

Abstract
Based on a 3D-QSAR pharmacophore derived from a diverse set of known cyclin-dependent kinase 9 (CDK9) inhibitors and a composite pharmacophore extracted from the complex structure of flavopiridol (FVP)-CDK9, thirty novel 5-fluoro-N2,N4-diphenylpyrimidine-2,4-diamine derivatives were designed and synthesized. Initial tests against four tumor cell lines with the sulforhodamine B (SRB) assay identified a series of potent compounds with GI50 values at lower micromolar or submicromolar level. Most of the highly cytotoxic compounds exhibited potent inhibitory activities against both CDK2/cyclin E1 and CDK9/cyclin T1. Notably, inhibitions against the two enzymes were generally correlated well with the cytotoxicity of these compounds. Appreciable inhibition was also observed for selected compounds in the anti-HIV-1 assay. Docking studies on compounds 6d and 9g provided conducive clues to further structural optimization.
Keywords: 5-fluoro-N2; N4-diphenylpyrimidine-2, 4-diamines; cyclin-dependent kinases; pharmacophore; cytotoxicity; anti-HIV-1
1. Introduction
Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases involved in the control of cell cycle and the regulation of gene transcription. Once bound to the regulatory subunit cyclins, CDKs can phosphorylate the downstream substrates and activate various cellular processes. Due to their intense involvement in many physiological processes and diseases, CDKs have attracted extensive research interests1. Several of the CDKs play critical roles in cell cycle progression and a number of CDK inhibitors are currently under clinical trials for the treatment of various cancers2. As a member of the CDK family, CDK9 plays a pivotal role in transcriptional regulation and is regarded as a potential target in oncology, virology and cardiology3. CDK9 complexes with cyclin T to form transcription elongation factor b (P-TEFb) and regulate gene transcription by phosphorylating the C-terminus of the large subunit of RNA polymerase II. It has been reported that differential effects on HIV replication and T-cell activation were observed upon direct inhibition of the P-TEFb kinase activity in primary human peripheral blood lymphocytes4. Therefore, selective CDK9 inhibitors have been actively pursued5–9 and evaluated as potentially effective agents to block HIV-1 replication5–7. The X-ray crystal structures of these inhibitors complexed with CDK9 as well as CDK2 have been resolved and analyzed to investigate the structural basis for isotype selectivity10–12. Nevertheless, recent studies13,14 have also suggested implication of CDK2 in HIV-1 transcription regulation through functional link with CDK9 (for example, by phosphorylating CDK9 14). Therefore, compounds with good potency against CDK2 and CDK9 might be therapeutically useful in the treatment of cancer and HIV infection.
To search for new CDK inhibitors, especially CDK9 inhibitors as anti-cancer and anti-HIV agents, we have previously established quantitative pharmacophore models based on a diverse set of CDK9 inhibitors15. The most representative model depicted a pharmacophore with two hydrogen-bond acceptor features and two hydrophobic aromatic features. As illustrated in Figure 1a, such pharmacophore matches well with the chemical features of flavopiridol (FVP), a flavonoid pan-CDK inhibitor and the most potent inhibitor against P-TEFb identified to date16. The interaction mode of FVP with CDK9 has been revealed by the crystal structure of the FVP-CDK9 complex17. As shown in Figure 1b, the FVP molecule occupies the ATP-binding site of CDK9. The O4 oxygen and the O5 hydroxyl of FVP form hydrogen bonds with the CDK9 hinge residues Cys106 and Asp104, respectively. An additional hydrogen bond is observed between the O3 hydroxyl and the Asp167 side chain. Moreover, the chloro-phenyl ring of FVP makes hydrophobic contacts with Ile25, and the protonated N1 of the piperidinyl group presents electrostatic interaction with Asp167. Therefore, a composite pharmacophore with three hydrogen bond forming atoms, one hydrophobic aromatic center and one positively charged center was constructed (Figure 1c). Remarkably, the quantitative and the composite pharmacophore models are in good consistency.
Figure 1.
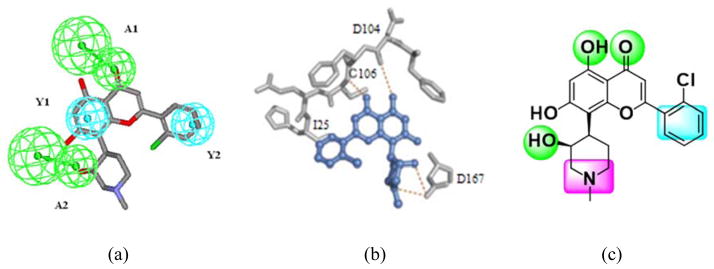
Pharmacophore models for CDK9 inhibitors. (a) a 3D-QSAR pharmacophore (with hydrogen-bond acceptors represented in green and hydrophobic aromatic features represented in cyan) derived from diverse CDK9 inhibitors and its mapping with FVP; (b) Key interactions between FVP and CDK9 revealed by their co-crystal structure; (c) a composite pharmacophore extracted from the interaction mode shown in (b). The hydrogen bond forming atoms are represented in green, the hydrophobic aromatic center is represented in cyan and the positively charged center is represented in purple.
We report herein the design, synthesis and biological evaluation of a series of novel 5-fluoro-N2,N4-diphenylpyrimidine-2,4-diamine derivatives. This novel class of compounds were designed by following a pharmacophore-oriented scaffold-hopping strategy. The hybrid pharmacophore combining key features in the 3D-QSAR and composite models demonstrated in Figure 1 was used to guide the molecular design (Figure 2). To ensure kinase inhibition and convey structural novelty, the 2-aminopyrimidine core structure, which is a privileged scaffold for CDK inhibitors8,18,19, was utilized for the design. 5-Fluoro substitution was introduced according to previous structure-activity relationship studies on CDK9 inhibitors with the 2-anilino-4-heteroarylpyrimidine scaffold8,18. The general formula and the pharmacophoric features of the target compounds are shown in Figure 2.
Figure 2. General formula of target compounds and their pharmacophoric features.

R1 contains a hydrogen bond forming atom and/or a positively charged center. Various R2 and R3 substituents are incorporated on the two phenyl rings to meet the pharmacophoric requirements.
2. Results and discussion
2.1 Chemistry
The general synthetic route for compounds 6a-6f was shown in Scheme 1. Compound 2a was prepared through a cross-coupling reaction between 1-bromo-3-nitrobenzene (1a) and morpholine20, and then followed by catalytic hydrogenation to afford compound 3a. Compounds 3b and 3c were synthetized similarly. Compounds 5a-5d were prepared by regio-selective substitution of the appropriate amines to 2,4-dichloro-5-fluoropyrimidine21. Further substitutions between 3a-3c and 5a-5d in alcoholic solvent afforded the target compounds 6a-6f 22.
Scheme 1.

Reagents and conditions: a. morpholine or N-methyl piperazine, CuI, L-proline, K2CO3, DMSO, N2, 100–110°C; b. Pd/C, H2, CH3OH or THF; c. the appropriate amine, Et3N, EtOH; d. CF3COOH, i-PrOH or 2-BuOH, reflux; e. 3-Nitroaniline (for 7) or 3-aminobenzoic acid (for 10), CF3COOH, i-PrOH or 2-BuOH, reflux; f. the appropriate acid or amine, DIEA, HATU, DMF.
