

Abstract
Rhodopsin, a G-protein coupled receptor, most abundant protein in retinal rod photoreceptors, is glycosylated at asparagines-2 and 15 on its N-terminus. To understand the role of rhodopsin's glycosylation in vivo, we generated and characterized a transgenic mouse model that expresses a non-glycosylated form of rhodopsin. We show that lack of glycosylation triggers a dominant form of progressive retinal degeneration. Electron microscopic examination of retinas at postnatal day 17 revealed the presence of vacuolar structures that distorted rod photoreceptor outer segments and became more prominent with age. Expression of non-glycosylated rhodopsin alone showed that it is unstable and is regulated via ubiquitin-mediated proteasomal degradation at the base of outer segments. We observed similar vacuolization in outer segments of transgenic mice expressing human rhodopsin with a T17M mutation (hT17M), suggesting that the mechanism responsible for the degenerative process in mice expressing the non-glycosylated rhodopsin and the RHOhT17M mice is likely the cause of phenotype observed in retinitis pigmentosa patients carrying T17M mutation.
Introduction
The retinal photoreceptor cell, with its two subclasses (rods and cones), responds to light stimulation by initiating visual transduction. The sub-classification is based upon a photoreceptor's differential sensitivity to light intensities and wavelengths and the physical appearance of the cell. Each photoreceptor cell is composed of four cellular compartments: the synaptic terminal, the cell body, the inner segment (IS) and the outer segment (OS). The rod photoreceptor OS, a modified cilia, is composed of a stack of membranous discs that are light sensitive due to the presence of the visual pigment rhodopsin (Rho), which consists of a single polypeptide apo-protein (opsin) and its covalently bound chromophore, 11-cis retinal, the latter a derivative of vitamin A.
Retinitis pigmentosa (RP) is an inherited progressive retinal degenerative disorder that is clinically and genetically heterogeneous [for review, see (1)]. RP, the most widespread inherited form of retinal degeneration, is characterized by initial night blindness followed by visual field constriction, and may progress to complete blindness (2). Mutations in RHO account for over 30% of autosomal dominant retinitis pigmentosa (ADRP) cases in humans (https://sph.uth.tmc.edu/retnet/disease.htm#03.202d). Therefore, deciphering the mechanisms of RHO-associated RP will impact the development of both gene-based and pharmacological therapies.
Rho undergoes several post-translational modifications, including N-linked glycosylation at two asparagine residues (Asn2, Asn15) at its N-terminus. Mutations of these residues are associated with ADRP (https://sph.uth.tmc.edu/retnet/disease.htm#03.202d). Animal models for both mutations are available, but each involves only one of the glycosylation sites (3,4). While the phenotypes support an important role for glycosylation in the overall function of Rho, the fact that the second site is not mutated makes it difficult to assess the role of glycosylation in the molecule's overall function. To address this need, we generated transgenic mice that express a NOn-Glycosylated (NOG) Rho. On a wild-type genetic background (Rho+/+), NOG mice exhibited a marked reduction in retinal function and number of photoreceptors, as well as vacuolization and disorganized rod OSs. To specifically detect the NOG protein on immunoblots, NOG mice were bred into the Rho−/− background. Despite the robust expression of intact NOG transcripts, the protein levels were low. However, the small amount that was detected reached the OS, the normal destination for Rho, indicating that its intracellular trafficking was not compromised. The presence of a small amount of the NOG protein was determined to be the result of rapid turnover of NOG Rho via the ubiquitin-proteasome system (UPS) at base of OS. The degenerative phenotype of the NOG retina highlights the deleterious influence of even slight amounts of the NOG protein. Findings from NOG mice indicate that N-linked glycosylation of Rho is necessary for the protein's incorporation into the OS and the proper morphogenesis and maintenance of rod OSs. Investigation of the degenerative phenotype in RHOhT17M transgenic mice revealed the presence of vacuoles similar to those observed in NOG mice, suggesting that the mechanism underlying photoreceptor degeneration in NOG retinas is likely at play in the RHOhT17M transgenic mice and in ADRP patients carrying the T17M RHO mutation. These observations provide novel insights into the pathobiology of retinal degeneration in this form of RP.
Results
The injection of the NOG construct produced five potential founders. Three of the five potential founders (designated NOG A, B and C) passed the transgene to their offspring in a Mendelian fashion and were further analyzed.
Retinal function is reduced in NOG mice
The functional consequences of NOG expression were determined by ERG between postnatal day (P) 30 and 120. At P30, rod-mediated photoreceptor function, measured by the scotopic a-wave, of NOG ARho+/+ mice (Fig. 1A, open diamonds) was reduced by ∼25% in comparison to non-transgenic (Rho+/+) littermates (cf. Fig. 1A, closed circles). By P60, ERG a-waves of NOG ARho+/+ mice were reduced to ∼50% of the wild-type levels (Fig. 1A, closed diamonds); by P120, NOG ARho+/+ a-waves were only ∼10% of the wild-type level (Fig. 1B, open diamonds). In comparison, ERG a-waves of NOG CRho+/+ mice were dramatically reduced at P30 (Fig. 1A, open triangle). The results from NOG BRho+/+ mice were similar to those from the NOG CRho+/+ mice (data not shown).
Figure 1.

Retinal function in NOG mice. Graphical representations of light-evoked retinal responses from NOG animals as obtained by ERG. (A) Rod-mediated ERG a-wave response functions elicited from P30 NOG A and C transgenic mice (open diamonds, open triangles, respectively), P60 NOG A and C (closed diamonds, closed triangles, respectively), and from non-transgenic Rho+/+ mice (closed circles) to strobe flashes presented to dark-adapted mice. (B–D) Age-dependent changes in the maximum ERG a-wave (B), b-wave (C) or cone ERG (D) for NOG A mice on Rho+/+ or Rho+/− backgrounds. Statistics used: 2-way ANOVA with Bonferroni's post-test. Data points indicate average ± SEM for the following number of animals: P30: Rho+/+ & Rho+/− = 17, NOG ARho+/+ = 4, NOG ARho+/− = 8, Rho−/− = 5, NOG ARho−/− = 6; P60: Rho+/+ & Rho+/− = 12, NOG ARho+/+ = 7, NOG ARho+/− = 3; P120: Rho+/+ & Rho+/− = 6, NOG ARho+/+ = 3, NOG ARho+/− = 4.
ERG b-waves, generated by depolarizing bipolar cells (5) of NOG ARho+/+ mice were unaffected at P30 (Fig. 1C, open diamonds). At later ages, b-wave amplitudes declined, secondary to the observed reduction in rod function (compare Fig. 1B with C).
Cone function of NOG ARho+/+ mice measured by the photopic b-wave amplitude was unaffected at P30 and P60 (Fig. 1D). A significant decline in cone function was observed at P120, when rod function was severely reduced.
The functional loss in NOG A is not due to rhodopsin overexpression
Total relative Rho transcript levels (endogenous plus transgenic) were examined in NOG retinas at P17 using qRT–PCR. The levels of Rho transcripts were normalized first to the Hprt transcript levels to account for sample-to-sample or experimental variability, then to the levels of Rds transcript to account for potential morphologic changes associated with the ERG a-wave reductions. Quantification of relative transcripts revealed that NOG A, B and C expressed Rho transcripts at 1.32, 0.15 and 1.77 folds, respectively, over Rho transcript levels in non-transgenic Rho+/+ mice (Fig. 2A). However, immunoblot analysis of NOG retinal extracts revealed that the NOG ARho+/+ retina expressed only glycosylated opsin (Fig. 2B). No opsin or Rds was detected in NOG BRho+/+ or NOG CRho+/+retinas, consistent with an early onset severe loss of photoreceptors.
