

Abstract
Addition of the trinucleotide cytosine/cytosine/adenine (CCA) to the 3′ end of transfer RNAs (tRNAs) is essential for translation and is catalyzed by the enzyme TRNT1 (tRNA nucleotidyl transferase), which functions in both the cytoplasm and mitochondria. Exome sequencing revealed TRNT1 mutations in two unrelated subjects with different clinical features. The first presented with acute lactic acidosis at 3 weeks of age and developed severe developmental delay, hypotonia, microcephaly, seizures, progressive cortical atrophy, neurosensorial deafness, sideroblastic anemia and renal Fanconi syndrome, dying at 21 months. The second presented at 3.5 years with gait ataxia, dysarthria, gross motor regression, hypotonia, ptosis and ophthalmoplegia and had abnormal signals in brainstem and dentate nucleus. In subject 1, muscle biopsy showed combined oxidative phosphorylation (OXPHOS) defects, but there was no OXPHOS deficiency in fibroblasts from either subject, despite a 10-fold-reduction in TRNT1 protein levels in fibroblasts of the first subject. Furthermore, in normal controls, TRNT1 protein levels are 10-fold lower in muscle than in fibroblasts. High resolution northern blots of subject fibroblast RNA suggested incomplete CCA addition to the non-canonical mitochondrial tRNASer(AGY), but no obvious qualitative differences in other mitochondrial or cytoplasmic tRNAs. Complete knockdown of TRNT1 in patient fibroblasts rendered mitochondrial tRNASer(AGY) undetectable, and markedly reduced mitochondrial translation, except polypeptides lacking Ser(AGY) codons. These data suggest that the clinical phenotypes associated with TRNT1 mutations are largely due to impaired mitochondrial translation, resulting from defective CCA addition to mitochondrial tRNASer(AGY), and that the severity of this biochemical phenotype determines the severity and tissue distribution of clinical features.
Introduction
Disorders of the oxidative phosphorylation (OXPHOS) system are both clinically and genetically heterogeneous. OXPHOS diseases are often designated collectively as mitochondrial encephalomyopathies, owing to the frequent involvement of brain and skeletal muscle, two tissues with a high rate of aerobic metabolism. This term is misleading because other organs are often implicated including liver, heart, blood, kidney, eye, pancreas, digestive tract, either alone or in various combinations. Mutations in mitochondrial tRNAs are greatly over-represented in human OXPHOS diseases, likely because such mutations do not appear to be subject to the same stringent purifying selection that eliminates pathogenic mutations in protein coding genes in the female germ line (1).
Mitochondrial tRNAs undergo a considerable number of base modifications, including two mitochondria-specific modifications of the wobble-base position: 5-formylcytidine and 5-taurinomethyl-(2-thio)uridine (reviewed in (2)). Pathogenic mutations have been reported in four genes encoding enzymes involved in the post-transcriptional modifications of mitochondrial tRNAs: PUS1 encoding the pseudouridine synthase, associated with myopathy and sideroblastic anemia (3); MTO1 and TRMU encoding the enzymes involved in the synthesis of the 5-taurinomethyluridine and 2-thiouridine at the wobble position of certain mt tRNAs (4,5), associated, respectively, with hypertrophic cardiomyopathy and transient liver failure; and TRIT1, which codes for tRNA isopentenyltransferase, responsible for the modification of the anticodon loops of a small subset of cytosolic and mitochondrial tRNAs, and which is associated with encephalopathy and myoclonic epilepsy (6).