The preparation of compounds 9a-9h and 11a-11p was also illustrated in Scheme 1. Compound 5b was reacted with m-nitroaniline and m-aminobenzoic acid to give compounds 7 and 10, respectively. Catalytic hydrogenation of compound 7 in the presence of Pd/C produced compound 8. Both 8 and 10 were subjected to condensation reactions under the treatment of 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIEA) to afford the target amide compounds 9a-9h and 11a-11p 23.
2.2 Biological evaluation
All the target compounds were initially tested against the tumor cell lines of A594, DU145, KB and KBvin with the sulforhodamine B (SRB) assay. The inhibitory activities of the compounds in terms of GI50 values were shown in Tables 1, 2 and 3.
Table 1.
Inhibitory activities of compounds 6a-6f against tumor cell growth.
| Compd. | R2 | GI50 (μM) a
|
|||
|---|---|---|---|---|---|
| A549 b | DU145 | KB | KBvin | ||
| 6a |
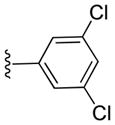
|
11.59 ± 1.03 | > 10 c | 14.80 ± 1.02 | 22.47 ± 2.46 |
| 6b |
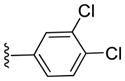
|
12.02 ± 0.90 | > 10 | > 10 | > 10 |
| 6c |
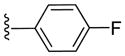
|
3.83 ± 1.18 | > 10 | > 10 | > 10 |
| 6d |
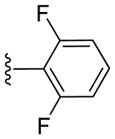
|
1.09 ± 0.46 | 4.24 ± 1.05 | > 10 | > 10 |
| 6e |
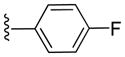
|
18.28 ± 3.34 | > 10 | > 10 | > 10 |
| 6f |
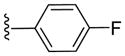
|
1.13 ± 0.61 | 0.91 ± 0.12 | 0.96 ± 0.21 | 0.91 ± 0.30 |
| FVP | - | 0.14 ± 0.007 | 0.15 ± 0.007 | 0.16 ± 0.007 | 0.18 ± 0.005 |
The GI50 values are the average of three independent measurements;
Cell lines: A549 (lung cancer), DU-145 (prostate cancer), KB (nasopharyngeal carcinoma) and KBvin (vincristine-resistant KB subline);
No significant inhibition against tumor cell growth was observed under the concentration of 10 μM.
Table 2.
Inhibitory activities of compounds 9a-9h against tumor cell growth
| Compd. | R1 | GI50 (μM) a
|
|||
|---|---|---|---|---|---|
| A549 b | DU145 | KB | KBvin | ||
| 9a |
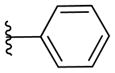
|
37.82 ± 4.88 | > 10 c | > 10 | > 10 |
| 9b |
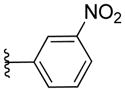
|
7.25 ± 0.96 | 12.62 ± 0.70 | 15.10 ± 1.16 | 11.35 ± 2.08 |
| 9c |
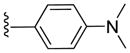
|
> 10 | > 10 | > 10 | > 10 |
| 9d |
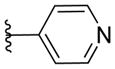
|
1.65 ± 0.67 | 4.33 ± 2.39 | 5.28 ± 3.49 | 4.57 ± 2.58 |
| 9e |
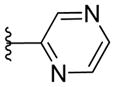
|
10.29 ± 1.98 | 26.38 ± 4.57 | 23.55 ± 3.92 | 20.10 ± 2.95 |
| 9f |
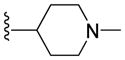
|
1.33 ± 0.10 | 1.15 ± 0.33 | 1.70 ± 0.80 | 11.93 ± 1.92 |
| 9g |
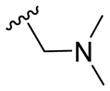
|
1.18 ± 0.13 | 1.21 ± 0.08 | 1.26 ± 0.05 | 1.81 ± 0.60 |
| 9h |
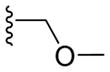
|
2.05 ± 0.52 | 4.46 ± 0.70 | 6.25 ± 1.38 | 3.94 ± 1.04 |
| FVP | - | 0.14 ± 0.007 | 0.15 ± 0.007 | 0.16 ± 0.007 | 0.18 ± 0.005 |
The GI50 values are the average of three independent measurements.
Cell lines: A549 (lung cancer), DU-145 (prostate cancer), KB (nasopharyngeal carcinoma) and KBvin (vincristine-resistant KB subline);.
No significant inhibition against tumor cell growth was observed under the concentration of 10 μM.
Table 3.
Inhibitory activities of compounds 11a-11p against tumor cell growth
| Compd. | R1 | GI50 (μM) a
|
|||
|---|---|---|---|---|---|
| A549 b | DU145 | KB | KBvin | ||
| 11a |
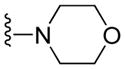
|
11.21 ± 2.06 | 11.94 ± 0.81 | 14.58 ± 2.93 | 13.05 ± 2.24 |
| 11b |
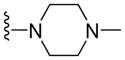
|
9.07 ± 1.30 | 10.89 ± 0.82 | 12.58 ± 3.05 | 12.27 ± 1.90 |
| 11c |
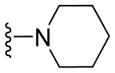
|
13.21 ± 1.45 | 13.02 ± 0.65 | 14.78 ± 2.53 | 13.81 ± 0.87 |
| 11d |
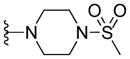
|
12.22 ± 1.95 | 17.91 ± 1.09 | 20.29 ± 4.09 | > 10 c |
| 11e |
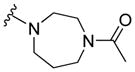
|
9.96 ± 0.60 | 15.65 ± 1.44 | 14.88 ± 2.20 | 16.68 ± 1.46 |
| 11f |
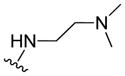
|
1.45 ± 0.07 | 4.34 ± 1.40 | 4.16 ± 0.69 | 7.26 ± 3.65 |
| 11g |
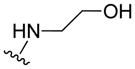
|
11.85 ± 2.08 | 13.38 ± 2.60 | 12.39 ± 2.48 | 21.40 ± 2.38 |
| 11h |
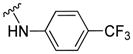
|
> 10 | > 10 | > 10 | > 10 |
| 11i |
|
> 10 | > 10 | > 10 | > 10 |
| 11j |
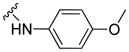
|
11.03 ± 0.52 | > 10 | > 10 | 15.71 ± 1.38 |
| 11k |
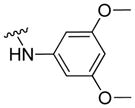
|
13.85 ± 0.15 | 13.67 ± 1.72 | 15.47 ± 1.58 | 12.74 ± 1.65 |
| 11l |
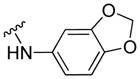
|
12.86 ± 0.71 | 11.58 ± 0.18 | 14.08 ± 0.79 | 12.01 ± 1.66 |
| 11m |
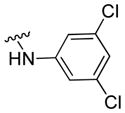
|
17.45 ± 2.49 | 17.50 ± 3.03 | 20.08 ± 1.06 | 20.73 ± 3.14 |
| 11n |
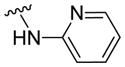
|
15.30 ± 0.86 | 15.06 ± 1.54 | 20.03 ± 10.96 | 13.67 ± 1.59 |
| 11o |
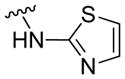
|
> 10 | > 10 | > 10 | > 10 |
| 11p |
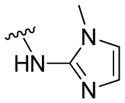
|
10.12 ± 0.80 | 15.10 ± 2.27 | 14.57 ± 1.70 | 13.12 ± 0.96 |
| FVP | - | 0.14 ± 0.007 | 0.15 ± 0.007 | 0.16 ± 0.007 | 0.18 ± 0.005 |
The GI50 values are the average of three independent measurements.