Figure 2.

Rhodopsin expression in NOG retinas. Quantitative RT–PCR analysis of NOGRho+/+ mice at P17. (A) The steady-state Rho transcript levels relative to Rds, n = 3. The Bouse A sample is a positive control and represents a transgenic mouse model with Rho overexpression (6). (B) Immunoblot analysis of P17 retinal extracts. Top blot is probed with 1D4, while the bottom blot is probed with anti-RDS. * = P < 0.0001, relative to the Rho+/+ sample, n = 3. Error bars = SEM.
The drastically reduced levels of total Rho transcripts in the NOG B retinas (Fig. 2A) suggest that the transgene integrated in a locus that is either essential for expression of the Rho gene or for proper retinal development. Regardless of the cause, the lack of any transcript at P17 rendered NOG B useless for this investigation. Therefore, it was excluded from the rest of the study.
The Rho transcript levels in NOG C were considerably higher than those in the non-transgenic retinas (Fig. 2A). Previous studies showed that RHO overexpression in the Bouse mouse leads to retinal functional and structural defects (6). To determine whether the observed phenotype was due to NOG or endogenous Rho overexpression, NOG mice were mated to Rho−/− mice to allow for the expression of NOG in the presence of half or no endogenous Rho.
There was no statistically significant difference in scotopic a-wave ERG responses of the NOG ARho+/− rods compared with those of NOG ARho+/+, at any time point (Fig. 1B, open squares). However, scotopic b-wave responses from NOG ARho+/− mice were larger than those from NOG ARho+/+ at P120 (P < 0.01, Fig. 1C, open squares). Both a and b wave scotopic functions were significantly reduced in the NOG ARho+/− retinas compared with the non-transgenic controls. We observed no differences in photopic responses. Elimination of half the endogenous Rho did not alter the phenotype in NOG C (data not shown). These findings suggest that the observed functional deficit is not influenced by the amount of opsin in the NOG retina, but instead a direct effect of NOG expression.
The NOG retina exhibits outer segment malformations and retinal degeneration
We performed histology to determine whether the age-dependent decline in ERG responses in the NOG ARho+/+ mice was due to a retinal degenerative process. At P17, the NOG ARho+/+ retinas possessed the full complement of photoreceptor nuclei in the outer nuclear layer (ONL; compare Fig. 3C with A), indicating that no photoreceptor death had occurred. However, examination of NOG ARho+/+ retinal ultrastructure revealed vacuole-like structures (Fig. 3D, asterisks) and disc disorganization (Fig. 3D, arrow) in the OS layer that were not observed in Rho+/+ retinas (cf. Fig. 3B). At P30, the vacuole-like structures were more obvious in the NOG ARho+/+ retinas (Fig. 3E). Ultrastructural examination revealed more extensive OS disorganization at P30 than at P17 (Fig. 3F, arrow and asterisks). Closer inspection of the vacuoles suggests that they were filled with membranous structures likely originating from OS discs (Fig. 3F’, asterisk and arrow). The OS disorganization advances as the animals age, as evident from examination of the transgenic retinas at P60 (Fig. 3G and H, respectively). Interestingly, although the OS are structurally disorganized, ERG a-waves of the P60 retinas were reduced by only ∼50% compared with P30 (Fig. 1). An age-dependent decline between P30 and P60 was also observed in non-transgenic Rho+/+ retinas. The slope of this decline was steeper in the NOG A retinas than in the non-transgenic retinas. Upon histologic examination of the NOG CRho+/+ retinas, it became evident that the lack of ERG responses results from the complete degeneration of the photoreceptor layers by P17 (data not shown).
Figure 3.

Reduced ERG responses reflect the morphologic changes as determined by light (LM) and electron (EM) microscopy. A, C, E and G are LM sections; B, D, F, F’ and H are EM images. (A and B) Non-transgenic Rho+/+ retinal sections at P17. (C and D) The NOG ARho+/+ EM section at P17 revealed vacuoles in the OS (asterisks). (E and F) P30 NOG ARho+/+ sections. When evaluated by EM (F), these vacuoles seem to contain membranous material similar to that of disorganized OS (asterisks). Boxed area in (F) is enlarged in (F’). (G and H) P60 NOG ARho+/+ sections. Scale bar in (A) (light micrographs) is 50 microns and in (B) (EM micrographs) is 5 microns. RPE, pigment epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Rhodopsin localization is normal in the NOG retina
RHO mutations within the N-terminus (Class II mutants) are characterized by their low protein expression, loss of ability to bind with the opsin chromophore (11-cis retinal) and an accumulation of the mutant protein in the endoplasmic reticulum (ER) (7). Furthermore, these mutants can interfere with the maturation of endogenous normal RHO (8,9).
To determine whether NOG protein expression could lead to degeneration due to Rho mislocalization and/or sequestration, we examined Rho localization in P40 NOG ARho+/+ retinas. Immunohistochemical examination at the light (Fig. 4C) and electron microscopic (Fig. 4D and D’) levels showed that Rho was localized in the OS of the NOG ARho+/+ retinas, similar to the non-transgenic controls (Fig. 4A, B and B’). In the Rho+/+ and NOG ARho+/+ retinas, Rho was localized to the disc and plasma membrane (Fig. 4B’ and D’, arrows). There was no accumulation of Rho in any other part of the photoreceptor cell. These results show that the degenerative process in the NOG retinas is not due to mislocalization of NOG or endogenous Rho.
Figure 4.

Immunolocalization of rhodopsin to the outer segments of transgenic mice. Immunohistochemical analyses at the light (A and C) and immunogold (B and D) levels were conducted to determine whether Rho localized properly to the OS. The micrographs represent a cross section of a P40 Rho+/+ (A and B) and NOG A Rho+/+ (C and D) retina labeled with polyclonal anti-Rho. Scale bar in (A) (light micrograph) is 50 microns; scale bar in (B) and (D) (immunogold) is 2 microns. Boxed areas in (B) and (D) are enlarged in (B’) and (D’), respectively. Arrows point to presence of Rho in plasma membrane besides its presence in OS discs.
Lack of ER stress in NOG-Expressing retinas
RHOhT17M transgenic mice exhibit progressive retinal degeneration (10) with induced endoplasmic reticulum (ER) stress (11). The T17M mutation eliminates N15-linked glycosylation of RHO. Mutant RHO was localized to the ONL of the RHOhT17M mice and several markers for unfolded protein response (UPR) were up-regulated, including Atf4, Eif2a, Xbp1, Bip, Canx, Hsp90, autophagy genes and pro-apoptotic Bcl2 family members (11). To examine for potential ER stress in the NOG retina, we used electron microscopy to investigate histological signs of distress, such as ER swelling; however, wild-type and NOG retinas did not show ER swelling (Supplementary Material, Fig. S1A–C).
To confirm the histological findings, we examined transcript levels of Chop (alternatively, Ddit3) and Grp78 (alternatively, Hspa5), two UPR-associated genes that are upregulated during ER stress, using qRT–PCR in NOG CRho−/− retinas at P8 and P10. Relative Chop and Grp78 expression levels in the NOG CRho−/− retinas were not significantly different at any time point, when compared with the Rho+/+ or Rho−/− retinas (Supplementary Material, Fig. S1D and E). Also, levels of activated cathepsins B and D were similar to those of the controls (Supplementary Material, Fig. S1F), showing that the autophagy system was not activated, further confirming that the phenotype observed in the NOG retinas is due to processes other than ER stress.
