

Abstract
Aggregation of the amyloid-β protein (Aβ) contributes to the neurodegeneration characteristic of Alzheimer’s disease. Of particular importance are the early stages of aggregation, which involve the formation of soluble oligomers and protofibrils. In these studies, we demonstrate the potential for CE with UV detection using a polyethylene oxide separation matrix to identify the evolution of various oligomeric species of Aβ1–40. To demonstrate the efficacy of this technique, UV-CE was utilized to compare two methods commonly used to prepare Aβ for aggregation experiments and their effect on the formation of early aggregates. SEC-purified Aβ1–40 initially contained more small species, including monomer, than did freshly dissolved Aβ1–40 pretreated with hexafluoroisopropanol. Strikingly, the lag time to oligomer formation for SEC-isolated Aβ1–40 samples was ~23 h shorter compared to freshly dissolved Aβ1–40 samples. Furthermore, oligomers formed from the aggregation of SEC-purified Aβ1–40 persisted within solution for a longer period of time. These results indicate that the initial sample preparation has a drastic influence on the early stages of Aβ1–40 aggregation. This is the first report of the use of UV-CE with a separation matrix to study the effect of sample preparation on early aggregation of Aβ1–40. UV-CE was also used in parallel with dot blot analysis and inhibitory compounds to discern structural characteristics of individual oligomer peaks, demonstrating the capacity of UV-CE as a complimentary technique to further understand the aggregation process.
Keywords: Amyloid beta, Capillary electrophoresis, Dot blot, Oligomer
1 Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that currently affects 5.4 million Americans and is the sixth leading cause of death [1]. Amyloid-β (Aβ) is a partially folded protein hypothesized to contribute to the neurodegenerative effects of Alzheimer’s disease. In its monomer form, this protein is harmless [2]. However, this monomer can self-assemble into Aβ oligomers, soluble protofibrils, and eventually fibrillar aggregates that deposit as amyloid plaques in the brain [3]. Controversy currently exists over the direct effect Aβ has on neurodegeneration, but it appears likely that soluble Aβ aggregates (oligomers or protofibrils), rather than large, insoluble fibrils, may be the most physiologically active Aβ species [4,5]. This hypothesis is supported by experimental observations in vitro, which show that soluble aggregates formed by synthetic Aβ1–40 and Aβ1–42 can induce toxicity in cultured cells [6,7] and in vivo where soluble Aβ aggregates generated in cell culture drastically inhibit hippocampal long-term potentiation in rats [8]. Most recently, the direct neuron-to-neuron transfer and corresponding toxicity of soluble oligomers formed by synthetic Aβ1–42 have been demonstrated [9–12]. To better understand these pathogenic species that present a therapeutic target for AD, research efforts have focused upon elucidating the size and structure of oligomers formed along the aggregation pathway.
The study of Aβ oligomers has been impeded by the challenges associated with isolation and detection of soluble Aβ oligomers, specifically those formed from Aβ1–40, because these species are transient [13, 14], present at low concentrations [15], and cover a range of small sizes and potential confirmations. Many techniques have been examined for their ability to detect the smallest oligomeric Aβ species. These include SDS-PAGE and/or Western blotting [16, 17], MS [18–20], and SEC [16]. The capabilities of these techniques for detection of Aβ oligomers have been comprehensively summarized in a recent review by Pryor et al. [21]. One particular technique, CE, has potential for the detection of Aβ species formed throughout aggregation. A study by Sabella et al. pretreated Aβ1–40 and Aβ1–42 with ACN/sodium carbonate followed by dilution into phosphate buffer and used UV-CE to detect small (3–50 kDa) aggregate species with limited resolution [22]. Colombo et al. used a similar sample preparation to examine the effects of inhibitor compounds on Aβ1–42 and found that pixantrone inhibited >100 kDa oligomeric species [23]. In addition, the separation of large fibrillar species from monomer has been achieved for both Aβ1–40 and Aβ1–42 using UV-CE for samples pretreated with TFA/hexafluoroisopropanol (HFIP) before solubilization in sodium hydroxide and dilution into physiological buffer [24]. Picou et al. used a similar sample preparation and were able to show that when UV-CE detected both monomer and an additional peak in “monomer” samples aggregation kinetics were accelerated [25]. These studies demonstrate the ability of UV-CE to detect Aβ monomer, oligomer, and fibrillar aggregates and highlight the need to monitor the aggregation process over time with higher resolution of early, oligomeric species. Alternatively, CE with LIF has also been explored. Picou et al. used LIF-CE to detect multiple thioflavin T labeled fibrils of Aβ1–40 and Aβ1–42 that were pretreated with TFA/HFIP [26].