Cell lines: A549 (lung cancer), DU-145 (prostate cancer), KB (nasopharyngeal carcinoma) and KBvin (vincristine-resistant KB subline);.
No significant inhibition against tumor cell growth was observed under the concentration of 10 μM.
The cytotoxic results suggested that fluoro-substitution on 4-anilino might be important for the anti-proliferative effects (6a and 6b versus 6c and 6d). Therefore, p-fluoroanilino was retained at R2, while methoxyl substitution was introduced at R3 (6e), and morpholino was further replaced with 4-methylpiperazin-1-yl (6f) to explore their effects on cytotoxicity. Although the introduction of methoxyl group at R3 decreased the inhibitory activity (6c versus 6e), the presence of 4-methylpiperazin-1-yl instead of morpholino at R1 significantly increased potency (6e versus 6f). Such observation implicated that the electrostatic interaction between the residue Asp167 and the protonated nitrogen in the piperidinyl group is more advantageous over the hydrogen bonding interaction between the oxygen atom in the morpholino moiety and Asp167. Therefore, positively charged atoms should be preferred in the close proximity to R1.
Compounds 9a-9h and 11a-11p retained the p-fluoroanilino at R2, while incorporating diverse R1 substituents with different sizes, electronic effects and hydrogen bond forming capability to further investigated the structure-activity relationships at this molecular area. For both compounds 9 and 11, aliphatic substituents, either linear or cyclic, were generally favored over aromatic substituents at R1. The results implied that the R1 substituents might serve as a counterpart of the sugar moiety in the ATP structure, therefore, aliphatic substituents might be more likely to meet the requirements for optimal pharmacophoric orientation and molecular shape of CDK inhibitors. In addition, compounds with a positively charged center were more potent than their congeners with only hydrogen bond forming atoms (9g versus 9h; 11a versus 11b; 11f versus 11g), which suggests the importance of electrostatic interactions in this molecular area. The series 9 were generally more potent than the series 11. For compounds with aromatic substituents at R1 of 9, electron-withdrawing groups might be preferred over electron-donating moieties for cytotoxicity (9b versus 9c; 9a versus 9d). However, electrostatic properties showed no definite effects on the cytotoxicity of compounds in series 11.
Nine of the most cytotoxic compounds were further evaluated against CDK2/cyclin E1 and CDK9/cyclin T1. Most of these compounds exhibited potent inhibitory activities against both systems (Table 4). Due to limited data, it is difficult to deduce informative SAR from the results. However, for most compounds, the molecular potency generally correlated well with the cellular cytotoxicity, which indicated that CDK inhibition might be responsible for the cytotoxicity of these compounds. Compound 6f was the only exception. The discrepancy between cellular and molecular activities of compound 6f implicated the involvement of alternative molecular mechanisms other than CDK inhibition. For all the compounds tested at molecular level, only compounds 6d and 9d showed moderate selectivity to CDK9 over CDK2.
Table 4.
Inhibitory activities of selected compounds against CDK2/cyclin E1 and CDK9/cyclin T1
| Compd. | IC50 (μM)*
|
|
|---|---|---|
| CDK2/CyclinE1 | CDK9/CyclinT1 | |
| 6d | 5.85 | 0.33 |
| 6f | > 10 # | > 10 |
| 9d | 15.30 | 2.57 |
| 9f | 1.24 | 2.45 |
| 9g | 1.71 | 0.78 |
| 9h | 3.27 | 2.02 |
| 11b | 22.10 | 11.30 |
| 11e | 60.60 | 15.60 |
| 11f | 4.65 | 1.72 |
| FVP | 0.13 | 4.59 (nM) |
The IC50 value was interpolated from dose-response data with at least six different concentrations. Each test was performed in duplicate. Data represent the average of duplicate measurements.
No significant inhibition was observed at the concentration of 10 μM.
To determine whether selective CDK9 inhibition correlates with anti-HIV activity, three compounds (6d, 11e and 11f) with different level of selectivity to CDK9 were tested in an anti-HIV-1 assay. Appreciable inhibition was observed for all three compounds. Notably, the results did reveal a trend that higher selectivity to CDK9 might lead to better therapeutic index (Table 5). Further validation with more CDK9 inhibitors is undergoing.
Table 5.
Anti-HIV-1 activities of selected compounds.
| Compd. | EC50* | IC50* | Selectivity CDK2/CDK9 |
TI# |
|---|---|---|---|---|
|
| ||||
| (μM) | ||||
| 6d | 1.37 ± 0.42 | 10.22 ± 1.27 | 17.7 | 7.45 |
| 11e | 7.08 ± 1.54 | 15.45 ± 1.72 | 3.9 | 2.18 |
| 11f | 3.15 ± 0.66 | 3.40 ± 0.27 | 2.7 | 1.08 |
| FVP | 0.015 ± 0.005 | 0.067 ± 0.007 | 28.3 | 4.47 |
The EC50 and IC50 values are the average of three independent measurements;
TI, therapeutic index, the ratio of the IC50 value to the EC50 value.
2.3 Molecular modeling
Docking studies on selected compounds were performed to recognize their interaction modes with CDKs. As exemplified by the docking poses of compound 9g in the ATP-binding sites of CDK9, two alternative binding poses were suggested (Figure 3). For pose A (Figure 3a), the molecule assumed an orientation similar to that of FVP. Besides the hydrogen bond between N-1 atom of the pyrimidine ring and Cys106, additional hydrogen bonds were observed between the 2-amino and Asp104, as well as the carboxyl oxygen and Asp167. Electrostatic interaction between the nitrogen in N,N-dimethyl glycine and Asp167 were also monitored. Alternatively, compound 9g could also adopt pose B (Figure 3b). It was a turnover of pose A, where the N,N-dimethyl glycine moiety occupied the hydrophobic subpocket engaged by the chloro-phenyl of FVP. The N-1 atom and 2-amino moiety on the pyrimidine ring interacted with the hinge residue Cys106 of CDK9 through hydrogen bonding, while the 4-anilino group protruded toward Asp167. The electron-withdrawing fluoro-substitution on N4-phenyl made it electron deficient and presented electrostatic interaction with Asp 167. The disclosure of alternative binding poses of this compound class might provide new clues for further structural optimization.
Figure 3.


Alternative interaction modes of 9g as suggested by the docking study.
Compound 6d, the inhibitor with the highest selectivity toward CDK9, was docked to both CDK2 and CDK9 to provide insights on structural basis for selective CDK9 inhibition. As shown in Figure 4, compound 6d bound to the ATP sites of CDK2 and CDK9 with an orientation different from that of FVP. It adopted a pose similar to pose B in Figure 3b. The prevalence of pose B for 6d could be attributed to its structural features in R1 and R2. Due to the absence of protonable nitrogen in R1, the electrostatic interaction between R1 and Asp145 in CDK2 or Asp167 in CDK9 was missed. On the other hand, the electron-withdrawing difluoro-substitution (R2) on N4-phenyl strengthened its interaction with Asp145 (CDK2) or Asp167 (CDK9).