NOG expression leads to the extrusion and accumulation of vacuoles in OS
The retinas of NOG mice are filled with vacuole-like structures in the OS layer (Fig. 3). We examined NOG ARho+/+ and wild-type OS cross sections to determine the location of these structures and define their origin. Examination of ultrastructural cross sections showed that in the P30 NOG ARho+/+ retinas, the vacuoles (asterisks in Fig. 5C and D) are present within the OS surrounded by the plasma membrane (PM, Fig. 5C and D), but not in the Rho+/+ retinas (Fig. 5A and B). Thus, we conclude that the vacuole-like structures are associated with NOG expression. It is also reasonable to conclude that the disruptions observed in Figure 3H result from at least the accumulation of these structures within the OS plasma membrane as the animals age.
Figure 5.

Outer segment cross sections of the NOG A retina. (A and B) Two examples of electron microscopic images of cross sections of a Rho+/+ retina at P30. (C and D) Two examples of electron microscopic images of cross sections of a P30 NOG ARho+/+ retina. The asterisks denote vacuole formation in the OS. Scale bar is 500 nm. CC, connecting cilium; PM, plasma membrane; OS, outer segment.
The NOGRho−/− retina is non-functional and degenerates
Although the NOG protein was undetectable on immunoblots of retinal extracts (Fig. 2B), a clear transgene-associated phenotype was observed. To determine how NOG was exerting its effects, we examined the function and structure of NOG ARho−/− and NOG CRho−/− retinas.
Examination of the retinal function in P30 NOG ARho+/− mice showed that elimination of half of the complement of Rho did not improve the ERG responses (Fig. 1B and C, open squares). Testing the functional competence of P30 NOG ARho−/− retinas showed that the ERG responses mirrored those of Rho−/− mice and provided no improvement over the ERG responses observed for NOG CRho+/+ and NOG CRho+/− (data not shown). This finding suggests that NOG, if present, cannot activate the phototransduction cascade. Consistent with the NOG A findings, elimination of endogenous Rho did not alter the rapid degeneration exhibited by NOG CRho+/+ and NOG CRho+/− retinas (data not shown). NOG CRho−/− retinas degenerated faster than Rho−/− retinas (12), supporting the idea that NOG expression is harmful to the retina.
To determine whether the absence of the NOG protein is due to transcriptional and/or post-transcriptional regulation, we performed Northern blot analysis. The commonly observed multiple Rho transcripts (cf. the Rho+/+ and Rho+/− lanes; (13)) were produced by the NOG transgene in both NOG families, except the 5.1 Kb transcript (Fig. 6); the poly-A signal responsible for the generation of the longest transcript (13) was not included in the construction of the transgene. Examination of total transcript levels in NOG CRho−/− versus NOG ARho−/− retinas revealed that the steady-state levels of the transgenic transcript are much higher in NOG CRho−/− retinas (115% over Rho+/+) than in NOG ARho−/− retinas (32% of Rho+/+). Taken together, the data show that the absence of detectable NOG protein in immunoblots is not due to low or unstable transcripts.
Figure 6.

Expression and localization of NOG at P10. (A) Northern blot analysis of NOG steady-state transcripts in total RNA prepared from P10 NOG ARho−/− and NOG CRho−/− retinas (lanes are marked accordingly). A probe recognizing all of the five transcripts of Rho was used. The 28S rRNA band is shown as a loading control. (B) Immunoblot analysis of Rho in P10 NOG ARho−/− and NOG CRho−/− retinas. Rho was immunoprecipitated from retinal extracts from the indicated genotypes using 1D4. As a control, the non-transgenic Rho+/+ sample was treated with PNGase F to remove the N-linked glycosylation. The bands at 25 and 50 kDa are the immunoglobulin light and heavy chains, respectively. (C) Immunogold labeling of Rho in non-transgenic (Rho+/+) (C and C’), NOG ARho−/− (D) and NOG CRho−/− (E and E’) P10 retinas using polyclonal anti-Rho (6). Arrows in (C) and (D) designate the connecting cilium (CC). Arrows in (E) designate Rho immunoreactivity. The boxed area is enlarged in (C’) or (E’). Scale bar is 2 µm.
To examine if there is any NOG protein synthesis, NOG was immunoprecipitated (IP) from P10 NOG ARho−/− and NOG CRho−/− retinas (Fig. 6B). Similar IPs were performed on retinal extracts from Rho+/+, Rho+/− and Rho−/− mice. Extracts were immunoblotted for Rho. A small amount of NOG was detected in the NOG CRho−/− retinas (Fig. 6B). We used immunogold labeling for Rho to determine whether this protein was transported to the OS. The results showed that the NOG protein is transported to the base of the budding OS in the NOG CRho−/− retinas, suggesting that NOG is properly transported out of the IS (Fig. 6E and E’). Consistent with the lower levels of expression in NOG A, as shown by Northern blot analysis, NOG immunoreactivity was undetectable by IP (Fig. 6B) or immunogold (Fig. 6D) analyses in the NOG ARho−/− retina.
The ubiquitin-proteasome regulates the steady-state levels of NOG in OS
We used radiolabeling to determine whether the low NOG protein level is due to translational and/or post-translational regulation. After explanted retinas were incubated with radiolabeled (35S) methionine (Met) and cysteine (Cys), groups of retinas were either collected immediately (Time 0) or incubated in regular media for up to 3 h. Protein extracts were prepared and total NOG or Rho was immunoprecipitated from the extracts. Autoradiography of the subsequent SDS–PAGE gel (Fig. 7A) revealed that, although the amount of labeled newly synthesized Rho remained the same in the Rho+/+ retinas throughout the 3 h of post-label, NOG levels were reduced by ∼35% by the 3 h time point in the NOG CRho−/− retina (Fig. 7A and Graph below). These data also show that the elevated amounts of nascent NOG protein detected reflect the relative levels of NOG transcripts observed earlier on Northern blot and qRT–PCR, and suggest a potential post-translational regulation of the NOG.
Figure 7.

NOG protein regulation. (A) Radiolabeling of Rho in P10 NOG CRho−/− retinas. The stability of newly synthesized NOG protein was examined using ex vivo radiolabeling of retinal extracts. After labeling for 5 h, retinas were incubated for 0, 0.5, 2 or 3 h in media containing cold amino acids. Protein extracts were prepared and Rho (100 µg and 200 µg of protein from extracts of Rho+/+ and NOG CRho−/−) was immunoprecipitated and immunoblotted. The top panel is an autoradiography image of the SDS–PAGE gel of designated retinal samples. The asterisk denotes the molecular weight of the glycosylated Rho monomer (in the Rho+/+ samples). The arrow designates the molecular weight of the NOG Rho monomer (in the NOG CRho−/− samples). The number sign designates the band to which the total lane intensity was normalized for quantification. The graph below the autoradiograph is quantification of results (in arbitrary units) of the total lane intensity (normalized by the ‘#’ band) from the autoradiography image (n = 2). (B) Immunoblot analysis of P10 explanted retinas treated with the proteasome inhibitor MG-132 (in DMSO), or retinas treated with DMSO only (controls) for 6 h. Three samples (2 retinas/sample) were treated with the MG-132 for each genotype (Rho+/+, Rho−/− and NOG CRho−/−). After the MG-132 or DMSO incubation, the samples were SDS–PAGE-fractionated and immunoblotted with 1D4. The bottom actin blot is a loading control. The asterisk marks Rho immunoreactivity observed upon MG-132 treatment of NOG CRho−/− retinas.