As illustrated by the studies above, sample preparation can vary widely among researchers and may have a strong influence on Aβ aggregate sizes formed at various points throughout aggregation. It is widely accepted that the initial solvent environment influences the starting conformation of the protein and thus the temporal evolution of aggregate sizes. Wang et al. examined the effect of three different initial solvent environments, namely TFA, DMSO, and urea, on the aggregation kinetics of Aβ [27]. They found that a “seeding effect” produced by the TFA and DMSO environments led to a faster rate of aggregation for Aβ previously exposed to these two solvents. Barrow et al. studied four different Aβ peptides and found that when dissolved in HFIP an α-helix structure, indicative of a monomeric state, is favored [28]. Previous studies using the inorganic solvent NaOH indicated both a higher presence of monomer/dimer species and the stability of a 2–6 nm aggregate formed over a 24-h time period [29,30]. Because of these differences, yet other studies have employed SEC to obtain monomeric species for aggregation studies without the use of nonnative solvents [31]. However, the lack of a high-resolution technique for studying the temporal evolution of early, oligomeric species has prohibited a complete understanding of how these sample preparations differ and thus influence the aggregation process.
In this study, we report the use of UV-CE with a polymer separation matrix to detect both oligomeric Aβ1–40 species (10–30 kDa) and the evolution of intermediate aggregates (100–300 kDa and >300 kDa) from Aβ1–40. To our knowledge, this is the first study to investigate Aβ1–40 oligomerization using a polymer matrix to separate oligomer species on CE. The efficacy of this novel detection approach is illustrated by applying CE to compare the oligomeric species formed during aggregation of Aβ1–40 samples prepared using two distinct techniques: SEC purification vs. fresh dissolution via dilution from a NaOH-solubilized stock that was pretreated with HFIP.
2 Materials and methods
2.1 Aβ preparation
Lyophilized Aβ1–40 (Anaspec, Fremont, CA) was stored desiccated at −80°C until preparation of freshly dissolved or SEC-purified Aβ1–40. Freshly dissolved Aβ1–40 was prepared by pretreating Aβ1–40 in HFIP to dissociate preexisting aggregates by reconstituting (4.33 mg/mL) in HFIP. This stock solution was aliquoted (0.0625 mg Aβ1–40), and HFIP was allowed to evaporate overnight. Vials were stored at −80°C until reconstitution in 5 mM NaOH just prior to experimentation, at which time the NaOH stock was diluted into 40 mM Tris-HCl (pH 8.0). SEC-purified Aβ1–40 was prepared as described previously [32]. Briefly, peptide was reconstituted (2 mg/mL) in 50 mM NaOH, and preexisting aggregates were removed by SEC (Superdex 75 HR10/30, GE Healthcare, Piscataway, NJ, USA) using 40 mM Tris-HCl (pH 8.0) running buffer. SEC-purified Aβ1–40 was flash frozen, shipped overnight on dry ice, and used immediately or stored at −80°C.
