

Abstract
NSDHL is a 3β-hydroxysterol dehydrogenase that is involved in the removal of two C-4 methyl groups in one of the later steps of cholesterol biosynthesis. Mutations in the gene encoding the enzyme are responsible for the X-linked, male lethal mouse mutations bare patches and striated, as well as most cases of human CHILD syndrome. Rare, hypomorphic NSDHL mutations are also associated with X-linked intellectual disability in males with CK syndrome. Since hemizygous male mice with Nsdhl mutations die by midgestation, we generated a conditional targeted Nsdhl mutation (Nsdhltm1.1Hrm) to investigate the essential role of cholesterol in the early postnatal CNS. Ablation of Nsdhl in radial glia using GFAP-cre resulted in live-born, normal appearing affected male pups. However, the pups develop overt ataxia by postnatal day 8–10 and die shortly thereafter. Histological abnormalities include progressive loss of cortical and hippocampal neurons, as well as deficits in the proliferation and migration of cerebellar granule precursors and subsequent massive apoptosis of the cerebellar cortex. We replicated the granule cell precursor proliferation defect in vitro and demonstrate that it results from defective signaling by SHH. Furthermore, this defect is almost completely rescued by supplementation of the culture media with exogenous cholesterol, while methylsterol accumulation above the enzymatic block appears to be associated with increased cell death. These data support the absolute requirement for cholesterol synthesis in situ once the blood-brain-barrier forms and cholesterol transport to the fetus is abolished. They further emphasize the complex ramifications of cholesterogenic enzyme deficiency on cellular metabolism.
Introduction
Cholesterol is an essential component of all mammalian cell membranes and is a major determinant of plasma membrane fluidity. It is enriched in the mammalian central nervous system (CNS) where myelin contains ∼80% of the cholesterol of the adult brain (1). The cholesterol biosynthetic pathway is also intimately tied to a variety of important cellular functions, including signaling in lipid rafts and the formation of steroid hormones, bile acids, vitamin D, meiosis-activating sterols and oxysterols. Isoprenoid intermediates in the first half of the pathway serve as precursors for the synthesis of modified tRNAs, dolichol, ubiquinone and farnesyl and geranylgeranyl moieties (reviewed in 2). Finally, active hedgehog proteins are modified by the covalent addition of cholesterol during their intracellular processing and are involved in numerous developmental pathways (3). Perturbations of cholesterol metabolism have been implicated in a variety of human CNS disorders ranging from autism (4) to Alzheimer disease (5,6).
Cholesterol is synthesized in a series of ∼30 enzymatic reactions (7,8). The condensation of the 30 carbon isoprenoid squalene forms the first sterol intermediate, lanosterol; subsequent enzymatic reactions define the ‘post-squalene’ half of the pathway (Fig. 1A). Human disorders and/or mouse models have now been described for each step in post-squalene cholesterol biosynthesis (9) and serve as a unique resource to help to understand the role of cholesterol in the developing CNS. All of the disorders are associated with major malformations and intellectual disability (ID), providing further evidence for the essential role of cholesterol in the developing fetus. However, the pathogenic mechanisms responsible for the defects in these disorders remain unclear. In particular, it has been suggested by us and others that cholesterol deficiency during critical periods of embryonic or early postnatal development and/or accumulation of toxic sterol intermediates above an enzymatic block may be responsible, although convincing evidence remains lacking (9–11).
Figure 1.

Generation of a conditional Nsdhl allele. (A) Schematic diagram of the cholesterol biosynthesis pathway from lanosterol to cholesterol, with sterols listed in shaded boxes and the enzymes that catalyze each step shown next to the arrows. NSDHL, along with SC4MOL and HSD17B7, is required for the removal of two C-4 methyl groups from 4,4-dimethylcholesta-8,24-dien-3β-ol to generate zymosterol. Reduction of the C-24 double bond by DHCR24 can occur at multiple points along the pathway, but is shown only as the last step for simplicity. Ketoconazole inhibits CYP51A1 in the demethylation of lanosterol at C-14. Abbreviations: CYP51A1, cytochrome P450 lanosterol 14α-demethylase; DHCR14, 3β-hydroxysterol-Δ14-reductase; LBR, lamin B receptor; SC4MOL, sterol C-4 methyloxidase-like; NSDHL, NADH steroid dehydrogenase-like; HSD17B7, hydroxysteroid 17β-dehydrogenase 7; EBP, emopamil binding protein (3β-hydroxysteroid-Δ8,Δ7-sterol isomerase) ; SC5D, 3β-hydroxysteroid-Δ5-desaturase; DHCR7, 7-dehydrocholesterol reductase; DHCR24, 3β-hydroxysterol Δ24-reductase; T-MAS, 4,4-dimethylcholesta-8,24-dien-3β-ol. (B) Experimental strategy for generating the Nsdhlflx5 allele. The top line represents the mouse wild type Nsdhl gene from exon 4 to exon 8, indicating the position of diagnostic restrictions sites for Asp718 (A) and Bgl1 (B). The exons and introns are not drawn to scale. The position of probe sequences A and B used on Southern blots to detect homologous integration of the targeting construct are indicated. Below is a simplified map of the targeting construct generated in the vector pL451, showing loxP sites (black arrowheads) flanking Nsdhl exon 5, the neomycin-resistance gene (Neo) flanked by FRT sites (white arrowheads) for positive selection, and the thymidine kinase gene (TK) for negative selection. Homologous integration of the Nsdhlflx5 construct into the Nsdhl locus following electroporation into ES cells results in altered sizes of the Asp718 and Bgl1 restriction fragments, as shown in the third diagram. Finally, the Neo cassette was excised by FLPo-mediated recombination in ES cell clones to generate the Nsdhlflx5 allele (bottom diagram). (C) Southern blots of genomic DNA from a WT and representative Nsdhlflx5/Y neo-resistant ES clone digested with Asp718 and BglI, and hybridized with probes A and B, respectively, that lie outside of the sequence in the flx5 construct. The targeted clone showed the expected changes in size of the diagnostic restriction fragments, demonstrating homologous integration into the Nsdhl locus. (D) A Western blot of total protein prepared from E9.5 male embryos from a Nsdhlflx5/flx5 x Sox2-cre cross probed with antibodies against NSDHL (top) and β-tubulin (bottom) as a loading control. The Nsdhlflx5 control sample was from pooled cre-negative male embryos, and showed the expected 38 kDa wild type band for NSDHL. No NSDHL signal was detectable in the NsdhlΔ5 sample from pooled cre-positive male embryos. The higher level of β-tubulin signal in the NsdhlΔ5 sample is due to more total protein loaded than in the Nsdhlflx5 lane.
The CNS may be particularly susceptible to perturbations of cholesterol synthesis because of its high cholesterol content and complete dependence on de novo synthesis postnatally. Since cholesterol does not cross the blood-brain-barrier, a functional cholesterol biosynthetic pathway is necessary within the brain itself (starting at ∼E10–11 in the mouse). At birth, ∼90% of the cholesterol in the mouse brain is made within the CNS (12,13). During pre- and early postnatal neurogenesis, cholesterol synthesis occurs in both glia and neurons. Subsequently, it is generally accepted that most cholesterol destined for neurons is synthesized in astrocytes, with efflux from these cells mediated via apoE and ABCA1 and/or ABCG1 transporters. Neurons internalize the cholesterol as lipoprotein particles via LDL and related receptors (14).
NSDHL (NADH steroid dehydrogenase-like) is a 3β-hydroxysterol dehydrogenase involved in the removal of two C4 methyl groups from 4,4-dimethylcholesta-8,24-dien-3β-ol, a sterol intermediate derived from lanosterol (see Fig. 1A). Mutations in the murine Nsdhl gene are responsible for the X-linked dominant, male lethal mouse mutations bare patches (Bpa) and striated (Str) (15). Subsequently, mutations in the orthologous human NSDHL gene were identified in females with CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) (16,17), a rare X-linked male lethal disorder with limb reduction defects and unilateral ichthyosiform skin lesions that often have a sharp midline demarcation (reviewed in 9,18).
Recently, males in two families with X-linked syndromic ID were determined to have hypomorphic NSDHL mutations, consistent with their long-term survival (10,19,20). The disorder has been given the eponym of CK syndrome; affected males exhibit microcephaly, seizures, facial dysmorphisms, mild skeletal abnormalities and significant behavior problems. Brain magnetic resonance imaging of the three affected males studied demonstrated cerebral cortical malformations that were most consistent with polymicrogyria and/or pachygyria (10,19). Analyses of mutant male mouse embryos for one Bpa allele at E10.5 revealed a thin and disorganized cerebral cortex with increased proliferation, as well as increased apoptosis noted (10). However, the male lethality of all known murine Nsdhl alleles prohibited further studies of the role of the enzyme in the developing CNS.
To enable such studies, as well as to further investigate the role of cholesterol and its biosynthetic intermediates in CNS development, behavior and neurodegeneration, we generated a conditional targeted Nsdhl allele using homologous recombination in embryonic stem (ES) cells. We demonstrate here the utility of this mouse model in the study of cholesterol metabolism in the CNS and describe a novel early postnatal cerebellar phenotype that occurs when the conditional allele is crossed with mice expressing cre recombinase under the control of the human glial fibrillary protein (GFAP) promoter (21). We also were able to begin to address the cellular consequences of cholesterol deficiency versus methylsterol accumulation using this model system.
Results
Generation of a conditional Nsdhl allele
To generate a conditional Nsdhl allele, exon 5 that contains the predicted enzymatic active site was flanked by LoxP sites in the vector pL451 that also contains a neomycin (Neo) selection cassette flanked by Frt sites [(22); see Methods and Fig. 1B and C)]. Four properly targeted, Flp-treated ES clones were injected into C57BL/6J blastocysts, and multiple high percentage chimeras (>90%) were obtained. Three of the four male chimeras that were bred demonstrated germline transmission to multiple female offspring (only F1 females can receive a targeted allele from a male chimera given an X chromosomal localization). The ‘floxed’ animals (official name: Nsdhltm1.1Hrm, MGI:5581334, to be designated as Nsdhlflx5 here) are fertile and without any obvious phenotype.
To verify the null phenotype following excision of exon 5, we mated homozygous Nsdhlflx5/Nsdhlflx5 females to B6N. Cg-Tg(Sox2-cre)1Amc/J (Sox2-cre) males that induce efficient recombination in all cells of the epiblast (23). We noted efficient deletion in embryonic lineages at E8.5–10.5, using a PCR assay (not shown). Furthermore, we found no detectable NSDHL protein by western blotting or immunohistochemistry (IHC) in Sox2-cre deleted E9.5 or E10.5 male embryos, respectively, suggesting that the protein lacking exon 5 is unstable and degraded (Fig. 1D, Supplementary Material, Fig. S1A). The phenotype in heterozygous deleted (NsdhlΔ5/+) females was indistinguishable from that in surviving Bpa1H/+ females carrying a nonsense allele (K103X) (15) (Supplementary Material, Fig. S1B and C). We conclude that the phenotype in mice lacking exon 5 replicates the original Bpa1H null allele.
Ablation of Nsdhl in radial glia using GFAP-cre produces a dramatic cerebellar phenotype in affected males
Mice with the Nsdhlflx5 allele were mated to transgenic animals that express cre-recombinase under the control of the human glial fibrillary acidic protein (GFAP) promoter, (FVB-Tg(GFAP-cre)25Mes, called hGFAP-cre) (21). In these mice, beginning at ∼E13.5, cre-recombinase is expressed in radial glia in the ventricular zone of the developing CNS. These cells give rise to most differentiated neurons and glia of the cerebral cortex and cerebellum, although GFAP and, hence, cre-recombinase are not expressed in cerebellar Purkinje cells or epithelial cells of the choroid plexus. After postnatal day (P) 7, cre expression occurs mostly in astrocytes.
