

Abstract
Neurons in the brain produce lamin C but almost no lamin A, a consequence of the removal of prelamin A transcripts by miR-9, a brain-specific microRNA. We have proposed that miR-9-mediated regulation of prelamin A in the brain could explain the absence of primary neurological disease in Hutchinson-Gilford progeria syndrome, a genetic disease caused by the synthesis of an internally truncated form of farnesyl–prelamin A (progerin). This explanation makes sense, but it is not entirely satisfying because it is unclear whether progerin—even if were expressed in neurons—would be capable of eliciting neuropathology. To address that issue, we created a new Lmna knock-in allele, LmnaHG-C, which produces progerin transcripts lacking an miR-9 binding site. Mice harboring the LmnaHG-C allele produced progerin in neurons, but they had no pathology in the central nervous system. However, these mice invariably developed esophageal achalasia, and the enteric neurons and nerve fibers in gastrointestinal tract were markedly abnormal. The same disorder, achalasia, was observed in genetically modified mice that express full-length farnesyl–prelamin A in neurons (Zmpste24-deficient mice carrying two copies of a Lmna knock-in allele yielding full-length prelamin A transcripts lacking a miR-9 binding site). Our findings indicate that progerin and full-length farnesyl–prelamin A are toxic to neurons of the enteric nervous system.
Introduction
Hutchinson–Gilford progeria syndrome (HGPS) is caused by a point mutation in exon 11 of LMNA, the gene for lamin C and prelamin A (the precursor to mature lamin A) (1,2). The HGPS mutation leads to the synthesis of a mutant form of prelamin A, progerin, with an internal deletion of 50 amino acids (1,2). This deletion does not affect protein farnesylation but eliminates the ZMPSTE24-mediated endoproteolytic cleavage step that would ordinarily convert farnesyl–prelamin A to mature lamin A (3). The absence of the final cleavage step means that progerin retains its carboxyl-terminal farnesyl lipid anchor and cannot be converted to mature lamin A (3). The HGPS mutation has no effect on lamin C.
The synthesis of progerin is toxic to multiple tissues and is responsible for all of the disease phenotypes associated with HGPS (e.g. osteolytic lesions, slow growth, loss of subcutaneous adipose tissue, atherosclerosis in large arteries) (4–6). Several of these phenotypes resemble those that occur with aging, but some features of physiologic aging, notably senile dementia, are absent in children with HGPS; consequently, HGPS is often referred to as a ‘segmental’ aging syndrome (7–9). Several years ago, we developed Lmna knock-in mice (LmnaHG/+) (see Table 1) that produce progerin (10,11) and found that they develop osteolytic lesions and other phenotypes found in children with HGPS. The very same phenotypes occur in Zmpste24-deficient mice (14,15), which accumulate the farnesylated form of full-length prelamin A. The fact that Zmpste24−/− and LmnaHG/+ mice develop similar phenotypes suggests that the toxic effects of progerin and full-length farnesyl–prelamin A are similar (3,16). LmnaHG/+ and Zmpste24−/− mice have an abnormal gait (due to bone disease), but they lack obvious pathology in the central nervous system (CNS).
Table 1.
Properties of mutant alleles
| Mutant allele | Ref. | Mutation | Protein product | Expression in neurons of the CNS | Comments |
|---|---|---|---|---|---|
| LmnaHG-C | This paper | A Lmna knock-in mutation that yields progerin transcripts with lamin C's 3′ UTR. Lacks introns 10 and 11 and therefore does not produce lamin C transcripts. The progerin transcript lacks a miR-9 binding site and therefore is expressed in neurons. | Progerin | Yes | This allele is expressed at high levels in neurons of the CNS and in peripheral tissues. This allele invariably causes achalasia but results in only mild progeria-like disease phenotypes. |
| LmnaHG | (10,11) | A Lmna knock-in mutation that yields progerin transcripts containing prelamin A's 3′ UTR. Lacks introns 10 and 11 and therefore does not produce lamin C transcripts. Progerin transcripts from this allele are widely expressed in peripheral tissues. However, this transcript contains a miR-9 binding site and is not expressed in neurons. | Progerin | No | Expressed at high levels in peripheral tissues but in negligible amounts in neurons of the brain. This allele invariably causes severe progeria-like disease phenotypes. |
| LmnaPLAO-C | (12) (in the initial paper, this allele was called LmnaPLAO-UTR) | A Lmna knock-in mutation that yields full-length prelamin A transcripts containing lamin C's 3′ UTR. Lacks introns 10 and 11 and therefore does not produce lamin C transcripts. The mutant prelamin A transcript lacks a miR-9 binding site and is therefore expressed in neurons. | Wild-type prelamin A | Yes | Expressed at high levels in peripheral tissues and in the brain. In the presence of ZMPSTE24, the prelamin A produced by this allele is converted to mature lamin A. Mice homozygous for this allele appear normal. |
| Lmna5NT | (12) | A Lmna knock-in mutation that yields full-length prelamin A transcripts containing a 5-bp mutation in prelamin A's 3′ UTR. Lacks introns 10 and 11 and therefore does not produce lamin C transcripts. The mutant prelamin A transcript lacks a miR-9 binding site and is therefore expressed in neurons. | Wild-type prelamin A | Yes | Expressed at high levels in peripheral tissues and in the brain. In the presence of ZMPSTE24, the prelamin A produced by this allele is converted to mature lamin A. Mice homozygous for this allele appear normal. |
| LmnaPLAO | (13) | A Lmna knock-in mutation that yields full-length wild-type prelamin A transcripts. Lacks introns 10 and 11 and therefore does not produce lamin C transcripts. | Wild-type prelamin A | No | Expressed at high levels in peripheral tissues but is expressed in negligible amounts in the brain. In the presence of ZMPSTE24, the prelamin A produced by this allele is converted to mature lamin A. Mice homozygous for this allele appear normal. |
| Zmpste24− | (14,15) | Deletion of exon 8 of Zmpste24 | Zmpste24 expression is eliminated. | No | ZMPSTE24 is a zinc metalloprotease that converts farnesyl–prelamin A to mature lamin A. Homozygosity for this knockout allele eliminates the production of mature lamin A and results in an accumulation of farnesyl–prelamin A in cells and tissues. Zmpste24−/− mice manifest severe progeria-like disease phenotypes and death by 6–8 months of age. These phenotypes are very similar to those in LmnaHG/+ mice. |
We hypothesized that the absence of neuropathology in children with HGPS (and Zmpste24−/− and LmnaHG/+ mice) could relate to low levels of prelamin A expression in the brain. We quickly found support for this hypothesis. Most cell types and tissues produce roughly equal amounts of lamin A and lamin C, but neurons in the brain produce large amounts of lamin C but little prelamin A/lamin A (17). Indeed, the only cells in the brain that produce significant amounts of lamin A are meningeal cells and endothelial cells (17). Because lamin C and prelamin A are splice isoforms of the same gene, we initially suspected that the preferential synthesis of lamin C in neurons would be caused by alternative splicing, but this was not the case. Prelamin A transcripts in the brain are eliminated by a microRNA (miR-9) (17). The 3′ UTR of prelamin A's transcript contains a single miR-9-binding site; lamin C's transcript does not contain a miR-9-binding site.
MiR-9 is expressed at high levels in the brain and regulates neurogenesis by influencing the expression of multiple transcription factors (18). When miR-9 expression is eliminated, the pathology in the CNS is severe. MiR-9 has many targets and has potent effects on cells; transfecting fibroblasts with miR-9 transforms the cells into neurons (19). The ‘physiologic rationale’ for regulating prelamin A expression in the brain with miR-9 is not clear. Of note, microRNA expression surveys have revealed that miR-9-1, miR-9-2 and miR-9-3 are expressed at high levels in the brain, with only negligible expression in peripheral tissues (http://mirnamap.mbc.nctu.edu.tw/php/mirna_entry.php?acc=MI0000467).We confirmed these findings in a recent paper (12).