An essential post-transcriptional modification is the addition of the cytosine/cytosine/adenine (CCA) trinucleotide to the 3′end of all newly synthesized tRNAs. CCA addition is required for amino-acid attachment, proper tRNA positioning at the ribosome and translation termination, and is catalyzed by the enzyme tRNA nucleotidyltransferase (TRNT1, or CCA-adding enzyme) (reviewed in (7)). A single gene encodes both cytoplasmic and mitochondrial TRNT1 in eukaryotes (8,9). CCA-adding enzymes possess a number of defining characteristics, specifically they recognize tRNA structures independent of their sequence by interacting almost exclusively with the sugar-phosphate backbone of the top half of the tRNA (10–12), selectively use cytidine triphosphate and adenosine triphosphate (ATP) rather than guanosine triphosphate and uridine triphosphate (13), switching from CTP to ATP after the first two residues without the help of a nucleic acid template (13) and finally, stop polymerization after incorporation of exactly three bases (14). TRNT1 performs only one round of CCA addition because unlike typical polymerases, TRNT1 does not move along the tRNA during synthesis. Consequently, the growing 3′end of the tRNA must refold in the active site for further nucleotide incorporation, and due to the limited size of the binding pocket, only one round of CCA addition is possible (11). We report two unrelated subjects with different clinical phenotypes, in whom we identify mutations in the TRNT1 gene and characterize the mechanisms of pathogenesis.
Results
Subjects
The first, a girl (subject N) was born at term to consanguineous Turkish parents, after an uneventful pregnancy. Fetal growth was normal, as was the initial postnatal period. She had a first crisis of lactic acidosis at 18 days of age. Thereafter she had chronic mild metabolic acidosis, hyperlactatemia and hyperalaninemia, punctuated by acute episodes of severe acidosis. Sideroblastic anemia and renal Fanconi syndrome were detected in the neonatal period and persisted. She developed global developmental delay, neurosensory deafness, acquired microcephaly with cortical atrophy on cerebral magnetic resonance imaging (MRI) and marked failure to thrive. Seizures and central apnea were documented at 18 months of age. She died of cardiac arrest during an episode of fever and acute acidosis at 21 months of age. Muscle, liver and kidney biopsies showed increased numbers of enlarged mitochondria on electron microscopy. Detailed clinical details are presented in Supplemental Material, Tables S1 and S2. The initial molecular investigations were negative for mitochondrial DNA mutations including the absence of mitochondrial DNA deletions.
The second subject (P) was born after 8 months gestation to unrelated healthy parents of Greek-Cypriot origin. His motor development was delayed but cognitive development appeared normal. By 3.5 years he had developed gait ataxia, dysarthria, gross motor regression, hypotonia, ptosis and horizontal ophthalmoplegia and had abnormal signals in brainstem and dentate nucleus on MRI. At 5 years 8 months he can stand with support. Hypotonia, ptosis and horizontal ophthalmoplegia persist and his speech is slow and dysarthric. His condition has remained unchanged at 8 years of age.
Identification of mutation in TRNT1 by whole-exome sequencing
We performed exome sequencing in both patients. Blood genomic DNA was captured with the Agilent SureSelect Human All Exon Capture V4 Kit and sequenced (two paired-end 100 bp reads, three exomes per lane) with the Illumina HiSeq2000 system. Briefly, gapped alignment to reference sequences (Hg19 and GRCh37) was performed with genomic short-read nucleotide alignment program and the genome analysis toolkit. A total of ∼400 variants, including 25 variants in nuclear-encoded mitochondrial genes, were retained as candidates according to our filtering criteria; variants were filtered to 0.5% minor allele frequency in an internal database of >1000 samples, rare or absent from the NHLBI Exome Sequencing Project (EVS) and NCBI dbSNP (<1%), then prioritized by online mendelian inheritance in man identity, human gene mutation database and mitochondrial filters. Each potential variant was visualized by integrative genomics viewer. No diagnostic genotypes were found in previously reported disease genes. Subject N had 25 variants in nuclear-encoded mitochondrial genes; only one variant was homozygous; TRNT1: NM_182916:exon4:c.C443T:p.A148V (coverage of 139X). No compound heterozygous variants were uncovered in mitochondrial genes and no second variant was identified in low coverage regions. Essentially similar methods were employed to identify the candidate variants in subject P except that compound heterozygous variants were retained as the parents were unrelated. Approximately 300 heterozygous variants survived the filtering process; however, only TRNT1 contained two variants, and Sanger analysis of DNA from the parents disclosed compound heterozygosity for the mutations c.383A>G (p.D128G) and c.518A>T (p.Y173F) in TRNT1. All three mutations are predicted damaging by several algorithms (SIFT, Polyphen2, MutationTaster and PhyloP/LRT), occur at evolutionarily conserved amino-acid residues (Fig. 1) and were confirmed by Sanger sequencing of the subject's DNA.