We established matings of homozygous Nsdhlflx5 females and heterozygous hGFAP-cre males and monitored the pups for the presence of the cre transgene by PCR genotyping. The experiments described below were performed on progeny from homozygous Nsdhlflx5 females derived by brother-sister mating of mice from 2 of the founder lines, derived from different ES cell clones, at the N2 or N3 backcross generation onto C57BL/6J. No phenotypic differences have been observed between the founder lines or using mice from different backcross generations. We performed our phenotypic analyses using deleted male mice (NsdhlΔ5/Y), to avoid variability related to random X-inactivation in heterozygous females. Floxed (Nsdhlflx5/Y), but not deleted, male littermates served as controls, avoiding potential sex differences that could be related to steroid hormones.
NsdhlΔ5/Y males from the cross of (Nsdhlflx5/Nsdhlflx5) females X (hGFAP-cre/+) males were present in expected numbers (284/1114 or 25.5% of live-born pups) and appeared normal at birth. However, by P8 to P10, they exhibited overt ataxia, difficulty with ambulation, and subsequent weight loss (Supplementary Material, Fig. S1D). They died between P12–20, probably secondary to defects in the cerebral cortex. On routine histology, at P0, we noted no NSDHL expression by IHC in most cortical and hippocampal neurons (Supplementary Material, Fig. S2E–J). The mutant hippocampus appeared disorganized with fewer hippocampal neurons present, particularly in the dentate gyrus (Supplementary Material, Fig. S2A and B). At P7, the NsdhlΔ5/Y brains showed obvious defects in the cerebellum, hippocampus and cerebral cortex (Supplementary Material, Fig. S3). The cerebellum and hippocampus were smaller than normal, with reduced numbers and organization of granule neurons. More pycnotic nuclei were evident in the outer layer of the NsdhlΔ5/Y cerebral cortex, suggesting elevated cell death.
In the mouse, most cerebral cortical and hippocampal neuronal proliferation, differentiation and migration are complete prior to birth. Cerebellar GCPs migrate from the rhombic lip adjacent to the fourth ventricle beginning at ∼E13.5 and cover the entire cerebellar surface prior to birth (24,25). However, unlike cortical neurons, they undergo extensive proliferation postnatally, as well as subsequent differentiation and migration. We focused our phenotypic and mechanistic analyses on the cerebellum because much of its well-studied growth and development occurs synchronously within the first 3 weeks after birth and involves relatively few cell types that are arranged in morphologically distinct layers. Moreover, relatively large, pure populations of GCPs can be isolated for in vitro analysis of both their proliferation and differentiation into neurons. These properties enabled us to perform studies examining the relative roles of cholesterol depletion and methylsterol accumulation in disease pathogenesis.
In the cerebellar cortex of NsdhlΔ5/Y males, early postnatal development (P0–P4) appeared normal by routine histology, although Nsdhl expression remained only in Purkinje cells, which do not express GFAP (21,26) (Supplementary Material, Fig. S3I and J). However, by P5, the external granule cell layer (EGL) in NsdhlΔ5/Y males appeared thinner with fewer granule cell precursors (GCPs) present (Fig. 2A and B). The magnitude of this difference progressed such that by P8, there was a single layer of GCPs in the EGL in the most severely affected mutant pups. Although mutant cerebellar Purkinje cells expressed NSDHL, they did not form extensive dendritic trees compared with their wild-type littermates, and they eventually degenerated (Fig. 2C, Supplementary Material, Fig. S4A). Bergmann glia, which are specialized astrocytes of the cerebellar cortex, were fewer in number, shorter and appeared disorganized in cerebella from mutant males (Fig. 2D). These results are consistent with previous studies demonstrating that contact with (normal) granule cells and Bergmann glia is required for the proper alignment and orientation of the Purkinje cells, as well as for their dendritic arborization (26,27).
Figure 2.

Early postnatal cerebellar phenotype of NsdhlΔ5 males. (A) Histological comparison of cerebellar lobule 3 between Nsdhlflx5/Y and NsdhlΔ5/Y brains at P4, P6 and P8. All samples were sagittal sections through the vermis. The relative thickness of the darkly-staining EGL (marked in upper left panel) was similar at P4. At P6, the EGL of the NsdhlΔ5/Y sample was substantially thinner than in the Nsdhlflx5/Y cerebellum. At P8, the EGL of the NsdhlΔ5/Y cerebellum was reduced to a single layer of GCPs, while the IGL was disorganized with numerous pycnotic nuclei. Scale bars: 100 μm. (B) Quantitation of the EGL area in lobule 3 on histological samples from Nsdhlflx5/Y and NsdhlΔ5/Y cerebella from P4 to P8. EGL area was significantly reduced in NsdhlΔ5/Y versus Nsdhlflx5/Y cerebella beginning at P5. N = number of cerebella examined. (C) Visualization of the Purkinje cell layer by anti-calbindin staining of Nsdhlflx5/Y and NsdhlΔ5/Y cerebella at P6 and P7. Purkinje cell dendrites are indicated by arrows. Note the almost complete lack of dendrites in the P7 mutant image. Scale bars: 100 μm. (D) Z-stack confocal images of Bergmann glia visualized by immunofluorescent staining for GFAP in Nsdhlflx5/Y and NsdhlΔ5/Y P6 cerebella. Scale bars: 50 μm. Abbreviations: BG, Bergmann glia; EGL, external granule layer; IGL, inner granule cell layer; PC, Purkinje cell, PS, pial surface. (E) Cholesterol, desmosterol and methylsterol concentrations in isolated GCPs from Nsdhlflx5/Y and NsdhlΔ5/Y cerebella at P3, P5 and P7. N = number of independent samples analyzed.
To determine whether the developmental timing of the EGL phenotype corresponds to a concurrent decrease in cholesterol levels, gas chromatograph-flame ionization detection (GC-FID) sterol analysis was performed on freshly isolated GCPs from NsdhlΔ5/Y and Nsdhlflx5/Y pups at P3, P5 and P7 (Fig. 2E). In P3 samples, cholesterol levels were not significantly different between mutants and controls. However, the NsdhlΔ5/Y samples showed a 25% reduction in cholesterol concentration relative to controls at P5 and P7. At all three stages, the mutant GCPs showed greatly elevated concentrations of sterol intermediates, identified using full scan MS data as 4α-methylcholest-7(8)-en-3β-ol and 4α-methylcholesta-8(9),24-dien-3β-ol (see Supplementary Material, Fig. S5A for sample GC-FID chromatograms and Fig. S5D for sample mass spectrometry scan data), consistent with loss of NSDHL function. Both compounds were nearly undetectable in control samples. The relative content of desmosterol, which contains a residual C24 double bond and is present in high concentrations in the developing CNS (28), was similar in control and mutant samples at all three stages (Fig. 2E). This discrepancy may reflect differences in the turnover of pre-existing, maternally derived cholesterol versus desmosterol in vivo. Indeed, Jansen et al. demonstrate that desmosterol accumulation in the mouse brain peaks in the first postnatal week; they suggest that post-transcriptional repression of 3β-hydroxysterol 24-reductase may be responsible for this accumulation (28).
Proliferation and migration defects in mutant cerebellar granule cells
GCPs in the outer EGL proliferate extensively between P0 and P14. One granule cell generates ∼250 progeny, and they account for ∼50% of all CNS neurons in the adult [reviewed in 25]. This proliferation peaks at ∼P6–8 and requires sonic hedgehog (SHH) secreted by Purkinje cells (29,30). Post-mitotic granule cells begin to migrate inward from the surface and establish an inner EGL. Migration requires additional secreted factors, including BNDF (31). The mechanisms responsible for the switch of cerebellar GCPs from proliferation to differentiation and migration are not completely understood. They do require and are guided by the Bergmann glia, which also differentiate in response to SHH signaling (29,32).
The presence of a thinner EGL after P4 in NsdhlΔ5/Y males suggested that there could be reduced proliferation and/or early differentiation and migration of mutant cerebellar GCPs, as found in several other mouse mutants with cerebellar phenotypes (26,33,34). As measured by phosphohistone H3 (PHH3) IHC (Fig. 3A and B) and BrdU incorporation (see Supplementary Material, S4C and D), we found progressively reduced proliferation of GCPs in mutant cerebella starting at P5. When apoptosis was measured using the TUNEL assay, a significant increase in TUNEL(+) cells was first observed at P7 in the NsdhlΔ5/Y males relative to controls (Fig. 3C). Thus, the diminished EGL in the mutant cerebella was the result of reduced proliferation beginning at ∼P5 followed by increased cell death at ∼P7. By immunofluorescence with antibodies to marker proteins of differentiating neurons, such as TAG-1 (contactin 2) (35) (Supplementary Material, Fig. S4B), and DCX (doublecortin) (36) (not shown), some mutant GCPs did become post-mitotic, began to differentiate, and migrated to reside within the inner layer of the EGL and inner granule cell layer. However, NsdhlΔ5/Y males also had a defect in GCP migration as shown by BrdU pulse-labeling experiments (Fig. 4). Mice were injected with BrdU at P6, and brains were collected either 2 or 48 h later and stained for BrdU incorporation. Labeling in the 2-h samples was mainly in the outer EGL (oEGL) of both Nsdhlflx5/Y and NsdhlΔ5/Y cerebella, as expected (Fig. 4A). In the samples collected after 48 h, a majority of the labeled cells had migrated to the inner EGL (iEGL) or IGL in the Nsdhlflx5/Y samples, while most of the BrdU(+) cells remained in the oEGL in the NsdhlΔ5/Y cerebella. Plotting the ratio of labeled cells in the IGL versus the oEGL in mutant and wild type cerebella at different labeling intervals showed that granule cell migration was not significantly different for cells born on P4, but migration of NsdhlΔ5/Y cells was reduced at P5 and P6 (Fig. 4B).
Figure 3.

Proliferation and apoptosis in the EGL of NsdhlΔ5/Y cerebella during early postnatal development. (A) Detection of mitotic cells by PHH3 immunostaining in lobule 3 of Nsdhlflx5/Y and NsdhlΔ5/Y cerebella at P6. Scale bars: 100 μm. (B) Quantitation of PHH3-positive cells in the EGL of all lobules in sections from the cerebella of Nsdhlflx5/Y and NsdhlΔ5/Y mice from P4 to P8. N = number of cerebella counted. (C) Quantitation of TUNEL-positive cells in the EGL of lobule 3 in Nsdhlflx5/Y and NsdhlΔ5/Y cerebella from P5 to P8. N = number of cerebella analyzed.
Figure 4.

Migration of BrdU-labeled GCPs in Nsdhlflx5/Y and NsdhlΔ5/Y cerebella. (A) Distribution of BrdU-labeled cells in the EGL of lobule 3 in Nsdhlflx5/Y and NsdhlΔ5/Y cerebella at 2 and 48 h after BrdU injection on P6. The lighter staining of cells in the Nsdhlflx5/Y sample at P8 was likely due to dilution of incorporated BrdU by cell division that followed the initial labeling 48 h earlier. Note that a large majority of the labeled GCPs in the NsdhlΔ5/Y sample remained in the oEGL in the P8 sample, 48 h post-injection. Abbreviations: oEGL, outer external granule layer; iEGL, inner external granule layer; ML, molecular layer; IGL, inner granule cell layer. Scale bars: 100 μm. (B) The migration efficiency of Nsdhlflx5/Y and NsdhlΔ5/Y GCPs at different ages. Migration was quantified by injecting pups with BrdU at P4, P5 or P6 and collecting cerebella for IHC 48 h later at P6, P7 or P8, respectively. The ratio of BrdU-labeled cells in the IGL to those remaining in the oEGL of lobule 3 was calculated for each sample (see Methods).