To study the in vivo relevance of miR-9 regulation of prelamin A, we recently created two Lmna knock-in alleles that produce prelamin A transcripts lacking an miR-9-binding site (12). The first of the two knock-in alleles, Lmna5NT, yielded prelamin A transcripts with a 5-bp mutation in prelamin A's miR-9-binding site; the second, LmnaPLAO-UTR (here renamed LmnaPLAO-C), encoded a prelamin A transcript in which prelamin A's 3′ UTR was replaced with lamin C's 3′ UTR (Table 1) (12). Mice homozygous for the Lmna5NT or LmnaPLAO-C alleles produced abundant prelamin A transcripts and lamin A protein in neurons, but neither mouse exhibited neuropathology (12).
Mir-9-mediated removal of prelamin A transcripts in neurons provides a plausible explanation for the absence of primary brain pathology in children with HGPS, but this explanation is not complete because it is uncertain whether progerin—even if were expressed in the brain—would elicit neurotoxicity. It is noteworthy that the liver and kidney of LmnaHG/+ mice produce abundant amounts of progerin yet are free of pathology (10,11).
The LmnaHG/+ mice described earlier (10,11) produce progerin transcripts containing the miR-9-binding site. For that reason, progerin expression in the brain of LmnaHG/+ mice was negligible (17), making it impossible to use these mice to assess the toxicity of progerin in neurons.
In the current study, we sought to determine whether the expression of progerin in neurons would elicit neuropathology, and if so, which neurons are most affected. To address that issue, we created a new Lmna knock-in allele, LmnaHG-C, which is identical to the original LmnaHG allele except that it produces progerin transcripts harboring lamin C's 3′ UTR (and lacking the miR-9 binding site) (Table 1). We anticipated that this new Lmna knock-in allele would yield robust expression of progerin in neurons.
Results
Lmna knock-in mice that express progerin in neurons
We used a sequence-replacement gene-targeting vector to create a new Lmna knock-in allele, LmnaHG-C, that yields progerin transcripts containing lamin C's 3′ UTR (Fig. 1). The LmnaHG-C allele is identical to the LmnaHG allele described earlier (10,11) except that it contains lamin C's 3′ UTR and therefore lacks a miR-9-binding site. The LmnaHG-C allele is also identical to the LmnaPLAO-UTR allele (here designated LmnaPLAO-C) (12) except that the LmnaHG-C allele encodes progerin rather than full-length prelamin A. All of the Lmna knock-in alleles lack introns 10 and 11 and therefore do not produce lamin C. The properties of the mutant alleles that we mention in this manuscript are described in Table 1.
Figure 1.

A new Lmna knock-in allele, LmnaHG-C, that yields progerin transcripts with lamin C's 3′ UTR. (A) Targeting vector to generate the LmnaHG-C allele. Intron 10, intron 11 and the last 150 bp of exon 11 were deleted; consequently, this allele produces only progerin (and no lamin C). Prelamin A's 3′ UTR was replaced with lamin C's 3′ UTR (LC UTR; grey), starting immediately after prelamin A's stop codon in exon 12. Primer locations for the 5′ (primers A and NeoR) and 3′ (primers NeoF and B) long-range PCR reactions as well as the genotyping PCR reactions (primers C and D, E and F) are indicated. (B) 5′ and 3′ long-range PCRs to detect the LmnaHG-C allele. The 5′ and 3′ PCRs yield fragments of 4.3 and 6.2 kb, respectively. In the 5′ PCR, a 5.2-kb fragment was amplified from the LmnaHG allele. No product was amplified from wild-type (+/+) DNA. NTC, no template control. (C) A PCR with primers C and D was used to confirm the targeting event and the introduction of lamin C's 3′ UTR. The length of the PCR product in the wild-type allele was 1488 bp versus 249 bp in the LmnaHG-C allele. (D) PCR with primers E (specific to progerin) and F (specific to lamin C's 3′ UTR) was used to confirm the introduction of lamin C's 3′ UTR (and the deletion of the last 150 bp of exon 11); the length of the product in the LmnaHG-C allele was 89 bp.
The LmnaPLAO-C allele yielded high levels of prelamin A transcripts and mature lamin A protein in both peripheral tissues and in the brain (reflecting an absence of miR-9-mediated regulation of prelamin A expression) (12). The LmnaHG-C allele shared these properties; progerin was expressed in all tissues of LmnaHG-C/+ mice, including in the brain (Fig. 2A). The amount of progerin in the cerebral cortex was more than 10-fold greater in LmnaHG-C/+ mice than in LmnaHG/+ mice (where miR-9 regulation of progerin is intact) (Fig. 2B and D). In contrast, the expression of progerin was robust in the lung of both LmnaHG-C/+ and LmnaHG/+ mice (Fig. 2B and D). The levels of progerin in LmnaHG-C/+ and LmnaHG/+ fibroblasts were similar (Fig. 2C and D). We also observed similar percentages of misshapen nuclei in LmnaHG-C/+ and LmnaHG/+ fibroblasts—and in LmnaHG/HG and LmnaHG-C/HG-C fibroblasts (Fig. 2E).
Figure 2.

Western blots, using a polyclonal antibody against lamin A/C, to assess the expression of progerin in cells and tissues. (A) Western blot analysis of lamin A/C expression in several tissues (including several regions of the brain) in a LmnaHG-C/+ mouse and a littermate wild-type (Lmna+/+) mouse. All samples are from 1-month-old mice. Actin was used as a loading control. (B) Western blot study of lamin A/C expression in the cerebral cortex and lung of LmnaHG/+ and LmnaHG-C/+ mice (n = 3/group). All samples were from 4-month-old mice. (C) Western blot analysis of lamin A/C expression in LmnaHG/+ fibroblasts and LmnaHG-C/+ fibroblasts. (D) Quantification, with an Li-Cor scanner, of progerin/actin ratios in the cerebral cortex and lung of LmnaHG/+ and LmnaHG-C/+ mice (B) and in the cultured fibroblasts (from a total of 4–5 fibroblast lines/genotype) (C). The progerin/actin ratios in the lung of LmnaHG/+ and LmnaHG-C/+ mice were 1.31 ± 0.02 and 1.08 ± 0.03, respectively. The ratios in the cerebral cortex were ∼20-fold different (0.02 ± 0.01 in LmnaHG/+ mice versus 0.62 ± 0.01 in LmnaHG-C/+ mice; P < 0.0001). (E) Percentages of misshapen cell nuclei in Lmna+/+, LmnaHG-C/+, LmnaHG/+, LmnaHG-C/HG-C and LmnaHG/HG fibroblasts (four cell lines/genotype; >300 cells examined/cell line). NS, not significant.
Lamin A expression in the cerebral cortex of wild-type mice was negligible (except in endothelial cells and meningeal cells) (Fig. 3A). In contrast, the expression of progerin in neurons of the cerebral cortex, cerebellum and hippocampus of 1-month-old LmnaHG-C/HG-C mice was robust (in these mice, the only ‘Lmna protein’ is progerin) (Fig. 3B–E).
Figure 3.
