
Selected Mendelian disorders of the histone machinery caused by alterations of writers (highlighters) and erasers. Acetylation is a binary mark (present or not), and histone lysine methylation is a quaternary mark (mono-, di-, tri-, or unmethylated). The diagram illustrates these two types of modifications (see key) on two of the N-terminal histone tails, histone H3 and histone H4. The writers (highlighters) and erasers place and remove the modifications, respectively; some of these are associated with open, permissive chromatin (green), and others are associated with closed, repressive chromatin (red). Based on the enzymatic component of the epigenetic machinery involved and the predicted consequence of the reported mutations for each disorder, the diagram shows conditions that would be expected to shift the balance toward closed chromatin states at target loci (top) and those that would be expected to shift the balance toward open chromatin states at target loci (bottom). The former category includes Rubinstein–Taybi syndrome (RTS) (93, 101), Kabuki syndrome (KS) (71, 86), Wiedemann–Steiner syndrome (WSS) (61), and possibly Weaver syndrome (WS) and Sotos syndrome (SS) (41, 69, 116); the latter category includes brachydactyly–mental retardation (BDMR) syndrome (128), Kleefstra syndrome (KLFS) (66), Claes–Jensen syndrome (CJS) (60), and possibly Sotos syndrome (SS) (69). For EZH2 in WS and NSD1 in SS (marked with asterisks), the epigenetic consequences are currently unclear.
Methylated lysines are read and interpreted by proteins containing chromodomains, plant homeodomain finger domains, and others. The particular reader involved determines the downstream consequences of the marks. For example, heterochromatin protein 1 alpha (HP1-alpha) binding to H3K9me3 via its chromodomain functions to propagate this silencing mark by recruiting additional copies of the same histone methyltransferase writers to pericentromeric heterochromatin (7). Other readers serve to link the distinct epigenetic systems. For example, chromatin-remodeling complexes are targeted to active promoters as readers of H3K4me; once recruited, they perform their main function as remodelers. H3K9me2/3 may target various genomic sites for DNA methylation (111). The subsequent role of DNA cytosine methylation may be to lock in silent states. Importantly, in addition to histone marks targeting DNA methylation, there is some evidence to the contrary that suggests that DNA methylation may also direct particular histone marks (89).
Once the genomic sites are targeted, the DNA methyltransferases (DNMTs) are the writers of DNA cytosine methylation. DNMTs come in two types: the de novo DNMTs (DNMT3a and DNMT3b) and the maintenance DNMT (DNMT1), the latter of which targets hemimethylated DNA (111). Cytosine methylation is read and interpreted by specific proteins containing methyl-CpG-binding domains (MBDs) (20, 62). More recently, other readers of CpG methylation have been identified, including the SET and RING finger–associated (SRA) family of proteins (UHRF1 and UHRF2) and certain zinc finger–containing family members, which bind preferentially or exclusively to methylated DNA (20). Others, like CTCF, bind exclusively to unmethylated DNA (9). These proteins perform a variety of functions and appear to have additional binding specificities, including particular histone marks and binding sites within the DNA sequence (20). In the case of MBDs, they bind preferentially to methylated DNA and recruit protein complexes containing transcriptional repressors, including histone deacetylases, to the regions, and this leads to gene silencing (64). This well-established idea has been called into question in the case of MBD3 and MBD5, which may not exhibit binding specificity toward methylated DNA (20). Furthermore, in the case of MeCP2, its role may not be exclusively repressive. As expected, MeCP2-null animals show increased acetylation levels; rather surprisingly, however, they also show an abundance of decreased rather than increased target gene expression (45). Interestingly, MeCP2 has recently been shown to displace histone H1 from nucleosomes and to be extremely abundant in the brain. In fact, the number of molecules of MeCP2 appears to be roughly equal to the number of nucleosomes in isolated neuronal nuclei, suggesting that MeCP2 may have a more global role in chromatin organization in addition to (or instead of) its previously suggested targeted role at individual promoters (45).
Until recently, enzymes capable of removing DNA cytosine methylation, so-called erasers of the 5-methylcytosine mark, have remained elusive. It was known that one mechanism of removing DNA cytosine methylation from the genome is to inhibit the maintenance DNA methylation machinery (95). Upon DNA replication, then, the methyl marks would be diluted out passively. However, more recently, ten-eleven translocation (TET) enzymes have been identified and found to play a major role in this process (58, 114). TETs catalyze the conversion of 5-methylcytosine to 5-hydroxymethylcytosine (114). The cytosine demethylation process is then completed by further biochemical conversion by the TET proteins and direct removal of the modified bases enzymatically (92, 95).
MENDELIAN DISORDERS OF THE DNA METHYLATION MACHINERY
Mendelian disorders of the epigenetic machinery, particularly involving the DNA methylation machinery, have been described for writers and readers of DNA methylation but not yet for erasers of DNA methylation. Importantly, most currently reported Mendelian disorders of the DNA methylation machinery have predominant neurobehavioral phenotypes. For instance, Rett syndrome is an X-linked disorder affecting mostly females and resulting from loss-of-function mutations in a reader of CpG methylation, MeCP2 (83). Individuals with Rett syndrome exhibit mostly normal development until 6–18 months of age, at which time they have developmental arrest and begin to lose previously acquired skills. They develop microcephaly, seizures, and stereotypic hand wringing and lose purposeful hand movements and speech (83).
Heterozygous disruption of another MBD protein–encoding gene, MBD5, in the form of intragenic or contiguous gene deletions (52, 115), chromosomal duplications (85), or point mutations (52, 85, 115), results in strikingly similar phenotypes characterized by developmental delays (including speech and motor delays) and abnormal behaviors with autistic features. Interestingly, individuals with either a deletion or a duplication at 2q23.1 involving MBD5 exhibit additional overlapping neurobehavioral characteristics (namely hypotonia and sleep disturbances) as well as characteristic dysmorphic features, including a broad forehead, thick/highly arched eyebrows, and hand and foot abnormalities (52, 85, 115). However, the deletion and duplication can be distinguished on the basis of certain growth characteristics, namely short stature, microcephaly, and small hands and feet, which tend to occur in individuals with the deletion. Seizures appear to be more common in the setting of deletions or point mutations (52, 85, 115).
In addition to mutations affecting the readers of DNA methylation, those affecting the writers have been implicated in distinct Mendelian disorders of the epigenetic machinery. The first, immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome (47), is caused by homozygous or compound heterozygous hypomorphic mutations in the de novo DNA methyltransferase gene DNMT3B. Cells from individuals with this autosomal recessive condition exhibit hypomethylation of particular DNA sequences as well as a predisposition to chromosomal rearrangements (80). The immunologic dysfunction, which presents early in life, includes hypo- or agammaglobulinemia and a less well-characterized defect in T cell immunity (80).