A decreased response to SHH underlies the proliferation defect in mutant granule cell precursors
A clear advantage to studies involving the early postnatal cerebellum is the ability to replicate most aspects of neuronal proliferation, differentiation, and migration in vitro (37,38), including the proliferation of primary GCPs in response to exogenous SHH (39–41). GCPs isolated from mutant NsdhlΔ5/Y mice at P4 and cultured for 48 h without added cholesterol demonstrated reduced proliferation in the presence of exogenous SHH compared with control Nsdhlflx5/Y littermates as measured by BrdU incorporation (Fig. 5A and B). Furthermore, there were many fewer and thinner neurite extensions in the mutant cells compared with the controls after 24 h (not shown) or 48 h in culture (Fig. 5A). The addition of cholesterol to the primary GCP cultures led to almost complete restoration of the proliferative response to SHH (Fig. 5B) and much improved cell morphology (Fig. 5A).
Figure 5.
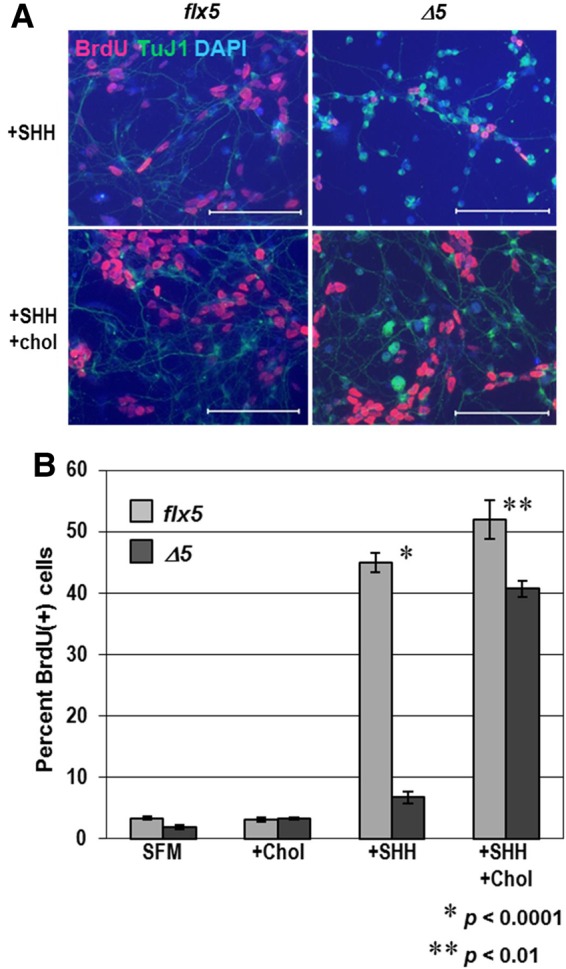
Response of cultured GCPs to SHH and exogenous cholesterol. (A) Immunofluorescent staining for BrdU (red) and neuronal marker TuJI (green), with DAPI (blue) counterstaining of Nsdhlflx5/Y and NsdhlΔ5/Y GCPs that were cultured for 48 h with either 1 μg/ml recombinant SHH alone or SHH plus 15 μg/ml cholesterol. BrdU was added to the samples 4 h before fixing. Note that there are fewer and thinner neurite extensions in the mutant NsdhlΔ5/Y cells than in the Nsdhlflx5/Y controls. Scale bars: 100 μm. (B) Four aliquots of isolated GCPs from individual Nsdhlflx5/Y and NsdhlΔ5/Y P4 cerebella were cultured for 48 h in either serum free medium (SFM) alone, SFM with 15 μg/ml cholesterol (chol), SFM with 1 μg/ml recombinant SHH, or SFM with 15 μg/ml cholesterol and 1 μg/ml SHH. BrdU was added to the cultures 4 h before fixing the cells. Cells were immunostained for BrdU and counterstained with DAPI. Values are the percentage of BrdU-labeled cells from the total (DAPI-stained) number of cells. Each bar represents the mean ± SEM of results from three independent experiments.
Hedgehog signaling in recipient cells via the canonical pathway involves binding of ligand to the transmembrane receptor PTCH1, relieving its inhibition of smoothened (SMO). Release of SMO inhibition is associated with movement of the protein into the primary cilium and subsequent activation of GLI transcription factors (42,43). The major effector of cerebellar granule cell proliferation via canonical hedgehog signaling is GLI2 (32,44), although induction of Gli1 gene expression is more robust in in vitro assays (45). SHH signaling also induces Nmyc expression which in turn activates D-type cyclins and progression through the G1 phase of the cell cycle (39,40,46). Nmyc induction is independent of Gli expression, but downstream of smoothened (SMO) (40). Phosphorylation of multiple sites on NMYC via GSK-3 in vivo promotes its degradation, enabling cell cycle exit in the presence of continuous SHH (47,48).
SHH treatment of Nsdhlflx5/Y cultured GCPs resulted in >500-fold and ∼8-fold induction of Gli1 and Gli2 gene expression, respectively, while their induction was greatly reduced in NsdhlΔ5/Y cultured cells (Fig. 6A). Growth of the SHH-treated mutant cells in the presence of exogenous cholesterol resulted in substantially increased Gli transcription, although the recovery of Gli1 expression was less dramatic than the proliferation response (compare Figs 5B and 6A). Transcription of Gli3 in GCPs is not responsive to SHH (49) and remained unchanged under all conditions in both wild type and mutant cells (not shown). Transcription of Ptch1 and Nmyc was induced ∼2 and 1.3-fold, respectively, by SHH in control cells, but remained unchanged in mutant cultured cells. NMYC targets Ccnd1 and Ccnd2, encoding cyclins D1 and D2, respectively, were also induced upon SHH treatment with a significantly greater response in wild type versus mutant cells (Fig. 6A).
Figure 6.

Expression of SHH target genes and genes for cholesterogenic enzymes in Nsdhlflx5/Y and NsdhlΔ5/Y cultured GCPs and cerebella. (A) qPCR analysis of gene expression in cultured GCPs isolated from Nsdhlflx5/Y and NsdhlΔ5/Y P4 cerebella that were cultured as described in Figure 5B. The relative quantity of mRNA from each gene was calculated from real-time PCR analysis of cDNA. Values were normalized to the untreated Nsdhlflx5 sample set equal to 1 for each gene. Each bar represents the mean ± SEM of results from three independent RNA samples that were each assayed in triplicate. (B) qPCR analysis of gene expression in Nsdhlflx5/Y and NsdhlΔ5/Y whole cerebella at P6, performed as in (A). Values represent the mean ± SEM of results for RNA samples from 3 cerebella that were each assayed in triplicate.
Addition of cholesterol to wild type or mutant cultured cells by itself did not affect the levels of transcription of any of the hedgehog-responsive genes tested (Fig. 6A). However, in the presence of added SHH, cholesterol resulted in variable, but uniformly, increased transcription of SHH and NMYC-induced genes in the mutant GCPs. The most dramatic response was found for N-Myc itself, where there was no longer a significant difference in levels of transcription between control and mutant cultured cells.
As with other cholesterol biosynthetic enzymes, NSDHL protein levels are high in neuronal cells in late gestation and the early postnatal period and then drop ∼6-fold by 10–12 weeks of age (50,51). There is also a surge in cholesterol synthesis in oligodendrocytes between 2 and 3 weeks of age, corresponding with axon myelination. As expected, the precise regulation of the cholesterol biosynthetic pathway resulted in upregulation of the two cholesterol biosynthetic genes examined, Hmgcr and Sc4mol, in mutant cells cultured without added cholesterol (Fig. 6A). Expression of these genes was significantly reduced in both WT and mutant cells with cholesterol supplementation (43% reduction in Sc4mol expression in WT and 26% in mutant cells; 26 and 17% reduction in Hmgcr expression in WT and mutant cells, respectively), indicating that the cells were able to incorporate and respond to free cholesterol. However, expression of Hmgcr and Sc4mol in the NsdhlΔ5/Y GCPs remained well above that of WT cells, suggesting that their cellular cholesterol homeostasis had not been completely restored. Unexpectedly, SHH treatment alone resulted in lower expression of Hmgcr and Sc4mol in both WT and mutant cells compared with growth in serum-free media (SFM) alone. We attribute this finding to the differentiation of a majority of cultured GCPs into TuJ1-positive neurons that occurs in the absence of SHH, with more newly-formed membranes that require cholesterol as they form neurite extensions (not shown).
Although less dramatic, significant decreases in transcript levels of hedgehog response genes were also seen in experiments using whole cerebella isolated at P6 (Fig. 6B). The smaller in vivo effect sizes may be related to the presence of residual cholesterol available to the cells, as well as the presence of non-deleted cells within deep structures in mutant cerebella. Using mice that express lacZ under the control of the Gli1 promoter, we were also able to demonstrate variable, but reduced, induction of this SHH response gene in vivo (Supplementary Material, Fig. S6).
Assessment of the effects of NSDHL deficiency on cholesterol homeostasis in cerebellar GCPs in vitro
To gauge the relative contributions of cholesterol depletion and methylsterol accumulation to the cerebellar phenotypes associated with NSDHL deficiency, additional experiments were performed in which GCP cultures were treated with ketoconazole to block endogenous cholesterol biosynthesis. Ketoconazole inhibits lanosterol 14-alpha-demethylase (CYP51A1) in the conversion of lanosterol to 4,4-dimethylcholesta-8(9),14,24-trien-3β-ol, in a step upstream of NSDHL in the cholesterol biosynthetic pathway (see Fig. 1A). Thus, ketoconazole reduces both cholesterol synthesis and the accumulation of methylsterols associated with NSDHL inactivation. Human LDL was used as a soluble and easily measured source of exogenous cholesterol.
Initially, we performed dose response assessments to determine the minimal concentrations of LDL and ketoconazole necessary to observe an effect on the Gli1 response to an exogenous SHH signal. Gli1 transcript level was used as a sensitive measure of responsiveness to SHH because, of the genes that were tested, it showed the greatest range of response to exogenous SHH in cultured GCPs (Fig. 6A). For the experiments below, we used a concentration of ketoconazole of 2 μM, that results in >90% suppression of Gli1 expression and only minor alterations of morphology in isolated WT GCPs, and 5 µg/ml LDL, that fully restores Gli1 expression in mutant cells to WT levels (Fig. 7A) and produces much improved mutant cell morphology (Supplementary Material, Fig. S7). Conversely, treatment of WT GCPs with ketoconazole reduced Gli1 RNA by 10-fold, to a level similar to that of mutant cells. The presence of LDL completely abrogated the effect of ketoconazole on Gli1 levels in both WT and mutant cells. Ketoconazole alone at a concentration of 2 μM had no effect on the response of mutant cells to SHH, while higher doses of ketoconazole produced significant cellular toxicity even in WT cells (not shown).
Figure 7.

Effects of LDL and ketoconazole treatment on Nsdhlflx5/Y and NsdhlΔ5/Y GCPs in vitro. GCPs were isolated from individual cerebella at P4, split into 4 culture wells with the indicated treatments and analyzed after 48 h. All samples received 1 µg/ml recombinant SHH throughout the 48 h culture. Values represent the mean ± SEM. (A) The relative expression levels of Gli1 were measured by qPCR in Nsdhlflx5/Y and NsdhlΔ5/Y GCPs treated with LDL (5 μg/ml) and/or ketoconazole (2 μM). The results represent 4 independent samples that were each assayed in triplicate with Gapdh used as the endogenous control and normalized to the untreated Nsdhlflx5/Y sample set equal to 1. (B) Percentage of viable cells, as measured by propidium iodide staining of live GCP cultures in situ. Unstained cells were counted as viable. The results are from 4 Nsdhlflx5/Y and 5 NsdhlΔ5/Y independent cultures. (C) Lysed cell cholesterol concentration (μg/mg protein) for aliquots of the cultured GCPs used for qPCR analysis in (A). None of the pairwise comparisons between Nsdhlflx5/Y and NsdhlΔ5/Y samples in the four culture conditions reached statistical significance (P < 0.05) (see text). (D) Lysed cell total methylsterol concentration for the samples used in (C). The concentration of methylsterols was significantly higher in NsdhlΔ5/Y GCPs than Nsdhlflx5/Y GCPs (P < 0.002) in all four culture conditions. (E) Lysed cell total desmosterol concentration for samples used in (A). Desmosterol levels were significantly lower in NsdhlΔ5/Y versus Nsdhlflx5/Y GCPs in both untreated and LDL-treated cultures. Ketoconazole treatment reduced the desmosterol concentration in Nsdhlflx5/Y cells to a level equivalent to that of NsdhlΔ5/Y cells, while addition of LDL to the ketoconazole-treated cultures had no effect on desmosterol.