Figure 1.

Mutational analysis of TRNT1. (A) Schematic representation of the TRNT1 gene showing the position of the mutations identified in this study. (B) Alignment of the amino-acid sequences of TRNT1 homologs in different species showing high degree of evolutionary conservation of the amino-acid residues mutated in the subjects.
Fibroblasts from the two subjects contain different residual levels of mutant TRNT1 protein and have near normal mitochondrial translation
To investigate the functional consequences of these mutations we first examined the level of mutant TRNT1 in fibroblasts of both subjects. Immunoblot analysis revealed normal levels of TRNT1 in subject P but a reduction to ∼10% of control levels in subject N, who also had the most severe clinical phenotype (Fig. 2). To evaluate the effects of the mutations in TRNT1 on mitochondrial protein synthesis, we pulse-labeled the mitochondrial translation products in fibroblasts from subjects and controls for 1 h with [35S]-methionine/cysteine in the presence of emetine, a cytosolic translation inhibitor (15). The synthesis of the mitochondrial translation products was moderately reduced as compared with controls for both subjects (Fig. 2). Although the global rate of translation in subject P was 57 ± 9% (SD, N = 7) of control, Blue-native polyacrylamide gel electrophoresis (PAGE) analysis confirmed normal levels of assembled OXPHOS complexes in fibroblasts from both subjects (data not shown), indicating that any differences in the rates of translation did not have functional consequences. Thus mitochondrial translation can proceed normally with mutant TRNT1 at 10% of control levels (Fig. 2). Retroviral expression of a wild-type TRNT1 cDNA had no significant effect on mitochondrial translation in fibroblasts from either subject (Fig. 2).
Figure 2.

Mitochondrial translation and levels of TRNT1 in the subjects. (A) Pulse-labeling of the mitochondrial translation products in fibroblasts from the subjects and controls, before and after over-expression of wild-type TRNT1. Upper panel. The 13 mitochondrial translation products are indicated at the left of the upper panels: seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX) and two subunits of complex V (ATP). Middle panel: duplicate samples were used for immunoblotting with antibodies against the 70 kDa subunit of complex II to confirm equal loading. Lower panel: the levels of TRNT1 in fibroblasts from the two subjects, and the over-expression of TRNT1 in control and subject fibroblasts were evaluated by immunoblotting with polyclonal antibodies against TRNT1. (B) Quantification of the rate of synthesis of the individual polypeptides in the pulse translation experiments.
Knockdown of TRNT1 below immunologically detectable levels abolishes mitochondrial translation
Next, we investigated the minimum level of TRNT1 required to sustain normal mitochondrial translation, in other words, the point at which a destabilizing mutation in the TRNT1 protein results in a defect of mitochondrial protein synthesis. To this end, we used siRNA to suppress TRNT1 in control and subject fibroblast lines. Stealth RNA interference duplexes (Invitrogen) were transiently transfected into subject fibroblasts, transfection was repeated on day 3 and on day 6, and cells were analyzed on day 10. TRNT1 levels were decreased in control and subject P fibroblasts to the levels present in fibroblasts from subject N (∼10% of control, Fig. 3). This had a very modest effect on mitochondrial protein synthesis, confirming that this level of TRNT1 is sufficient for normal mitochondrial translation. In contrast, TRNT1 was undetectable by immunoblot in subject N fibroblasts treated with siRNA, and mitochondrial translation was markedly reduced (Fig. 3). Strikingly, both ND4L and ATP8 were synthesized at near control rates (Fig. 3B). The abnormal bands seen in this translation gel likely represent premature translation products.
Figure 3.