Figure 4.

Interaction modes of 6d with CDK2 (a) and CDK9 (b) as suggested by the docking study.
As indicated in Figure 4, the key interactions of 6d in CDK2 and CDK9 were similar. Hydrogen bonds existed between N-1 atom/2-amino moiety of 6d and the hinge residue (Leu83 for CDK2 and Cys105 for CDK9). The difluoro-substituted N4-phenyl ring extended to the proximity of Asp145 (CDK2) or Asp167 (CDK9). The most obvious difference between the interaction modes of 6d with CDK2 and CDK9 was the orientation of the morpholino ring in R1. Compound 6d assumed a condensed U-conformation in the ATP-binding site of CDK2, and the morpholino ring was virtually parallel to the N4-phenyl ring (Figure 4a). While, it was more extended in the binding site of CDK9 and the morpholino ring was basically vertical to the N4-phenyl ring. The changing of the Lys89 in CDK2 to Gly122 in CDK9 was responsible for the discrepancy in the interaction modes. Due to such a difference, the binding site of CDK9 is relatively larger and can accommodate more steric moieties in R1. The lone pair interaction between the nitrogen atom in morpholino and the Lys89 in CDK2 might cause further shift of the morpholino ring toward the N4-phenyl. The structural insights gained from docking studies might guide further molecular design of selective CDK9 inhibitors.
3. Experimental
3.1 Materials and methods
All reactions were monitored by thin-layer chromatography with pre-coated silica gel F254 plates purchased from Merck, Inc. All melting points were determined on Yanaco Melting point apparatus and are uncorrected. Mass spectra (MS) were taken in HR-ESI mode on Exactive Plus™ LC/MSD Orbitrap from Thermo Fisher Scientific. 1H-NMR spectra were recorded on Varian Mercury 300-MHz spectrometer. Chemicals were obtained from local suppliers and were used without further purification.
3.2 Chemistry
3.2.1 Preparation of compound 3
2-Nitro-4-bromoanisole (2.32 g, 10 mmol), morpholine (3.58 g, 40 mmol), CuI (0.19 g, 0.1 mmol), L-proline (0.23 g, 2 mmol) and K2CO3 (2.76 g, 20 mmol) were dissolved in 20 ml DMSO and stirred under nitrogen for 48 h at 100 °C. After cooling to room temperature, the reaction mixture was poured into water and extracted with ethyl acetate, the combined organic layers were successively washed by water and saturated solution of Na2CO3. The organic layer was dried over Na2SO4, and filtered. The solvents were removed under reduced pressure and the crude material was purified by column chromatography on silica gel, eluted with petroleum ether/ethyl acetate (10:1) to afford compound 2b as a yellow oil (1 g, 42%).
Compound 2b (1 g, 4.8 mmol) and Pd/C (0.1 g) were added in methanol and stirred under hydrogen atmosphere for 24 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure. The crude material was purified by column chromatography on silica gel, eluted with petroleum ether/ethyl acetate (2:1) to afford compound 3b as a white solid (0.3 g, 34%).
Compounds 3a and 3c were synthetized from different start materials using the same procedure as described.
3.2.2 Preparation of compound 5
2,4-Dichloro-5-fluoro-uricpyrimidine (4, 1.67 g, 10 mmol), p-fluoro-aniline (1.11 g, 10 mmol) and trifluoroacetic acid (1.52 g, 15 mmol) were mixed in 10 ml ethanol and stirred for 24 h at room temperature. The precipitation was filtered and then washed by ethanol, dried over Na2SO4, and filtered. The solvents were removed under reduced pressure getting 5b as a white solid (2.4 g, 99%).
Compound 5a, 5c and 5d were synthetized from different starting materials with the same procedure as described.
3.2.3 Preparation of compound 6
To a solution of compound 3a (122 mg, 0.687 mmol) and compound 5d (200 mg, 0.687 mmol) in 10 ml isopropanol, one drop of acetic acid was added. The reaction mixture was stirred at 110 °C and was refluxed for 24 h. After cooling to room temperature, the precipitation was filtered to get compound 6d as a white solid (200 mg, 40%).
Compounds 6a-6c and 6e-6f were synthetized from different starting materials using the same procedure as described.
Compound 6a: yield: 67 %, mp: 189–190°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.07 (s, 1H), 9.74 (s, 1H), 8.28 (d, J = 3.9 Hz, 1H), 7.87 (s, 2H), 7.31 (s, 1H), 7.15–7.18 (m, 3H), 6.74 (s, 1H), 3.74 (s, 4H), 3.08 (s, 4H). HR-ESI-MS: m/z = 434.0942 [M+H]+, calcd for C20H19N5OFCl2: 434.0945.
Compound 6b: yield: 90%, mp: 196–198°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.40 (s, 1H), 10.14 (s, 1H), 8.32 (d, J = 4.2 Hz, 1H), 8.04 (d, J = 1.8 Hz, 1H), 7.72 (d, J = 7.2 Hz, 1H), 7.56 (d, J = 8.7 Hz, 1H), 7.18–7.25 (m, 2H), 7.09 (d, J = 7.5 Hz, 1H), 6.81 (d, J = 6.6 Hz, 1H), 3.73 (s, 4H), 3.06 (s, 4H). HR-ESI-MS: m/z = 434.0952 [M+H]+, calcd for C20H19N5OFCl2: 434.0945.
Compound 6c: yield: 47%, mp: 220–222°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.36 (s, 1H), 9.03 (s, 1H), 8.08 (d, J = 3.6 Hz, 1H), 7.78 (dd, J = 5.1, 9.3 Hz, 2H), 7.21 (s, 1H), 7.12 (t, J = 6.6 Hz, 3H), 7.05 (t, J = 8.1 Hz, 1H), 6.50 (d, J = 7.5 Hz, 1H), 3.68 (t, J = 4.8 Hz, 4H), 2.96 (t, J = 4.8 Hz, 4H). HR-ESI-MS: m/z = 384.1625 [M+H]+, calcd for C20H20N5OF2: 384.1630.
Compound 6d: yield: 40%, mp: 181–183°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.19 (s, 1H), 8.92 (s, 1H), 8.10 (d, J = 3.3 Hz, 1H), 7.36–7.43 (m, 1H), 7.22 (t, J = 8.1 Hz, 2H), 7.02 (t, J = 8.4 Hz, 2H), 6.85 (t, J = 8.1 Hz, 1H), 6.41 (d, J = 6.9 Hz, 1H), 3.66 (t, J = 4.8 Hz, 4H), 2.90 (t, J = 4.8 Hz, 4H). HR-ESI-MS: m/z = 402.1534 [M+H]+, calcd for C20H19N5OF3: 402.1536.
Compound 6e: yield: 23%, mp: 195–198°C, 1H NMR (400MHz, DMSO-d6): δ 9.40 (s,1H), 8.09(d, J = 3.6 Hz,1H), 7.70-7.67 (m,4H), 7.12 (t, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 1H), 6.51 (dd, J = 2.8, 8.8 Hz, 1H), 3.76 (s, 3H), 3.64 (t, J = 4.4 Hz, 4H), 2.82 (t, J = 4.4 Hz, 4H). HR-ESI-MS: m/z = 414.1728 [M+H]+, calcd for C20H21N2O5: 414.1736.