2.2 Aβ aggregation
To observe the time course for Aβ1–40 oligomer formation, freshly dissolved Aβ1–40 was prepared by diluting HFIP/NaOH-treated Aβ1–40 to a final concentration of 0.22 mg/mL. To compare the effects of sample preparation, SEC-isolated Aβ1–40 was diluted in parallel to a final concentration of 0.22 mg/mL. Both dilutions were made into 40 mM Tris-HCl (pH 8.0) containing 5 mM NaCl. Both sample preparations were incubated at 25°C under continuous agitation (800 rpm). Evolution of amyloid aggregates was evaluated by removing aliquots both prior to the onset of aggregation and at times between 5 and 48 h following the onset of aggregation and analyzing via UV-CE and dot blot analysis.
2.3 Aβ aggregation in the presence of inhibitors
To identify oligomer and fibril peaks within CE electropherograms, Congo Red and Orange G, reported to inhibit oligomer and fibril formation, respectively, were employed [33]. Congo Red and Orange G were solubilized in DMSO and combined, individually, with freshly dissolved Aβ1–40, prepared via dilution into 40 mM Tris-HCl (pH 8.0) containing 5 mM NaCl for final concentrations of 0.22 mg/mL Aβ1–40 and 0.15 mg/mL Congo Red or 0.23 mg/mL Orange G. Freshly dissolved Aβ1–40 devoid of inhibitor was also prepared as a control. Solutions were incubated at 25°C under continuous agitation (800 rpm). Both prior to the onset of aggregation and at times of 24 and 28 h following the onset of aggregation, an aliquot was removed and analyzed by UV-CE.
2.4 UV-CE measurements
Aliquots of 15 μL were separated via CE in 0.5% w/v polyethylene oxide (PEO; 2 MDa, Sigma-Aldrich, St. Louis, MO) coated capillaries (Lt = 31 cm; Ld = 10 cm) with a 0.5% PEO (300 kDa, Sigma-Aldrich) separation matrix and a capillary temperature of 25°C. Capillaries were dynamically coated by rinsing with water (10 min at 20 psi), 0.1 M HCl (15 min at 20 psi), 0.5% PEO (2 MDa) in water (20 min at 50 psi), and with water (15 min at 20 psi). CE separations using UV detection (214 nm) were performed using a P/ACE MDQ Glycoprotein System from Beckman Coulter, (Fullerton, CA). Samples were pressure injected at 0.5 psi for 8 s and separated at 7 kV. Between each run, the capillary was rinsed with deionized water for 10 min.
To correlate electrophoresis peaks with approximate molecular weights, 50 μL aliquots of aggregated Aβ1–40 were removed at selected times and ultrafiltrated (20 min, 14,100 × g) through Amicon (Millipore, Billerica, MA) filters with molecular weight cutoff values of 10, 30, 50, 100, and 300 kDa. The filtrate and retentate were analyzed via UV-CE to verify elution times for oligomers within each molecular weight range.
2.5 Dot blot analyses
Aliquots of 3 μL were spotted on nitrocellulose membranes with a pore size of 0.2 μm (VWR) and allowed to dry (1 h). Membranes were blocked (1 h) using 5% skim milk in TBS containing 0.01% Tween 20 (TBS-T). After washing three times with TBS-T, membranes were incubated (1 h) at 4°C with gentle shaking with either Aβ sequence specific 6E10 antibody (1:2000 dilution), Aβ oligomer specific A11 antibody (1:2000 dilution), or Aβ fibril specific OC antibody (1:4000 dilution). Membranes were again washed with TBS-T and bound antibody was detected via incubation (1 h) at 4°C with alkaline phosphatase conjugated anti-mouse IgG (6E10, 1:2000 dilution) or alkaline phosphatase conjugated anti-rabbit IgG (A11/OC, 1:3000 dilution). All antibodies were diluted in 5% skim milk in TBS-T. After washing with 1× TBS-T/MgCl2, membranes were developed using a substrate solution containing 0.17 mg/mL 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and 0.33 mg/mL nitroblue tetrazolium chloride (NBT). Dot blots were analyzed using ImageJ (http://imagej.nih.gov/ij/) and a dot was considered positive when it produced a signal at least three times higher than background.