Mitochondrial translation is severely impaired following knockdown of TRNT1 to immunologically undetectable levels. (A) Mitochondrial translation products synthesized in fibroblasts from subjects and controls, before and after transient transfection with a siRNA construct specific to TRNT1. The 13 mitochondrial translation products are indicated at the left of the upper panel as in Figure 1. Middle panel: duplicate samples were used for immunoblotting with antibodies against prohibitin to confirm equal loading. Lower panel: the knockdown of TRNT1 in fibroblasts from the two patients and controls was confirmed by immunoblotting with polyclonal antibodies against TRNT1. (B) Quantification of the rate of synthesis of the individual polypeptides in the pulse translation experiments.
A destabilizing mutation in the TRNT1 enzyme interferes preferentially with the handling of a non-canonical mitochondrial tRNA
To determine the pathogenic mechanism of a destabilizing mutation in TRNT1, we tested whether different specific mitochondrial and cytoplasmic tRNAs were affected to the same extent. We hypothesized that tRNAs that deviate from the canonical cloverleaf structure would be more prone to a defect in CCA tail addition by a mutant TRNT1 enzyme, present at very low levels, as they may represent sub-optimal substrates for the enzyme. Several mitochondrial tRNAs have non-canonical structures, the most atypical being the mitochondrial tRNA for Ser(AGY), which completely lacks the D arm (16). High-resolution northern blotting for small RNAs was carried out with RNA isolated from control and subject fibroblasts, and from those overexpressing TRNT1 or in which TRNT1 was suppressed. In cells from both subjects, as well as in control cells treated with siRNA, mt tRNASer(AGY) appeared as a doublet, the distinct lower band likely corresponding to tRNA species with a partial CCA tail. The ratio of the upper–lower band was about 70:30 in subject P, 55:45 in subject N and 60:40 in the control knockdown cells. In addition, the total amount of tRNASer(AGY) was reduced in subject N cells by about 40% (Fig. 4). Importantly, the migration patterns of the other mitochondrial and cytosolic tRNAs examined were no different in subject and control cells. In siRNA-treated fibroblasts from subject N, in which TRNT1 was immunologically undetectable, mitochondrial tRNASer(AGY) was rendered undetectable. Two other mitochondrial tRNAs were unaffected as compared with the corresponding controls, and the two cytoplasmic tRNAs tested were decreased to 20–30% of control (Fig. 4). We conclude that the non-canonical mitochondrial tRNASer(AGY) is particularly sensitive to loss of function mutations in TRNT1. Interestingly, only two mitochondrial mRNAs, those coding for ND4L and ATP8, do not contain Ser(AGY) codons, and as noted above their synthesis was virtually unaffected in the subject N cells in which TRNT1 was knocked down to immunologically undetectable levels (Fig. 3).
Figure 4.

Differential effect of a destabilizing mutation in TRNT1 on individual tRNAs. Total RNA isolated under acidic conditions from the subject and control fibroblast lines indicated at the top of the figure (12 μg per sample) was separated through a 12% polyacrylamide/8 m urea gel, transferred to a positively charged membrane and hybridized with probes complementary to the mitochondrial and cytoplasmic tRNAs indicated at the right of the figure.
Muscle is particularly vulnerable to a destabilizing mutation in TRNT1
We next sought to determine whether muscle is particularly susceptible to a destabilizing mutation in TRNT1, as this tissue was clinically affected in both subjects. To this end we immunoblotted mitochondrial fractions isolated from fibroblasts or muscle biopsy specimens of controls with antibodies against TRNT1, the 70 kDa subunit of Complex II or prohibitin. This analysis revealed that endogenous levels of TRNT1 in muscle are ∼10% of the levels present in fibroblasts (Fig. 5), which could cause muscle to be particularly vulnerable to destabilizing TRNT1 mutations. We did not have adequate material to assess the levels of tRNASer(AGY) in muscle from subject N.
Figure 5.