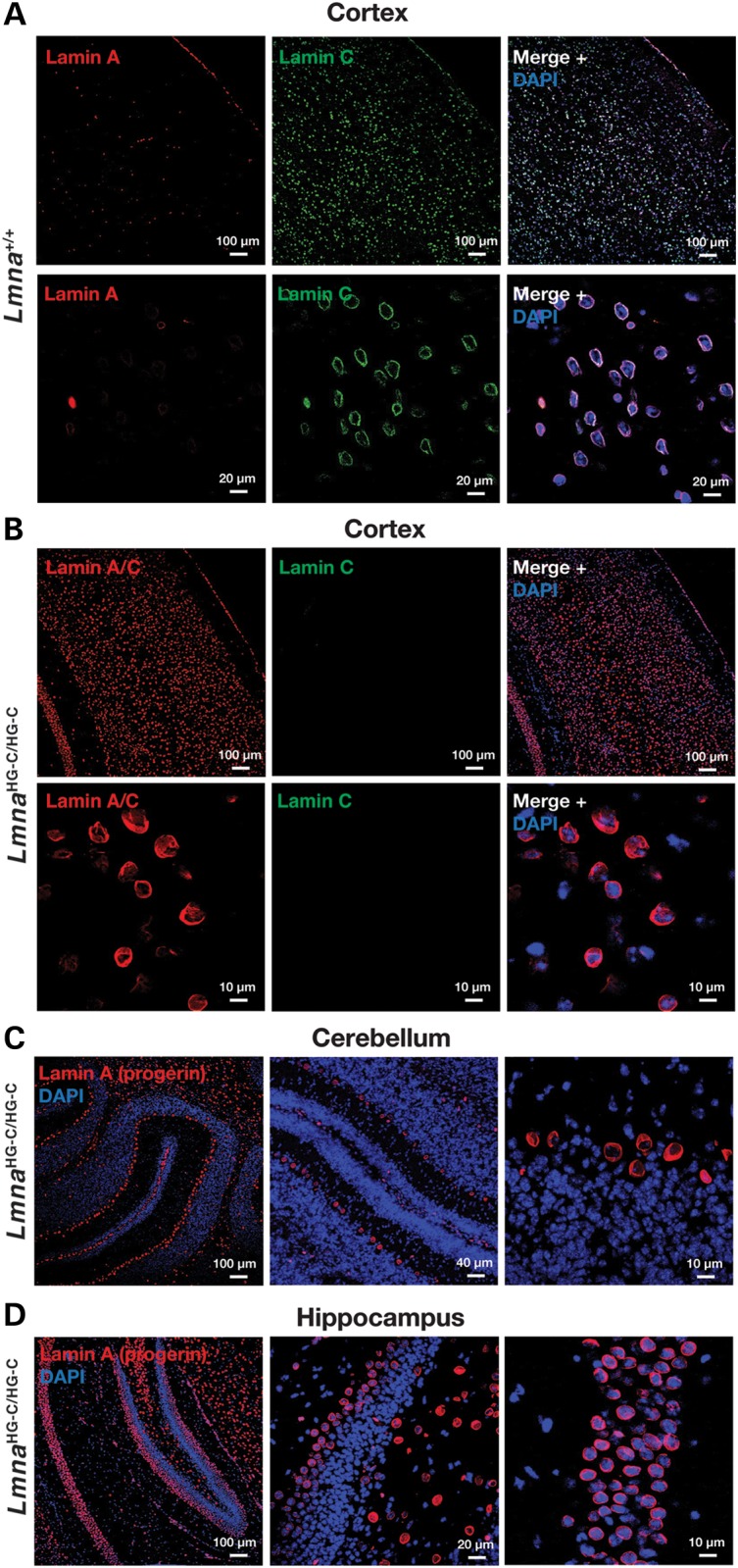
Confocal immunofluorescence microscopy of the brain of 1-month-old wild-type (Lmna+/+) and LmnaHG-C/HG-C mice. (A and B) Low- and high-magnification images of the cerebral cortex stained for lamin C (green) and either lamin A or lamins A/C (red). The scattered lamin A-positive cells in the cortex of the Lmna+/+ mouse are endothelial cells or meningeal cells (17). (C and D) Cerebellum (C) and hippocampus (D) of an LmnaHG-C/HG-C mouse stained with a lamin A-specific antibody (red).
LmnaHG-C/+ and wild-type mice were similar in size and appearance at birth. By 6 weeks of age, the average weights of male and female LmnaHG-C/+ mice (n = 20) were 22.1 ± 1.01 and 20.2 ± 1.06 g, respectively. During the next 4 months, several LmnaHG-C/+ mice gained an additional 2–3 g in body weight, but the majority of the mice (n > 15) lost ∼20% of their body weight during this time span. LmnaHG-C/+ mice died in a sporadic fashion, beginning at 3–4 months of age. One-half of the mice died by 5.5 months of age, but some of the mice lived 11–12 months. In some cases, we were required to euthanize LmnaHG-C/+ mice because of weight loss and inanition. Homozygous mice (LmnaHG-C/HG-C) were viable but were invariably quite small (∼10 g at 4 weeks of age); all homozygotes died between 3 and 8 weeks of age (n = 21).
We showed previously that the migration of cortical neurons during embryonic development is defective in Lmnb1−/− and Lmnb2−/− embryos, resulting in neuronal layering abnormalities in the cerebral cortex (20–23). No such pathology was present in 2-week-old LmnaHG-C/+ or LmnaHG-C/HG-C mice (Fig. 4A and B). The cellularity of the cortex in LmnaHG-C/+ mice appeared normal (Fig. 4B), and we did not find increased numbers of apoptotic neurons in the cerebral cortex or cerebellum of LmnaHG-C/HG-C mice (Fig. 4C). There was no obvious pathology in the brain of 5-month-old LmnaHG-C/+ mice (Supplementary Material, Fig. S1).
Figure 4.

Brains from 2-week-old wild-type (Lmna+/+), LmnaHG-C/+ and LmnaHG-C/HG-C mice. (A) Sagittal brain sections stained with H&E. CC, cerebral cortex; H, hippocampus; C, cerebellum. Images were recorded with a ×2 objective. (B) H&E-stained sections of the cerebral cortex. Images were recorded with a ×10 objective. (C) Caspase 3 (green) staining of the cerebral cortex of LmnaHG-C/HG-C mice (left panels) and cerebellum of LmnaHG-C/HG-C mice (right panels).
Achalasia in LmnaHG-C/+ and Zmpste24−/−LmnaPLAO-C/PLAO-C mice
LmnaHG-C/+ and LmnaHG-C/HG-C mice displayed obvious pathology in the gastrointestinal tract. By 4 months of age, all LmnaHG-C/+ mice (n > 24) had a dilated esophagus (Fig. 5) that often contained food. This phenotype was never observed in wild-type mice. The esophagus in LmnaHG-C/+ mice appeared yellow, reflecting markedly reduced amounts of muscle within the muscularis externa. In contrast, the esophagus in wild-type mice was pink-red, similar to the color of the intercostal muscles (Fig. 5). Stained sections of the esophagus in LmnaHG-C/+ mice revealed a cornified squamous epithelium and thinning of the inner circular and outer longitudinal layers of the muscularis externa (Fig. 6). At 3 months of age, the thickness of the muscularis externa of the mid-esophagus in LmnaHG-C/+ mice was 0.08 ± 0.04 mm (versus 0.19 ± 0.04 mm in wild-type mice); at 7 months, the thickness of the muscularis externa was 0.04 ± 0.03 mm (versus 0.18 ± 0.03 mm in wild-type mice). This pathology is similar to that of achalasia in humans. In achalasia, the failure of relaxation in the lower esophageal sphincter leads to dilatation of the esophagus, thinning of the muscularis externa and a cornified epithelial lining (24). In addition, LmnaHG-C/+ mice invariably had a dilated cecum and proximal colon (Fig. 7).
Figure 5.