Disruption of another writer of DNA methylation, DNMT1, can cause two related conditions, both of which result from mutations affecting the targeting-sequence domain of the protein. Mutations within exon 20 cause hereditary sensory and autonomic neuropathy with dementia and hearing loss (HSAN1E) (67). Individuals with this disorder are healthy until young adulthood, when they develop sensorineural hearing loss and sensory neuropathy. They often eventually exhibit debilitating ataxia, seizures, cognitive and behavioral decline, and dementia (67). At the molecular level, mutations within the targeting-sequence domain lead to misfolding and impaired targeting of DNMT1 to heterochromatin (67). In addition, cells exhibit a global loss of 5-methylcytosine with increased focal areas of cytosine methylation at some gene promoters, a cellular phenotype reminiscent of that seen in cancer cells (67). The importance of DNMT1 in the brain is further exemplified by a related late-onset disorder, autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCADN), which results from mutations in exon 21 of the DNMT1 gene (129).
It is interesting that, unlike most Mendelian disorders of the epigenetic machinery, which present in infancy or childhood and are nonprogressive (with the exception of Rett syndrome), DNMT1-related disorders present later in adulthood and have a progressive course. This is perhaps related to the fact that there is only one enzyme known that serves as a maintenance DNA methyltransferase (DNMT1). When a particular component of the epigenetic machinery that exhibits redundancy is disrupted, other related components can partially compensate for the loss and can tip the balance back toward a normal state, partially restoring chromatin and cellular homeostasis and thereby preventing a progressive disease state. However, when DNMT1 is similarly disrupted, there is no other maintenance methyltransferase to compensate and restore the balance; only DNMT1 encoded by the normal allele remains. This is insufficient to compensate fully, and thus the DNA cytosine methylation is diluted and permanently lost over time, possibly owing to an imbalance between maintenance DNA methylation on the one hand and TET-dependent (and replication-dependent) DNA demethylation on the other. It is possible that the onset of symptoms occurs once the progressive loss of DNA cytosine methylation reaches a critical threshold. This idea of age-dependent degeneration of epigenetic patterns was previously suggested as a mechanism for the late onset of common complex diseases (17).
Another interesting and unique feature of the DNMT1-related Mendelian disorders of the epigenetic machinery is that the manifestations are essentially limited to the nervous system. Although many of the Mendelian disorders of the epigenetic machinery exhibit neurologic manifestations, few are exclusively neurologic disorders, instead having multisystemic manifestations. This suggests that in other tissues, one functional copy of DNMT1 can maintain appropriate DNA methylation levels, but in the central and peripheral nervous systems, two copies are required.
MENDELIAN DISORDERS OF THE HISTONE MACHINERY
Mendelian disorders of the histone machinery have been described for writers, erasers, readers, and chromatin remodelers. The histone writer and eraser system is unique because it involves opposing players that must achieve a balance of activity and subsequently of histone marks at particular target genes in any given cell state (Figure 1). This idea is illustrated by Kabuki syndrome (KS), which can be caused by a defect in either a writer or an eraser (Figure 2). KS is an autosomal dominant or X-linked intellectual disability syndrome with specific dysmorphic features, including a flattened facial appearance with characteristic eyes exhibiting long palpebral fissures, eversion of the lower lids, highly arched eyebrows, and long eyelashes, as well as short stature. KS is caused by mutations in either of two genes with complementary functions, i.e., mixed lineage leukemia 2 (MLL2) (86) or lysine-specific demethylase 6A (KDM6A) (71) (Figure 2). These defects lead to indistinguishable conditions (KS1 and KS2). It may seem counterintuitive that loss-of-function mutations in a writer and an eraser lead to similar phenotypes; however, this makes sense when the specific functions are examined. MLL2 is a histone H3K4 methyltransferase that adds trimethylation to H3K4 (56), a mark exclusively seen in open chromatin (32). KDM6A is a demethylase that removes trimethylation from H3K27, a closed chromatin mark (32, 109). MLL2 is a transcriptional coactivator that interacts with transcriptional machinery at the promoters of target genes to facilitate gene expression (56). Decreased MLL2 (or KDM6A) levels would therefore be expected to interfere with the upregulation of numerous critical target genes in susceptible cell types. Interestingly, MLL2 is a Trithorax ortholog, but in Drosophila the opposing Trithorax/Polycomb systems are known to compete to establish a dynamic balance that determines the gene expression levels of particular target genes (108).
Bjornsson and colleagues recently characterized a novel mouse model of KS with deletion of the SET domain of MLL2 (KMT2D) that appears to have a significant deficiency of H3K4me3 in the granule cell layer of the dentate gyrus, which is associated with defective neurogenesis and hippocampal memory defects (H.T. Bjornsson, J.S. Benjamin, J. Weissman, L. Zhang, E.E. Gerber, et al., manuscript in review). This work indicates that specific cell populations may have particular sensitivity to decreased levels of histone-modifying enzymes and the marks that they place. The fact that a deficiency in either removing a closed chromatin mark or placing an open chromatin mark leads to the same disease state suggests that the balance between systems that place open and closed chromatin marks at particular loci may be central to the pathogenesis of KS.
Another disorder, Rubinstein–Taybi syndrome (RTS), is inherited in an autosomal dominant manner and is characterized by specific dysmorphic features, including talon cusps, broad thumbs and great toes with angulation, a grimacing smile, short stature, and intellectual disability (93, 101). The condition is caused by haploinsufficiency of either of two histone acetyltransferase enzyme genes (CREBBP and EP300) (93, 101), which have overlapping functions (6, 88) (Figure 2). RTS is therefore a deficiency of two different writers, both targeting critical sites on histone tails and leading to the same phenotype. Histone acetylation is a binary mark (present or not) and is seen exclusively in open chromatin. The fact that a defect in either CREBBP or EP300 leads to highly overlapping phenotypes might indicate that both histone acetyltransferases are targeted to an overlapping set of genes. Lymphoblastic cell lines from patients with mutations in CREBBP demonstrate global deficiency of histone acetylation (74), and mouse models with targeted CREBBP alleles have demonstrated hippocampal memory defects associated with reduced histone acetylation (1, 68, 122). A deficiency of histone acetylation might lead to a deficiency of open chromatin states in critical cell populations, with correspondingly lowered levels of target gene expression. Although both KS and RTS might be expected to tilt the balance toward less open chromatin states, the phenotypes are quite different. This probably reflects different target gene specificities for the MLL2/KDM6A (44) and CREBBP/EP300 (132) systems.