2.6 Statistical analysis
The area of CE peaks were determined using Origin (V. 8.0) software from OriginLab (Northampton, MA). A Gaussian fit was applied to calculate the peak area and migration time. Differences in peak area were evaluated via an unpaired t-test using GraphPad QuickCalcs (GraphPad Software, San Diego, CA).
3 Results and discussion
3.1 Analysis of Aβ1–40 oligomers using CE with UV detection
To explore the utility of CE for the detection of Aβ1–40 oligomers that appear during early stages of Aβ1–40 aggregation, freshly dissolved Aβ1–40 at a concentration of 0.22 mg/mL in 40 mM Tris (pH 8.0) containing 5 mM NaCl was agitated at 800 rpm to promote amyloid assembly. The reaction was analyzed using UV-CE to assess the appearance of oligomers and progression into larger aggregate species. At 0 h, UV-CE demonstrated the presence of an early, sharp peak at an elution time of ~9 min (Fig. 1B) in addition to a broader peak migrating at 220 min (Fig. 1A). The size of these species was estimated using a filtration analysis similar to that performed by Sabella et al. who used molecular weight cutoff membranes to size early Aβ aggregates detected via UV-CE [22]. The sharp peak eluting at ~9 min exhibited a molecular weight of 10–30 kDa (~2–6 mer). The broad peak at 220 min exhibited a molecular weight of 100–300 kDa (~23–70 mer), indicating the presence of larger species before the onset of aggregation. A similar peak pattern for the small species was obtained after 5, 10, and 24 h of aggregation (Fig. 1B), while the peak corresponding to the larger species became broader and exhibited a progressively shorter migration time (Fig. 1A). At 28 h, a set of sharp peaks with migration times of 8–8.5 min appeared (Fig. 1B). The size of these species was estimated by filtration analysis to be >300 kDa (>70 mer). By 36 and 48 h, the intensity of the sharp peak at ~8.5 min increased and produced a single peak.
Figure 1.

Detection of freshly dissolved Aβ1–40 early aggregation into oligomers and higher molecular weight aggregates using UV-CE. (A) shows peaks for the total analysis time while (B) highlights the fastest migrating oligomer peaks. Results are representative of three independent experiments.
Using UV-CE with an SDS rinse for the detection of Aβ1–40 oligomers formed in PBS (pH 7.4) at room temperature, Sabella et al. observed a decrease in the intensity of a 10–30 kDa peak over an incubation period of 24 h with the disappearance of all peaks after 48 h [22]. However, in contrast to our results, no new peaks were observed over an aggregation period of 48 h. Our results add to the evidence that UV-CE is an effective means of detecting Aβ oligomers by further utilizing UV-CE to detect the formation of Aβ1–40 species ranging from 100 to 300 kDa and >300 kDa with the use of a polymer matrix and coated capillary.
3.2 Effect of sample preparation on Aβ1–40 aggregation
The procedure used to solubilize lyophilized Aβ1–40 has been reported to have an influence on the initial conformation and subsequent aggregation of this peptide [27]. In particular, organic solvents accelerate Aβ aggregation and misrepresent the “native” aggregation of the protein [34]. Furthermore, the presence of aggregates can serve as “seeds” that promote the aggregation process [35, 36]. Since a range of Aβ1–40 sizes was detected at the onset of aggregation (Fig. 1A) following a commonly used sample preparation method for freshly solubilized protein, we explored the aggregation ofAβ1–40 purified via SEC.
SEC-purifiedAβ1–40 was diluted to 0.22 mg/mL in 40mM Tris (pH 8.0) containing 5 mM NaCl and agitated at 800 rpm to promote amyloid assembly. The appearance of oligomers and larger aggregates was again analyzed via UV-CE coupled with filtration analysis. At 0 h, a single peak migrating at ~9.5 min was observed, which was estimated to range in size from 10 to 30 kDa (Fig. 2A). This observation is in contrast to freshly dissolved peptide, for which a second peak of higher molecular weight was also observed (Fig. 1A). In addition, after just 5 h of aggregation, the ~9.5 min peak was no longer observed, while a sharp peak with a faster migration time between 8 and 9 min appeared and persisted throughout the aggregation (Fig. 2B). Thus, these large species are formed ~23 h earlier than for the freshly dissolved sample. Again, the size of these species was estimated as >300 kDa (>70 monomer units). In addition, a later peak appeared that became broader and exhibited a progressively shorter migration time (Fig. 2A), which corresponded to a size of 100–300 kDa.