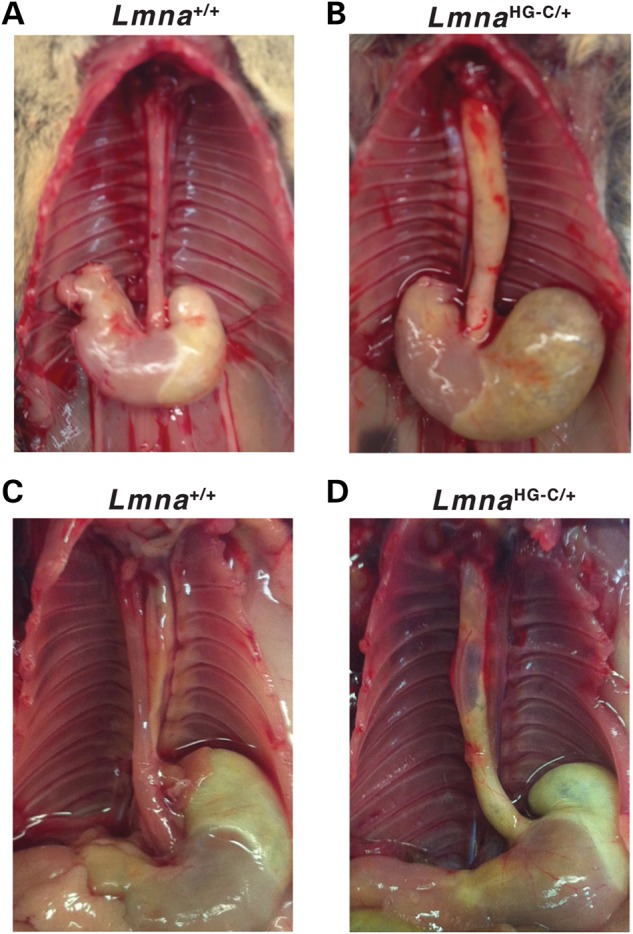
Megaesophagus in LmnaHG-C/+ mice. (A and B) A dilated, yellow esophagus in a 6-month-old LmnaHG-C/+ mouse. The esophagus of a wild-type littermate mouse (Lmna+/+) is pink-red, reflecting the presence of a muscularis externa near the surface of the esophagus. (C and D) A dilated, yellow esophagus in a 7-month-old LmnaHG-C/+ mouse. A segment of the mid-esophagus in that mouse lacked any muscularis externa and was so thin that it was nearly transparent. The esophagus of a wild-type littermate is shown for comparison.
Figure 6.

Stained sections of the mid-esophagus of 6-month-old wild-type mice (left panels) and LmnaHG-C/+ mice (right panels). The esophagus in the LmnaHG-C/+ mice contained a cornified epithelium, and the longitudinal and circular muscle layers of the muscularis externa were thin. (A) Trichrome-stained sections. (B) Hematoxylin and eosin-stained sections. The black brackets denote the epithelial layer; the yellow arrowheads mark the two layers of muscle in the muscularis externa.
Figure 7.
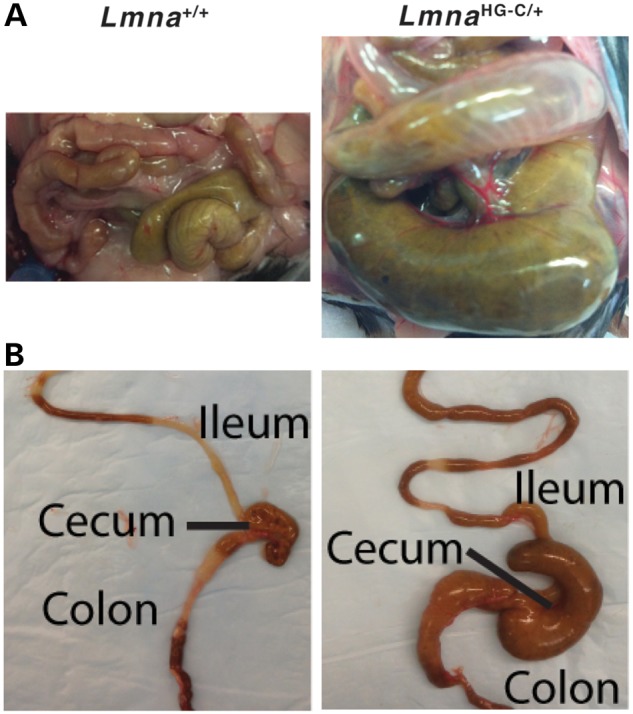
Dilated ileum, cecum and proximal colon in LmnaHG-C/+ mice. (A) Photographs, taken at the same magnification, of the intestines of a wild-type (Lmna+/+) mouse and a LmnaHG-C/+ mouse. (B) Photographs of the explanted intestinal tract from 4-month-old Lmna+/+ and LmnaHG-C/+ mice.
Finding intestinal pathology in LmnaHG-C/+ mice was unexpected, and for that reason we considered it essential to validate our findings with a related but genetically distinct mouse model. As noted earlier, we documented very similar disease phenotypes in LmnaHG/+ mice (which accumulate progerin) and Zmpste24−/− mice (which accumulate a farnesylated version of full-length prelamin A) (11,14,15,25). Given those findings, we suspected that Zmpste24−/− mice might develop the same gastrointestinal pathology observed in LmnaHG-C/+ mice if they were capable of producing prelamin A in neurons. To test this idea, we bred Zmpste24−/− mice harboring two copies of the LmnaPLAO-C allele (Zmpste24−/−LmnaPLAO-C/PLAO-C). As we had predicted, Zmpste24−/−LmnaPLAO-C/PLAO-C mice developed a markedly dilated, yellow esophagus (indistinguishable from the esophagus in LmnaHG-C/+ mice) (Fig. 8A and B). By microscopy, the pathology in Zmpste24−/−LmnaPLAO-C/PLAO-C mice was similar: the esophagus was dilated, lined with a cornified epithelium, and manifested thinning and fibrosis of the muscularis externa. Zmpste24−/−LmnaPLAO-C/PLAO-C mice also had a dilated cecum and proximal colon. There was no pathology in the CNS of adult Zmpste24−/−LmnaPLAO-C/PLAO-C mice (Supplementary Material, Fig. S2), despite the fact that the neurons in these mice accumulate farnesyl–prelamin A (12).
Figure 8.

Esophageal pathology in Zmpste24−/−LmnaPLAO-C/PLAO-C mice. (A and B) A markedly dilated esophagus in 4- (A) and 12-month-old (B) Zmpste24−/−LmnaPLAO-C/PLAO-C mice. The esophagus in both mice was mainly yellow in color, reflecting an extremely thin layer of muscle. (C) Normal-sized esophagus in a 2-month-old Zmpste24−/−LmnaPLAO/PLAO mouse; this esophagus was pink-red, reflecting the presence of a normal muscularis externa. Also shown are H&E-stained sections of the esophagus. Scale bar, 100 μm (A and B), and 50 μm (C). In sections of esophagus from Zmpste24−/−LmnaPLAO-C/PLAO-C mice, scattered myocytes and fibrosis were observed in the muscular layer, and the esophagus was so dilated that only a low percentage of the circumference could be photographed with a ×20 objective. The esophagus in the Zmpste24−/−LmnaPLAO/PLAO mouse was smaller and had a normal muscular layer with little fibrosis. In the H&E-stained sections, the black brackets denote the epithelial layer; the arrowheads in the section from the Zmpste24−/−LmnaPLAO/PLAO mouse denote the two muscle layers of the muscularis externa.
No intestinal pathology was present in Zmpste24−/− mice (where prelamin A expression in neurons is prevented by miR-9 regulation) or in LmnaPLAO-C/PLAO-C mice (n > 10) (where prelamin A in neurons is converted to mature lamin A) (12). Also, no intestinal pathology was observed in Zmpste24−/− mice carrying one or two copies of a Lmna knock-in allele, LmnaPLAO, which yields prelamin A transcripts containing prelamin A's miR-9 binding site (Fig. 8C) (13).