Many Rho mutants are thought to be regulated by the UPS (8,14,15). The presence of an active UPS in the OS environment (15) supports a role for that system in the post-synthesis regulation of proteins in the OS. To examine whether the UPS is regulating the levels of the NOG protein, explanted NOG CRho−/− retinas were treated with the proteasome inhibitor, MG-132. Following incubation, immunoblot analysis revealed an increase in a high molecular weight protein in the NOG CRho−/− samples (Fig. 7B, asterisk) that was not observed in the vehicle treated control or the Rho−/− samples.
The accumulation of NOG when MG132 was added (Fig. 7B), the transport of NOG protein to the base of OS (Fig. 6E’), the vacuoles in the OS (Fig. 5), and the detection of potentially ubiquitinated rhodopsin in the OS (8,14,15) suggest the presence of a functional proteasome system. To investigate this, IHC colocalization of prominin 1 and the 20S subunit of the proteasome was performed. Figure 8 shows that the 20S subunit of the proteasome is present above prominin 1 (red label above the yellow label, arrows in Fig. 8), which resides at the base of the OS. Some prominin 1 and 20S labeling is also observed in the OS. This pattern of OS labeling of prominin 1 has been reported before (16,17). The presence of proteasome components at the base of OS is further confirmed by immunogold localization of 20S subunit of the proteasome in the outer segments of normal mouse photoreceptors. The 20S subunit of the proteasome is localized in clusters in the outer segments of photoreceptors (examples marked with arrowheads in Fig. 9A, C and D), inner segments (examples are marked by black and white arrowheads in Fig. 9A and D) and at the base of the connecting cilium (arrow in Fig. 9B) or the apical portion of the connecting cilium (arrow in Fig. 9C).
Figure 8.
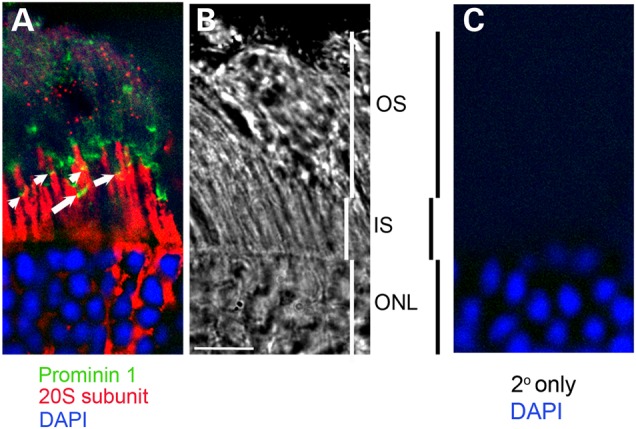
Immunohistologic localization of the 20S subunit of the proteasome to the base of the outer segments in the wild-type retina. (A) A single plain of a de-convolved image showing the presence of the 20S subunit of the proteasome (red) in inner segments and at the base of the outer segments. Some label extends further up into the OS and punctate labeling is also observed higher in the OS. Prominin 1 (green) is at base of OS and colocalizes with 20S labeling (yellow, arrows). (B) A bright-field image of the corresponding area in (A) showing the different layers of the retina. (C) Retinal sections probed with the secondary antibody alone. Scale bar is 10 μM.
Figure 9.

Immunogold localization of 20S subunit of the proteasome in the outer segments of normal mouse photoreceptors. The 20S subunit of the proteasome is localized in clusters in the outer segments of photoreceptors (e.g. arrowheads in A, C and D), inner segments (e.g. black and white arrowheads in A and D) and at the base of the connecting cilium (arrow in B) or the apical portion of the connecting cilium (arrow in C). OS, outer segment; IS, inner segment; CC, connecting cilium. Scale bar in (D) is 500 nm.
The retinal phenotype in NOG mice resembles that observed in RHOhT17M transgenic mice
RHOhT17M transgenic mice were previously established and analyzed (4). ERG responses of these mice were reduced at early ages and declined further with age. Vitamin A supplementation to RHOhT17M mice on rhodopsin wild-type background reduced the rate of functional decline and led to a >40% increase in the thickness of the ONL and the appearance of longer OS with more, better-packed discs, when compared with mice fed control diet (4). A later study, however, showed that hT17M RHO was mislocalized to the ONL (11). We hypothesized that the absence of hT17M from the OS of transgenic mice is due to its degradation at base of the OS, similar to NOG leading to the generation of vacuoles. To test this idea, we performed ultrastructural analysis of the OS of RHOhT17M/+ mice to search for vacuoles. The vacuolar structures are abundant in RHOhT17M/+ retinas (examples are marked by black arrows, Fig. 11A–H). The structures may be tubular in nature (Fig. 11C, white arrow), but appear circular because they are cut transversely upon processing. There may also be a combination of small vesicles as well as tubular structures that are the result of the fusion of smaller vesicular structures. These small vesicles (Fig. 11G, asterisk) may represent nascent discs that are unable to further develop as a result of the degradation of partially glycosylated RHO. More importantly, these vesicular structures are encapsulated within the OS plasma membrane, similar to those in NOG (Fig. 11I, PM).
Figure 11.

A model describing a mechanism for the phenotype in retinas expressing NOG opsin. Newly synthesized rhodopsin (glycosylated or NOG) is transported via vesicles to the base of the OS, where its glycosylation status (at least at N15) is checked. If properly glycosylated, it will be allowed to incorporate into OS discs. If not glycosylated, or possibly improperly glycosylated, an E2 class ubiquitin-conjugating enzyme with an E3 ubiquitin ligase, will add ubiquitin moieties and direct the protein toward the proteasome for degradation. Due to the absence of a recycling mechanism, the empty vesicles are released into the OS, where they aggregate generating larger vesicles as the retina ages, eventually leading to full disruption of the OS, which in turn leads to cell death. Since some OS discs are being assembled and a very small amount of NOG is detected, it is clear that the system is very selective in only degrading NOG. Mitochondrion image was obtained from http://penrules.com/portfolio/illustration/mitochondria.html. Dotted lines represent the two disputed positions of the plasma membrane at base of the OS.
Discussion
Thousands of people in the USA suffer from RP, a retinal disorder resulting from mutations in RHO, among other genes (RetNet, https://sph.uth.edu/retnet/). Identification and characterization of RHO mutants has revealed the necessity of RHO's glycosylation to maintain the health and viability of the photoreceptors. However, little has been proposed to explain the correlation between the degenerative phenotype and the mutations in RHO that lead to changes in glycosylation.
In this report, we show that lack of glycosylation at the N-terminus of Rho leads to a retinal degenerative phenotype in mice similar to that observed in patients carrying RHO mutations leading to alterations in RHO's glycosylation. We demonstrate that NOG is properly transcribed, but the protein is degraded hours post-synthesis. We also show that NOG reaches the OS, similar to newly synthesized Rho (18,19). Finally, we illustrate that NOG is degraded via the UPS. The results presented here show that NOG is transported to the base of the OS, and, due to lack of glycosylation, is degraded in the OS (Fig. 11). Due to the presence of a proteasome component in the cilia (Fig. 8 and 9), it is also possible that NOG is degraded in the cilia and the empty vesicles are then released at the base of the OS. Regardless, the degradation of NOG suggests that glycosylation is of critical importance for post-translational stability of Rho in the OS. This is further supported by the fact that the rate of degeneration increased in direct correlation with steady-state levels of the NOG transcript. Since Rho is transported to the OS in membranous vesicles, we propose that NOG degradation leads to the accumulation of empty vesicles in the OS (Figs 3, 10 and 11). These vesicles fuse with each other and eventually lead to complete disruption of the OS disc structures, causing the degenerative phenotype. The expression of the NOG protein and subsequent vesicle formation is obviously more harmful to the cell than the expression of half the amount of normal Rho, as Rho+/− mice do not exhibit progressive retinal degeneration (12).