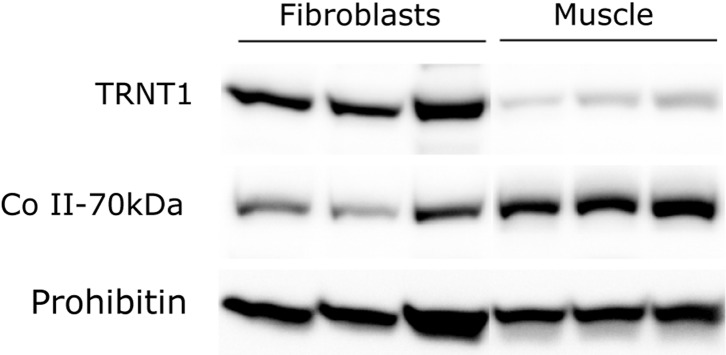
Endogenous levels of TRNT1 are considerably lower in muscle mitochondria than in fibroblast mitochondria. Immunoblot analysis of TRNT1 in mitochondrial extracts from three control muscle biopsy specimens and three control fibroblast lines. The 70 kDa subunit of complex II and prohibitin were used as loading controls.
Discussion
The results we present here show that very low levels of TRNT1 can support normal mitochondrial translation, at least in fibroblasts. However, as TRNT1 is essential for CCA addition to all tRNAs, and therefore necessary for all cellular protein synthesis, complete loss of TRNT1 function would cause embryonic lethality. Therefore, symptomatic TRNT1-deficient subjects have markedly decreased but not absent TRNT1 activity. Furthermore, the non-canonical mitochondrial tRNASer(AGY) is more sensitive to reduction of TRNT1 function of all the tRNAs tested to date. Together, these results suggest defective mitochondrial translation as the most plausible explanation of the clinical features in TRNT1 deficiency.
The marked clinical differences between the TRNT1-deficient subjects reported here are reminiscent of the Pearson Syndrome (PS)–Kearns–Sayre Syndrome (KSS) continuum. Both PS and KSS are usually caused by heteroplasmic large mtDNA deletions producing mitochondrial protein synthesis defects. PS is characterized by sideroblastic anemia, exocrine pancreatic deficiency, myopathy, variable hepatic and renal failure and is associated with a high load of mutant mtDNA in most if not all tissues including hematopoietic stem cells (17). Patients with KSS have ptosis, chronic progressive external ophthalmoplegia and myopathy, associated with lower levels and a more restricted distribution of mutant mtDNAs (18,19), which are usually undetectable in blood. The different level and different tissue distribution of mutant mtDNA heteroplasmy in PS and KSS are important determinants of the severity of the mitochondrial translation defect and of clinical presentation. Tissue-specific involvement in the subjects with TRNT1 mutations likewise appears to reflect the magnitude of mitochondrial translation impairment. Although we studied only two patients with TRNT1 deficiency, all of our data are consistent with the notion that the biochemical severity of mitochondrial translation relates to the severity of neurological signs and to the development of multiple organ dysfunction.
While this manuscript was in preparation, compound heterozygous and homozygous mutations in TRNT1 were reported in a cohort of patients with a syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fever and developmental delay (SIFD) (20), similar to many features of subject N. Together with our observations, this establishes sideroblastic anemia and neurological dysfunction as features of TRNT1 deficiency in humans. The other features of our patients (renal tubulopathy, later onset neurological difficulties) may expand the clinical spectrum.
Sideroblastic anemia is a rare condition for which mitochondrial disease is a major diagnostic consideration. All known causes of congenital sideroblastic anemia involve mitochondrial iron, iron–sulfur cluster or protein synthesis pathways (21). Although the pathogenic mechanism is obscure, it is intriguing that in addition to TRNT1, two other mt-tRNA-related genes, encoding the mitochondrial tyrosine aminoacyl tRNA synthetase YARS2 (22) and the mitochondrial pseudouridine synthase PUS1(3), are also linked to sideroblastic anemia.