Both LmnaHG-C/+ and Zmpste24−/−LmnaPLAO-C/PLAO-C mice lacked the capacity to suppress neuronal expression of prelamin A; both produced a toxic version of prelamin A; and both had striking disease in the gastrointestinal tract. The parsimonious explanation for the intestinal pathology is that neuronal expression of progerin and farnesyl–prelamin A is toxic to enteric neurons. To explore this possibility, we examined whole mounts of the esophagus of wild-type and LmnaHG-C/+ mice stained with an antibody against the neuronal marker Protein gene product 9.5 (PGP9.5) (Fig. 9). In LmnaHG-C/+ mice, PGP9.5 staining revealed few enteric neurons and nerve fibers in the esophagus (or elsewhere in the gastrointestinal tract), whereas PGP9.5 staining in wild-type mice was robust. Consistent with these findings, we observed reduced levels of the vesicular acetylcholine transporter (a marker for cholinergic neurons) in the esophagus of LmnaHG-C/+ mice (Fig. 10). Interestingly, we observed reduced staining for c-Kit (a receptor tyrosine kinase that serves as a marker for interstitial cells of Cajal; ICC) in the gastrointestinal tract of LmnaHG-C/+ mice (Fig. 10). A similar reduction in c-Kit was observed in the stomach, small intestines, and colon (not shown).
Figure 9.

Abnormal enteric neurons and nerve fibers in the gastrointestinal tract of an LmnaHG-C/+ mouse. The neuronal marker Protein gene product 9.5 (PGP9.5; red) was used to visualize enteric neurons and nerve fibers in esophagus (A), stomach (B) and colon (C) of a 6-month-old wild-type (Lmna+/+) mouse and a littermate LmnaHG-C/+ mouse. In the LmnaHG-C/+ mouse, only occasional neurons and nerve fibers were detected in the esophagus. Scale bar in (C; right) applies to all panels.
Figure 10.

Enteric nerves and ICCs are disrupted in the lower esophageal sphincter of LmnaHG-C/+ mice. (A) Excitatory motor nerve fibers detected with antibodies against the vesicular acetylcholine transporter (vAChT, red) in a 5-month-old wild-type (Lmna+/+) mouse and a littermate LmnaHG-C/+ mouse. (B) Spindle-shaped intramuscular ICCs detected with an antibody against c-Kit (green). (C) Merged images. Scale bar in (C; right) applies to all panels.
Progerin is expressed in peripheral tissues of both LmnaHG-C/+ and LmnaHG/+ mice. However, the progeria-like disease phenotypes were more severe in LmnaHG/+ mice (11,26,27) than in LmnaHG-C/+ mice. For example, the body weights of adult LmnaHG/+ mice (11,26,27) were far lower than those of LmnaHG-C/+ mice in this study. Also, osteolytic lesions and rib fractures were never observed in LmnaHG-C/+ mice, whereas they were frequent in LmnaHG/+ mice (11). Finally, some of the LmnaHG-C/+ mice survived for 12 months (despite the intestinal pathology), whereas LmnaHG/+ mice invariably died by 6–8 months of age (11). Similarly, the progeria-like disease phenotypes were less severe in Zmpste24−/−LmnaPLAO-C/PLAO-C mice than in Zmpste24−/− LmnaPLAO/PLAO mice. Zmpste24−/−LmnaPLAO/PLAO mice were very small and died by ∼8–12 weeks of age (13), whereas some Zmpste24−/−LmnaPLAO-C/PLAO-C mice lived for >11 months.
Discussion
We reported that miR-9 regulates prelamin A expression in neurons, and we proposed that miR-9 regulation of prelamin A expression might explain the absence of primary neuropathology in HGPS (12,17,23,28). This explanation was plausible but incomplete, simply because it remained unclear whether progerin—even if expressed in neurons—would elicit neuropathology. In the current study, we addressed that issue by creating a new Lmna knock-in allele, LmnaHG-C, which yields progerin transcripts lacking the miR-9-binding site. We predicted that mice harboring the LmnaHG-C allele would express progerin in neurons. This prediction was upheld; LmnaHG-C/+ mice produced large amounts of progerin in the brain (>10-fold more than in LmnaHG/+ mice, where neuronal expression of progerin is suppressed by miR-9). LmnaHG-C/+ mice did not develop pathology in the CNS, but 100% of the mice developed an enlarged esophagus by 3–4 months of age. The esophageal pathology in LmnaHG-C/+ mice resembled that in humans with achalasia (29–31), an esophageal motility disorder caused by impaired relaxation of the lower esophageal sphincter. The esophagus in LmnaHG-C/+ mice had a cornified epithelial lining and severe thinning of the muscularis externa. Immunohistochemical studies also revealed reduced numbers of enteric neurons and associated nerve fibers. Similar pathology has been observed in humans with achalasia and in other genetic models of achalasia (30,32,33). In contrast to our findings with LmnaHG-C/+ mice, we never encountered a dilated esophagus in LmnaHG/+ mice (11), where the production of progerin in neurons is suppressed by miR-9 regulation (17).
In our study, progerin levels in the cerebral cortex were much higher in LmnaHG-C/+ mice than in LmnaHG/+ mice, reflecting the importance of miR-9 regulation of prelamin A expression in the brain (12,17). We did not observe large differences in progerin expression in other tissues (12), consistent with the fact that miR-9 expression levels are low in most peripheral tissues (http://mirnamap.mbc.nctu.edu.tw/php/mirna_entry.php?acc=MI0000467). We suspect that the high levels of progerin expression in neurons of LmnaHG-C/+ mice are responsible for the achalasia phenotype that we observed. However, we cannot exclude the possibility that miR-9 is expressed in specific subpopulations of cells within selected peripheral tissues. If that were the case, it conceivably could be relevant to the disease phenotypes that we observed.
Because the esophageal pathology in LmnaHG-C/+ mice was completely unexpected, we considered it essential to verify our findings with a related but genetically distinct mouse model. We reasoned that if the expression of progerin by neurons in LmnaHG-C/+ mice were responsible for the gastrointestinal tract pathology, then we should find the same pathology in mice that expressed full-length farnesyl–prelamin A in neurons. Indeed, this was the case; achalasia was invariably found in Zmpste24-deficient mice harboring two copies of the LmnaPLAO-C allele (12), which lacks a miR-9-binding site and therefore allows expression of farnesyl–prelamin A in neurons. Like LmnaHG-C/+ mice, Zmpste24−/−LmnaPLAO-C/PLAO-C mice had a dilated, yellow esophagus with a cornified epithelium and thinning of the muscularis externa. The achalasia in Zmpste24−/−LmnaPLAO-C/PLAO-C mice depended both on the capacity to produce prelamin A in neurons and on a mutation that leads to the accumulation of a farnesylated prelamin A. Achalasia was not observed in Zmpste24−/− or Zmpste24−/−LmnaPLAO/PLAO mice (13,15) (where miR-9 regulation prevents the production of farnesyl–prelamin A in neurons), nor was it observed in LmnaPLAO-C/PLAO-C mice (where the farnesyl–prelamin A in neurons is converted to mature lamin A). The fact that neuronal expression of both progerin and full-length farnesyl–prelamin A led to the same pathology, achalasia, is consistent with earlier studies showing that the toxicities of the two proteins are similar in peripheral tissues (11,15,25,34).