We then determined the effect of ketoconazole and/or LDL on GCP viability by propidium iodide (PI) staining of live cells in situ after 48 h in culture. Mutant cells treated with SHH alone showed significantly lower survival (65%) than WT cells (90%) (Fig. 7B). LDL supplementation increased the survival of mutant cells to a level comparable with that of WT GCPs. As was seen with pure cholesterol, LDL treatment also resulted in greatly improved cell morphology in the mutant GCPs (Supplementary Material, Fig. S7). Ketoconazole treatment did not affect the viability of WT cells, but it significantly increased mutant cell viability (from 65 to 78%; Fig. 7B). Moreover, in the presence of ketoconazole, the mutant GCPs again showed a markedly improved morphology, with larger cell bodies and more extensive neurite outgrowth (Supplementary Material, Fig. S7). These data suggest that elevated methylsterol levels in mutant cells may directly affect cell viability.
In serum free media (SFM), in the presence of added SHH, measured cholesterol levels after 48 h in culture were 19.75 ± 2.9 SEM µg/mg protein in WT cell lysates and 15.05 ± 0.93 µg/mg protein in mutant GCPs lysates (74% of wild type, P = 0.13) (Fig. 7C). The lack of a significant difference in cholesterol levels may be attributable to the state of differentiation of WT versus mutant cells after SHH treatment and concomitant differences in cellular morphology. Specifically, since sterol levels were calculated relative to the total protein content of the samples and the dramatic reduction in neurites, cell surface membranes, and the size of the cell bodies of the mutant cells grown in SFM is accompanied by reduced levels of total proteins, the ratio of cholesterol levels to protein may underestimate the extent of cholesterol depletion (compare Supplementary Material, Fig. S7A and B, for example). Cholesterol levels for all of the cultures dropped significantly with ketoconazole treatment (Fig. 7C). As expected, ketoconazole-treated cultures showed increased levels of lanosterol as a result of CYP51A1 inhibition (Supplementary Material, Fig. S5C). The improved morphology in mutant cells (Supplementary Material, Fig. S7F) may reflect further reduced levels of methylsterols, since treatment with either cholesterol or ketoconazole resulted in significant decreases in the methylsterol levels (Fig. 7D). Finally, treatment with both LDL and ketoconazole produced the lowest levels of methylsterols in the mutant GCPs, similar to those in WT cells grown without either additive.
In contrast to the relatively small differences in desmosterol levels in freshly isolated WT versus mutant GCPs (Fig. 2E), we noted much more dramatically reduced desmosterol levels in cultured mutant cells grown with or without LDL in comparison with cultured WT cells (Fig. 7E). WT desmosterol levels in the absence of exogenous LDL-cholesterol were similar to those seen in freshly isolated GCPs, suggesting a deficiency of desmosterol in the cultured mutant cells. We believe this deficiency reflects the high demand for cholesterol as the cultured mutant GCPs attempt to differentiate.
Taken together, these results demonstrate that cholesterol depletion is sufficient to account for virtually all of the reduced response of mutant GCPs to SHH in vitro. Although the reduction of methylsterol levels by ketoconazole treatment apparently improved the viability and cell morphology of the mutant GCPs, it had no effect on Gli1 expression (Fig. 7A).
Since sterols have been implicated in modulating the initial steps of hedgehog signaling in target cells by binding to PTCH1 and SMO (52,53), we asked whether the HH signaling defect in NsdhlΔ5/Y GCPs could be bypassed by treating the cells with the smoothened agonist SAG, a synthetic small molecule that activates the HH signaling pathway by direct binding to SMO (54). Gli1 expression levels were measured in cultured GCPs that were isolated from individual cerebella at P4, split into 6 wells and treated with either SHH or SAG (Fig. 8). WT GCPs treated with SAG (100 nm) showed the same level of Gli1 expression as SHH-treated cells. Gli1 levels were reduced by 42% with ketoconazole treatment of SAG-stimulated WT GCPs, compared with a >10-fold reduction in SHH-stimulated cells. Thus, signaling mechanisms at or upstream of SMO appear to be more sensitive to depletion of cholesterol and/or oxysterols by ketoconazole than downstream steps. The response of NsdhlΔ5/Y cells to SAG was more than10-fold lower than that of wild type cells, similar to their diminished response to SHH. However, ketoconazole increased Gli1 levels by ∼4-fold in SAG-treated mutant cells (P = 0.029), in marked contrast to the lack of an effect of ketoconazole on their response to SHH. Note that the Gli1 response of the mutant cells to stimulation with SAG was inversely proportional to the levels of C4 methylsterols that were measured in ketoconazole treated cultures (Fig. 7D). This suggests that the deleterious effects of methylsterols, that include reduced viability and reduced neurite outgrowth, may also affect HH signaling at steps downstream of SMO. However, in the presence of ketoconazole, Gli1 levels were still more than 2-fold lower in mutant cells than WT cells. This may be due to the fact that in the presence of ketoconazole, while methylsterol levels were reduced in the mutant cells, they were still ∼10-fold higher than in ketoconazole-treated WT cells (Fig. 7D). Finally, LDL treatment increased Gli1 expression in SAG-treated mutant cells to levels similar to those in WT cells, likely due to both restoring normal cholesterol levels and further reducing methyl sterols to the levels seen in untreated wild type cells (Fig. 7D). In summary, a comparison of results from SHH versus SAG stimulation of the HH signaling pathway in GCPs confirmed that steps at or above SMO are more sensitive to sterol depletion than downstream steps. Furthermore, by circumventing the sterol-sensitive steps with SAG treatment, we uncovered an effect of elevated methylsterol levels in NsdhlΔ5/Y cells that also reduces the efficiency of HH signaling by unknown mechanisms downstream of SMO.
Figure 8.

Comparison of Gli1 relative expression levels in response to SHH or SAG between Nsdhlflx5/Y and NsdhlΔ5/Y GCPs cultured in vitro. GCPs were isolated from individual cerebella at P4, split into 6 culture wells with the indicated treatments and analyzed after 48 h in culture. Concentrations of ketoconazole and LDL were as in Figure 7. The results are from 4 independent samples of each genotype that were each assayed in triplicate by qPCR with Gapdh used as the endogenous control and normalized to the untreated Nsdhlflx5 sample set equal to 1.
Finally, we asked whether commercially available testicular meiosis-activating sterol (T-MAS; 4,4-dimethyl-5α-cholesta-8(9),24-dien-3β-ol) would affect HH signaling in WT GCPs. T-MAS and the related follicular fluid meiosis-activating sterol (FF-MAS; 4,4-dimethyl-5α-cholesta-8(9),14,24-trien-3β-ol) are naturally occurring C4-dimethyl sterols that accumulate upon inhibition of the C4-demethylase complex (see Fig. 1A). They were originally identified as compounds that produced resumption of meiosis in oocytes in vitro (55,56). Pure T-MAS was mixed with fatty acid-depleted BSA as a carrier to form a water-soluble complex that was added to the culture medium. T-MAS had no effect on the level of Gli1 expression in response to SHH (Fig. 9A). GC-FID sterol analysis of cell lysates indicated that T-MAS was incorporated by the cells in a dose-dependent manner (Fig. 9B) although its intracellular localization relative to endogenous sterol intermediates was not determined. The 2.5 μg/ml treatment resulted in a cellular T-MAS level similar to that of methylsterols in untreated NsdhlΔ5/Y GCPs (3–4 μg/mg protein, Fig. 7D). The GCPs treated with 10 μg/ml T-MAS showed an abnormal morphology, with smaller cell bodies and thinner neurites, reminiscent of NsdhlΔ5/Y cells (Supplementary Material, Fig. S8). Furthermore, T-MAS treatment at 10 ug/ml produced significantly reduced cell viability in WT GCPs (Fig. 9D). This effect was less dramatic than in mutant cells with elevated levels of endogenous methylsterols (Fig. 7B), perhaps because WT cells should be able to convert accessible T-MAS to less toxic intermediates, as well as cholesterol itself. In addition, the mutant cells that are cholesterol deficient may be more sensitive to methylsterol toxicity.
Figure 9.

Effect of exogenous T-MAS on WT (Nsdhlflx5/Y) GCPs in vitro. (A) Relative Gli1 expression levels measured by qPCR in cultured Nsdhlflx5/Y GCPs treated with T-MAS. All samples were cultured with 1 μg/ml SHH. Bars represent the mean ± SEM of 4 independent samples that were each assayed in triplicate with GAPDH as the endogenous control and normalized to the untreated sample set equal to 1. (B) Lysed cell pellet T-MAS concentration and (C) cholesterol concentration determined for aliquots of the cultured Nsdhlflx5/Y GCPs treated with exogenous T-MAS used in panel A. Values indicate the mean ± SEM of 3 independent GCP cultures. (D) Percentage of viable cells in T-MAS-treated cultures, measured by propidium iodide staining, performed as in Figure 7B. The results are the mean ± SEM from 3 independent Nsdhlflx5/Y GCP cultures.
Discussion
We have developed the first conditional mouse model for a disorder of post-squalene cholesterol biosynthesis, although a conditional Fdft1 allele encoding squalene synthase (SQS) has been reported (57). In our study, we tested the utility of the conditional Nsdhl allele by investigating the effects of defective cholesterol synthesis in vivo in the developing CNS. Ablation of Nsdhl in radial glia at E13.5 using the hGFAP-cre transgene resulted in postnatal lethality between P12 and P20. The expected loss of NSDHL in neuronal lineages and astrocytes of the cerebrum, hippocampus and cerebellum was confirmed by IHC. Histologic differences among affected brain regions were evident at birth, with the mutant hippocampus already disorganized, while the cerebellum showed no obvious phenotype (Supplementary Material, Fig. S2). Subsequently, between P5 and P8, cerebellar GCPs underwent a rapid sequence of reduced proliferation, impaired migration and, finally, massive cell death. Abnormalities were also noted in the Bergmann glia (Fig. 2D) and in Purkinje cells that continue to express NSDHL. The latter displayed deficient arborization and dendrite outgrowth and eventual cell death (Fig. 2C and Supplementary material, Fig. S4A). We also noted some delays in cerebellar lobule development by P7 (see Supplementary Material, Fig. S4C and D and Fig. 6), although we did not observe any obvious patterning defects in mutant cerebella. Folia number and patterning are SHH dependent; however, they are determined in late gestation (44,58), when sufficient cholesterol should still be available in the mutant CNS.