Compound 6f: yield: 21%, mp: 71–73°C, 1H-NMR ( 300 MHz, DMSO-d6): δ 9.40 (s, 1H), 8.09 (d, J = 5.4 Hz, 1H), 7.71-7.64 (m, 4H), 7.11 (t, J = 8.5 Hz, 2H), 6.86 (d, J = 9.0 Hz, 1H), 6.52 (dd, J= 3.9, 5.4 Hz, 1H), 3.75 (s, 3H), 2.87 (m, 4H), 2.38 (m, 4H), 2.20 (s, 3H). HR-ESI-MS: m/z = 427.2040 [M+H]+, calcd for C22H25N6O F2: 427.2052.
3.2.4 Preparation compound 8
Compound 5b (2.42 g, 10 mmol), m-nitroaniline (1.38 g, 10 mmol) and trifluoroacetic acid (1.14 g, 10 mmol) were mixed in 10 ml isopropanol and stirred under reflux for 24 h. After cooling to room temperature, the precipitation was filtered and recrystallized in CH2Cl2 to afford a light yellow solid 2.5 g (7).
Compound 7 (2.5 g, 7.2 mmol) and Pd/C (0.25 g) were added in a mixture of methanol and THF, and stirred under hydrogen atmosphere for 24 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure and the crude material was recrystallized in CH2Cl2 to afford compound 8 as a light yellow solid (2.0 g, yield for two steps: 64%).
3.2.5 Preparation compound 9
Compound 8 (150 mg, 0.479 mmol) and pyrazine-2-carboxylic acid (65.3 mg, 0.527 mmol) were dissolved in a mixture of CH2Cl2 and THF. HATU (218.5 mg, 0.576 mmol) and DIEA (379 mg, 2.874 mmol) were then added. The reaction mixture was stirred at room temperature for 24 h, and was then poured into icy water. The precipitation was filtered and washed by dry methanol for several times to afford compound 9e as a white solid (101 mg, 51%).
Compounds 9a-9d and 9f-9h were synthetized from different start materials using similar procedure as described.
Compound 9a: yield: 43%, mp: 253–255°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.16 (s, 1H), 9.37 (s, 1H), 9.25 (s, 1H), 8.10 (d, J = 3.3 Hz, 1H), 8.04 (s, 1H), 7.93 (d, J = 6.9 Hz, 2H), 7.85 (dd, J = 5.7, 8.7 Hz, 2H), 7.49–7.58 (m,3H), 7.41 (d, J = 8.1Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.08 (t, J = 9.0 Hz, 2H). HR-ESI-MS: m/z = 418.1478 [M+H]+, calcd for C23H18N5OF2: 418.1474.
Compound 9b: yield: 33%, mp: 242–243°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.50 (s, 1H), 9.39 (s, 1H), 9.30 (s, 1H), 8.77 (s, 1H), 8.41 (dd, J = 9.0, 12.9 Hz, 2H), 8.09 (d, J = 10.2 Hz, 2H), 7.84 (t, J = 6.9 Hz, 3H), 7.42 (d, J = 7.2 Hz, 1H), 7.20–7.30 (m, 2H), 7.07 (t, J = 8.4 Hz, 3H). HR-ESI-MS: m/z = 463.1329 [M+H]+, calcd for C23H17N6O3F2: 463.1325.
Compound 9c: yield: 99%, mp: > 285°C, 1H NMR (300MHz, DMSO-d6): δ 9.77 (s, 1H), 9.34 (s, 1H), 9.18 (s, 1H), 8.08 (d, J = 3.6 Hz, 2H), 7.98 (s, 1H), 7.86-7.81 (m, 3H), 7.35 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.17-7.04 (m, 3H), 6.73 (d, J = 9 Hz, 2H), 2.98 (s, 6H). HR-ESI-MS: m/z = 461.1885[M+H]+, calcd for C20H21N2O5: 461.1896.
Compound 9d: yield: 48%, mp: 280°C (dec.), 1H NMR (400MHz, DMSO-d6): δ 10.40 (s, 1H), 9.38 (s, 1H), 9.28 (s, 1H), 8.77 (d, J = 5.2 Hz, 2H), 8.10 (d, J = 3.6 Hz, 1H), 8.04 (s, 1H), 7.84-7.81 (m, 4H), 7.43 (d, J = 8 Hz, 1H), 7.28-7.19 (m, 2H), 7.10-7.06 (m, 2H). HR-ESI-MS: m/z = 419.1414[M+H]+, calcd for C20H21N2O5: 419.1426.
Compound 9e: yield: 51%, mp: 265–266°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.47 (s,1H), 9.39 (s, 1H), 9.28 (d, J = 10.2 Hz, 2H), 8.92 (s, 1H), 8.79 (s, 1H), 8.12 (dd, J = 3.6, 10.2 Hz, 2H), 7.09 (m, 2H), 7.38 (d, J = 7.5 Hz, 2H), 7.22 (t, J = 8.1 Hz, 1H), 7.07 (t, J = 8.7 Hz, 2H). HR-ESI-MS: m/z = 420.1381 [M+H]+, calcd for C21H16N7OF2: 420.1379.
Compound 9f: yield: 35%, mp: 272°C (dec.), 1H-NMR (300 MHz, DMSO-d6): δ 9.73 (s, 1H), 9.36 (s, 1H), 9.17 (s, 1H), 8.08 (d, J = 3.6 Hz, 1H), 7.81–7.87 (m, 3H), 7.32 (d, J = 8.4 Hz, 1H), 7.07–7.18 (m, 4H), 2.82 (d, J = 11.1 Hz, 2H), 2.17 (s, 3H), 1.88 (t, J = 9.6 Hz, 2H), 1.60–1.69 (m, 4H). HR-ESI-MS: m/z = 439.2055 [M+H]+, calcd for C23H25N6OF2: 439.2052.
Compound 9g: yield: 48%, mp: 206–208°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.50 (s, 1H), 9.36 (s, 1H), 9.19 (s, 1H), 8.08 (d, J = 3.6 Hz, 1H), 7.85-7.82 (m, 3H), 7.36 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.20-7.10 (m, 3H), 3.04 (s, 2H), 2.26 (s, 6H). HR-ESI-MS: m/z = 399.1743 [M+H]+, calcd for C20H21N6OF2: 399.1739.
Compound 9h: yield: 24%, mp: 211–214°C, 1H NMR (300MHz, DMSO-d6): δ 9.57 (s, 1H), 9.37 (s, 1H), 9.21 (s, 1H), 8.08 (d, J = 3.9 Hz, 1H), 7.85-7.81 (m, 3H), 7.38-7.35 (m, 1H), 7.16-7.11 (m, 4H), 3.96 (s, 2H), 3.36 (s, 3H). HR-ESI-MS: m/z = 386.1421[M+H]+, calcd for C20H21N2O5: 386.1423.
3.2.6 Preparation compound 10
Compound 5d (2.42 g, 10 mmol), m-aminobenzoic acid (1.37 g, 10 mmol) and trifluoroacetic acid (1.14 g, 10 mmol) were mixed in 15 mL isopropanol and was stirred under reflux for 24 h. After cooling to room temperature, the precipitation was filtered and washed by CH2Cl2 for several times to afford compound 10 as a white solid (2.5 g, 73%).