Materials and Methods
Pulse-labeling of mitochondrial translation products
Fibroblasts were incubated in Dulbecco's Modified Eagle Medium (DMEM) lacking methionine and cysteine and supplemented with 10% dialyzed serum, in the presence of 100 µg emetine and 200 µCi/ml [35S]-methionine and cysteine mix (Perkin Elmer) for 1 h at 37°C. Radiolabelled mitochondrial translation products (50 µg/sample) were then separated through a 15–20% polyacrylamide gradient gel, which was subsequently dried and exposed for autoradiography (15).
Cell culture, RNA interference and over-expression experiments
Fibroblasts were grown in DMEM supplemented with 10% fetal bovine serum at 37°C and 5% CO2. To knockdown TRNT1, Stealth RNA interference duplexes (Invitrogen) were transiently transfected at a concentration of 10 nm according to the manufacturer's instructions for reverse transfection. Transfection was repeated on day 3 and on day 6, and cells were analyzed on day 10.
The TRNT1 cDNA was amplified by RT-PCR (OneStep kit, QIAGEN) with primers adapted for Gateway cloning (Life Technologies), and then cloned in the Gateway-compatible, pBABE-Puro retroviral vector. The retroviral construct was subsequently transiently transfected into the Phoenix packaging line by the calcium phosphate method and viral particles were collected 48 h later and used to infect fibroblasts in the presence of 4 µg/ml polybrene as in (23). Selection was started 48–72 h later in 2.5 µg/ml puromycin.
Immunoblot analysis
Whole cells or mitochondrial fractions isolated from fibroblasts or muscle were extracted on ice for 40–45 min in 1.5–1.75% dodecyl maltoside/PBS, mixed with Laemmli sample buffer and run on 12% polyacrylamide gels. Following transfer to nitrocellulose membranes, immunoblotting was preformed with antibodies against TRNT1 (rabbit polyclonal from Sigma Prestige), the 70 kDa subunit of Complex II (mouse monoclonal from Abcam) or prohibitin (rabbit polyclonal from Abcam).
Acid-urea PAGE and northern blotting
Total RNA from fibroblasts was isolated under acidic conditions using TRIzol (Invitrogen, Carlsbad, CA) as instructed by the manufacturer. RNA from one control sample was also incubated at alkaline pH (0.1 m Tris–HCl, pH 9.0), 37°C, for 1 h to promote deacylation of aminoacyl-tRNAs. Twelve micrograms of total RNA per sample were mixed with 1:1 loading buffer containing 8 m urea, 0.1 m sodium acetate, pH 5, 0.05% bromophenol blue, 0.05% xylene cyanol, then boiled for 3 min, placed on ice and loaded on a 0.75-mm thick, 12% acrylamide (19:1, acrylamide–bisacrylamide) gel containing 8 m urea in 0.1 m sodium acetate buffer (pH 5.0). Electrophoresis was carried out at 200 V for 24 h, at 4°C, followed by transfer onto Hybond N+ membrane (GE Healthcare). Prehybridization and hybridization were performed using the EXPRESSHyb solution (Clontech) according to the manufacturer's instructions. The oligonucleotides used for the generation of the 32P-labeled probes by the end-labeling technique were complementary to the 3′-end of the tRNAs of interest, 24-mer in length.
Supplementary Material
Funding
This work was supported by grants from the CIHR (MT-15460) to E.A.S. and CIHR (178978) to G.M. and by a joint grant from la Fondation GO to C.B.G. and E.A.S.
Supplementary Material
Acknowledgements
Exome sequencing of Patient N was performed at the CHU Sainte-Justine and Génome Québec Center for Pediatric Clinical Genetics.
Conflict of Interest statement. The authors declare no conflict of interest.