All of the known human disorders and mouse models of post-squalene cholesterol biosynthesis are associated with major somatic and/or CNS malformations, suggesting that perturbations of the pathway have potent teratogenic effects. Microcephaly and ID are also features in surviving infants with all of the human sterol synthesis pathway disorders, except for affected heterozygous females with X-linked CHILD syndrome and X-linked dominant chondrodysplasia puncata (9,18). We and others have proposed that decreased cholesterol or total sterols; accumulation of toxic sterol intermediates proximal to the enzymatic block; abnormal feedback regulation of earlier steps in the pathway; generation of abnormal bioactive oxysterols; and/or abnormal signaling by hedgehog proteins that normally contain bound cholesterol, may all play a role in disease pathogenesis, although exact mechanism(s) remain to be determined. It is very likely that multiple mechanisms occurring at different stages of development and/or in different tissues are involved in disease pathogenesis, and data are beginning to accumulate to support some of these mechanisms. Some of the teratogenic effects in these disorders likely result from a lack of cholesterol for incorporation into membranes. Methylated sterol intermediates, in particular, are unable to fulfill the role of cholesterol within membranes. They result in dramatic changes in fluidity within the lipid bilayers (59–61), and they are not incorporated well into lipid rafts (60,61). However, while there is some overlap in the phenotypes among all of the human and murine disorders, unique features of each would argue against cholesterol deficiency as the only mechanism.
Cholesterol levels in isolated NsdhlΔ5/Y GCPs were normal at P3, but became progressively lower than WT in P5 and P7 samples. We believe this reduction largely reflects depletion of maternal and fetal cholesterol supplied to the CNS prior to birth. The timing of cholesterol depletion in these cells corresponded closely to the appearance of defects in the EGL, with the earliest cerebellar phenotype detected in NsdhlΔ5/Y pups being decreased GCP proliferation at P5 that preceded increased apoptosis in these cells by approximately two days.
Cerebellar GCP proliferation is driven largely by SHH secreted by underlying Purkinje cells (29,30). NSDHL was not deficient in Purkinje cells of NsdhlΔ5/Y pups, and these cells appeared morphologically normal until P7. Thus, the proliferation defect is more likely to result from reduced response to SHH in the GCPs rather than impaired signaling from Purkinje cells. Indeed, isolated NsdhlΔ5/Y GCPs in vitro showed a markedly reduced response to exogenous SHH with regard to both proliferation and expression of SHH target genes, and addition of exogenous cholesterol restored this proliferative response to SHH to near wild-type levels (Fig. 5).
The hypothesis that reduced hedgehog signaling contributes to the pathology associated with genetic disorders of cholesterol biosynthesis was originally based on the fact that proper signaling by hedgehog ligands in vivo is, in part, dependent on their covalent modification by cholesterol (3). However, to date, no evidence has emerged to directly demonstrate that processing and modification of hedgehog ligands are affected in these disorders. Rather, Cooper et al. (62) demonstrated defects in the response of mutant cells to hedgehog signaling, likely at the level of SMO, in mouse embryo fibroblasts (MEFs) derived from Dhcr7−/− and Sc5d−/− mice that are murine models of Smith-Lemli-Opitz syndrome and lathosterolosis, respectively. In addition, 7-dehydrocholesterol can substitute for cholesterol in SHH processing and activation (62). Furthermore, an active N-terminal hedgehog peptide, which is not modified by cholesterol, can be secreted and signal in recipient cells, although the signal may spread further extracellularly (63,64). We have also found normal processing of SHH in NSDHL-deficient MEFs, as well as in mutant Nsdhl− male embryos at E10.5 (F. Jiang and G.E. Herman, unpublished results). A reduced response to hedgehog signaling in recipient cells in vivo has been demonstrated in NSDHL-deficient placentas by our group (65), as well as in the rudolph (rud) mutant mouse that carries a hypomorphic allele of Hsd17B7 (66), encoding a 3β-ketosterol reductase that functions with NSDHL and a methyloxidase, SC4MOL, in the sterol C4 demethylation complex (see Fig. 1A). Rud embryos display severe CNS and skeletal defects, and do not survive past birth. Cultured MEFs derived from rud embryos show a reduced response to exogenous SHH in both proliferation and expression of Gli1 and Ptch1. In vivo, rud embryos display lower expression of a Ptc-LacZ reporter in the forebrain and developing long bones. PTCH1 and SMO show normal translocation to the primary cilium of rud MEFs in response to SHH, and cholesterol levels in rud embryos are reported to be normal. Thus, the authors propose that the hedgehog-related defects result from impaired activation of SMO, most likely due to a deficiency of activating oxysterols or to inhibitory effects of accumulating sterol intermediates. Indeed, selected oxysterols can activate hedgehog signaling in vitro (67), and cause translocation of PTCH1 and SMO to the cilium in the absence of ligand (68).
The exact mechanism by which inactivation of Nsdhl disrupts hedgehog signaling has not been conclusively established. However, the lack of GCP proliferation or the lack of a Gli1 response to SHH in cultured mutant cells that is rescued by exogenous cholesterol (Figs 5 and 6) suggests that product deficiency is the primary abnormality. In addition, significant reduction of methylsterols using ketoconazole (Fig. 7A and D) did not restore the Gli1 response. Further evidence against accumulation of methylsterols being the primary mechanism is the lack of an effect of the 4-methylsterol T-MAS on Gli1 expression in cultured wild-type cells (Fig. 9). However, methylsterol accumulation is clearly associated with altered cell morphology and cell death and may be responsible for the massive apoptosis seen in the cerebellum in vivo. These abnormalities are also unlikely to be SHH-related. Although previous studies have shown that blocked hedgehog signaling can induce cell death in neuronal lineages in in vitro model systems (69,70), conditional knockout of several genes involved in hedgehog signaling in the cerebellum causes reduced GCP proliferation without a subsequent increase in apoptosis (26,44). Moreover, apoptosis in NsdhlΔ5/Y cerebella occurred throughout the inner granule layer (IGL) as well, where hedgehog signaling is not thought to be active.
We were also able to demonstrate differences in the response of cultured mutant GCPs to SHH versus the SMO agonist SAG. Ketoconazole could partially restore the Gli1 response of mutant cells treated with SAG, while it had no effect on the SHH-stimulated signal (Fig. 8). These data separate, at least partially, the cellular response to cholesterol depletion and methylsterol accumulation and are consistent with the model proposed by Stottmann et al. (66) for rud embryos of an effect at the level of SMO, possibly resulting from methylsterol accumulation.
Methylsterol accumulation has also been implicated in the pathogenesis of other cholesterol synthesis, as well as acquired, disorders. In CK syndrome, all of the affected males tested had normal sterol profiles, total cholesterol and steroid hormone levels, and lipoprotein profiles in plasma (10,19). However, when mutant lymphoblasts were cultured in lipid-depleted media, accumulation of 4-methylsterols, similar to that seen in CHILD syndrome, was found. Furthermore, in the only affected male tested, the CSF cholesterol level was normal, while corresponding 4-methylsterol levels were increased (10). These data led the authors to speculate that accumulation of methylsterols, rather than primary deficiency of cholesterol, caused CK syndrome. In the first patient described with autosomal recessive SC4MOL deficiency, another disorder with C4 methylsterol accumulation (11), the phenotype included severe psoriasiform dermatitis, arthralgias, immune dysfunction, congenital cataracts, short stature, microcephaly, and ID. The serum cholesterol was low at 85 mg/dl (normal 140–176 mg/dl) with markedly elevated levels of 4,4′-dimethyl and 4α-monomethyl sterols detected in plasma, as well as in cultured skin fibroblasts grown in complete or lipid-depleted media. There was also increased proliferation of affected skin fibroblasts grown in lipid-depleted media that the authors attributed to the ‘meiosis-activating effects’ of the sterol metabolites that accumulate (55,56). They postulated that the hyperproliferative skin phenotype in disorders of the C4 demethylase complex results from elevations of these sterol intermediates. Finally, Astsaturov and colleagues identified SC4MOL and NSDHL in an siRNA screen for proteins that sensitize tumor cells to EGFR inhibitors, with the goal of improving cancer therapy (71). Depleting either enzyme in the presence of EGFR inhibitors led to increased apoptosis in several cancer cell lines, while other enzymes in the pathway did not produce similar effects. Depletion of the upstream enzyme CYP51A1 (see Fig. 1A) using siRNA or ketoconazole eliminated the effect, while added cholesterol did not. These authors also proposed that accumulation of 4-methylsterols is specifically responsible for this sensitization and further demonstrated that the loss of SC4MOL or NSDHL led to altered EGFR trafficking with higher degradation rates within the cell lines examined.
The reduced migration of NsdhlΔ5/Y GCPs that was observed beginning around P5, is also unlikely to be related to abnormal hedgehog signaling since it has not been implicated in this process in GCPs. The tyrosine kinase receptor TrkB and its ligand, BDNF, are both expressed by GCPs; both are required for proper migration, among several other factors. Recent data suggest that, in rodent cortex and hippocampus, BDNF recruits TrkB to intracellular lipid rafts upon binding, while a small pool of TrkB in plasma membrane rafts is unchanged (72,73). Trafficking of TrkB into early endosomes, with subsequent activation of PI3K/AKT and Rac/cdc42, is necessary for the GCP chemotactic response to brain-derived neurotrophic factor (BDNF) (74). Given the recent finding by Sukhanova et al. (71) that NSDHL depletion alters trafficking of another receptor tyrosine kinase (EGFR), it is possible that a similar mechanism is causative here. Future studies examining the migration of control and mutant GCPs in vitro in the presence of a BDNF gradient could address this issue. It should be noted that, while disruption of several signaling pathways is possible, and, perhaps likely, we do not believe that there is a general disruption of all membrane signaling functions, since expression of Hes1, a Notch gene target involved in GCP proliferation in the cerebellum (75), was unaffected in cultured GCPs from mutant cells (not shown).
It is also possible that the migration abnormalities could arise from defects in the Bergmann glia. Postmitotic GCPs of the inner EGL first migrate tangentially, parallel to the pial surface, and then migrate radially inward along Bergmann glia processes to reach the IGL (25). During tangential migration, GCPs extend leading and trailing processes. For radial migration, a third process extends inward through the developing molecular layer, while the parallel fibers are retained and form synapses with Purkinje cell dendrites. Given the greatly reduced neurite outgrowth of NsdhlΔ5/Y GCPs during differentiation in vitro, it is possible that extension of processes from the mutant cells is likewise restricted in vivo, thus preventing normal migration. Neuronal growth cone motility is dependent on cholesterol-rich lipid rafts that are thought to organize signaling molecules that regulate adhesion and cytoskeletal dynamics during cell migration (76). Likewise, the Bergmann glial processes that appeared abnormal and fewer in number in NsdhlΔ5/Y P6 cerebella may provide a poor substrate for radial migration of GCPs. Migration of GCPs along Bergmann glia also involves the formation of gap junctions and intercellular signaling, that may be affected by disruption of normal membrane organization of lipid rafts in cholesterol-deficient cells (77,78).
In summary, we have demonstrated a novel, early postnatal lethal phenotype in a conditional allele of the cholesterol biosynthetic enzyme NSDHL. Our studies of the early postnatal cerebellum in vivo and in vitro demonstrate severe defects in GCP proliferation and hedgehog signaling that we believe are due primarily to cholesterol deficiency or depletion. In vitro studies in cultured GCPs also demonstrate a likely role for methylsterol accumulation in cell death, consistent with the work of others in studies of rud embryos (66) and EGFR receptor sensitive cancer cells (71). Further studies will be necessary to explore the pathogenesis of the migration defects. The Nsdhlflx5 conditional allele should be extremely valuable to investigate perturbations in cholesterol homeostasis in other regions of the brain, as well as in adult and aging mice.