In contrast, brachydactyly–mental retardation (BDMR) syndrome provides an example of the opposite scenario (128): a disorder whose molecular abnormality tips the balance toward open chromatin at target loci (Figure 2). In this condition, caused by haploinsufficiency of a histone deacetylase gene (HDAC4), patients have skeletal abnormalities, including brachycephaly and brachydactyly, as well as intellectual disability (128). This condition also demonstrates dosage sensitivity, as the severity of the phenotype appears to be dictated by the amount of residual function of HDAC4 (84). HDAC4 is an eraser of the same marks deposited by CREBBP/EP300 (127), tilting the balance toward closed chromatin states. For instance, decreased amounts of HDAC4 appear to upregulate the MEF2 gene in neurons, offering a possible explanation for the intellectual disability seen in the syndrome (102). In addition, HDAC4−/− mice have skeletal abnormalities resembling those of humans with BDMR, as well as increased expression of RUNX2, a known target of HDAC4 repression (124). Therefore, either too much or too little open chromatin at target genes can lead to disease, indicating that the balance between open and closed chromatin states at those sites must be tightly regulated.
Although the deposition of epigenetic modifications can directly affect expression, reading of those marks must also be important to coordinate members of the epigenetic machinery (97), link chromatin-modifying systems to other systems (136), and ultimately affect downstream gene expression. In that regard, some readers contain distinct domains that confer the ability to place histone modifications (writers) or remodel chromatin (98). Börjeson–Forssman–Lehmann syndrome (BFLS), however, is an X-linked recessive intellectual disability syndrome caused by missense mutations in PHF6, encoding a protein that appears to be a reader without other functional domains (75), although PHF6 is known to bind to the NuRD complex (28, 118) and may help to lock in repression through this interaction. BFLS patients have intellectual disability, dysmorphic features, and obesity (120). Loss-of-function mutations in the same gene have also been found in a number of leukemias (118).
In contrast to the pure reader PHF6, ATRX is a component of the epigenetic machinery with multiple functions. It acts as both a chromatin remodeler and a reader to coordinate the various epigenetic systems (26). As a reader, ATRX exhibits tripartite binding via its ATRX-DNMT3-DNMT3L (ADD) domain, interacting with H3K9me3 and H3K4me0 on a single histone H3 tail while simultaneously binding HP1-alpha (33, 57). This elaborate combinatorial binding interaction targets ATRX to specific genomic sites, where it acts as an ATP-dependent chromatin remodeler. ATRX also interacts with MeCP2 and DAXX, the latter interaction suggesting a role for ATRX in replacing the H3.3 variant (81, 137).
Unlike many Mendelian disorders of the epigenetic machinery, for alpha-thalassemia/mental retardation X-linked (ATRX) syndrome, a disease-relevant target gene is known. Mutations in ATRX lead to downregulation of alpha-globin gene expression, resulting individuals with ATRX syndrome having features of the autosomal recessive disease alpha-thalassemia. This supports the broader hypothesis that the pathogenesis of Mendelian disorders of the epigenetic machinery involves downstream epigenetic consequences affecting particular target genes. Furthermore, for ATRX, these trans epigenetic effects then interact with cis variation (genetically determined repeat length) within the alpha-globin locus itself (26) to further modify gene expression. The phenotypic features of ATRX syndrome are more extensive than those of isolated alpha-thalassemia, though, indicating the presence of additional disease-relevant target genes for the former. Features of ATRX syndrome include severe intellectual disability, genital anomalies, characteristic facial appearance, short stature, microcephaly, and brachydactyly in addition to alpha-thalassemia (38). Interestingly, most individuals with this disorder have mutations in the ADD domain or in the ATPase domain of ATRX (40). Mutations in the ADD domain correlate with more severe intellectual disability, whereas mutations in the C terminus lead to more severe genital anomalies (40).
IS AN IMBALANCE OF CHROMATIN STATES CENTRAL TO PATHOGENESIS?
Multiple lines of evidence suggest that Mendelian disorders of the epigenetic machinery result from perturbations of a delicate balance between open and closed chromatin states at particular target genes. Many of these conditions can result from both point mutations within a gene and chromosome microdeletions containing the same gene, suggesting haploinsufficiency as a common disease mechanism (e.g., 19, 66, 69, 71). The majority of the writer/eraser systems, which are composed of enzymes that either add or remove chromatin modifications, demonstrate autosomal dominant inheritance (Table 1). This is intriguing because, in contrast, the majority of other Mendelian enzyme deficiencies are autosomal recessive conditions. Given the prolific efficacy of most enzymes, biallelic mutations with a concomitant reduction of enzymatic activity to <5% of control levels are necessary to cause the expression of disease phenotypes; heterozygotes retaining approximately 50% of enzymatic activity are typically asymptomatic.
The dosage sensitivity for Mendelian disorders of the epigenetic machinery is intriguing in light of the enormous redundancy of some of the protein components with apparently overlapping functions. For instance, there are dozens of enzymes described with H3K4me3 activity, many of which are ubiquitously expressed (31). Despite this seemingly evident redundancy, loss of a single allele of one of these enzymes is sufficient to cause the multisystem disease KS. However, perhaps this redundancy limits the number of affected cell types and thereby leads to a more benign phenotype that is compatible with life despite the wide expression of this component of the epigenetic machinery. Although alternative explanations exist for dosage sensitivity in humans, we favor the hypothesis that the total activity of writers and erasers, the epigenetic marks that they place, and thus the resulting open (active) and closed (silent) chromatin states must remain tightly balanced in cells at particular gene promoters (Figure 1). The dosage sensitivity of components of the epigenetic machinery could suggest that levels of coactivators are carefully controlled in cells to fine-tune gene expression levels. For instance, cellular systems with CREBBP deficiency reveal dose-dependent effects on gene expression, which are dependent on the diversity of modification systems available for a particular target site and/or the strength of recruitment to a particular site (65). Alternatively, given that each individual epigenetic player within a cell acts on multiple downstream target genes and genomic regions and that each works in close cooperation with transcription factors, which are also known to be finely titrated, disruption of just one allele of one of these genes is sufficient to alter this delicate balance.