In our studies, whole-mount immunohistochemistry assays of the gastrointestinal tract of LmnaHG-C/+ mice revealed reduced numbers of enteric neurons and associated nerve fibers in the muscularis externa. Given that progerin is toxic for cells and tissues and given that the LmnaHG-C mutation was specifically designed to permit progerin expression in neurons, it seems quite likely that the production of progerin in neurons is responsible for the enteric nerve pathology that we encountered. In an earlier study, achalasia was observed in mice lacking Sprouty2 (35), a negative regulator of multiple receptor tyrosine kinases. In that case, whole-mount immunohistochemistry studies revealed hyperplasia of enteric neurons and disorganization of nerve fibers in the esophagus (35). Recent studies have suggested that interstitial cells of Cajal (ICCs) play a role in the relaxation of the esophagus (36,37). [ICCs are pacemaker cells that control the contraction of smooth muscle cells in the gastrointestinal tract and mediate enteric motor responses.] In our studies, we found evidence of disease in ICCs (reduced expression of c-Kit in the gastrointestinal tract in LmnaHG-C/+ mice). Reduced esophageal ICC staining has also been observed in biopsy material from humans undergoing surgery for achalasia (38,39).
In LmnaHG-C/+ and Zmpste24−/−LmnaPLAO-C/PLAO-C mice, we observed dilated intestines, cecum and colon in addition to the esophageal pathology. It is conceivable that changes in intestinal flora brought on by the achalasia contribute to the enlargement of the cecum/proximal colon in mice (40–42). However, our immunohistochemistry studies showed that the pathology in the enteric nervous system was present in several segments of the gastrointestinal tract. The majority of humans with achalasia do not have significant pathology in the colon, but there have been examples of cases in which megaesophagus and megacolon co-exist (29,43). In some cases, the coexistence of achalasia and megacolon has been familial (29).
In the current study, we showed that eliminating miR-9 regulation in the new progerin knock-in allele (LmnaHG-C) resulted in large amounts of progerin expression in the CNS; however, progerin expression in neurons of LmnaHG-C/+ mice did not lead to obvious CNS pathology. Similarly, we found no CNS pathology in Zmpste24−/−LmnaPLAO-C/PLAO-C mice. Consistent with these findings, we did not encounter pathology in forebrain-specific Zmpste24 knockout mice carrying two copies of LmnaPLAO-C allele; those mice produce farnesyl–prelamin A in forebrain neurons (Jung et al. unpublished). At this point, we do not understand why the expression of progerin in neurons has negligible effects in the CNS but striking effects in the gastrointestinal tract, but we suspect that neurons of the CNS are less susceptible to the toxicity of progerin. Our inability to find significant CNS pathology in LmnaHG-C/+ mice appears to be consistent with a recent manuscript from Baek et al. (44); they found negligible effects of human progerin in the brain of tetracycline-regulated transgenic mice with aberrant expression of the progerin open-reading frame in the brain, skin and bone. In those studies, the gastrointestinal tract was not examined. On the other hand, Miller et al. (45) found that overexpression of progerin in neurons derived from induced pleuripotent stem cells led to breakdown of neurites.
Could our mouse models of achalasia be relevant to the human disease? It would be reasonable to be skeptical, simply because the development of achalasia in our models required two types of mutations (one eliminating neuronal regulation of prelamin A by miR-9, and a second that led to the accumulation of a farnesylated version of prelamin A). The coexistence of both types of mutations in a human patient would be quite unlikely. In addition, achalasia in humans is not accompanied by disease phenotypes of progeria. Nevertheless, one could still argue that our findings could be relevant to human achalasia. At this point, no one understands why mammals evolved an efficient strategy for suppressing the expression of prelamin A transcripts and lamin A protein in neurons; however, this regulatory mechanism is conserved and presumably is important. One could speculate that mature lamin A, which is normally absent in neurons, could be toxic for enteric neurons (although to a lesser degree than progerin or farnesyl–prelamin A). While we never identified overt intestinal pathology in mice that produce mature lamin A in neurons (i.e. LmnaPLAO-C/PLAO-C mice), it is conceivable that the low-level toxicity of mature lamin A in enteric neurons is simply not apparent within the short life span of a laboratory mouse. In humans, achalasia is generally not detected until the 4th or 5th decade of life, and in most cases the underlying cause is utterly mysterious. If our speculation about low-level toxicity of mature lamin A in enteric neurons is accurate, then it would be quite reasonable to speculate that some cases of achalasia in humans could be caused by point mutations in prelamin A's miR-9-binding site.
Both LmnaHG-C/+ and LmnaHG/+ mice express progerin in peripheral tissues, but the progeria-like disease phenotypes were less severe in LmnaHG-C/+ mice (11). Similarly, progeria-like disease phenotypes were less severe in Zmpste24−/−LmnaPLAO-C/PLAO-C mice than in Zmpste24−/−LmnaPLAO/PLAO mice. We suspect, but have not proved, that the milder progeria-like disease phenotypes in LmnaHG-C/+ and Zmpste24−/−LmnaPLAO-C/PLAO-C mice relate to lower amounts of progerin and farnesyl–prelamin A in certain cell types. In the current study, we found roughly similar levels of progerin in LmnaHG-C/+ and LmnaHG/+ fibroblasts, but it remains quite possible that LmnaHG-C/+ and LmnaHG/+ mice differ with respect to progerin expression in cell types that underlie osteolytic lesions, bone fractures and progressive inanition. Such expression differences need not to be largely significant; genetic and pharmacologic studies have shown that modest differences in progerin and prelamin A expression levels translate into substantial differences in quantifiable disease phenotypes and survival (11,25,27,34).
Materials and Methods
Generation of a new Lmna knock-in allele, LmnaHG-C
To create the LmnaHG-C allele, we used a gene-targeting vector identical to the one used to create the LmnaHG allele (10,11), except that the sequences for prelamin A's 3′ UTR in the 5′ arm were replaced with lamin C's 3′ UTR. Lamin C's 3′ UTR was amplified with primers 5′-TGCAGCATCATGTAAGGCCCAGCCCACAAGGGTA-3′ and 5′-GACACCACAGCATCTGGCATTCCAAAACAT. The new 3′ UTR sequences were introduced into the 5′ arm of the vector with the In-Fusion Advantage PCR cloning kit (Clontech). The integrity of the gene-targeting vector was verified by restriction endonuclease digestion and DNA sequencing. The gene-targeting vector was linearized with NotI and electroporated into embryonic stem (ES) cells (strain 129/OlaHsd). To identify targeted clones, long-range PCR on the 5′ end was performed with primers 5′-TCGAATCCGCATTGACAGCCTCT-3′ (primer A in Fig. 1A) and 5′-AAGCGAAGGAGCAAAGCTGCTA-3′ (primer NeoR). Long-range PCR on the 3′ end was performed with primers 5′-TGCTCCTGCCGAGAAAGTAT-3′ (primer NeoF in Fig. 1A) and 5′-AAAGTTCAGGCCCTTCTGGT-3′ (primer B).
Targeted ES cells were used to create male chimeric mice, which were bred with C57BL/6 females to produce heterozygous mice (LmnaHG-C/+). LmnaHG-C/+ mice were intercrossed to generate homozygotes (LmnaHG-C/HG-C). Mice were genotyped by PCR with genomic DNA from tail biopsies and oligonucleotide primers 5′-AGTACAACCTGCGCTCACGC-3′ (primer C) and 5′-ATGTGTCTGCCCCTGAAAAC-3′ (primer D) (Fig. 1A). The wild-type Lmna allele yields a 1488-bp PCR product, whereas the LmnaHG-C allele yields a 249-bp product.
Additional genetically modified mice
Zmpste24 knockout mice (Zmpste24−/−) and mice harboring other Lmna knock-in alleles (LmnaHG, LmnaPLAO, LmnaPLAO-UTR) have been described previously (10–13) (Table 1). In this study, the LmnaPLAO-UTR allele (12) was renamed the LmnaPLAO-C allele. All mice in this study were weaned at 21 days of age and housed in a virus-free barrier facility with a 12-h light–dark cycle. Mice were fed a chow diet containing 4.5% fat.