3.2.7 Preparation compound 11
Compound 10 (200 mg, 0.585 mmol) and morpholine (62 mg, 0.702 mmol) were dissolved in DMF, HATU (267 mg, 0.702 mmol) and DIEA (302 mg, 2.34 mmol) were then added. The reaction mixture was stirred at room temperature for 24 h and then was poured into icy water. The precipitation was filtered to afford compound 11a as a white solid (188 mg, 78%).
Compound 11b-11p were synthetized from different start materials using similar procedure as described.
Compound 11a: yield: 78%, mp: 174–176°C, 1H NMR (300MHz, DMSO-d6): δ 9.42 (s, 1H), 9.37 (s, 1H), 8.12 (d, J = 3.6 Hz, 1H), 7.78-7.74 (m, 3H), 7.65 (d, J = 7.5 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.15 (t, J = 8.7 Hz, 2H), 6.89 (d, J = 7.8 Hz, 1H), 3.55 (m, 6H), 3.31 (m, 2H). HR-ESI-MS: m/z = 412.1574[M+H]+, calcd for C20H21N2O5: 412.1580.
Compound 11b: yield: 24%, mp: 184–185°C, 1H NMR (300MHz, DMSO-d6): δ 9.42 (s, 1H), 9.37 (s, 1H), 8.12 (d, J = 3.6 Hz, 1H), 7.78-7.74 (m, 3H), 7.65 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.15 (t, J = 8.7 Hz, 2H), 6.86 (d, J = 7.8 Hz, 1H), 3.57 (s, 2H), 3.31 (s, 2H), 2.26 (s, 4H), 2.16 (s, 3H). HR-ESI-MS: m/z = 425.1883[M+H]+, calcd for C20H21N2O5: 425.1896.
Compound 11c: yield: 73%, mp: 197–199°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.41 (s, 1H), 9.35 (s, 1H), 8.12 (d, J = 3.6 Hz, 1H), 7.74–7.79 (m, 3H), 7.64 (d, J = 8.1 Hz, 1H), 7.25 (t, J = 8.1 Hz, 1H), 7.15 (t, J = 8.4 Hz, 2H), 6.84 (d, J = 7.5 Hz, 1H), 3.54 (brs, 2H), 3.24 (brs, 2H), 1.39–1.58 (brs, 6H). HR-ESI-MS: m/z = 410.1794 [M+H]+, calcd for C22H22N5OF2: 410.1787.
Compound 11d: yield: 78%, mp: 263–265°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.42 (s, 1H), 9.38 (s, 1H), 8.12 (d, J = 3.6 Hz, 1H), 7.76 (t, J = 5.1 Hz, 3H), 7.68 (d, J = 8.4 Hz, 1H), 7.28 (t, J = 8.1 Hz, 1H), 7.16 (t, J = 8.7 Hz, 2H), 6.92 (d, J = 7.2 Hz, 1H), 3.55–3.80 (brs, 2H), 3.35–3.50 (brs, 2H), 3.00–3.20 (brs, 4H), 2.89(s, 3H). HR-ESI-MS: m/z = 489.1518 [M+H]+, calcd for C22H23N6O3F2S: 489.1515.
Compound 11e: yield: 39%, mp:161–163°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.42 (s, 1H), 9.37 (s, 1H), 8.12 (d, J = 3.0 Hz, 1H), 7.74–7.79 (m, 3H), 7.63 (d, J = 7.2 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.13 (t, J = 4.2 Hz, 2H), 6.76–6.83 (m, 1H), 3.50–3.61 (m, 3H), 3.40–3.45 (m, 3H), 1.97–2.02 (m, 3H), 1.79–1.85 (m, 2H), 1.63 (s, 1H), 1.49 (s, 1H). HR-ESI-MS: m/z = 467.2015 [M+H]+, calcd for C24H25N6O2F2: 467.2002.
Compound 11f: yield: 34%, mp: 174–176°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.39 (s, 1H), 9.32 (s, 1H), 8.20 (m, 1H), 8.10 (d, J = 3.6 Hz, 1H), 8.02 (s, 1H), 7.83-7.75 (m, 3H), 7.34-7.25 (m, 2H), 7.11 (t, J = 8.7 Hz, 2H), 2.36 (t, J = 7.2 Hz, 4H), 2.16 (m, 6H). HR-ESI-MS: m/z = 413.1899 [M+H]+, calcd for C21H23N6OF2: 413.1896.
Compound 11g: yield: 67%, mp: 212–214°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.39 (s, 1H), 9.32 (s, 1H), 8.26 (t, J = 5.4 Hz, 1H), 8.11 (d, J = 3.9 Hz, 1H), 8.08 (s, 1H), 7.79–8.83 (m, 2H), 7.34 (t, J = 7.8 Hz, 1H), 7.29 (t, J = 7.8 Hz, 1H), 7.11 (t, J = 8.7 Hz, 2H), 4.69 (t, J = 5.7 Hz, 1H), 3.48 (dd, J = 6.3, 9.0 Hz, 2H), 3.29 (t, J = 6.0 Hz, 1H). HR-ESI-MS: m/z = 386.1431 [M+H]+, calcd for C19H18N5O2F2: 386.1423.
Compound 11h: yield: 5.7%, mp:247–248°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.51 (s, 1H), 9.42 (d, J = 5.7 Hz, 1H), 9.41 (s, 1H), 8.13–8.17 (dd, J = 3.9, 12.0 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 6.9 Hz, 1H), 7.79 (dd, J = 4.8, 9.0 Hz, 2H), 7.71 (d, J = 9.3 Hz, 2H), 7.47 (d, J = 8.1 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 8.7 Hz, 2H). HR-ESI-MS: m/z = 486.1362 [M+H]+, calcd for C24H17N5OF5: 486.1348.
Compound 11i: yield: 50%, mp: 269–271°C, 1H-NMR (300 MHz, DMSO-d6): δ 9.98 (s, 1H), 9.37 (s, 1H), 8.11 (t, J = 3.6 Hz, 2H), 7.81 (t, J = 8.7 Hz, 3H), 7.61 (d, J = 8.7 Hz, 2H), 7.44 (d, J = 8.4 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.09 (t, J = 8.7 Hz, 2H), 6.92 (d, J = 8.7 Hz, 2H), 3.72 (t, J = 4.8 Hz, 4H), 3.06 (t, J = 4.8 Hz, 4H). HR-ESI-MS: m/z = 503.2010 [M+H]+, calcd for C27H25N6O2F2: 503.2002.
Compound 11j: yield: 69%, mp: 227–228°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.05 (s, 1H), 9.39 (d, J = 3.6 Hz, 1H), 8.13 (d, J = 3.6 Hz, 2H), 7.78–7.83 (m, 3H), 7.65 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 7.5 Hz, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.09 (t, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H). HR-ESI-MS: m/z = 448.1591 [M+H]+, calcd for C24H20N5O2F2: 448.1580.
Compound 11k: yield:47 %, mp:214–216°C, 1H-NMR (300 MHz, DMSO-d6): δ 10.09 (s, 1H), 9.40 (s, 1H), 8.13 (s, 2H), 7.78–7.81 (m, 3H), 7.42 (t, J = 7.5 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 7.07–7.13 (m, 4H), 6.25 (s, 1H), 3.72 (s, 6H). HR-ESI-MS: m/z = 478.1697 [M+H]+, calcd for C25H22N5O3F2: 478.1685.