Materials and Methods
Nsdhlflx5 allele
The Nsdhl flx5 targeting vector was constructed by inserting three PCR products with engineered restriction sites into the vector PL451 that contains a neomycin selection cassette (Neo) flanked by frt sites (22). The PCR products were amplified from BAC clone 440B9, that includes the Nsdhl gene and was obtained from the RPC-22 BAC library of mouse 129S6/SvEvTac genomic DNA (79). First, an 824 bp PCR product that included Nsdhl exon 5 with flanking intronic sequence and an engineered LoxP site was inserted into the BamH1 site downstream of the vector LoxP site. Second, a 1.7 kb fragment from intron 4 was inserted into the HindIII site upstream of the Neo cassette to provide a 5′ homology arm. Third, a 3.7 kb fragment that included exons 6 and 7 with flanking intronic sequence was inserted into the NotI site downstream of the floxed exon 5 fragment to provide a 3′ homology arm. Finally, a viral thymidine kinase (HSV-tk) expression cassette was inserted into the ClaI site upstream of the 5′ homology arm to provide negative selection against non-homologous recombination events. The wild type sequence of the Nsdhl exons and loxP sites in the final construct was verified by DNA sequencing. The construct was linearized with SalI and electroporated into TC1 (129/SvEvTac) ES cells by our institutional ES Cell Core laboratory. G418-resistent clones were isolated and replicate cultures were grown in the presence of ganciclovir to identify HSV-tk(-) clones. Southern blot analysis of DNA from selected ES cell clones digested with Asp718 or BglI was used to identify clones with homologous integration of the Flx5 construct. Three properly targeted clones were electroporated with a plasmid (FLPo) that expresses a codon-optimized Flp recombinase (80) to remove the Neo cassette. Two Neo(-) clones were injected into C57BL/6 blastocysts to generate chimeric founder animals. Three of the resulting male founders with a high percentage ES cell contribution transmitted the Nsdhlflx5 allele to female offspring.
Mice
Mice homozygous for the Nsdhlflx5 allele (Nsdhltm1.1Hrm) were maintained on a mixed B6;129-Nsdhlflx5 background and also backcrossed onto C57BL/6J. GFAP-cre [FVB-Tg(GFAP-cre)25Mes/J, stock #004600] and Gli1LacZ (Gli1tm2Alj/J, stock #008211) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). GFAP-cre animals were maintained as heterozygotes on an FVB background by brother-sister mating. GFAP-cre heterozygous males were crossed to Nsdhlflx5/Nsdhlflx5 females to produce the NsdhlΔ5/GFAP-cre and Nsdhlflx5/Y males that were used for experiments. Gli1LacZ animals were backcrossed to C57BL/6 and maintained as heterozygotes. GFAP-cre males were crossed to Gli1LacZ females to generate GFAP-cre/Gli1LacZ compound heterozygous males that were then crossed to Nsdhlflx/Nsdhlflx5 females to produce NsdhlΔ5/GFAP-cre/Gli1LacZ and Nsdhlflx5/Gli1LacZ males that were used for Gli1LacZ reporter expression analysis (see below). Sox2-cre mice were obtained from the Jackson Laboratory (stock #014094) and were maintained in the Research Institute ES Cell Core. For studies described here, the Sox2-cre males were on a mixed background (immunohistochemistry) or at N5 generation of a backcross onto C57BL/6J (Western blotting). Genotyping of mice was performed by PCR on DNA from toe clips using primers and PCR conditions as recommended by the Jackson Laboratory or as shown in Supplementary Material, Table S1. The housing and handling of mice conformed to protocols approved by the Institutional Animal Care and Use Committee of the Research Institute at Nationwide Children's Hospital.
Antibodies
Primary antibodies used for immunoblotting and immunohistochemistry were: NSDHL [rabbit polyclonal, (50)], BrdU (rat monoclonal, AbD Serotec, Raleigh NC), GFAP (mouse monoclonal, Thermo-Fisher Scientific, Fremont CA.), TUJ1 (mouse monoclonal, Covance, Emeryville, CA), phosphohistone H3 (PHH3) (rabbit polyclonal, Cell Signaling, Danvers, MA) calbindin (rabbit polyclonal, Millipore, Temecula CA), TAG-1 (Developmental Hybridoma Bank, Iowa City, IO) and β-tubulin (rabbit polyclonal, Thermo Scientific, Fremont, CA). Secondary antibodies were: HRP-linked goat anti-rabbit IgG (Cell Signaling), DyLight 488-conjugated donkey anti-mouse IgG, DyLight 549-conjugated donkey anti-rat IgG, and DyLight 549-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA).
Histology and immunohistochemistry
Preparation of mouse tissues for histology, immunohistochemistry and hematoxylin/eosin staining was performed as previously described (50). Briefly, tissues were fixed in either Bouin's fixative or 4% paraformaldehyde (PFA) in PBS by whole-body transcardial perfusion. Primary antibody dilutions were: NSDHL (1:1000), PHH3 (1:250), calbindin (1:3000), BrdU (1:1000), GFAP (1:250), TAG-1 (1:10), and TUJ1 (1:1000). All immunohistochemistry samples were counterstained with hematoxylin. For BrdU labeling in vivo, mice received an intraperitaneal injection of BrdU (100 μg/ml in PBS) at a dose of 1 μg/g body weight at designated times. For immunodetection of BrdU, brains fixed with Bouin's fixative were embedded in paraffin and 5 μm sections were mounted on Fisherbrand Superfrost Plus slides (Fisher Scientific). Sections were deparaffinized and incubated in 3% hydrogen peroxide, 10% methanol in PBS for 30 min, followed by a 1 h incubation in 200 μg/ml pepsin A (Worthington, Lakewood, NJ), 0.01N HCl in PBS. The samples were denatured in 2N HCl for 1 h at 37°C, followed by 10 min of incubation in 0.1 M sodium borate, pH 8.5. After rinsing in PBS, samples were incubated in blocking solution [2% normal goat serum in TBST (50 mm Tris, 150 mm NaCl, 0.25% Triton X-100)] for 2 h. Sections were incubated overnight at 4°C with rat anti-BrdU antibody in blocking solution. After washing with PBS, samples were incubated for 4 h with biotinylated anti-rat IgG secondary antibody, followed by HRP-conjugated streptavidin using a Vectastain Elite ABC kit (Vector Laboratories). Signal was detected using VECTOR NovaRED peroxidase substrate kit (Vector Laboratories, Burlingame, CA). For TUNEL assays, brains were fixed by perfusion with 4% PFA, and sections were assayed using the ApopTag Peroxidase In Situ Apoptosis Detection kit according to the manufacturer's instructions (Millipore). Stained samples were photographed using SPOT RT Color digital cameras operated by SPOT version 3.5 software (Diagnostic Instruments, Sterling Heights, MI) mounted on a Nikon SMZ-10A dissecting microscope or a Nikon Eclipse E800 microscope. Areas of the EGL and IGL were measured using SPOT software. Quantitation of PHH3-positive cells and BrdU-labeled cells was performed by manual counting on photomicrographs. For GCP migration analysis using BrdU pulse-labeling, the outer external granule layer (oEGL) on stained sections was defined, using SPOT software, as a zone 20 μm in width adjacent to the pial surface around the perimeter of the lobule. The inner granule layer was defined as the remaining area of the lobule, excluding the white matter at the center of the lobule.
Lacz staining
Gli1LacZ reporter expression analysis was performed on brains from NsdhlΔ5/GFAP-cre/Gli1LacZ and Nsdhlflx5/Gli1LacZ males as previously described (81). Briefly, brains were fixed in 0.2% glutaraldehyde, 2% formalin for 2 h following transcardial perfusion, infused with 30% sucrose and floating sections (60 μm) were cut from frozen tissue using a sliding microtome. Floating sections were incubated in X-gal staining solution for 4 h at room temperature, rinsed in PBS and fixed in 4% PFA. Stained samples were photographed using a Nikon SMZ-10A dissecting microscope as described above.
Immunoblotting
Total protein extracts were made by homogenizing pooled E9.5 control or mutant embryos in RIPA buffer with 1× Protease Inhibitor Cocktail (Sigma, St. Louis, MO). Immunoblots to detect NSDHL and β-tubulin were performed as previously described (50). Approximately 5–20 μg of protein from each sample was resolved by SDS-PAGE. Blots were probed sequentially with anti-NSDHL and anti-β-tubulin primary antibodies at 1:2000 and 1:4000 dilution, respectively.
Confocal microscopy
Floating sections (40 μm) were cut from frozen brains that had been fixed with 4% PFA and infused with 30% sucrose. Sections were stained with anti-GFAP primary antibody and DyLight 488-conjugated donkey anti-mouse IgG. Z-stack fluorescence images using 2 μm optical sections were acquired on a Zeiss LSM 510 Meta confocal microscope using LSM software.
GCP culture
Granule cell precursors were isolated from individual cerebella at P4 and cultured as previously described (37) except that Percoll gradient separation and preplating were omitted. Briefly, freshly dissected cerebella were dissociated using the Papain Dissociation System Kit according to the manufacturer's instructions (Worthington Biochemical, Lakewood, NJ). GCPs were plated at a density of 2.5 × 105 cells/cm2 on poly-D-lysine coated 6-well or 12-well tissue culture plates (Corning, Corning, NY) in 3 or 1 ml respectively of serum-free culture medium (SFM), consisting of Neurobasal-A medium (Life Technologies, Carlsbad, CA) supplemented with GlutaMAX I (Life Technologies), 250 μm KCl, 50 I.U./ml penicillin, 50 μg/ml streptomycin and B-27 supplement (Life Technologies) and cultured at 37°C, 5% CO2 for 48 h. Where indicated, SFM was supplemented with 15 μg/ml cholesterol (Sigma) from a 15 mg/ml stock solution in 95% ethanol, 1 μg/ml recombinant mouse sonic hedgehog N-terminus (SHH-N) (R&D Systems, Minneapolis, MN) from a 100 μg/ml stock solution in 1% BSA in PBS, 5 μg/ml human LDL (Biomedical Technologies, Stoughton, Ma) from a 5 mg/ml stock solution, 2 μm ketoconazole (Santa Cruz Biotechnology, Santa Cruz, CA) from a 1 mm stock in DMSO, and/or 100 nm smoothened agonist, SAG (EMD Millipore, Billerica, MA) from a 100 mm stock in DMSO. Equal volumes of vehicle alone were added to all untreated wells.
For BrdU labeling of GCPs in vitro, BrdU was added to each well to a final concentration of 10 μg/ml during the last 4 h of incubation. The cells were fixed with 2% PFA and BrdU immunostaining was performed as described above, except that BrdU primary antibody incubation was followed by an 8 h incubation with DyLight 549-conjugated donkey anti-rat IgG secondary antibody. Coverslips were mounted on slides with ProLong Gold antifade reagent with DAPI (Life Technologies). Quantitation of BrdU labeling was performed by manually counting the number of BrdU-positive cells from a total of at least 1500 total cells from five random fields on each coverslip. Results represent the combined data from three independent experiments.
In preliminary experiments using cultured GCPs, we occasionally noted highly variable or discrepant results. In subsequent experiments, we saved a small portion of the freshly isolated GCPs and performed RT-PCR on isolated RNA using primers that distinguished between the Nsdhlflx5 allele, that produces WT message, and the recombined NsdhlΔ5 allele. In approximately 5% of NsdhlΔ5/Y samples analyzed (6/124 samples examined), the Nsdhlflx5 allele constituted ≥5% of the PCR product, indicating incomplete cre-mediated excision. The amount of WT mRNA present correlated with the discrepant experimental results. For data presented here, any mutant samples in which WT product was detected were excluded from the analyses. This included in vitro proliferation experiments, sterol analyses, qPCR studies, and studies examining the effects of ketoconazole, SAG, or T-MAS on cultured GCPs.
For treatment of cell cultures, T-MAS (4,4-dimethyl-cholest-8(9),24-dien-3ß-ol) (Avanti Polar Lipids, Alabaster, AL) was complexed with low fatty acid bovine serum albumin (CellMaxx bovine albumin, catalog #199899, MP Biochemicals, Solon, OH) as previously described (82). Pure T-MAS was stored in dessicated aliquots under nitrogen gas in glass vials at −20°C. Lipid-protein complexes were freshly prepared for each experiment by dissolving 100 μg of T-MAS in 50 μl ethanol, adding 50 μl of PBS (pH 7.4) and finally adding 100 μl of BSA solution (66 mg/ml in PBS). The mixture was vortexed and placed on a rotating platform overnight at 4°C. This stock solution was added to cell culture medium to a final T-MAS concentration of either 2.5 or 10 μg/ml.