Analysis of mouse embryonic fibroblasts
Primary mouse embryonic fibroblasts (MEFs) were prepared from E14.5 embryos (46,47) and plated onto 6-well plates. To gauge the frequency of nuclear shape abnormalities in MEFs, passage 2–3 MEFs were grown on coverslips and then fixed and permeabilized (10,25,26,48,49). The slides were incubated with antibodies against lamin A (1:200, sc-20680, Santa Cruz Biotechnology) for 2 h. Binding of the primary antibody was detected with Alexa Fluor 568-conjugated anti-rabbit antibody (1:800, Jackson ImmunoResearch Laboratories), and DNA was stained with DAPI. Images were obtained on a Zeiss LSM700 laser-scanning microscope, and nuclear shape abnormalities were assessed in a blinded fashion (4 fibroblast cell lines/genotype; >300 cells examined/cell line).
Protein extraction and western blots
Urea-soluble cell extracts from early-passage MEFs and tissues (10,25,34,48,49) were size fractionated on 4–12% gradient polyacrylamide Bis–Tris gels (Invitrogen), and the proteins were transferred to nitrocellulose membranes for western blotting. Antibody dilutions were 1:200 for anti-lamin A/C goat IgG (sc-6215, Santa Cruz Biotechnology); 1:1000 for anti-actin goat IgG (sc-1616, Santa Cruz Biotechnology); and 1:5000 for IRDye 700- and 800-anti-goat IgG (Rockland Immunochemicals). The IRDye-conjugated antibodies were detected with an Odyssey infrared imaging scanner.
Histology and immunofluorescence microscopy
Mouse tissues were fixed for 4 h in 10% paraformaldehyde (PFA), dehydrated for 24 h in 70% ethanol and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin or with trichrome. For immunofluorescence microscopy, tissues were embedded in Sakura Tissue-Tec OCT compound, frozen on dry ice and 10-μm-thick sections were cut with a cryostat. Slides were first fixed in ice-cold methanol followed by a quick rinse with acetone and permeabilization with PBS containing 0.1% Tween-20. In some cases, sections were incubated in M.O.M. Mouse Ig Blocking Reagent (Vector Laboratories). Slides were incubated overnight at 4°C with primary antibodies [a mouse monoclonal antibody-specific for lamin A (Millipore, 1:400); a goat polyclonal antibody against lamin A/C (Santa Cruz, 1:400); a rabbit monoclonal antibody against caspase 3, (Cell Signaling, 1:100)] diluted in blocking buffer containing 0.1% Triton X-100 (12). Alexa Fluor-labeled secondary antibodies were incubated in a similar fashion for 0.5–1 h. After washing the slides, cells were fixed with 4% PFA for 10 min, incubated in PBS/DAPI for 10 min to stain DNA and mounted with Prolong-Gold Antifade reagent (Invitrogen). Light microscopy images were captured with a Leica MZ6 dissecting microscope [a Plan 0.5× objective (air)] with a DFC290 digital camera (Leica) and a Nikon Eclipse E600 microscope (12). Confocal microscopy was performed with a Zeiss LSM700 laser-scanning confocal microscope (12).
Whole-mount studies and confocal immunofluorescence microscopy
To prepare whole mounts of intestine for immunohistochemistry, tissues were pinned with the mucosa facing upward to the Sylgard elastomer base of a dissecting dish containing fresh KRB, and the mucosa was removed by sharp dissection. Strips of tunica muscularis were stretched to 110% of the resting length and width; the tissues were fixed in either acetone (4°C, 30 min) or 4% (w/v) paraformaldehyde (room temperature, 1 h). After fixation, tissues were washed overnight in phosphate-buffered saline (PBS; 0.01 M, pH 7.2) and rewashed with fresh PBS the following day (4 times for 1 h). Tissues were subsequently incubated in 1% bovine serum albumin for 1 h and then incubated for 48 h at 4°C with antibodies against Protein gene product 9.5 (Ultraclone), vesicular acetylcholine transporter (Millipore), or c-Kit (R&D Systems). Binding of primary antibodies was detected with Alexa Fluor-labeled secondary antibodies (Molecular Probes). Tissues were examined with a Zeiss LSM 510 Meta confocal microscope (Zeiss, Germany). Final images were constructed using Zeiss LSM 5 Image Examiner software and processed in Adobe Photoshop 7.0 and Corel Draw 7.0. For the whole-mount studies, mice with achalasia (and control mice) were shipped to the University Nevada in four shipments between June 2013 and April 2014 (4–7 mice/shipment).
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by the National Institutes of Health Grants (HL86683 to L.G.F., HL089781 to L.G.F., and AG035626 to S.G.Y.; and DK57236 to S.M.W.). Some of the confocal microscopy studies were performed in a core laboratory supported by P01 DK41315 and NIH1 S10 RR16871.
Supplementary Material
References
- 1.Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., et al. (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature, 423, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Le Merrer M., Lévy N. (2003) Lamin A truncation in Hutchinson–Gilford progeria. Science, 300, 2055. [DOI] [PubMed] [Google Scholar]
- 3.Young S.G., Fong L.G., Michaelis S. (2005) Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—New evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res., 46, 2531–2558. [DOI] [PubMed] [Google Scholar]
- 4.Debusk F.L. (1972) The Hutchinson-Gilford progeria syndrome. J. Pediatr., 80, 697–724. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Palazzi F., McLaren A.T., Slowie D.F. (1992) Report on a case of Hutchinson-Gilford progeria, with special reference to orthopedic problems. Eur. J. Pediatr. Surg., 2, 378–382. [DOI] [PubMed] [Google Scholar]
- 6.Gordon L.B., McCarten K.M., Giobbie-Hurder A., Machan J.T., Campbell S.E., Berns S.D., Kieran M.W. (2007) Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics, 120, 824–833. [DOI] [PubMed] [Google Scholar]
- 7.Martin G.M. (1982) Syndromes of accelerated aging. Natl. Cancer Inst. Monogr., 60, 241–247. [PubMed] [Google Scholar]
- 8.Gordon C.M., Gordon L.B., Snyder B.D., Nazarian A., Quinn N., Huh S., Giobbie-Hurder A., Neuberg D., Cleveland R., Kleinman M., Miller D.T., Kieran M.W. (2011) Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Miner. Res., 26, 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro C.L., Cau P., Levy N. (2006) Molecular bases of progeroid syndromes. Hum. Mol. Genet., 15, R151–R161. [DOI] [PubMed] [Google Scholar]
- 10.Yang S.H., Bergo M.O., Toth J.I., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P., Gelb M.H., Young S.G., Fong L.G. (2005) Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl Acad. Sci. USA, 102, 10291–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang S.H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M.O., Young S.G., Fong L.G. (2006) A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest., 116, 2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung H.J., Tu Y., Yang S.H., Tatar A., Nobumori C., Wu D., Young S.G., Fong L.G. (2014) New Lmna knock-in mice provide a molecular mechanism for the ‘segmental aging’ in Hutchinson-Gilford progeria syndrome. Hum. Mol. Genet., 23, 1506–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies B.S., Barnes R.H., 2nd, Tu Y., Ren S., Andres D.A., Spielmann H.P., Lammerding J., Wang Y., Young S.G., Fong L.G. (2010) An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum. Mol. Genet., 19, 2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung G.K., Schmidt W.K., Bergo M.O., Gavino B., Wong D.H., Tam A., Ashby M.N., Michaelis S., Young S.G. (2001) Biochemical studies of Zmpste24-deficient mice. J. Biol. Chem., 276, 29051–29058. [DOI] [PubMed] [Google Scholar]