Our balance hypothesis is supported by the observation that KS exhibits identical phenotypes upon disruption of either a writer of H3K4me3 (an active mark) or an eraser of H3K27me3 (a silencing mark). This indicates that the transition between open and closed chromatin states is critical to disease pathogenesis. Similarly, defects in either deposition of histone acetylation (RTS) or removal of histone acetylation marks (BDMR) lead to disease phenotypes. A one-sided deficiency might be expected to have genome-wide effects, but, perhaps similarly to ATRX, there are a limited number of disease-relevant target genes that are particularly sensitive in each case. At target gene promoters in cells haploinsufficient for MLL2, for example, one might expect to see decreased H3K4me3 and thus increased H3K4me0. Although most genes might tolerate this deficiency, perhaps a small number of disease-relevant target genes in key cell populations are critically affected by this disrupted balance, resulting in altered gene expression. Alternatively, this might promote placement of other marks—possibly H3K9me3, DNA methylation, and H3K27me3—which also could affect transcription.
We now know of a number of Mendelian disorders of the epigenetic machinery that involve disruption of histone writers and erasers (Table 1). If the delicate balance of histone marks and the subsequent effects on the predisposition toward open or closed chromatin states are central to pathogenesis, then it might be critical to determine whether the effect of disruption of each component of the epigenetic machinery would be expected to lead to a defect in open chromatin (likely leading to downregulation of gene expression) or a defect of closed chromatin (likely leading to upregulation of gene expression) at downstream target genes. Figure 2 categorizes disorders based on expected effects on chromatin states.
This idea that a delicate balance of chromatin marks and target gene expression is important in the pathogenesis of Mendelian disorders of the epigenetic machinery is not limited to the histone modification system. Disruption of a dosage-sensitive reader of epigenetic marks, MBD5, can lead to similar phenotypic differences if MBD5 is deleted or duplicated. Indeed, MBD5 expression is reduced in individuals with haploinsufficiency of the predicted reader of epigenetic marks, regardless of mutation mechanism (115), and MBD5 expression is increased when the region is duplicated (85). However, downstream epigenetic and cellular consequences and particular target loci have not been identified. Similarly, a protein closely related to MBD5 and a known reader of DNA cytosine methylation, MeCP2, exhibits marked dosage sensitivity, which is illustrated nicely by the varying phenotypic severity of the corresponding X-linked disorder, Rett syndrome, based on the number of copies of the MeCP2 gene expressed (45). The disorder is more severe in males, who have no functional copies of the gene; for females, the phenotype can vary greatly depending on the pattern of X inactivation as well as on the type and severity of mutation. The more skewed the X inactivation pattern is toward the normal X, the less severe is the disease phenotype (45). That both MeCP2 and MBD5, two readers of epigenetic marks, appear to be so exquisitely dosage sensitive suggests the importance of maintaining not only a critical balance of epigenetic marks and chromatin states within cells but also a critical number of interpreters of those marks. Furthermore, the phenotypic findings in these disorders along with the distinct neurobehavioral-predominant pathogenic features of each suggest that this balance is particularly delicate within the central nervous system.
CAN IMBALANCE INFORM OTHER DISEASE STATES?
In light of the themes that have emerged as we have learned more about Mendelian disorders of the epigenetic machinery, we may be able to hypothesize about the pathogenic sequence in certain less well-elucidated disorders within this category. For instance, Wiedemann–Steiner syndrome (WSS) results from haploinsufficiency of MLL (61), an H3K4 histone methyltransferase gene closely related to MLL2, mutations of which causes KS (86) (Figure 2). Both WSS and KS result in intellectual disability and short stature as well as common distinct dysmorphic features, including long eyelashes and highly arched eyebrows. Interestingly, the causative molecular changes for WSS and KS would both be expected to tip the balance toward closed chromatin states (Figure 2). Thus, their related molecular etiologies and phenotypic similarities are in line with our idea that balance is critical in these disorders.
Two additional related disorders where imbalance may inform pathogenic sequence are Weaver syndrome (WS) and Sotos syndrome (SS). Both are inherited disorders characterized by generalized somatic overgrowth and intellectual disability, and each has its own characteristic dysmorphic features that somewhat overlap with the other (91). Their phenotypic similarity suggests a common pathogenic consequence of their distinct genetic etiologies. Therefore, in the context of our balance hypothesis, we would predict that both would shift the balance of chromatin in the same direction. Making progress in understanding one condition may therefore inform the other.
WS results from mutations in Enhancer of Zeste 2 (EZH2), which encodes a writer of the silencing mark H3K27me3 (41, 116). Most are missense changes, although a few truncating mutations have been described in the penultimate exon, which are expected to escape nonsense-mediated mRNA decay and possibly result in the production of truncated proteins (117). These observations hint at a gain-of-function mutation mechanism (Figure 2) and are indirectly supported by constitutional EZH2 ablation in mice, which does not recapitulate the WS phenotype but rather leads to early embryonic lethality with mutant embryos that are smaller than wild-type littermates (87). Confounding studies of EZH2 expression in cancer cells reveal both upregulation and downregulation, depending on the type of malignancy (51).
SS results from heterozygous loss-of-function mutations (69) or whole-gene deletions (63) in NSD1, which encodes a histone methyltransferase with dual specificity for H3K36 and H4K20 (12, 99, 126, 133). As expected, both H3K36me3 and H4K20me3 appear to be reduced in lymphoblastoid cell lines derived from individuals with SS (12), and expression of one target gene (MEIS1) is induced in association with an increase in an activating mark (H3K4me3) and a decrease in repressive chromatin marks (H3K9me3, H3K27me3, and H4K20me3). However, depletion of NSD1 using RNA interference led to both upregulation and downregulation of distinct subsets of genes (12). Here, identification of disease-relevant target genes might inform the pathogenic sequence and assist with the development of rational therapeutic strategies.
Placing WS and EZH2 in the context of our balance hypothesis seems straightforward. If WS mutations are activating, then the silencing mark H3K27me3 would increase, favoring closed chromatin states (Figure 2); however, if mutations result in loss of function of EZH2 and decreased H3K27me3, then the balance would shift toward open chromatin. For SS, predictions are less straightforward owing to NSD1’s dual specificities for H4K20 and H3K36 and the roles of the latter in both transcriptional activation and silencing. Loss of H4K20me3, a repressive mark, would be expected to shift the balance of chromatin toward a more open state (Figure 2), whereas loss of H3K36me3 (a traditionally active mark), could shift the balance either way and may depend on the presence of other nearby marks, making predictions about overall chromatin states in SS difficult (Figure 2).