Sterol analysis
GCPs were isolated from cerebella at P3, P5 and P7 by papain dissociation as described above. The cells were rinsed by suspending and pelleting two times in ice-cold PBS. Cell pellets were frozen and stored at −80°C. The yield of GCPs from each cerebellum was 2–5 × 106 cells, depending on the age of the animals. For analysis of cultured cells, GCPs were isolated from P4 cerebella and cultured in SFM with the indicated treatments for 48 h as described above. The cells were rinsed two times with ice-cold PBS and frozen at −80°C in the tissue culture plate. Cell pellets or frozen cells in 6-well plates were solubilized in 1.0 ml of 0.05% SDS. An aliquot of the lysate was used for protein measurement (BCA™ Protein Assay Kit, Pierce, Rockford, IL) and another for measurement of sterols. Sterols were extracted from the lysate by Folch extraction with chloroform/methanol (2:1), after internal standard [epicoprostanol, EPIC, Sigma C-2882 or cholestane (Steraloids, Newport, RI, C3300-000)] was added. The organic phase was removed and evaporated under nitrogen at 40°C. Sterols were then saponified by the addition of ethanol/ KOH and incubation at 37°C for I hour. Sterols were extracted twice with hexane. The combined hexane extracts were evaporated to dryness and derivatized with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA, Sigma 33027) at 80°C for 30 min. Concentrations of the trimethylsilyl ethers of sterols were measured according to standard methods developed by Kelley (83) using gas chromatography with a ZB1701 column (Phenomenex, Torrance, CA: 30 m, 0.25 mm ID, 0.25 mm film thickness) and FID. In brief, cholesterol (Sigma C-8667) or desmosterol (Steraloids C3151-000) and EPIC or cholestane internal standards were used to generate calibration curves for cholesterol or desmosterol quantification, respectively. Replicate calibration curves demonstrated satisfactory linearity over the range 0.2–10 mg and 20–800 ng for cholesterol using 1 μg cholestane internal standard and 20–800 ng for cholesterol and desmosterol using 100 ng EPIC internal standard (with R2 values > 0.995). Concentrations of other sterols were calculated by comparison of their peak area to that of internal standard assuming a 1:1 weight ratio via GC-FID sterol detection (83). Using this technique within-run precision for replicate methyl sterol analysis (n = 3) in representative P3 and P5 samples was determined to be <21% relative standard deviation (RSD). Sterol peaks were identified by comparison against authentic standards for desmosterol, lanosterol, and the methylsterol T-MAS. Derivitization of lanosterol and T-MAS were not complete under the conditions used (see Supplemental Material, Fig. S5C and data not shown). Quantitation of T-MAS in Figure 9 was based on GC-FID peak area detected for derivatized plus underivatized T-MAS. For other methylsterols, peak identification was by re-analysis of samples using GC-mass spectrometry and mass spectrum matching against National Institutes of Standards and Technology (NIST) data in the Wiley 7th edition library [with a quality score of 90 obtained for 4α-methylcholest-7(8)-en-3β-ol and 92 for 4α-methylcholesta-8(9),24-dien-3β-ol on isolated GCPs from P7 (see Supplementary Material, Fig. S5D)].
qPCR
Total RNA was extracted from 2 × 106 cultured GCPs or whole cerebella using Trizol reagent (Life Technologies), followed by clean-up on RNeasy Mini Columns (Qiagen, Valencia, CA). RNA quality was assessed using a Bioanalyzer (Agilent, Santa Clara, CA) and all samples had a RIN >8. cDNA was synthesized from 1 μg of RNA for each sample using the SuperScript III First-strand Synthesis kit (Life Technologies) according to the manufacturer's instructions. The volume of each cDNA sample was brought up to 100 μl with water and stored in aliquots at −80°C. Real-time PCR reactions were performed on 2 μl of cDNA using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) with 0.4 μM of each primer. Primer sequences are listed in Supplementary Material, Table S1. Amplification was carried out in MicroAmp optical 96-well plates (Applied Biosystems) on a 7500 Real-time PCR System instrument (Applied Biosystems). The amplification program was 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min, followed by a melt curve stage. The relative quantity of cDNA for each gene was calculated using the ΔΔCT method (84), with Gapdh used as the reference gene in all samples. Primers for Sc4mol and Hmgcr were previously validated for qPCR (85). The amplification efficiency of additional primer pairs used in this study was measured by generating a standard curve from a dilution series of cDNA derived from WT GCPs. The R2 values of the standard curves for all primer pairs were ≥0.992. The amplification efficiency (E = 10(−1/slope)) ranged from 95 to 98% for all primer pairs except Ccnd1 (92%) and Ccnd2 (87%). The ΔΔCt values were calculated using 95% as the efficiency. Melt curve analysis of products from each primer pair generated a single peak, indicating a unique amplicon for each. The results represent the combined data from at least three independent RNA samples, each assayed in triplicate.
Statistical analysis
For time course experiments in Figures 2–4, the data were analyzed by two way ANOVA using the R statistical environment (http://www.R-project.org), and P-values for pairwise differences in least square means were calculated using the Ismeans package (http://CRAN.R-project.org/package-Ismeans). For all other experiments, statistical differences between samples were determined by pair-wise comparison of the means of WT versus mutant sample data using two-tailed Student's t-test (SPSS version 19), with P < 0.05 considered significant. All bar graph values are the mean ± SEM.
Supplementary Material
Funding
This work was supported, in part, by the National Institutes of Health (HD038572 to G.E.H.); and by funds from The Research Institute at Nationwide Children’s Hospital. The Embryonic Stem Cell Core at Nationwide Children’s Hospital was supported, in part, by the National Institutes of Health (CA16058, CA097189).
Supplementary Material
Acknowledgements
The authors wish to thank Christopher Pierson, MD, PhD, Nationwide Children's Hospital and John Oberdick, PhD, The Ohio State University, for helpful discussions concerning the cerebellar pathology; Wesley Banks, III and Isain Zapata, PhD, Nationwide Children's Research Institute, for assistance with statistical data analysis; and Jean-Baptist Roullet, PhD, Oregon Health and Science University, and Igor Astsaturov, MD, PhD, Fox Chase Cancer Center, for useful suggestions regarding experimental procedures.
Conflict of Interest statement. None declared.
References
- 1.Saher G., Quintes S., Nave K.A. (2011) Cholesterol: a novel regulatory role in myelin formation. Neuroscientist, 17, 79–93. [DOI] [PubMed] [Google Scholar]
- 2.Herman G.E. (2003) Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum. Mol. Genet., 12 Spec No 1, R75–R88. [DOI] [PubMed] [Google Scholar]
- 3.Mann R.K., Beachy P.A. (2004) Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem., 73, 891–923. [DOI] [PubMed] [Google Scholar]
- 4.Tierney E., Bukelis I., Thompson R.E., Ahmed K., Aneja A., Kratz L., Kelley R.I. (2006) Abnormalities of cholesterol metabolism in autism spectrum disorders. Am. J. Med Genet. B Neuropsychiatr. Genet., 141, 666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjorkhem I., Leoni V., Meaney S. (2010) Genetic connections between neurological disorders and cholesterol metabolism. J. Lipid Res., 51, 2489–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wollmer M.A. (2010) Cholesterol-related genes in Alzheimer's disease. Biochim. Biophys. Acta, 1801, 762–773. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein J.L., Brown M.S. (1990) Regulation of the mevalonate pathway. Nature, 343, 425–430. [DOI] [PubMed] [Google Scholar]
- 8.Gaylor J.L. (2002) Membrane-bound enzymes of cholesterol synthesis from lanosterol. Biochem. Biophys. Res. Commun., 292, 1139–1146. [DOI] [PubMed] [Google Scholar]
- 9.Porter F.D., Herman G.E. (2011) Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res., 52, 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLarren K.W., Severson T.M., du Souich C., Stockton D.W., Kratz L.E., Cunningham D., Hendson G., Morin R.D., Wu D., Paul J.E., et al. (2010) Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am. J. Hum. Genet., 87, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He M., Kratz L.E., Michel J.J., Vallejo A.N., Ferris L., Kelley R.I., Hoover J.J., Jukic D., Gibson K.M., Wolfe L.A., et al. (2011) Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J. Clin. Invest., 121, 976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida S., Wada Y. (2005) Transer of maternal cholesterol to embryo and fetus in pregnant mice. J. Lipid Res., 46, 2168–2174. [DOI] [PubMed] [Google Scholar]
- 13.Tint G.S., Yu H., Shang Q., Xu G., Patel S.B. (2006) The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J. Lipid Res., 47, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benarroch E.E. (2008) Brain cholesterol metabolism and neurologic disease. Neurology, 71, 1368–1373. [DOI] [PubMed] [Google Scholar]
- 15.Liu X.Y., Dangel A.W., Kelley R.I., Zhao W., Denny P., Botcherby M., Cattanach B., Peters J., Hunsicker P.R., Mallon A.M., et al. (1999) The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat. Genet., 22, 182–187. [DOI] [PubMed] [Google Scholar]
- 16.Konig A., Happle R., Bornholdt D., Engel H., Grzeschik K.-H. (2000) Mutations in the NSDHL gene, encoding a 3b-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am. J. Med. Genet., 90, 339–346. [PubMed] [Google Scholar]
- 17.Bornholdt D., Konig A., Happle R., Leveleki L., Bittar M., Danarti R., Vahlquist A., Tilgen W., Reinhold U., Poiares Baptista A., et al. (2005) Mutational spectrum of NSDHL in CHILD syndrome. J. Med. Genet., 42, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman G.E., Kratz L.E. (2012) Disorders of Sterol Synthesis: Beyond Smith-Lemli-Opitz Syndrome. Am. J. Med. Genet. C, 160C, 301–321. [DOI] [PubMed] [Google Scholar]
- 19.du Souich C., Chou A., Yin J., Oh T., Nelson T.N., Hurlburt J., Arbour L., Friedlander R., McGillivray B.C., Tyshchenko N., et al. (2009) Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus. Am. J. Med. Genet. A, 149A, 2469–2478. [DOI] [PubMed] [Google Scholar]
- 20.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A., et al. (2009) A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet., 41, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuo L., Theis M., Alvarez-Maya I., Brenner M., Willecke K., Messing A. (2001) hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis, 31, 85–94. [DOI] [PubMed] [Google Scholar]
- 22.Liu P., Jenkins N.A., Copeland N.G. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res., 13, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi S., Lewis P., Pevny L., McMahon A.P. (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev., 119(Suppl 1), S97–S101. [DOI] [PubMed] [Google Scholar]
- 24.Carletti B., Rossi F. (2008) Neurogenesis in the cerebellum. Neuroscientist, 14, 91–100. [DOI] [PubMed] [Google Scholar]
- 25.Chedotal A. (2010) Should I stay or should I go? Becoming a granule cell. Trends Neurosci., 33, 163–172. [DOI] [PubMed] [Google Scholar]
- 26.Chizhikov V.V., Davenport J., Zhang Q., Shih E.K., Cabello O.A., Fuchs J.L., Yoder B.K., Millen K.J. (2007) Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J. Neurosci., 27, 9780–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baptista C.A., Hatten M.E., Blazeski R., Mason C.A. (1994) Cell-cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron, 12, 243–260. [DOI] [PubMed] [Google Scholar]