- 15.Bergo M.O., Gavino B., Ross J., Schmidt W.K., Hong C., Kendall L.V., Mohr A., Meta M., Genant H., Jiang Y., et al. (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl Acad. Sci. USA, 99, 13049–13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young S.G., Meta M., Yang S.H., Fong L.G. (2006) Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem., 281, 39741–39745. [DOI] [PubMed] [Google Scholar]
- 17.Jung H.J., Coffinier C., Choe Y., Beigneux A.P., Davies B.S., Yang S.H., Barnes R.H., 2nd, Hong J., Sun T., Pleasure S.J., Young S.G., Fong L.G. (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl Acad. Sci. USA, 109, E423–E431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata M., Nakao H., Kiyonari H., Abe T., Aizawa S. (2011) MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J. Neurosci., 31, 3407–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo A.S., Sun A.X., Li L., Shcheglovitov A., Portmann T., Li Y., Lee-Messer C., Dolmetsch R.E., Tsien R.W., Crabtree G.R. (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature, 476, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffinier C., Chang S.Y., Nobumori C., Tu Y., Farber E.A., Toth J.I., Fong L.G., Young S.G. (2010) Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc. Natl Acad. Sci. USA, 107, 5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coffinier C., Fong L.G., Young S.G. (2010) LINCing lamin B2 to neuronal migration: growing evidence for cell-specific roles of B-type lamins. Nucleus, 1, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coffinier C., Jung H.J., Nobumori C., Chang S., Tu Y., Barnes R.H., 2nd, Yoshinaga Y., de Jong P.J., Vergnes L., Reue K., Fong L.G., Young S.G. (2011) Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell, 22, 4683–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young S.G., Jung H.J., Coffinier C., Fong L.G. (2012) Understanding the roles of nuclear A- and B-type lamins in brain development. J. Biol. Chem., 287, 16103–16110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanowitz H.B., Kirchhoff L.V., Simon D., Morris S.A., Weiss L.M., Wittner M. (1992) Chagas’ disease. Clin. Microbiol. Rev., 5, 400–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fong L.G., Ng J.K., Meta M., Cote N., Yang S.H., Stewart C.L., Sullivan T., Burghardt A., Majumdar S., Reue K., Bergo M.O., Young S.G. (2004) Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl Acad. Sci. USA, 101, 1811–18116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S.H., Andres D.A., Spielmann H.P., Young S.G., Fong L.G. (2008) Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J. Clin. Invest., 118, 3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S.H., Qiao X., Farber E., Chang S.Y., Fong L.G., Young S.G. (2008) Eliminating the synthesis of mature lamin A reduces disease phenotypes in mice carrying a Hutchinson-Gilford progeria syndrome allele. J. Biol. Chem., 283, 7094–7099. [DOI] [PubMed] [Google Scholar]
- 28.Jung H.J., Lee J.M., Yang S.H., Young S.G., Fong L.G. (2013) Nuclear lamins in the brain - new insights into function and regulation. Mol. Neurobiol., 47, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gockel H.R., Schumacher J., Gockel I., Lang H., Haaf T., Nothen M.M. (2010) Achalasia: will genetic studies provide insights? Hum. Genetics, 128, 353–364. [DOI] [PubMed] [Google Scholar]
- 30.Goyal R.K., Chaudhury A. (2010) Pathogenesis of achalasia: lessons from mutant mice. Gastroenterology, 139, 1086–1090. [DOI] [PubMed] [Google Scholar]
- 31.Ghoshal U.C., Daschakraborty S.B., Singh R. (2012) Pathogenesis of achalasia cardia. World J. Gastroenterol., 18, 3050–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gockel H.R., Gockel I. (2011) [Of mice and men: Lsc/p115 and achalasia]. Z. Gastroenterol., 49, 369–370. [DOI] [PubMed] [Google Scholar]
- 33.van der Weyden L., Happerfield L., Arends M.J., Adams D.J. (2009) Megaoesophagus in Rassf1a-null mice. Int. J. Exp. Pathol., 90, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fong L.G., Frost D., Meta M., Qiao X., Yang S.H., Coffinier C., Young S.G. (2006) A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science, 311, 1621–1623. [DOI] [PubMed] [Google Scholar]
- 35.Taketomi T., Yoshiga D., Taniguchi K., Kobayashi T., Nonami A., Kato R., Sasaki M., Sasaki A., Ishibashi H., Moriyama M., et al. (2005) Loss of mammalian Sprouty2 leads to enteric neuronal hyperplasia and esophageal achalasia. Nat. Neurosci., 8, 855–857. [DOI] [PubMed] [Google Scholar]
- 36.Groneberg D., Zizer E., Lies B., Seidler B., Saur D., Wagner M., Friebe A. (2014) Dominant role of interstitial cells of Cajal in nitrergic relaxation of murine lower esophageal sphincter. J. Physiol., 593, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller M., Colcuc S., Drescher D.G., Eckardt A.J., von Pein H., Taube C., Schumacher J., Gockel H.R., Schimanski C.C., Lang H., Gockel I. (2014) Murine genetic deficiency of neuronal nitric oxide synthase and interstitial cells of Cajal: implications for achalasia? J. Gastroenterol. Hepatol., 29, 1800–1807. [DOI] [PubMed] [Google Scholar]
- 38.Gockel I., Bohl J.R., Eckardt V.F., Junginger T. (2008) Reduction of interstitial cells of Cajal (ICC) associated with neuronal nitric oxide synthase (n-NOS) in patients with achalasia. Am. J. Gastroenterol., 103, 856–864. [DOI] [PubMed] [Google Scholar]
- 39.Villanacci V., Annese V., Cuttitta A., Fisogni S., Scaramuzzi G., De Santo E., Corazzi N., Bassotti G. (2010) An immunohistochemical study of the myenteric plexus in idiopathic achalasia. J. Clin. Gastroenterol., 44, 407–410. [DOI] [PubMed] [Google Scholar]
- 40.Savage D.C., Dubos R. (1968) Alterations in the mouse cecum and its flora produced by antibacterial drugs. J. Exp. Med., 128, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Machado W.M., Moraes-Filho J.P., Santos M.A., Bettarello A. (1989) Jejunal flora of patients with megaoesophagus secondary to Chagas disease. Trans. R. Soc. Trop. Med. Hyg., 83, 199–201. [DOI] [PubMed] [Google Scholar]
- 42.Machado W.M. (1995) [Small intestine flora in chagasic patients with megaesophagus and/or megacolon: study using the H2 breath test]. Arq. Gastroenterol., 32, 19–23. [PubMed] [Google Scholar]
- 43.Martin E., Perez San Jose C., Emparan C., Aguinagalde M., Sabas J. (1998) Idiopathic megacolon associated with oesophageal achalasia. Eur. J. Gastroenterol. Hepatol., 10, 147–150. [DOI] [PubMed] [Google Scholar]
- 44.Baek J.H., Schmidt E., Viceconte N., Strandgren C., Pernold K., Richard T.J., Van Leeuwen F.W., Dantuma N.P., Damberg P., et al. (2014) Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum Mol Genet., 24, 1305–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller J.D., Ganat Y.M., Kishinevsky S., Bowman R.L., Liu B., Tu E.Y., Mandal P.K., Vera E., Shim J.W., Kriks S., et al. (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell, 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim E., Lowenson J.D., MacLaren D.C., Clarke S., Young S.G. (1997) Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl Acad. Sci. USA, 94, 6132–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim E., Ambroziak P., Otto J.C., Taylor B., Ashby M., Shannon K., Casey P.J., Young S.G. (1999) Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J. Biol. Chem., 274, 8383–8390. [DOI] [PubMed] [Google Scholar]
- 48.Toth J.I., Yang S.H., Qiao X., Beigneux A.P., Gelb M.H., Moulson C.L., Miner J.H., Young S.G., Fong L.G. (2005) Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl Acad. Sci. USA, 102, 12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fong L.G., Ng J.K., Lammerding J., Vickers T.A., Meta M., Cote N., Gavino B., Qiao X., Chang S.Y., Young S.R., et al. (2006) Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest., 116, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.