Figure 2.

Detection of SEC-isolated Aβ1–40 early aggregation into oligomers and higher molecular weight aggregates using UV-CE. (A) shows peaks for the total analysis time while (B) highlights the fastest migrating oligomer peaks. Results are representative of three independent experiments.
While the filtration analysis facilitates an estimate of aggregate size, it does not provide information about conformation of the aggregate species. The recent development of conformation-specific antibodies, some of which selectively recognize Aβ oligomers, has led to an increase in the application of dot blotting to study Aβ aggregation [37–39]. We utilized the conformation-specific antibodies A11 and OC, which have been demonstrated to recognize prefibrillar oligomers [37, 38] and fibrillar β-sheet structures [37], respectively. These analyses were coupled with the use of the sequence-specific antibody 6E10, known to recognize Aβ1–16 [37, 38], for detection of total Aβ protein.
Freshly dissolved and SEC-purified Aβ1–40 were aggregated under the same conditions used in UV-CE studies, and aggregates were analyzed via dot blot analysis (Fig. 3) with antibodies recognizing Aβ1–16 (A), oligomer conformations (A11, B), and fibrillar conformations (OC) (C). Both freshly dissolved and SEC-purified Aβ1–40 exhibit 6E10-positive stains for all aggregation times (Fig. 3A), indicating the steady presence of protein. A11- and OC-positive dots are detected in freshly dissolved Aβ1–40 samples after 24 h and in SEC-purified Aβ1–40 samples after just 5 h (Fig. 3B and C). These results indicate that both oligomeric and fibrillar species are formed much faster from SEC-purified protein than from freshly dissolved Aβ1–40. Furthermore, the observed pattern of oligomer and fibril conformations corresponds to the appearance of UV-CE peaks with an elution time of <8 min, indicating that these large (>300 kDa) species exhibit conformational characteristics. However, UVCE was able to detect both these species as well as broad peaks representing oligomeric species estimated to be 100–300 kDa (~23–70 mers) prior to the positive detection of either oligomeric or fibril species using dot blots. Together, these results indicate that conformation-specific antibodies may not recognize the smallest, earliest oligomers, but instead recognize larger aggregates.
Figure 3.

Freshly dissolved Aβ1–40 and SEC-purified Aβ1–40 aggregation monitored by dot blotting. Membranes were stained with sequence-specific antibody 6E10 (panel A), oligomer-specific antibody A11 (panel B), and fibril-specific antibody OC (panel C). Images were altered (brightness decreased by 15–20%, contrast increased by 50–55%, and converted to grayscale) to enhance visualization of dots and in some cases to align the dots.
The addition of inhibitors to the aggregating protein was used to further determine the nature of the UV-CE peaks eluting at ~8.5 min, which correlate with positive A11 (oligomer) and OC (fibril) dot blots. As shown in Fig. 4, these sharp peaks persisted when aggregations were performed in the presence of Orange G, known to inhibit Aβ fibril formation, but were absent after incubation with Congo Red, an inhibitor of Aβ oligomer formation [33]. Thus, these sharp peaks (>300 kDa) are oligomeric in nature. Interestingly, while the >300 kDa peaks are still present, their migration time appears to be faster, indicating that the Orange G may be affecting the oligomeric structure in some way. The appearance of conformation organization within these species may lead to an increased negative surface charge and thus the observed faster migration time. Petkova et al. have proposed a structural model for Aβ1–40 aggregates in which negatively charged N-terminal residues are exposed to solution, while hydrophobic residues are protected at the center of aggregates [40]. An increase in negative surface charge for Aβ1–42 aggregates has been experimentally observed by Wang et al. [41] using surface plasmon resonance tomonitor the absorption of Aβ1–42 to four model self-assembled monolayers (SAM) with varying degrees of hydrophobicity and charge. As aggregation progressed, the amount of protein absorbed onto positively charged NH2-SAM increased while the amount of protein absorbed onto negatively charged COOH-SAM decreased, suggesting an increase in negative surface charge of Aβ1–42 aggregates.