- 28.Jansen M., Wang W., Greco D., Bellenchi G.C., di Porzio U., Brown A.J., Ikonen E. (2013) What dictates the accumulation of desmosterol in the developing brain? FASEB J., 27, 865–870. [DOI] [PubMed] [Google Scholar]
- 29.Dahmane N., Ruiz i Altaba A. (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development, 126, 3089–3100. [DOI] [PubMed] [Google Scholar]
- 30.Lewis P.M., Gritli-Linde A., Smeyne R., Kottmann A., McMahon A.P. (2004) Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev. Biol., 270, 393–410. [DOI] [PubMed] [Google Scholar]
- 31.Borghesani P.R., Peyrin J.M., Klein R., Rubin J., Carter A.R., Schwartz P.M., Luster A., Corfas G., Segal R.A. (2002) BDNF stimulates migration of cerebellar granule cells. Development, 129, 1435–1442. [DOI] [PubMed] [Google Scholar]
- 32.Corrales J.D., Rocco G.L., Blaess S., Guo Q., Joyner A.L. (2004) Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development, 131, 5581–5590. [DOI] [PubMed] [Google Scholar]
- 33.Hagihara K., Zhang E.E., Ke Y.H., Liu G., Liu J.J., Rao Y., Feng G.S. (2009) Shp2 acts downstream of SDF-1alpha/CXCR4 in guiding granule cell migration during cerebellar development. Dev. Biol., 334, 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blank M.C., Grinberg I., Aryee E., Laliberte C., Chizhikov V.V., Henkelman R.M., Millen K.J. (2011) Multiple developmental programs are altered by loss of Zic1 and Zic4 to cause Dandy-Walker malformation cerebellar pathogenesis. Development, 138, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakurai K., Toyoshima M., Ueda H., Matsubara K., Takeda Y., Karagogeos D., Shimoda Y., Watanabe K. (2009) Contribution of the neural cell recognition molecule NB-3 to synapse formation between parallel fibers and Purkinje cells in mouse. Dev. Neurobiol., 69, 811–824. [DOI] [PubMed] [Google Scholar]
- 36.Takacs J., Zaninetti R., Vig J., Vastagh C., Hamori J. (2008) Postnatal expression of Doublecortin (Dcx) in the developing cerebellar cortex of mouse. Acta Biol. Hung., 59, 147–161. [DOI] [PubMed] [Google Scholar]
- 37.Lee H.Y., Greene L.A., Mason C.A., Manzini M.C. (2009) Isolation and culture of post-natal mouse cerebellar granule neuron progenitor cells and neurons. J. Vis. Exp., 23, e990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer D., Minichiello L. (2010) Cell culture of primary cerebellar granule cells. Methods Mol. Biol., 633, 233–239. [DOI] [PubMed] [Google Scholar]
- 39.Kenney A.M., Rowitch D.H. (2000) Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell Biol., 20, 9055–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kenney A.M., Cole M.D., Rowitch D.H. (2003) Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development, 130, 15–28. [DOI] [PubMed] [Google Scholar]
- 41.Knoepfler P.S., Kenney A.M. (2006) Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle, 5, 47–52. [DOI] [PubMed] [Google Scholar]
- 42.Hui C.C., Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell. Dev. Biol., 27, 513–537. [DOI] [PubMed] [Google Scholar]
- 43.Goetz S.C., Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corrales J.D., Blaess S., Mahoney E.M., Joyner A.L. (2006) The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development, 133, 1811–1821. [DOI] [PubMed] [Google Scholar]
- 45.Galvin K.E., Ye H., Wetmore C. (2007) Differential gene induction by genetic and ligand-mediated activation of the Sonic hedgehog pathway in neural stem cells. Dev. Biol., 308, 331–342. [DOI] [PubMed] [Google Scholar]
- 46.Hatton B.A., Knoepfler P.S., Kenney A.M., Rowitch D.H., de Alboran I.M., Olson J.M., Eisenman R.N. (2006) N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res., 66, 8655–8661. [DOI] [PubMed] [Google Scholar]
- 47.Kenney A.M., Widlund H.R., Rowitch D.H. (2004) Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development, 131, 217–228. [DOI] [PubMed] [Google Scholar]
- 48.Sjostrom S.K., Finn G., Hahn W.C., Rowitch D.H., Kenney A.M. (2005) The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev. Cell, 9, 327–338. [DOI] [PubMed] [Google Scholar]
- 49.Ruiz i Altaba A., Palma V., Dahmane N. (2002) Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci., 3, 24–33. [DOI] [PubMed] [Google Scholar]
- 50.Cunningham D., Spychala K., McLarren K.W., Garza L.A., Boerkoel C.F., Herman G.E. (2009) Developmental expression pattern of the cholesterogenic enzyme NSDHL and negative selection of NSDHL-deficient cells in the heterozygous Bpa1H/+ mouse. Molec. Genet. Metab., 98, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quan G., Xie C., Dietschy J.M., Turley S.D. (2003) Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res. Dev. Brain Res., 146, 87–98. [DOI] [PubMed] [Google Scholar]
- 52.Bidet M., Joubert O., Lacombe B., Ciantar M., Nehme R., Mollat P., Bretillon L., Faure H., Bittman R., Ruat M., et al. (2011) The hedgehog receptor patched is involved in cholesterol transport. PLoS ONE, 6, e23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nachtergaele S., Mydock L.K., Krishnan K., Rammohan J., Schlesinger P.H., Covey D.F., Rohatgi R. (2012) Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol., 8, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frank-Kamenetsky M., Zhang X.M., Bottega S., Guicherit O., Wichterle H., Dudek H., Bumcrot D., Wang F.Y., Jones S., Shulok J., et al. (2002) Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J. Biol., 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byskov A.G., Andersen C.Y., Leonardsen L. (2002) Role of meiosis activating sterols, MAS, in induced oocyte maturation. Mol. Cell Endocrinol., 187, 189–196. [DOI] [PubMed] [Google Scholar]
- 56.Acimovic J., Rozman D. (2013) Steroidal triterpenes of cholesterol synthesis. Molecules, 18, 4002–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saher G., Brugger B., Lappe-Siefke C., Mobius W., Tozawa R., Wehr M.C., Wieland F., Ishibashi S., Nave K.A. (2005) High cholesterol level is essential for myelin membrane growth. Nat. Neurosci., 8, 468–475. [DOI] [PubMed] [Google Scholar]
- 58.Sudarov A., Joyner A.L. (2007) Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev., 2, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bloch K.E. (1983) Sterol structure and membrane function. CRC Crit. Rev. Biochem., 14, 47–92. [DOI] [PubMed] [Google Scholar]
- 60.Xu X., Bittman R., Duportail G., Heissler D., Vilcheze C., London E. (2001) Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts). Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide. J. Biol. Chem., 276, 33540–33546. [DOI] [PubMed] [Google Scholar]
- 61.Wang J., Megha, London E. (2004) Relationship between sterol/steroid structure and participation in ordered lipid domains (lipid rafts): implications for lipid raft structure and function. Biochemistry, 43, 1010–1018. [DOI] [PubMed] [Google Scholar]
- 62.Cooper M.K., Wassif C.A., Krakowiak P.A., Taipale J., Gong R., Kelley R.I., Porter F.D., Beachy P.A. (2003) A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet., 33, 508–513. [DOI] [PubMed] [Google Scholar]
- 63.Porter J.A., Young K.E., Beachy P.A. (1996) Cholesterol modification of hedgehog signaling proteins in animal development. Science, 274, 255–259. [DOI] [PubMed] [Google Scholar]
- 64.Li Y., Zhang H., Litingtung Y., Chiang C. (2006) Cholesterol modification restricts the spread of Shh gradient in the limb bud. Proc. Natl. Acad. Sci. USA, 103, 6548–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang F., Herman G.E. (2006) Analysis of Nsdhl-deficient embryos reveals a role for Hedgehog signaling in early placental development. Hum. Mol. Genet., 15, 3293–3305. [DOI] [PubMed] [Google Scholar]
- 66.Stottmann R.W., Turbe-Doan A., Tran P., Kratz L.E., Moran J.L., Kelley R.I., Beier D.R. (2011) Cholesterol metabolism is required for intracellular hedgehog signal transduction in vivo. PLoS Genet., 7, e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dwyer J.R., Sever N., Carlson M., Nelson S.F., Beachy P.A., Parhami F. (2007) Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem., 282, 8959–8968. [DOI] [PubMed] [Google Scholar]
- 68.Rohatgi R., Milenkovic L., Scott M.P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science, 317, 372–376. [DOI] [PubMed] [Google Scholar]
- 69.Ahlgren S.C., Bronner-Fraser M. (1999) Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr. Biol., 9, 1304–1314. [DOI] [PubMed] [Google Scholar]
- 70.Cai C., Thorne J., Grabel L. (2008) Hedgehog serves as a mitogen and survival factor during embryonic stem cell neurogenesis. Stem Cells, 26, 1097–1108. [DOI] [PubMed] [Google Scholar]
- 71.Sukhanova A., Gorin A., Serebriiskii I.G., Gabitova L., Zheng H., Restifo D., Egleston B.L., Cunningham D., Bagnyukova T., Liu H., et al. (2013) Targeting C4-demethylating genes in the cholesterol pathway sensitizes cancer cells to EGF receptor inhibitors via increased EGF receptor degradation. Cancer Discov., 3, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suzuki S., Numakawa T., Shimazu K., Koshimizu H., Hara T., Hatanaka H., Mei L., Lu B., Kojima M. (2004) BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J. Cell Biol., 167, 1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pereira D.B., Chao M.V. (2007) The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J. Neurosci., 27, 4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou P., Porcionatto M., Pilapil M., Chen Y., Choi Y., Tolias K.F., Bikoff J.B., Hong E.J., Greenberg M.E., Segal R.A. (2007) Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron, 55, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chizhikov V., Millen K.J. (2003) Development and malformations of the cerebellum in mice. Mol. Genet. Metab., 80, 54–65. [DOI] [PubMed] [Google Scholar]
- 76.Guirland C., Zheng J.Q. (2007) Membrane lipid rafts and their role in axon guidance. Adv. Exp. Med. Biol., 621, 144–155. [DOI] [PubMed] [Google Scholar]
- 77.Wiencken-Barger A.E., Djukic B., Casper K.B., McCarthy K.D. (2007) A role for Connexin43 during neurodevelopment. Glia, 55, 675–686. [DOI] [PubMed] [Google Scholar]
- 78.Defamie N., Mesnil M. (2012) The modulation of gap-junctional intercellular communication by lipid rafts. Biochim. Biophys. Acta, 1818, 1866–1869. [DOI] [PubMed] [Google Scholar]
- 79.Osoegawa K., Tateno M., Woon P.Y., Frengen E., Mammoser A.G., Catanese J.J., Hayashizaki Y., de Jong P.J. (2000) Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res., 10, 116–128. [PMC free article] [PubMed] [Google Scholar]
- 80.Raymond C.S., Soriano P. (2007) High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE, 2, e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jukkola P.I., Rogers J.T., Kaspar B.K., Weeber E.J., Nishijima I. (2011) Secretin deficiency causes impairment in survival of neural progenitor cells in mice. Hum. Mol. Genet., 20, 1000–1007. [DOI] [PubMed] [Google Scholar]
- 82.Charbonneau D.M., Tajmir-Riahi H.A. (2010) Study on the interaction of cationic lipids with bovine serum albumin. J. Phys. Chem. B, 114, 1148–1155. [DOI] [PubMed] [Google Scholar]
- 83.Kelley R.I. (1995) Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin. Chim. Acta, 236, 45–58. [DOI] [PubMed] [Google Scholar]
- 84.Livak K.J., Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 85.Cunningham D., Swartzlander D., Liyanarachchi S., Davuluri R.V., Herman G.E. (2005) Changes in gene expression associated with loss of function of the NSDHL sterol dehydrogenase in mouse embryonic fibroblasts. J. Lipid Res., 46, 1150–1162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.