Figure 10.

Electron micrographs of retinal sections from RHOT17M transgenic mice. (A–H) Longitudinal retinal sections from P30 RHOhT17M/+ mice. Vacuolar structures were observed within the OS. Black arrows indicate representative vacuoles. White arrow in (C) points to a tubular structure. Asterisk in (G) points to the varying sizes of vacuoles present. (I) Representative cross section of an outer segment of the RHOT17M retina. Observe the vacuoles within the plasma membrane (PM). Scale bar is 500 nm.
Previous studies by Papermaster and colleagues in Xenopus showed that nascent rho exits the Golgi ∼1 h post-synthesis and reaches the OS within 3 h (18,19). The apparent degradation of NOG between 2 and 3 h post-synthesis, and its translocation to the base of the OS, suggests that NOG regulation occurs before its incorporation into new discs at the base of the OS. These findings are consistent with the prior observation that non-glycosylated opsin in Xenopus retinas (generated by treatment of retinas in short-term organ culture with tunicamycin) could traffic normally through the rod cell and translocate to the site of disc assembly, but could not form morphologically normal new discs (20). Rather, tubulo-vesicular membranes formed and accumulated in the space between the inner and outer segment compartments of rod cells (the ‘intersegmental space’), without becoming incorporated into the OS, per se. However, expression of the N2/15S non-glycosylated rho in transgenic Xenopus showed that the mutant protein was incorporated in the OS (21). This apparent disagreement with the current study points to a differential requirement for rhodopsin glycosylation between species. Consistent with this, glycosylation in Drosophila is only required for rho maturation (22). We observed that inhibition of the UPS, a system that has been shown to be present and active in the OS (8,14,15), led to stabilization of NOG. Although we cannot exclude the possibility that NOG is ubiquitinated in the IS, the observation of membranous vesicles in the OS supports the conclusion that NOG is transported to OS, where it is subsequently degraded.
In Xenopus retinas, as mentioned above, tunicamycin treatment in vitro led to the formation of membranous material that was blebbed off into the intersegmental space (23). This finding is consistent with the observation of vacuolar material in the OS of NOG mice. Therefore, we propose that these vacuolar structures within the plasma membrane of the OS in NOG retinas (Figs 3 and 5) result from the accumulation and fusion of the residual membranous vesicles after NOG degradation at the base of the OS (Fig. 10).
Our studies with the NOG animal model confirmed the structural findings from other models, including an in-depth examination of rho glycosylation in the Xenopus, and shed new light on the regulation of rhodopsin and its role in OS morphogenesis. The resilient nature of the mammalian retina and its capacity to respond to dramatic insults, as evident in the functional responses from the vacuole-ridden NOG retinas, was an interesting finding. Despite the severe disarray of the OS structure, the photoreceptors showed considerable functional competence.
The presence of vacuoles in the OS of the RHOhT17M mice was another key finding. These vacuoles are similar to those observed in NOG retinas and are also located within the OS. Therefore, this study provides evidence for the mechanism at work in retinal degeneration in patients with the T17M mutation. This is important when considering a therapeutic approach to T17M-associated RP. One possible therapeutic approach is the use of pharmacological agents that target the proteasome. Since the components of the proteasome may vary between different organs, further research is required to identify the specific components of the OS proteasome.
Vitamin A treatment of the RHOhT17M mice led to improved retinal function and structural appearance (10). This suggests that either vitamin A is an inhibitor of the OS proteasome, or that vitamin A, by binding to Rho, acts as a chaperone that alleviates the need for glycosylation at N15. Rho glycosylation may be important in the interaction of Rho with its chaperone(s). However, it is important to remember that mice lacking the chromophore 11-cis retinal due to the elimination of the Rpe65 gene have normal OS, and the observed degenerative phenotype of the rod photoreceptors is much slower than in NOG mice (24).
The English mastiff dog, expressing a spontaneous rhoT4R mutation, exhibits a progressive retinal degenerative disorder (25). The T4R mutation abolishes glycosylation at Asn2, but not at Asn15 (26). The T4R mutant protein was present in the OS, but was more prone to losing its chromophore than wild-type rho (3). This raises the question of whether glycosylation at both sites on the N-terminus of Rho is required for the protein to pass the checkpoint, or if glycosylation on only one site is required. It is clear from the work with the T4R dog that glycosylation on Asn2 may not be a requirement for passage through the checkpoint. This leaves us to suggest that glycosylation at Asn15 is more critical for the passage through the OS checkpoint.
Expression of hT17M in mice led to a progressive retinal degenerative phenotype associated with retention of the transgenic protein in a perinuclear compartment and induction of the ER stress response (11). However, we did not observe ER stress in the NOG retinas. This can be attributed either to the elimination of the first glycosylation site in NOG, or the expression of human RHO in the mouse retina. We favor the latter, since glycosylation of any protein is tissue/species specific. This may subsequently affect the binding of Rho to its chaperone, influencing its trafficking and incorporation into the OS disc membrane.
In summary, this report provides further evidence supporting a role for Rho glycosylation in OS disc morphogenesis, but not in Rho's synthesis, maturation or intracellular transport. It also provides evidence for the potential mechanism leading to RP in patients with mutations that lead to changes in RHO glycosylation. This report can aid in the development of a better therapeutic means of intervention for patients suffering from RP caused by mutations leading to changes in RHO glycosylation.
Materials and methods
Transgenic mice
The NOG transgenic construct was prepared from a 15 kb mouse genomic fragment containing the mouse opsin promoter sequence (MOPS), 6 kb of the 5′ flanking sequence, all exons and introns, and 3 kb of the 3′ flanking sequence. The N-terminal modifications to generate an Asn to Gln amino acid change at Asn-2 and 15 were introduced into the DNA sequence using PCR site-directed mutagenesis, as has been described before (6).
Transgenic mice were produced as stated previously (6). Potential transgenic founders (F0) and their offspring (F1 and later) were identified by PCR using a Rho-specific primer (5′-GCCCCAATTTCTACGTTCCTTTTTCCCAG-3′) and a transgene-specific primer (5′-TGCTCAAGATGAGATAGATGAATGTTCCGTGC-3′). NOG potential founders were mated to breeders (an in-house inbred strain produced from a cross between FVB/N and C57BL/6 mice) (27) to produce the first generation and to maintain subsequent generations. All mice were cross-bred to the in-house wild-type strain for over six generations to insure that background is the same across lines. The inbred strain was tested for and eliminated the rd1 and the rd8 mutations. Southern blot analysis was used to determine both the pattern of transgene integration and copy numbers in the transgenic lines (data not shown).
To study the effects of the NOG transgene in the presence of differing levels of endogenous RHO, each of the lines was mated either with wild-type (Rho+/+) or Rho knockout mice (Rho−/−, generously provided by Dr. Janis Lem, Tufts University Sackler School of Graduate Biomedical Sciences, Boston, MA, USA) (12). The subsequent offspring were screened by PCR for the presence of the transgene (see above) and the knockout construct (12). NOG mice in the different Rho backgrounds (Rho+/+, Rho+/− and Rho−/−) were used in this study. All experiments were approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee, and conformed to the National Institute of Health Guide for the Care and Use of Laboratory Animals and the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research.
Transgenic mice expressing human rhodopsin with the T17M mutation (RHOhT17M) (10) were maintained on the C57BL/6N genetic background and were crossed with Rho−/− mice. Mouse tails were collected, and a 2.0× Taq RED Master Mix Kit (Apex, San Diego, CA, USA) was used to extract the DNA. PCR was performed with human RHO-specific primers (sense: 5′-GAGTGCACCCTCCTTAGGCA-3′ and anti-sense: 5′-TCCTGACTCGAGGACCCTAC-3′) to identify a single 300 bp band.