Interestingly, there is evidence that some marks placed by EZH2 and NSD1 might be mutually exclusive (126). For example, Yuan et al. (135) showed that H3K27 methylation may preclude H3K36 methylation. Thus, if WS mutations in EZH2 are activating, then the predicted increase in H3K27me3 might lead to a concomitant reduction in H3K36me3, the latter of which has been demonstrated in SS (12). This potential shared pathogenic consequence of distinct mutations in two different histone methyltransferases could explain, at least in part, the striking phenotypic similarity between these two disorders of somatic overgrowth, particularly if the described changes affect common downstream target genes.
PHENOTYPIC OVERLAP
Many Mendelian disorders of the epigenetic machinery demonstrate significant phenotypic overlap, both with one another and with classical imprinting disorders. Both groups are characterized by neurobehavioral and/or growth abnormalities, highlighting the importance of the various epigenetic systems in neurologic development, continued brain function, and basic mechanisms of growth. This observation suggests common downstream targets for some Mendelian disorders of the epigenetic machinery and raises the question of whether at least a subset of those targets may be the very loci involved in classical imprinting disorders.
The features of intellectual disability, short stature, obesity, and brachydactyly and/or small hands might be considered cardinal features of epigenetic disease, as they are seen in many disorders, including PWS (22), MBD5 disruption syndromes (52, 85, 115), BDMR (128), KS (71, 86), WSS (61), and BFLS (75). Within this group, PWS and MBD5 disruption syndromes share additional similarities, including hypotonia, hyperphagia, and sleep disturbances. The overlap is especially fascinating when considering the diversity of molecular epigenetic systems affected within this subset of conditions, suggesting common molecular pathogenic mechanisms.
In contrast, although patients with BWS exhibit pre- and postnatal overgrowth and macroglossia (24) and patients with RSS exhibit pre- and postnatal undergrowth (29), neither condition typically manifests neurobehavioral abnormalities. Like BWS, WS and SS result in generalized overgrowth; however, they also result in intellectual disability. Patients exhibit common facial features characterized by a tall, broad forehead; hypertelorism; and downslanting palpebral fissures, but are distinguished by a characteristic “stuck-on” chin in WS and prognathism in SS (117).
Furthermore, subsets of the Mendelian disorders of the epigenetic machinery share overlapping malformations and distinct dysmorphic features. Those with cardiac malformations include RTS (93, 101), KS (71, 86), SS (63, 69), MBD5 disruption syndromes (52, 85, 115), and Kleefstra syndrome (KLFS) (66), whereas those with genital dysmorphisms include RTS (93, 101), ATRX (39), PWS (22), KLFS (66), and Claes–Jensen syndrome (CJS) (60). Additionally, many of these conditions have similar craniofacial features, including a broad, prominent forehead in WS, SS, MBD5 disruption syndromes, and RSS and a large or broad tongue in BWS, MBD5 disruption syndromes, and KLFS. Finally, MBD5 disruption syndromes appear to act as masqueraders of various epigenetic phenotypes in terms of minor dysmorphic features (52, 85, 115). In addition to the phenotypes mentioned above, individuals with this spectrum of disorders often have highly arched, thick eyebrows reminiscent of those in KS, and those with the duplication have broad great toes resembling those in RTS. Thus, the phenotypic overlap between MBD5 disruption disorders and disorders of histone acetylation, histone methylation, and classical imprinting disorders may indicate convergence of the multiple layers of the epigenetic marks onto partly shared subsets of disease-related target genes. This phenotypic overlap may inform our understanding of molecular pathogenesis and allow prediction of other novel related diseases.
THERAPEUTIC APPROACHES BASED ON RESTORING BALANCE
Herein, we have attempted to summarize the distinctive features of this emerging group of disorders, the Mendelian disorders of the epigenetic machinery. In an effort to stimulate discussion, we have proposed that the pathogenesis of many of these disorders relates to an ongoing dose-dependent deficiency of otherwise finely titrated components of the epigenetic machinery, causing improper expression levels of disease-relevant target genes, referred to here as the balance hypothesis. If the pathogenesis of disorders involving disrupted writers and erasers reflects the dynamic status of this balance rather than fixed developmental insults, this might have profound implications for therapeutic development (Figure 3).
Figure 3.
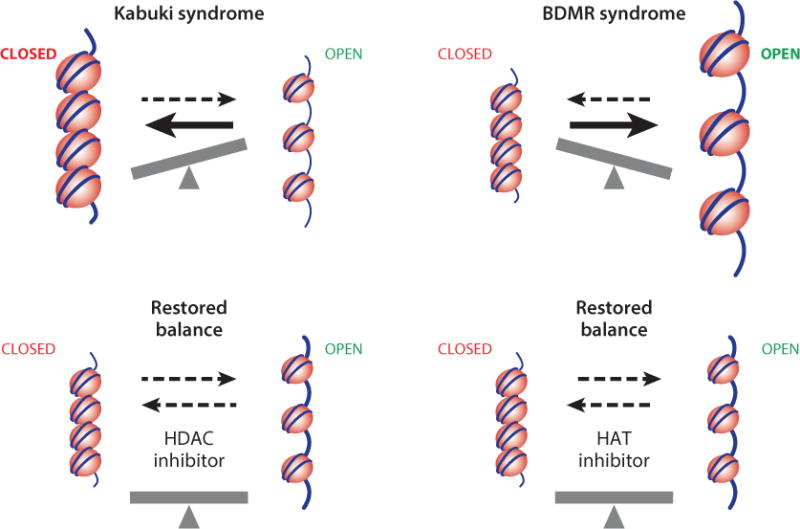
Therapeutic approaches based on understanding and restoring the balance of chromatin states. Our balance hypothesis offers a starting point for proof-of-principle studies for individual Mendelian disorders of the epigenetic machinery, which are potentially treatable causes of intellectual disability. If abnormalities of the expression of target genes are the culprit, then these disorders would be prime candidates for therapeutic development because the target genes would be expected to be fully functional, albeit improperly expressed, in patients with these disorders. For instance, Kabuki syndrome (KS) is related to a deficiency of trimethylation of lysine 4 on histone H3 (H3K4me3) or an inability to remove H3K27me3, marks that are predominantly seen in open and repressive chromatin, respectively. If the pathophysiology of KS is related to an imbalance between open and closed chromatin states (top left), with an inability to use critical gene transcripts, then this balance could be restored by inhibiting the transition to closed chromatin with a histone deacetylase (HDAC) inhibitor (bottom left). In contrast, brachydactyly–mental retardation (BDMR) syndrome would be expected to lead to an overrepresentation of open chromatin states (top right), with excessive transcription of disease-relevant target genes. Therefore, a histone acetyltransferase (HAT) inhibitor could be a useful therapeutic strategy (bottom right).