References
- 1.Stewart J.B., Freyer C., Elson J.L., Cansu Z., Trifunovic A., Larsson N-G. (2008) Strong purifying selection in transmission of mitochondrial DNA. PLOS Biol., 6, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki T., Nagao A., Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet., 45, 299–329. [DOI] [PubMed] [Google Scholar]
- 3.Bykhovskaya Y., Casas K., Mengesha E., Inbal A., Fischel-Ghodsian N. (2004) Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet., 74, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghezzi D., Baruffini E., Haack T.B., Invernizzi F., Melchionda L., Dallabona C., Strom T.M., Parini R., Burlina A.B., Meitinger T., et al. (2012) Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet., 90, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeharia A., Shaag A., Pappo O., Mager-Heckel A.M., Saada A., Beinat M., Karicheva O., Mandel H., Ofek N., Segel R., et al. (2009) Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet., 85, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yarham J.W., Lamichhane T.N., Pyle A., Mattijssen S., Baruffini E., Bruni F., Donnini C., Vassilev A., He L., Blakely E.L., et al. (2014) Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet., 10, e1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betat H., Rammelt C., Morl M. (2010) tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cell. Mol. Life Sci., 67, 1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J.Y., Joyce P.B., Wolfe C.L., Steffen M.C., Martin N.C. (1992) Cytoplasmic and mitochondrial tRNA nucleotidyltransferase activities are derived from the same gene in the yeast Saccharomyces cerevisiae. J. Biol. Chem., 267, 14879–14883. [PubMed] [Google Scholar]
- 9.Nagaike T., Suzuki T., Tomari Y., Takemoto-Hori C., Negayama F., Watanabe K., Ueda T. (2001) Identification and characterization of mammalian mitochondrial tRNA nucleotidyltransferases. J. Biol. Chem., 276, 40041–40049. [DOI] [PubMed] [Google Scholar]
- 10.Tomita K., Fukai S., Ishitani R., Ueda T., Takeuchi N., Vassylyev D.G., Nureki O. (2004) Structural basis for template-independent RNA polymerization. Nature, 430, 700–704. [DOI] [PubMed] [Google Scholar]
- 11.Xiong Y., Steitz T.A. (2004) Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature, 430, 640–645. [DOI] [PubMed] [Google Scholar]
- 12.Toh Y., Takeshita D., Numata T., Fukai S., Nureki O., Tomita K. (2009) Mechanism for the definition of elongation and termination by the class II CCA-adding enzyme. EMBO J., 28, 3353–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F., Xiong Y., Wang J., Cho H.D., Tomita K., Weiner A.M., Steitz T.A. (2002) Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell, 111, 815–824. [DOI] [PubMed] [Google Scholar]
- 14.Shi P.Y., Maizels N., Weiner A.M. (1998) CCA addition by tRNA nucleotidyltransferase: polymerization without translocation? EMBO J., 17, 3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasarman F., Shoubridge E.A. (2012) Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol. Biol., 837, 207–217. [DOI] [PubMed] [Google Scholar]
- 16.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F., et al. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 17.Rotig A., Bourgeron T., Chretien D., Rustin P., Munnich A. (1995) Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum. Mol. Genet., 4, 1327–1330. [DOI] [PubMed] [Google Scholar]
- 18.Holt I.J., Harding A.E., Morgan-Hughes J.A. (1988) Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature, 331, 717–719. [DOI] [PubMed] [Google Scholar]
- 19.Moraes C.T., DiMauro S., Zeviani M., Lombes A., Shanske S., Miranda A.F., Nakase H., Bonilla E., Werneck L.C., Servidei S., et al. (1989) Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N. Engl. J. Med., 320, 1293–1299. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty P.K., Schmitz-Abe K., Kennedy E.K., Mamady H., Naas T., Durie D., Campagna D.R., Lau A., Sendamarai A.K., Wiseman D.H., et al. (2014) Mutations in TRNT1, encoding the CCA-adding enzyme, cause congenital sideroblastic anemia with B cell immunodeficiency, periodic fevers and developmental delay (SIFD). Blood, 124, 2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bottomley S.S., Fleming M.D. (2014) Sideroblastic anemia: diagnosis and management. Hematol. Oncol. Clin. North Am., 28, 653–670, v. [DOI] [PubMed] [Google Scholar]
- 22.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giege R., et al. (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet., 87, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weraarpachai W., Sasarman F., Nishimura T., Antonicka H., Aure K., Rotig A., Lombes A., Shoubridge E.A. (2012) Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet., 90, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.