The Bouse mouse was previously described (6). This transgenic mouse over-expresses wild-type rhodopsin with two modifications at the C-terminus in presence of endogenous rhodopsin. The modifications permit differentiation, by an anti-human-rhodopsin specific antibody, of the transgenic protein in presence of the endogenous protein.
All mice were raised under ∼ 100 lux, 12 h light/12 h dark cycle.
Light, electron microscopy and immunogold cytochemistry
For light microscopy of retinas, the retinal sections were treated as described before (28). Images for histology were photographed with a Zeiss Axioskop using 20× objective and photographed with Zeizz Axiocam camera and Axiovison 3.1 software. Images were obtained at room temperature.
Electron microscopy (EM) was performed as described previously (28,29) by imbedding tissues in EMBED 812 Resin (Electron Microscopy Sciences, Hatfield, PA, USA) and then using a JEOL 1200EX transmission microscope with an AMT digital camera using AMTV601 software. Images were obtained at room temperature. Images were then processed for presentation using Adobe Photoshop CS5, Extended, V12.04 X32.
Immunohistochemistry
Immunohistochemistry was performed by fixing eyes in 4% for 2 h followed by cryopreservation in 10%, 20% and then 30% sucrose in phosphate-buffered saline. Tissue was then embedded in using M1 embedding medium (Thermo Scientific, Waltham, MA, USA), frozen and sections cut at 10 microns using Leica CM3050S cryostat (Leica Biosystems, Buffalo Grove, IL, USA). Sections were then probed with either anti-20S at 1:1000 (catalogue #ab3325, Abcam, Cambridge, MA, USA), anti-RP1 at 1:4000 (a generous gift from Dr. Eric A. Pierce, Massachusetts Eye and Ear, Harvard Medical School) or anti-prominin-1 at 1:250 (catalogue # 14-1331, eBioscience, San Diego, CA, USA) antibodies. All secondary antibodies were Alexa Fluor antibodies from Life Technologies (Grand Island, NY, USA). Sections were imaged at room temperature using Olympus BX-62 (Olympus, Tokyo, Japan) with a spinning disc confocal microscope. Images were obtained using UPlanSApo (Olympus) objective and Hamamatsu C-4742 (Hamamatsu Photonics K.K., Hamamatsu City, Japan) camera and Slidebook version 4.2 software. A no-neighbors paradigm was used for deconvolution.
Images were then processed for presentation using Adobe Photoshop CS5, Extended, V12.04 X32.
EM immunogold and silver-enhanced immunohistochemistry
For EM immunogold labeling, thin sections were cut on a Leica UC7 ultramicrotome (Leica Microsystems, Inc., Buffalo Grove, IL, USA) with a diamond knife (Diatome, Hatfield, PA, USA) and placed on 200 mesh Nickel formvar coated grids. Grids were incubated in 0.02 m glycine in PBS for 15 min, washed in PBS 4 × 3 min and blocked for 15 min RT in 5% normal goat serum, 1% BSA in PBS (blocking and antibody buffer). Sections were incubated in anti-opsin (1;100) or anti-20S subunit (1:10) at 4°C overnight in a humid chamber, washed in PBS 5 × 3 min and incubated in secondary antibody (goat anti-rabbit IgG:10 nm colloidal gold) for 2 h at room temperature. Grids were then washed in PBS 3 × 3 min, distilled water 2 × 3 min, fixed with 2.5% glutaraldehyde 5 min and washed 5 × 2 min in distilled water. Sections were post-stained with 2% uranyl acetate (10 min) and Reynold's lead citrate (5 min) and imaged with a JEOL 1200 EX transmission electron microscope at 80 kV.
For silver-enhanced immunohistochemistry, 0.75 μm thick sections on glass slides were treated essentially as described above for thin sections (EM), except the primary antibody (or non-immune control serum) was diluted 1:500 (v/v) with blocking buffer and incubation was carried out for 2 h at room temperature. After rinsing briefly with PBS, sections were treated for 2 h at room temperature with 1 nm colloidal gold-conjugated goat anti-rabbit IgG secondary antibody [AuroProbe, One GAR, GE Healthcare, Piscataway, NJ, USA; diluted 1:50 (v/v) with blocking buffer]. Sections were rinsed three times (15 min each) with PBS, followed by fixation for 10 min at room temperature with 2% (v/v) glutaraldehyde in PBS. Then sections were rinsed twice with distilled water (5 min each). Silver intensification was performed with an IntenSE [M Silver Enhancement Kit (GE Healthcare)], per the manufacturer's instructions. Sections were then rinsed with distilled water, counterstained with 1% (w/v) toluidine blue in 1% (w/v) sodium borate, rinsed again with distilled water, air dried and coverslipped using Permount (Fisher Scientific, Fairlawn, NJ, USA). Sections were viewed and photographed with an Olympus BH-2 photomicroscope in the auto-expose mode, using an X20 DPlanApo 20UV objective. Images were then processed for presentation using Adobe Photoshop CS5, Extended, V12.04 X32.
Electroretinography
Electroretinograms (ERGs) were recorded from the corneal surfaces of anesthetized mice as described previously (6). For the assessment of rod photoreceptor function, strobe flash stimuli were presented in a Nicolet GS-2000 Ganzfeld after overnight dark adaptation. To evaluate cone mediated functions, strobe flash stimuli were superimposed upon a steady adapting field (1.46 log cd/m2), following a 5-min light adaptation period.
Northern blot analysis
Total RNA was extracted from mouse retinas using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). 7 µg (Rho+/+), 14 µg (Rho+/−) and 50 µg (Rho−/− and NOGRho−/−) of total RNA were electrophoretically separated on a 1% (w/v) agarose gel containing 18% (v/v) formaldehyde. Images of the 18 and 28S ribosomal bands were captured using a Bio-Rad Molecular Imager® FX before transfer, to determine the integrity of the RNA in the samples and to quantify the bands. After electrophoresis, RNA was transferred to an S&S NYTRAN® SuPerCharge nylon membrane (Schleicher and Schuell, Keene, NH, USA) and hybridized with an RHO cDNA fragment that detected both the transgene and endogenous transcripts. Hybridization and washes were performed as described previously (13). After washing, the blots were exposed to a Molecular Imager® FX Imaging Screen K (Bio-Rad Laboratories, Hercules, CA, USA) for 5 h at room temperature. The screen was imaged and the bands quantified using the Bio-Rad Molecular Imager FX. Total Rho levels for each lane were normalized to the values obtained for their respective 28S rRNA bands.
Relative quantitative RT–PCR
Quantitative RT–PCR (qRT–PCR) was performed as previously described (28). Retinas were obtained and extracted from 3–5 animals (age and genotype indicated on figure) using TRIzol® reagent (Invitrogen). For each sample, the housekeeping gene Hypoxanthine-Guanine Phosphoribosyl Transferase (Hprt) was analyzed and the threshold cycle (cT) values for Rho were normalized to their respective Hprt or retinal degeneration slow (Rds) values. The PCR products were verified using gel electrophoresis. Results were obtained in triplicate and experiments were performed at least three times.