For instance, mouse models of RTS with loss-of-function mutations in CREBBP demonstrate hippocampal memory defects that respond to two different histone deacetylase inhibitors: SAHA (vorinostat) and trichostatin A (1, 68). Similarly, Bjornsson and colleagues have shown that a mouse model of KS with a severe deficiency of H3K4me3 and defects of neurogenesis within the granule cell layer of the dentate gyrus also demonstrates hippocampal memory defects, which respond postnatally to treatment with a histone deacetylase inhibitor (H.T. Bjornsson, J.S. Benjamin, J. Weissman, L. Zhang, E.E. Gerber, et al., manuscript in review). These data suggest that targeting the epigenetic machinery might ameliorate an ongoing imbalance of chromatin states, with effects on disease phenotypes (Figure 3). A number of US Food and Drug Administration–approved agents, including valproic acid (94), carbamazepine (15), and vorinostat (23), are known to have histone deacetylase inhibitory effects in addition to their other described functions. Treatment with these and other histone deacetylase inhibitors may be fruitful in these disorders, tipping the balance toward open chromatin (Figure 3). In contrast, BDMR, a disorder of excess open chromatin (Figure 3), is known to lead to upregulation of at least one target gene (102). If the balance hypothesis is correct, then one might expect this disorder and others that lead to more open chromatin to respond to histone acetyltransferase inhibitors (Figure 3), such as curcumin and anacardic acid (79). This also highlights the importance of trying to classify these disorders based on proposed effects on gene expression and chromatin states (Figure 2), although classifying some disorders (WS and SS) might be difficult without first uncovering disease-relevant target genes.
Epigenetic therapies have also been used with some success as cancer therapeutics (5, 134), and there is enormous interest in the development of epigenetic therapies for neurologic conditions (121). In fact, histone deacetylase inhibitors positively affect a number of diverse neurologic conditions, ranging from childhood disorders such as spinal muscle atrophy to adult-onset conditions such as Alzheimer’s disease (121). We would argue, however, that the Mendelian disorders of the histone machinery might be the perfect group for epigenetic therapeutic development, given the specific known deficiencies of particular components of the epigenetic machinery, which could allow for rational therapeutic design. Studies of this diverse group of diseases might offer valuable insights into how best to manipulate the epigenome, with the ultimate goal being the development of broad-based epigenetic therapeutic strategies.
EPIGENETIC MODIFICATIONS.
The term epigenetics refers to heritable changes in gene expression in the absence of alterations in the DNA sequence (104). Epigenetic modifications play a major role in tissue- and cell-type-specific differences in gene expression (100, 138) and many other cellular processes (73, 131, 137). The role of epigenetics in disease is well established in the field of cancer (10, 34) and imprinting disorders (8).
CpG methylation of DNA at promoters is associated with gene silencing, whereas gene body CpG methylation is associated with active transcription (50). The human genome is relatively depleted of CpG dinucleotides, which when present tend to be methylated (30). However, CpG islands—stretches of DNA sequence enriched for CpG dinucleotides—are protected from DNA methylation (16, 111). In contrast, CpG shores are seen in close proximity to CpG islands with comparatively reduced CpG densities (55). Shores are often methylated in association with tissue-specific transcription silencing (55).
The histone modification machinery is thought to integrate cellular signaling pathways, which converge on promoters of genes to create a single unified signal (2, 103). Currently the meaning of the combinatorial histone modification “language” (13, 37, 112) is incompletely understood. However, a deeper understanding will likely yield insights into gene expression regulation (37).
SUMMARY POINTS.
Mendelian disorders of the epigenetic machinery are caused by defects in various components of the machinery, which write, erase, read, and remodel epigenetic marks.
In contrast to most classical imprinting disorders, these genetic aberrations are expected to have global epigenetic consequences in trans.
Despite the redundancy of the components of the epigenetic machinery, loss or gain of a single allele is not tolerated, leading to disease states and indicating tight regulation of the amount of each component at disease-relevant target genes.
Specific examples suggest that the balance between systems that add and remove marks is critical to pathogenesis. For instance, Kabuki syndrome is caused by a deficiency of a writer of an open chromatin mark or an eraser of a closed chromatin mark. Similarly, Rubinstein–Taybi and brachydactyly–mental retardation syndromes affect the same marks but in opposite directions.
If the pathogenesis of these disorders relates to an ongoing dose-dependent deficiency of otherwise finely titrated components of the epigenetic machinery, leading to improper expression levels of disease-relevant target genes (referred to here as the balance hypothesis), then agents that favor either chromatin opening (histone deacetylase inhibitors) or chromatin closing (histone acetyltransferase inhibitors) might have therapeutic potential.
FUTURE ISSUES.
Can we find additional disease-relevant target genes for other Mendelian disorders of the epigenetic machinery (in addition to alpha-thalassemia/mental retardation X-linked and brachydactyly–mental retardation syndromes) for which disrupted expression leads to specific features of disease phenotypes?
Can disease-relevant genes act as biomarkers of prognosis and therapeutic response? Can expression analysis of disease-relevant target genes help us explain the phenotypic variation seen in these disorders and allow us to map modifier genes?
Why do defects of ubiquitously expressed components of the epigenetic machinery lead to cell- and tissue-specific disease phenotypes that differ from one another? Are there particular cell populations with sensitivity to these defects?
Can we use available agents that target epigenetic systems to restore abnormal gene expression of disease-relevant target genes in multiple conditions? Can we appropriately titrate those agents to treat ongoing disease phenotypes (such as intellectual disability) without causing toxicity?
Acknowledgments
We would like to acknowledge Professors Barbara Migeon, Ada Hamosh, and Richard Lee as well as Dr. Patrick Onyango for critical reading of the manuscript and valuable comments. We would also like to acknowledge Catherine Kiefe for her assistance with creating and editing the figures. This work was supported by a grant to H.T.B. by the NIH Director’s Early Independence Award (DP5OD017877).
Glossary
- Mendelian disorders of the epigenetic machinery
disorders caused by genetic disruption of components of the epigenetic machinery, including writers, erasers, readers, and remodelers
- Imprinting
the differential expression of alleles for a particular gene depending on the parental origin of the allele; each parental allele contains a distinctive set of epigenetic marks
- In cis
refers to the local regulation of a gene involving nearby regulatory elements or epigenetic marks on the same chromosome
- Trans-acting factor
a diffusible regulator of a gene encoded at a different chromosomal site and thereby distant from the gene being regulated
- Chromatin
DNA and its associated proteins (histones)
- Writer
a molecule that adds epigenetic modifications to either a DNA molecule or histone proteins
- Reader
a molecule that can read the modification state of a DNA molecule or histone proteins
- Remodeler
a molecule that uses ATPase activity to change the nucleosome composition at loci in trans
- Dosage sensitivity
an attribute of some genes in which the loss or gain of a copy of the gene leads to a disease state
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org
Contributor Information
Jill A. Fahrner, Email: jfahrne1@jhmi.edu.