Figure 4.

Freshly dissolved Aβ1–40 peak patterns obtained in the presence and absence of inhibitory compounds. Aβ1–40 was aggregated alone (no inhibitor) or in the presence of 0.15 mg/mL Orange G or 0.23 mg/mL Congo Red.
To further understand the aggregation process as monitored by UV-CE, the peak area for the initially present species (10–30 kDa peak migrating at ~9 min) was compared to the peak area for the >300 kDa oligomer peak migrating at ~8.5 min (Fig. 5). For freshly dissolved protein (Fig. 5A), the peak area of the 10–30 kDa peak initially increases over 24 h. This increase could indicate the breakdown of the larger species (100–300 kDa) present at 0 h into smaller oligomers. However after 28 h, the 10–30 kDa peak area decreases and the faster migrating oligomeric peak (>300 kDa) appears. Thus, these large species likely result from the coalescence or growth of 10–30 kDa oligomers. Finally, between 28 and 48 h of aggregation, both the 10–30 kDa and >300 kDa peak areas further decrease, suggesting the formation of an aggregate too large to elute via CE. For SEC-purified Aβ1–40, the 10–30 kDa peak (Fig. 5B) likewise begins to decrease at 5 h in conjunction with appearance of the faster migrating oligomeric peak (>300 kDa). This transition occurs much earlier than for the freshly purified sample. Interestingly, SEC-purified Aβ1–40 exhibits a significantly larger initial population of 10–30 kDa species compared to freshly dissolved Aβ1–40. In contrast, the population of 10–30 kDa species for freshly dissolved Aβ1–40 increases initially and then begins to decrease before forming larger oligomeric species (>300 kDa). These trends suggest that a critical concentration of 10–30 kDa species may be necessary for the formation of larger aggregates that exhibit conformation. It is also of interest that the >300 kDa species formed from SEC-purified protein has a longer lifetime, appearing at 5 h and decreasing at 48 h, than does this same species formed from freshly dissolved Aβ1–40, which appears later and begins to decrease after only 36 h. Clearly, there are notable differences in the aggregation of freshly dissolved and SEC-purified samples, indicating the importance of utilizing a technique such as UV-CE to assess both the species within prepared samples and the appearance of early aggregates to fully understand the aggregation process.
Figure 5.
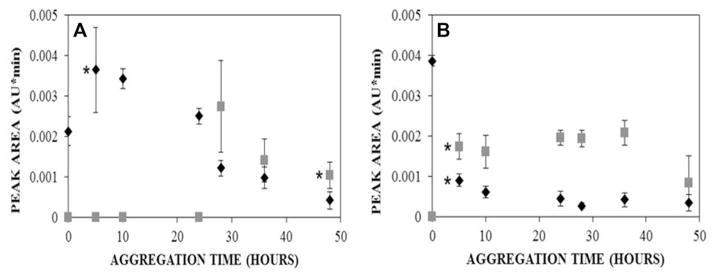
Effect of aggregation time on the peak areas obtained for the initially present 10–30 kDa species (◆, n = 3) and >300 kDa oligomer (
 , n = 3) for freshly dissolved Aβ1–40 (A) and SEC-purified Aβ1–40 (B). Peak area was determined using a Gaussian fit. (A) * represents the first time point in which peak areas are statistically different from the 0 h peak area with p < 0.05.