All primers for the PCR analysis were designed using Primer3 (Hprt-For: 5′-GCAAACTTTGCTTTCCCTGGTT-3′, Hprt-Rev: 5′-CAAGGGCATATCCAACAACA-3′; Rho-For: 5′-TCACCACCACCCTCTACACA-3′, Rho-Rev: 5′-TAGTCAATCCCGCATGAACA-3′; Rds-For: GTTCAAGTGCTGTGGGAACA, Rds-Rev: 5′-CTGTGTGGAGGTAGCGGAGT-3′). Primer sequences were selected to be oriented on exon-exon junctions to discourage possible amplification of any genomic DNA contaminants.
Immunoprecipitation and immunoblotting
For immunoprecipitation (IP), frozen retinas were homogenized and sonicated in a buffer containing 10 mm Tris–HCl (pH 7.4), 50 mm NaCl, 0.5 mm EDTA, 1 mm PMSF, and 1% (v/v) Triton X-100, and were incubated with rotation at 4°C for 2 h. The concentration of the protein samples was determined, and 100 μg (Rho+/+) or 200 μg (Rho−/− and NOGRho−/−) of the samples were placed into tubes. The volumes were brought to 200 μl with IP buffer. The solutions were treated as described previously (30). The anti-RHO monoclonal antibody, 1D4 (provided generously by Dr. Robert Molday, University of British Columbia, Vancouver BC, Canada) (31), was added at a 1:20 (v/v) dilution to the homogenate and allowed to incubate with gentle rotation at 4°C overnight. After IP with Protein G-Sepharose beads (Sigma-Aldrich, St. Louis, MO, USA) and washing, 2X loading buffer was added to the bead mixture and allowed to solubilize at 25°C for 30–45 min. After solubilization, the solution was centrifuged at 2000g for 5 min and the supernatant loaded on SDS–PAGE.
For immunoblotting, retinas from individual animals were homogenized and sonicated in buffer containing 50 mm Tris/HCl (pH 7.8), 100 mm NaCl, 5 mm EDTA, 1% (v/v) Triton X-100, 0.1% SDS, 2.5% (v/v) glycerol, and 1.0 mm PMSF. After solubilization at 4°C for 2 h, the protein extract was reduced in 2X loading buffer [100 mm Tris/HCl (pH 6.8), 4% (w/v) SDS, 197 mm Dithiothreitol and 20% (v/v) glycerol] for 30–45 min at room temperature (25°C). Protein extracts were resolved using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE), transferred to PVDF membrane (Bio-Rad, Hercules, CA, USA) and probed as described previously (6).
Primary antibody incubations were done in 5% milk/TTBS overnight at 4°C. 1D4 was used at a dilution of 1:5000 (v/v). Anti-actin (Abcam) was used at a 1:2000 (v/v) dilution. Anti-RDS (32) was used at a 1:1000 (v/v) dilution. SuperSignal® Dura West chemiluminescent substrate (Pierce, Rockford, IL, USA) was used for detection of the primary antibodies to their cognate antigens, and the blots were visualized by the Kodak Image Station 4000R Imager (Kodak, Rochester, NY, USA).
Cleavage of oligosaccharide chains with PNGase F
To deglycosylate retinal proteins, we followed the manufacturer's protocol for N-Glycanase (Peptide-N-Glycosidase F, PNGase F; Prozyme, San Leandro, CA, USA). The deglycosylated protein solutions were then treated with 2X loading buffer for western blot analysis.
Ex vivo labeling of retinal proteins
Freshly extracted retinas were placed in a 48-well plate (2 retinas/well) with Cys- and Met-free Dulbecco's Modified Eagle's Medium (DMEM-MCF, Invitrogen). Retinas were exposed to either media only (no-label) or Trans35S label (MP Biomedical, Solon, OH, USA) in DMEM-MCF for 5 h. After 5 h, one sample was removed (‘0 min post-label’), and along with the no-label sample, was extracted for IP. The rest of the retinas were washed three times with DMEM-MCF, fresh DMEM-MCF (minus the label) was added to the wells and samples were allowed to incubate for 0.5, 2 or 3 h. Then, retinas were processed for IP using 200 µg aliquot of the Rho+/+ and 400 µg of the Rho−/− retinal protein extracts. After IP, half of each sample was run on sister SDS–PAGE gels. After completion, one gel was transferred and probed with 1D4, while the sister gel was stained [0.25% (w/v) Coomassie Brilliant Blue (CBB; R-250), 50% (v/v) Methanol, 10% (v/v) Acetic Acid], destained [25% (v/v) Isopropanol, 10% (v/v) Acetic Acid] and treated with 1 M salicylate for 30 min. The gel was vacuum-dried for 1.5 h using a Bio-Rad Model 583 Gel Dryer and exposed to blue X-ray film (Phenix Research Products, Hayward, CA, USA) at −80°C overnight and for the following 3 days. Following exposure to the X-ray film, the gels were exposed to a Kodak Phosphoscreen (Kodak, Rochester, NY, USA) for 4 h at room temperature. The Phosphoscreen was developed using a Bio-Rad Molecular Imager® FX. Values of total RHO were quantified using the Bio-Rad Quantity One software. An arbitrary standard was selected across all lanes to normalize to their respective RHO value.
Ex vivo treatment of retinal explants with MG-132
Freshly extracted retinas were placed in a 48-well plate (2 retinas/well) containing Modified Eagle's Medium (MEM; Invitrogen). Retinas were either incubated in 10 µm of the proteasome inhibitor MG-132 (in DMSO) (Sigma-Aldrich) or vehicle only (DMSO); final DMSO = 0.2% (v/v). The MG-132 and DMSO-treated retinas were incubated at 37°C and 5% CO2 for 6 h. After the incubation, the retinas were washed with PBS three times and protein was extracted for western blot. Each of the genotypes was tested in triplicate.
Statistical analysis
To determine statistical significance, data were entered into GraphPad Prism® (GraphPad Software, Inc., La Jolla, CA, USA) and subjected to one-way analysis of variance (ANOVA) or two-way ANOVA (when applicable). A Bonferroni's post hoc test was used for both one-way and two-way ANOVAs. A P-value of <0.05 was chosen as the determinant for statistical significance, unless otherwise noted. Error bars indicate standard error of the mean (SEM).
Supplementary material
Supplementary Material is available at HMG online.
Conflict of Interest statement. All authors have no commercial affiliation or conflict of interests to declare.
Funding
This work was supported by grants from the National Eye Institute P30EY021725, R01 EY007361 (S.J.F.), R01 EY020905 (M.S.G.), R01 EY10609 (M.I.N.), R01 EY14052 and R01 EY018137 (M.R.A.); the Department of Veterans Affairs (N.S.P.); Foundation Fighting Blindness (M.I.N., N.S.P., M.R.A.); Hope For Vision, NY (M.R.A.); the Reynolds Oklahoma Center on Aging (M.R.A.); an Unrestricted Grant from Research to Prevent Blindness (S.J.F., N.S.P.), and by facilities and resources provided by the VA Western New York Healthcare System (S.J.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, the National Eye Institute, the National Institutes of Health, the Department of Veterans Affairs or the U.S. Government.