Hans T. Bjornsson, Email: hbjorns1@jhmi.edu.
LITERATURE CITED
- 1.Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–59. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–51. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–88. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 4.Arnaud P, Feil R. Epigenetic deregulation of genomic imprinting in human disorders and following assisted reproduction. Birth Defects Res C. 2005;75:81–97. doi: 10.1002/bdrc.20039. [DOI] [PubMed] [Google Scholar]
- 5.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours— lessons from the past. Nat Rev Clin Oncol. 2013;10:256–66. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–43. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 7.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–24. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 8.Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 9.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3:a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–83. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- 12.Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci USA. 2009;106:21830–35. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 15.Beutler AS, Li S, Nicol R, Walsh MJ. Carbamazepine is an inhibitor of histone deacetylases. Life Sci. 2005;76:3107–15. doi: 10.1016/j.lfs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–13. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 17.Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–58. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breuning MH, Dauwerse HG, Fugazza G, Saris JJ, Spruit L, et al. Rubinstein-Taybi syndrome caused by submicroscopic deletions within 16p13.3. Am J Hum Genet. 1993;52:249–54. [PMC free article] [PubMed] [Google Scholar]
- 20.Buck-Koehntop BA, Defossez PA. On how mammalian transcription factors recognize methylated DNA. Epigenetics. 2013;8:131–37. doi: 10.4161/epi.23632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campeau PM, Kim JC, Lu JT, Schwartzentruber JA, Abdul-Rahman OA, et al. Mutations in KAT6B, encoding a histone acetyltransferase, cause genitopatellar syndrome. Am J Hum Genet. 2012;90:282–89. doi: 10.1016/j.ajhg.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 23.Cea M, Cagnetta A, Gobbi M, Patrone F, Richardson PG, et al. New insights into the treatment of multiple myeloma with histone deacetylase inhibitors. Curr Pharm Des. 2013;19:734–44. [PMC free article] [PubMed] [Google Scholar]
- 24.Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C. 2013;163C:131–40. doi: 10.1002/ajmg.c.31363. [DOI] [PubMed] [Google Scholar]
- 25.Clayton-Smith J, O’Sullivan J, Daly S, Bhaskar S, Day R, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–81. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clynes D, Higgs DR, Gibbons RJ. The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem Sci. 2013;38:461–66. doi: 10.1016/j.tibs.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 27.Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesion acetylation cycle. Nature. 2012;489:313–17. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–38. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 29.Eggermann T, Begemann M, Spengler S, Schröder C, Kordass U, et al. Genetic and epigenetic findings in Silver-Russell syndrome. Pediatr Endocrinol Rev. 2010;8:86–93. [PubMed] [Google Scholar]
- 30.Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–21. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–49. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ENCODE Proj. Consort. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol. 2011;18:777–82. doi: 10.1038/nsmb.2070. [DOI] [PubMed] [Google Scholar]
- 34.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–40. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 35.Ficz G, Heintzmann R, Arndt-Jovin DJ. Polycomb group protein complexes exchange rapidly in living Drosophila. Development. 2005;132:3963–76. doi: 10.1242/dev.01950. [DOI] [PubMed] [Google Scholar]
- 36.Field M, Tarpey PS, Smith R, Edkins S, O’Meara S, et al. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am J Hum Genet. 2007;81:367–74. doi: 10.1086/520677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons RJ. Alpha thalassaemia-mental retardation, X linked. Orphanet J Rare Dis. 2006;1:15. doi: 10.1186/1750-1172-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with α-thalassemia (ATR-X syndrome) Cell. 1995;80:837–45. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 40.Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat. 2008;29:796–802. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- 41.Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110–18. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregor A, Oti M, Kouwenhoven EN, Hoyer J, Sticht H, et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet. 2013;93:124–31. doi: 10.1016/j.ajhg.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo C, Chang CC, Wortham M, Chen LH, Kernagis DN, et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc Natl Acad Sci USA. 2012;109:17603–8. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–52. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 46.Hackett JA, Surani MA. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond B. 2013;368:20110328. doi: 10.1098/rstb.2011.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–17. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harakalova M, van den Boogaard MJ, Sinke R, van Lieshout S, van Tuil MC, et al. X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J Med Genet. 2012;49:539–43. doi: 10.1136/jmedgenet-2012-100921. [DOI] [PubMed] [Google Scholar]
- 49.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–43. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 51.Hock H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev. 2012;26:751–55. doi: 10.1101/gad.191163.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hodge JC, Mitchell E, Pillalamarri V, Toler TL, Bartel F, et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry. 2014;19:368–79. doi: 10.1038/mp.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hood RL, Lines MA, Nikkel SM, Schwartzentruber J, Beaulieu C, et al. Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am J Hum Genet. 2012;90:308–13. doi: 10.1016/j.ajhg.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-A chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–72. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. The human colon cancermethylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2006;27:1889–903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat Struct Mol Biol. 2011;18:769–76. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 60.Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, et al. Mutationsin the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–36. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. 2012;91:358–64. doi: 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jørgensen HF, Bird A. MeCP2 and other methyl-CpG binding proteins. Ment Retard Dev Disabil Res Rev. 2002;8:87–93. doi: 10.1002/mrdd.10021. [DOI] [PubMed] [Google Scholar]
- 63.Kamimura J, Endo Y, Kurotaki N, Kinoshita A, Miyake N, et al. Identification of eight novel NSD1 mutations in Sotos syndrome. J Med Genet. 2003;40:e126. doi: 10.1136/jmg.40.11.e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kantor B, Makedonski K, Shemer R, Razin A. Expression and localization of components of the histone deacetylases multiprotein repressory complexes in the mouse preimplantation embryo. Gene Expr Patterns. 2003;3:697–702. doi: 10.1016/j.modgep.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, et al. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 2010;29:3660–72. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, et al. Loss-of-function mutations in euchromatin bistone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79:370–77. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–72. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002;30:365–66. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 70.Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet. 2005;42:780–86. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–24. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–23. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Kim J, Oberdoerffer P. Metabolic modulation of chromatin: implications for DNA repair and genomic integrity. Front Genet. 2013;4:182. doi: 10.3389/fgene.2013.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez-Atalaya JP, Gervasini C, Mottadelli F, Spena S, Piccione M, et al. Histone acetylation deficits in lymphoblastoid cell lines from patients with Rubinstein-Taybi syndrome. J Med Genet. 2012;49:66–74. doi: 10.1136/jmedgenet-2011-100354. [DOI] [PubMed] [Google Scholar]