Compound 11l: yield: 45%, mp: 224–226°C, 1H-NMR ( 300 MHz, DMSO-d6): δ 10.07 (s, 1H), 9.39 (s, 2H), 8.12 (t, J = 3.9 Hz, 2H), 7.78–7.82 (m, 3H), 7.41 (t, J = 1.8 Hz, 2H), 7.35 (t, J = 7.8 Hz, 1H), 7.16 (dd, J = 2.1, 8.4 Hz, 1H), 7.09 (t, J = 9.0 Hz, 2H), 6.87 (d, J = 8.4 Hz, 1H), 5.99 (s, 2H). HR-ESI-MS: m/z = 462.1383 [M+H]+, calcd for C24H18N5O3F2: 462.1372.
Compound 11m: yield: 25%, mp: 224–226°C, 1H-NMR (300 MHz, CD3OD): δ 8.13 (s, 1H), 7.87 (d, J = 3.0 Hz, 1H), 7.65 (d, J = 1.5 Hz, 2H), 7.56–7.60 (m, 3H), 7.38 (d, J = 7.2 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 7.10 (s, 1H), 6.88 (t, J = 8.7 Hz, 2H). HR-ESI-MS: m/z = 486.0710 [M+H]+, calcd for C23H16N5OF2Cl2: 486.0695.
Compound 11n: yield: 26%, mp: 131–133°C, 1H-NMR ( 300 MHz, DMSO-d6): δ 10.58 (s, 1H), 9.39 (d, J = 5.7 Hz, 2H), 8.36 (d, J = 3.6 Hz, 1H), 8.12–8.19 (m, 3H), 7.77–7.86 (m, 4H), 7.74 (d, J = 7.8 Hz, 1H), 7.30 (t, J = 8.1 Hz, 1H), 7.05–7.16 (m, 3H). HR-ESI-MS: m/z = 419.1426 [M+H]+, calcd for C22H17N6OF2: 419.1426.
Compound 11o: yield: 100%, mp: > 285°C, 1H NMR(300MHz, DMSO-d6): δ 9.32 (s, 1H), 9.18 (s, 1H), 8.23 (s, 1H), 8.09 (d, J = 3.6 Hz, 1H), 7.88-7.84 (m, 2H), 7.67 (d, J = 7.5 Hz, 2H), 7.20-7.18 (m, 2H), 7.15-7.07 (m, 3H), 6.60 (s, 1H). HR-ESI-MS: m/z = 425.0984[M+H]+, calcd for C20H21N2O5: 425.0991.
Compound 11p:yield: 48%, mp: 221–223°C, 1H-NMR ( 300 MHz, DMSO-d6): δ 10.06 (s, 1H), 9.36 (s, 1H), 9.23 (s, 1H), 8.10 (d, J = 3.6 Hz, 1H), 8.03 (s, 1H), 7.82 (dd, J = 4.8, 9.0 Hz, 2H), 7.07–7.43 (m, 7H), 3.96 (s, 3H). HR-ESI-MS: m/z = 422.1538 [M+H]+, calcd for C21H18N7OF2: 422.1535.
3.3 Biological evaluation
3.3.1 Cytotoxic activity assay
The following human tumor cell lines were used in the assay: A549 (lung cancer), DU-145 (prostate cancer), KB (nasopharyngeal carcinoma) and KBvin (vincristine-resistant KB subline). All cell lines were obtained from the Lineberger Comprehensive Cancer Center (UNC-CH) or from ATCC (Rockville, MD) and were cultured in RPMI-1640 medium supplemented with 25 mM HEPES, 0.25% sodium bicarbonate, 10% fetal bovine serum, and 100 mg/mL kanamycin.
The cytotoxicity assay was performed by following experimental protocols described in the reference24. Freshly trypsinized cell suspensions were seeded in 96-well microtiter plates at densities of 1500–7500 cells per well with compounds added from DMSO-diluted stock. After 3 days in culture, attached cells were fixed with cold 50% trichloroacetic acid and then stained with 0.4% sulforhodamine B (SRB). The absorbance at 562 nm was measured using a microplate reader after solubilizing the bound dye. The mean IC50 is the concentration of agents that reduces cell growth by 50% under the experimental conditions and it is the average from three independent determinations that are reproducible and statistically significant.
3.3.2 CDK2/Cyclin E1 kinase assay
The kinase assay against CDK2/cyclinE1 was performed by following experimental protocols described in the reference25. The compounds were mixed with the enzyme (2.28 ng) in a buffer containing 40 mM Hepes/Tris(pH 7.4), 0.8 mM EGTA/Tris, 8 mM MgCl2, 1.6 mM DTT and 0.008% Tween 20. Thereafter, the reaction was initiated by adding 100 nM of the substrate Ulight-CFFKNIVTPRTPPPSQGK-amide (MBP) and 10 μM ATP, and the mixture was incubated for 15 minutes at room temperature. For control basal measurements, the enzyme was omitted from the reaction mixture. Following incubation, the reaction was stopped by adding 13 mM EDTA. After 5 minutes, the anti-phospho-MBP antibody labeled with europium chelate was added. After 60 more minutes, the fluorescence transfer was measured at λex=337 nm, λem=620 nm and λem=665 nm using a microplate reader (Envision, Perkin Elmer). The enzyme activity was determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results were expressed as a percent inhibition of the control enzyme activity. The compounds were tested in each experiment at several concentrations to obtain an inhibition curve from which the IC50 values were calculated.
3.3.3 CDK9/Cyclin T1 kinase assay
The kinase assay against CDK9/cyclinT1 was performed by following experimental protocols described in the reference26. The compounds were mixed with the enzyme (21.72 ng) in a buffer containing 40 mM Hepes/Tris(pH 7.4), 0.8 mM EGTA/Tris, 8 mM MgCl2, 1.6 mM DTT and 0.008% Tween 20. Thereafter, the reaction was initiated by adding 100 nM of the substrate Ulight-CFFKNIVTPRTPPPSQGK-amide (MBP) and 10 μM ATP, and the mixture was incubated for 90 minutes at room temperature. For control basal measurements, the enzyme was omitted from the reaction mixture. Following incubation, the reaction was stopped by adding 13 mM EDTA. After 5 minutes, the anti-phospho-MBP antibody labeled with europium chelate was added. After 60 more minutes, the fluorescence transfer was measured at λex=337 nm, λem=620 nm and λem=665 nm using a microplate reader (Envision, Perkin Elmer). The enzyme activity was determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results were expressed as a percent inhibition of the control enzyme activity. The compounds were tested in each experiment at several concentrations to obtain an inhibition curve from which the IC50 values were calculated.
3.3.4 Anti-HIV-1 assay
The anti-HIV-1 assay was performed with NL4-3 virus by following experimental protocols described in the reference27. A 96-well microtiter plate was used to set up the HIV-1 NL4-3 replication assay. HIV-1 NL4-3 at a multiplicity of infection (MOI) of 0.01 was used to infect MT4 cells. Culture supernatants were collected on day 4 post infection for the p24 assay using an ELISA kit from ZeptoMetrix Corporation (Buffalo, New York).