Supplementary Material
References
- 1.Daiger S.P., Sullivan L.S., Bowne S.J. (2013) Genes and mutations causing retinitis pigmentosa. Clin. Genet., 84, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daiger S.P., Bowne S.J., Sullivan L.S. (2007) Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol., 125, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu L., Jang G.F., Jastrzebska B., Filipek S., Pearce-Kelling S.E., Aguirre G.D., Stenkamp R.E., Acland G.M., Palczewski K. (2004) A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G protein-coupled receptor. J. Biol. Chem., 279, 53828–53839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li T., Sandberg M.A., Pawlyk B.S., Rosner B., Hayes K.C., Dryja T.P., Berson E.L. (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17-methionine and proline-347-serine in transgenic mice and in cell cultures. Proc. Natl Acad. Sci. USA, 95, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kofuji P., Ceelen P., Zahs K.R., Surbeck L.W., Lester H.A., Newman E.A. (2000) Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci., 20, 5733–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan E., Wang Q., Quiambao A.B., Xu X., Qtaishat N.M., Peachey N.S., Lem J., Fliesler S.J., Pepperberg D.R., Naash M.I., Al Ubaidi M.R. (2001) The relationship between opsin overexpression and photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci., 42, 589–600. [PubMed] [Google Scholar]
- 7.Sung C.H., Davenport C.M., Nathans J. (1993) Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J. Biol. Chem., 268, 26645–26649. [PubMed] [Google Scholar]
- 8.Rajan R.S., Kopito R.R. (2005) Suppression of wild-type rhodopsin maturation by mutants linked to autosomal dominant retinitis pigmentosa. J. Biol. Chem., 280, 1284–1291. [DOI] [PubMed] [Google Scholar]
- 9.Huang P.C., Gaitan A.E., Hao Y., Petters R.M., Wong F. (1993) Cellular interactions implicated in the mechanism of photoreceptor degeneration in transgenic mice expressing a mutant rhodopsin gene. Proc. Natl Acad. Sci. USA, 90, 8484–8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li T., Sandberg M.A., Pawlyk B.S., Rosner B., Hayes K.C., Dryja T.P., Berson E.L. (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17 --> methionine and proline-347 --> serine in transgenic mice and in cell cultures. Proc. Natl Acad. Sci. USA, 95, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunte M.M., Choudhury S., Manheim J.F., Shinde V.M., Miura M., Chiodo V.A., Hauswirth W.W., Gorbatyuk O.S., Gorbatyuk M.S. (2012) ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci., 53, 3792–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lem J., Krasnoperova N.V., Calvert P.D., Kosaras B., Cameron D.A., Nicolo M., Makino C.L., Sidman R.L. (1999) Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc. Natl Acad. Sci. USA, 96, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al Ubaidi M.R., Pittler S.J., Champagne M.S., Triantafyllos J.T., McGinnis J.F., Baehr W. (1990) Mouse opsin. Gene structure and molecular basis of multiple transcripts. J. Biol. Chem., 265, 20563–20569. [PubMed] [Google Scholar]
- 14.Obin M.S., Jahngen-Hodge J., Nowell T., Taylor A. (1996) Ubiquitinylation and ubiquitin-dependent proteolysis in vertebrate photoreceptors (rod outer segments). Evidence for ubiquitinylation of Gt and rhodopsin. J. Biol. Chem., 271, 14473–14484. [DOI] [PubMed] [Google Scholar]
- 15.Obin M., Lee B.Y., Meinke G., Bohm A., Lee R.H., Gaudet R., Hopp J.A., Arshavsky V.Y., Willardson B.M., Taylor A. (2002) Ubiquitylation of the transducin betagamma subunit complex. Regulation by phosducin. J. Biol. Chem., 277, 44566–44575. [DOI] [PubMed] [Google Scholar]
- 16.Zacchigna S., Oh H., Wilsch-Brauninger M., Missol-Kolka E., Jaszai J., Jansen S., Tanimoto N., Tonagel F., Seeliger M., Huttner W.B.,et al. (2009) Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J. Neurosci., 29, 2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Z., Anderson D.W., Papermaster D.S. (2012) Prominin-1 localizes to the open rims of outer segment lamellae in Xenopus laevis rod and cone photoreceptors. Invest. Ophthalmol. Vis. Sci., 53, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papermaster D.S., Schneider B.G., Besharse J.C. (1985) Vesicular transport of newly synthesized opsin from the golgi apparatus toward the rod outer segment. Invest. Ophthalmol. Vis. Sci., 26, 1386–1404. [PubMed] [Google Scholar]
- 19.Papermaster D.S., Schneider B.G., DeFoe D., Besharse J.C. (1986) Biosynthesis and vectorial transport of opsin on vesicles in retinal rod photoreceptors. J. Histochem. Cytochem., 34, 5–16. [DOI] [PubMed] [Google Scholar]
- 20.Fliesler S.J., Rayborn M.E., Hollyfield J.G. (1985) Membrane morphogenesis in retinal rod outer segments: inhibition by tunicamycin. J. Cell Biol., 100, 574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tam B.M., Moritz O.L. (2009) The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J. Neurosci., 29, 15145–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Webel R., Menon I., O'Tousa J.E., Colley N.J. (2000) Role of asparagine-linked oligosaccharides in rhodopsin maturation and association with its molecular chaperone, NinaA. J. Biol. Chem., 275, 24752–24759. [DOI] [PubMed] [Google Scholar]
- 23.Fliesler S.J., Rayborn M.E., Hollyfield J.G. (1986) In Lark D.L., Normark S., Uhlin B.-E., Wolf-Watz H. (eds), Protein-Carbohydrate Interactions in Biological Systems. pp. 191–205. [Google Scholar]
- 24.Redmond T.M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J.X., Crouch R.K., Pfeifer K. (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet., 20, 344–351. [DOI] [PubMed] [Google Scholar]
- 25.Kijas J.W., Miller B.J., Pearce-Kelling S.E., Aguirre G.D., Acland G.M. (2003) Canine models of ocular disease: outcross breedings define a dominant disorder present in the English mastiff and bull mastiff dog breeds. J. Hered., 94, 27–30. [DOI] [PubMed] [Google Scholar]
- 26.Zhu L., Jang G.F., Jastrzebska B., Filipek S., Pearce-Kelling S.E., Aguirre G.D., Stenkamp R.E., Acland G.M., Palczewski K. (2004) A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G protein-coupled receptor. J. Biol. Chem., 279, 53828–53839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan E., Wang Q., Quiambao A.B., Xu X., Qtaishat N.M., Peachey N.S., Lem J., Fliesler S.J., Pepperberg D.R., Naash M.I., Al-Ubaidi M.R. (2001) The relationship between opsin over-expression and photoreceptor degeneration in the vertebrate retina. Invest. Ophthmol. Vis. Sci., 42, 589–600. [PubMed] [Google Scholar]
- 28.Sherry D.M., Murray A.R., Kanan Y., Arbogast K.L., Hamilton R.A., Fliesler S.J., Burns M.E., Moore K.L., Al-Ubaidi M.R. (2010) Lack of protein-tyrosine sulfation disrupts photoreceptor outer segment morphogenesis, retinal function and retinal anatomy. Eur. J. Neurosci., 32, 1461–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherry D.M., Kanan Y., Hamilton R., Hoffhines A., Arbogast K.L., Fliesler S.J., Naash M.I., Moore K.L., Al-Ubaidi M.R. (2012) Differential developmental deficits in retinal function in the absence of either protein tyrosine sulfotransferase-1 or -2. PLoS One, 7, e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding X.Q., Nour M., Ritter L.M., Goldberg A.F., Fliesler S.J., Naash M.I. (2004) The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum. Mol. Genet., 13, 2075–2087. [DOI] [PubMed] [Google Scholar]
- 31.Hicks D., Molday R.S. (1986) Differential Immunogold-dextran labeling of bovine and frog rod and cone cells using monoclonal antibodies against bovine rhodopsin. Exp. Eye Res., 42, 55–71. [DOI] [PubMed] [Google Scholar]
- 32.Nour M., Ding X.Q., Stricker H., Fliesler S.J., Naash M.I. (2004) Modulating expression of peripherin/rds in transgenic mice: critical levels and the effect of overexpression. Invest. Ophthalmol. Vis. Sci., 45, 2514–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.