- 75.Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, et al. Mutations in PHF6 are associated with Börjeson-Forssman-Lehmann syndrome. Nat Genet. 2002;32:661–65. doi: 10.1038/ng1040. [DOI] [PubMed] [Google Scholar]
- 76.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–51. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 79.Manzo F, Tambaro FP, Mai A, Altucci L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin Ther Pat. 2009;19:761–74. doi: 10.1517/13543770902895727. [DOI] [PubMed] [Google Scholar]
- 80.Matarazzo MR, De Bonis ML, Vacca M, Della Ragione F, D’Esposito M. Lessons from two human chromatin diseases, ICF syndrome and Rett syndrome. Int J Biochem Cell Biol. 2009;41:117–26. doi: 10.1016/j.biocel.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 81.Maze I, Noh KM, Allis CD. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology. 2013;38:3–22. doi: 10.1038/npp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mito Y, Henikoff JG, Henikoff S. Histone replacement marks the boundaries of cis-regulatory domains. Science. 2007;315:1408–11. doi: 10.1126/science.1134004. [DOI] [PubMed] [Google Scholar]
- 83.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–81. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 84.Morris B, Etoubleau C, Bourthoumieu S, Reynaud-Perrine S, Laroche C, et al. Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am J Med Genet A. 2012;158A:2015–20. doi: 10.1002/ajmg.a.35463. [DOI] [PubMed] [Google Scholar]
- 85.Mullegama SV, Rosenfeld JA, Orellana C, van Bon BW, Halbach S, et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet. 2014;22:57–63. doi: 10.1038/ejhg.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–93. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, et al. The Polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330–36. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–59. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 89.Okitsu CY, Hsieh CL. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol. 2007;27:2746–57. doi: 10.1128/MCB.02291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ollikainen M, Craig JM. Epigenetic discordance at imprinting control regions in twins. Epigenomics. 2011;3:295–306. doi: 10.2217/epi.11.18. [DOI] [PubMed] [Google Scholar]
- 91.Opitz JM, Weaver DW, Reynolds JF., Jr The syndromes of Sotos and Weaver: reports and review. Am J Med Genet. 1998;79:294–304. doi: 10.1002/(sici)1096-8628(19981002)79:4<294::aid-ajmg12>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 92.Pastor WA, Aravind L, Rao A. TET onic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional coactivator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 94.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–41. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 95.Piccolo FM, Fisher AG. Getting rid of DNA methylation. Trends Cell Biol. 2014;24:136–43. doi: 10.1016/j.tcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 96.Poole RL, Docherty LE, Al Sayegh A, Caliebe A, Turner C, et al. Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am J Med Genet A. 2013;161:2174–82. doi: 10.1002/ajmg.a.36049. [DOI] [PubMed] [Google Scholar]
- 97.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 98.Qiu J, Shi G, Jia Y, Li J, Wu M, et al. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 2010;20:908–18. doi: 10.1038/cr.2010.81. [DOI] [PubMed] [Google Scholar]
- 99.Rayasam GV, Wendling O, Angrand PO, Mark M, Niederreither K, et al. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003;22:3153–63. doi: 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–10. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 101.Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–80. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–59. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 104.Russo VEA, Martienssen RA, Riggs AD, editors. Epigenetic Mechanisms of Gene Regulation. Woodbury, NY: Cold Spring Harb. Lab. Press; 1996. [Google Scholar]
- 105.Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–80. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 106.Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, et al. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat. 2013;34:1519–28. doi: 10.1002/humu.22394. [DOI] [PubMed] [Google Scholar]
- 107.Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–84. doi: 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schwartz YB, Pirrotta V. Polycomb complexes and epigenetic states. Curr Opin Cell Biol. 2008;20:266–73. doi: 10.1016/j.ceb.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 109.Sengoku T, Yokoyama S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011;25:2266–77. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, et al. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65:1342–48. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 112.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 113.Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, et al. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet. 2009;46:68–72. doi: 10.1136/jmg.2008.060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–35. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–63. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127–33. doi: 10.18632/oncotarget.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tatton-Brown K, Rahman N. The NSD1 and EZH2 overgrowth genes, similarities and differences. Am J Med Genet C. 2013;163C:86–91. doi: 10.1002/ajmg.c.31359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Todd MA, Picketts DJ. PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J Proteome Res. 2012;11:4326–37. doi: 10.1021/pr3004369. [DOI] [PubMed] [Google Scholar]
- 119.Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–78. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- 120.Turner G, Gedeon A, Mulley J, Sutherland G, Rae J, et al. Börjeson-Forssman-Lehmann syndrome: clinical manifestations and gene localization to Xq26–27. Am J Med Genet. 1989;34:463–69. doi: 10.1002/ajmg.1320340402. [DOI] [PubMed] [Google Scholar]
- 121.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 122.Valor LM, Pulopulos MM, Jimenez-Minchan M, Olivares R, Lutz B, et al. Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation but does not affect cell viability. J Neurosci. 2011;31:1652–63. doi: 10.1523/JNEUROSCI.4737-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Van Houdt JKJ, Nowakowska BA, Sousa SB, van Schaik BDC, Seuntjens E, et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44:445–49. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- 124.Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–66. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 125.Vissers LELM, van Ravenswaaij CMA, Admiraal R, Hurst JA, de Vries BBA, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–57. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 126.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115–26. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, et al. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol. 1999;19:7816–27. doi: 10.1128/mcb.19.11.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–28. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012;21:2205–10. doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wolffe AP. Nucleosome positioning and modification: chromatin structures that potentiate transcription. Trends Biochem Sci. 1994;19:240–44. doi: 10.1016/0968-0004(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 131.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–86. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 132.Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–17. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang H, Pesavento JJ, Starnes TW, Cryderman DE, Wallrath LL, et al. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J Biol Chem. 2008;283:12085–92. doi: 10.1074/jbc.M707974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–46. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yuan W, Xu M, Huang C, Liu N, Chen S, et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–89. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011;21:564–78. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–66. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 138.Zuckerkandl E. A possible role of “inert” heterochromatin in cell differentiation. Action of and competition for “locking” molecules. Biochimie. 1974;56:937–54. doi: 10.1016/s0300-9084(74)80516-x. [DOI] [PubMed] [Google Scholar]