3.4 Molecular modeling
The structures of compounds 6d and 9g were generated and molecular docking was performed with the Discovery Studio 2.5 software package (Accelrys, San Diego, USA). The structures of CDK2 and CDK9 were obtained from the Protein Data Bank (PDB codes: 4BCP and 4BCG). The docking calculation was carried out with the CDOCK protocol. Default settings were used. All calculations were performed on a DELL Precision T5500 workstation.
4. Conclusions
Thirty novel 5-fluoro-N2,N4-diphenylpyrimidine-2,4-diamine derivatives were designed as potential CDK, especially CDK9 inhibitors. Cytotoxicity for all the target compounds was measured with the SRB assay. Compounds with GI50 values against tumor cells at lower micromolar to submicromolar level were identified. Some most cytotoxic compounds exhibited potent inhibitory activities against both CDK2/cyclin E1 and CDK9/cyclin T1. Good correlation between cellular and molecular activities was observed. Noticeable inhibition against HIV-1 replication was also detected in an anti-HIV-1 assay, and there appeared to be a correlation between selective CDK9 inhibition and the anti-HIV-1 therapeutic index. Docking studies on this compound class hinted the existence of alternative binding poses and explained the realization of isotype selectivity, which provided new clues for further structural optimization.
Acknowledgments
This investigation was supported by the National Natural Science Foundation of China (81172985 and 81261120391) awarded to Z. Xiao. Partial support was also provided by NIH grants CA177584 from the National Cancer Institute and AI 033066 from the National Institute of Allergy and Infectious Disease awarded to K.H. Lee.
Notes and references
- 1.Lim S, Kaldis P. Development. 2013;140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 2.Cicenas J, Valius M. J Cancer Res Clin Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Fischer PM. Trends Pharmacol Sci. 2008;29:302–313. doi: 10.1016/j.tips.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Salerno D, Hasham MG, Marshall R, Garriga J, Tsygankov AY, Graña X. Gene. 2007;405:65–78. doi: 10.1016/j.gene.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali A, Ghosh A, Nathans RS, Sharova N, O’Brien S, Cao H, Stevenson M, Rana TM. Chem Bio Chem. 2009;10:2072–2080. doi: 10.1002/cbic.200900303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Németh G, Varga Z, Greff Z, Bencze G, Sipos A, Szántai-Kis C, Baska F, Gyuris Á, Kelemenics K, Szathmáry Z, Minárovits J, Kéri G, Őrfi L. Curr Med Chem. 2011;18:342–358. doi: 10.2174/092986711794839188. [DOI] [PubMed] [Google Scholar]
- 7.Németh G, Greff Z, Sipos A, Varga Z, Székely R, Sebestyén M, Jászay Z, Béni S, Nemes Z, Pirat JL, Volle JN, Virieux D, Gyuris Á, Kelemenics K, Áy É, Minarovits J, Szathmary S, Kéri G, Őrfi L. J Med Chem. 2014;57:3939–3965. doi: 10.1021/jm401742r. [DOI] [PubMed] [Google Scholar]
- 8.Shao H, Shi S, Huang S, Hole AJ, Abbas AY, Baumli S, Liu X, Lam F, Foley DW, Fischer PM, Noble M, Endicott JA, Pepper C, Wang S. J Med Chem. 2013;56:640–659. doi: 10.1021/jm301475f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albert TK, Rigault C, Eickhoff J, Baumgart K, Antrecht C, Klebl B, Mittler G, Meisterernst M. Br J Pharmacol. 2014;171:55–68. doi: 10.1111/bph.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumli S, Endicott JA, Johnson LN. Chem Biol. 2010;17:931–936. doi: 10.1016/j.chembiol.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Baumli S, Hole AJ, Noble ME, Endicott JA. ACS Chem Biol. 2012;7:811–816. doi: 10.1021/cb2004516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hole AJ, Baumli S, Shao H, Shi S, Huang S, Pepper C, Fischer PM, Wang S, Endicott JA, Noble ME. J Med Chem. 2013;56:660–670. doi: 10.1021/jm301495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debebe Z, Ammosova T, Breuer D, Lovejoy DB, Kalinowski DS, Karla PK, Kumar K, Jerebtsova M, Ray P, Kashanchi F, Gordeuk VR, Richardson DR, Nekhai S. Mol Pharmacol. 2011;79:185–196. doi: 10.1124/mol.110.069062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breuer D, Kotelkin A, Ammosova T, Kumari N, Ivanov A, Ilatovskiy AV, Beullens M, Roane PR, Bollen M, Petukhov MG, Kashanchi F, Nekhai S. Retrovirology. 2012;9:94. doi: 10.1186/1742-4690-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang C, Xiao Z, Guo Z. J Mol Graph Model. 2011;29:800–8008. doi: 10.1016/j.jmgm.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH. J Biol Chem. 2000;275:28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- 17.Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN. EMBO J. 2008;27:1907–1918. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones CD, Andrews DM, Barker AJ, Blades K, Daunt P, East S, Geh C, Graham MA, Johnson KM, Loddick SA, McFarland HM, McGregor A, Mossa L, Rudge DA, Simpson PB, Swain ML, Tama KY, Tucker JA, Walker M. Bioorg Med Chem Lett. 2008;18:6369–6373. doi: 10.1016/j.bmcl.2008.10.102. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Griffiths G, Midgley CA, Barnett AL, Cooper M, Grabarek J, Ingram L, Jackson W, Kontopidis G, McClue SJ, McInnes C, McLachlan J, Meades C, Mezna M, Stuart I, Thomas MP, Zheleva DI, Lane DP, Jackson RC, Glover DM, Blake DG, Fischer PM. Chem Biol. 2010;17:1111–1121. doi: 10.1016/j.chembiol.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Ma D, Cai Q, Zhang H. Org Lett. 2003;5:2453–2455. doi: 10.1021/ol0346584. [DOI] [PubMed] [Google Scholar]
- 21.Wang T, Lamb ML, Scott DA, Wang H, Block MH, Lyne PD, Lee JW, Davies AM, Zhang HJ, Zhu Y, Gu F, Han Y, Wang B, Mohr PJ, Kaus RJ, Josey JA, Hoffmann E, Thress K, Macintyre T, Wang H, Omer CA, Yu D. J Med Chem. 2008;51:4672–4684. doi: 10.1021/jm800343j. [DOI] [PubMed] [Google Scholar]
- 22.Pease EJ, Williams EJ, Bradbury RH, Pearson SE. US6649608B2. US Patent. 2003
- 23.Wendt MD, Geyer A, McClellan WJ, Rockway TW, Weitzberg M, Zhao X. Bioorg Med Chem Lett. 2004;14:3063–3068. doi: 10.1016/j.bmcl.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 24.Reddy MVB, Shen YC, Ohkoshi E, Bastow KF, Qian K, Lee KH, Wu TS. Eur J Med Chem. 2012;47:97–103. doi: 10.1016/j.ejmech.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Meijer L, Borgne A, Mulner O, Chong JPJ, Blow J, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 26.de Falco G, Giordano A. J Cell Physiol. 1998;177:501–506. doi: 10.1002/(SICI)1097-4652(199812)177:4<501::AID-JCP1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Huang L, Ho P, Lee KH, Chen CH. Bioorg Med Chem. 2006;14:2279–2289. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]