, n = 3) for freshly dissolved Aβ1–40 (A) and SEC-purified Aβ1–40 (B). Peak area was determined using a Gaussian fit. (A) * represents the first time point in which peak areas are statistically different from the 0 h peak area with p < 0.05.
4 Concluding remarks
In the current study, we explored the potential of UV-CE with a PEO matrix to monitor the early stages of Aβ1–40 aggregation. Strikingly, we found that SEC-isolated Aβ1–40 initially contained larger quantities of smaller species and exhibited a lag time to oligomer formation that was ~23 h shorter compared to freshly dissolved Aβ1–40. In addition, SEC-isolated Aβ1–40 produced oligomers that persisted within solution for a longer period of time. These findings indicate that the aggregate composition of the initial sample has a drastic effect on the early stages of aggregation, highlighting the importance of sample preparation. Furthermore, we utilized conformation-specific antibodies to confirm the presence of prefibrillar oligomers and aggregates with fibrillar structure. Correlations between dot blots and UV-CE analyses identified oligomers >300 kDa as exhibiting conformational characteristics, while oligomer and fibril-specific inhibitors confirmed a prefibrillar conformation associated with these species that is hypothesized to yield alterations in surface charge that render their short elution time.
These studies are the first to utilize a polymer separation matrix to study the early stages of native Aβ1–40 aggregation using UV-CE. The results indicate that the presence of this matrix does not provide a purely sieving effect as the species do not elute in a linear molecular weight order and therefore, work should be done in the future to investigate that exact nature of its interaction with the oligomeric species. UV-CE is a powerful tool to monitor the disappearance of Aβ species initially present and the appearance of new oligomers throughout aggregation. Furthermore, when coupled with other oligomer-specific techniques, UV-CE can contribute to the characterization of individual oligomer species. Together, these findings highlight the potential of UV-CE as a complementary technique with which to provide a more thorough understanding of Aβ aggregation.
Acknowledgments
This work was supported by grant numbers 1P30RR031154-02 and P30 GM103450 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and by support provided by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
Abbreviations
- Aβ
amyloid-β
- AD
Alzheimer’s disease
- HFIP
hexafluoroisopropanol
- PEO
polyethylene oxide
- SAM
self-assembled monolayers
- TBS-T
TBS containing 0.01% Tween 20
Footnotes
The authors have declared no conflict of interest.
References
- 1.Alzheimer’s Association. Alzheimers Dement. 2012;8:1–72. [Google Scholar]
- 2.Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato M, Kinoshita H, Toyo’oka T. Anal Chem. 2007;79:4887–4891. doi: 10.1021/ac0701482. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Velasquez FJ, Kotarek JA, Moss MA. J Neurochem. 2008;107:466–477. doi: 10.1111/j.1471-4159.2008.05618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Velasquez FJ, Moss MA. J Neurochem. 2008;104:500–513. doi: 10.1111/j.1471-4159.2007.04988.x. [DOI] [PubMed] [Google Scholar]
- 6.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Drafft GA, Klein WL. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh D, Klyubin I, Fadeeva J, Cullen W, Anwyl R, Wolfe M, Rowan M, Selkoe D. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 10.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M. J Neurosci. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei G, Jewett AI, Shea J. Phys Chem Chem Phys. 2010;12:3622–3629. doi: 10.1039/c000755m. [DOI] [PubMed] [Google Scholar]
- 14.Kittner M, Knecht V. J Phys Chem B. 2010;114:15288–15295. doi: 10.1021/jp1065264. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto T, Adams KW, Fan Z, McLean PJ, Hyman BT. J Biol Chem. 2011;286:27081–27091. doi: 10.1074/jbc.M111.257378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Podlisny M, Walsh D, Selkoe D. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 17.Moore B, Rangachari V, Tay W, Milkovic N, Rosenberry T. Biochemistry. 2009;48:11796–11806. doi: 10.1021/bi901571t. [DOI] [PubMed] [Google Scholar]
- 18.Iurascu M, Cozma C, Tomczyk N, Rontree J, Desor M, Drescher M, Przybylski M. Anal Bioanal Chem. 2009;395:2509–2519. doi: 10.1007/s00216-009-3164-3. [DOI] [PubMed] [Google Scholar]
- 19.Bernstein S, Dupuis N, Lazo N, Wyttenbach T, Condron M, Bitan G, Teplow D, Shea J, Ruotolo B, Robinson C, Bowers M. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein S, Wyttenbach T, Baumketner A, Shea J, Bitan G, Teplow D, Bowers M. J Am Chem Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 21.Pryor E, Moss MA, Hestekin CN. Int J Mol Sci. 2012;13:3038–3072. doi: 10.3390/ijms13033038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabella S, Quaglia M, Lanni C, Racchi M, Govoni S, Caccialanza G, Calligaro A, Bellotti V, Lorenzi E. Electrophoresis. 2004;25:3186–3194. doi: 10.1002/elps.200406062. [DOI] [PubMed] [Google Scholar]
- 23.Colombo R, Carotti A, Catto M, Racchi M, Lanni C, Verga L, Caccialanza G, De Lorenzi E. Electrophoresis. 2009;30:1418–1429. doi: 10.1002/elps.200800377. [DOI] [PubMed] [Google Scholar]
- 24.Picou R, Kheterpal I, Wellman A, Minnamreddy M, Ku G, Gilman SD. J Chromatogr B. 2011;879:627–632. doi: 10.1016/j.jchromb.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 25.Picou R, Moses JP, Wellman AD, Kheterpal I, Gilman SD. Analyst. 2010;135:1631–1635. doi: 10.1039/c0an00080a. [DOI] [PubMed] [Google Scholar]
- 26.Picou RA, Schrum DP, Ku G, Cerqua RA, Kheterpal I, Gilman SD. Anal Biochem. 2012;425:104–112. doi: 10.1016/j.ab.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Wang SS, Chen Y, Chen P, Liu K. Biochem Eng J. 2006;29:129–138. [Google Scholar]
- 28.Barrow CJ, Yasuda A, Kenny PTM, Zagorski MG. J Mol Biol. 1992;225:1075–1093. doi: 10.1016/0022-2836(92)90106-t. [DOI] [PubMed] [Google Scholar]
- 29.Garai K, Sahoo B, Sengupta P, Maiti S. J Chem Phys. 2008;128:045102. doi: 10.1063/1.2822322. [DOI] [PubMed] [Google Scholar]
- 30.Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury PT, Jr, Fink AL, Teplow DB. Amyloid. 2000;7:166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 31.Nichols M, Moss M, Rosenberry T. Biochemistry. 2002;41:6115–6127. doi: 10.1021/bi015985r. [DOI] [PubMed] [Google Scholar]
- 32.Kotarek JA, Johnson KC, Moss MA. Anal Biochem. 2008;378:15–24. doi: 10.1016/j.ab.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 33.Necula M, Kayed R, Milton S, Glabe CG. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 34.Sureshbabu N, Kirubagaran R, Jayakumar R. Eur Biophys J. 2009;38:355–367. doi: 10.1007/s00249-008-0379-8. [DOI] [PubMed] [Google Scholar]
- 35.Kirkitadze M, Condron M, Teplow D. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 36.Terzi E, Hoelzemann G, Seelig J. Biochemistry. 1997;36:14845–14852. doi: 10.1021/bi971843e. [DOI] [PubMed] [Google Scholar]
- 37.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong HE, Qi W, Choi H, Fernandez EJ, Kwon I. ACS Chem Neurosci. 2011;2:645–657. doi: 10.1021/cn200056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ladiwala AR, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, Davis J, Vannostrand WE, Tessier PM. J Biol Chem. 2012;287:24765–24773. doi: 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Shah N, Zhao J, Wang C, Zhao C, Liu L, Li L, Zhou F, Zheng J. Phys Chem Chem Phys. 2011;13:15200–15210. doi: 10.1039/c1cp21156k. [DOI] [PubMed] [Google Scholar]