

Abstract
Physicians treating patients with the classic Philadelphia-negative myeloproliferative neoplasms (Ph-negative MPNs) (polycythemia vera [PV], essential thrombocythemia [ET] and primary myelofibrosis [PMF]) traditionally had few therapeutic drugs available. Spurred by the discovery of activating mutation of the JAK2 tyrosine kinase (JAK2 V617F mutation) in patients with Ph-negative MPNs several years ago, several JAK2 inhibitors were synthesized and are currently undergoing clinical trials in patients with PMF, PV and ET. Initial results from these studies have shown that these drugs can markedly reduce spleen size and alleviate constitutional symptoms, increase weight and improve exercise capacity in MF patients, thus improve quality of their life, which is significant clinical benefit. In ET and PV JAK2 inhibitor therapy may efficiently control blood cell count, as well as improve splenomegaly and control disease related symptoms. JAK2 inhibitors are a novel class of agents with promising results for treating patients with MF, PV and ET. In this article we will review the current evidence regarding the role of JAK2 mutations in the pathogenesis of Ph-negative MPNs and summarize results from the most recent clinical trials with JAK2 inhibitors in these disorders. JAK2 inhibitors are a novel class of agents with promising results for treating patients with MF, PV and ET.
Keywords: Polycythemia vera, Essential thrombocythemia, Myelofibrosis, JAK2 inhibitor, JAK2 V617F
Introduction
The classical Philadelphia chromosome–negative myeloproliferative neoplasms (Ph-negative MPNs) include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF); MF can also develop secondarily in patients with PV and ET (post-PV or -ET MF) (1). Ph-negative MPNs are chronic myeloid neoplasms which are thought to arise from a primitive hematopoietic stem cell which has undergone malignant transformation. Clinical manifestations include variable degrees of erythrocytosis, thrombocytosis, and leukocytosis, or cytopenias, extramedullary hematopoiesis (e.g. splenomegaly),, increased risk for thrombosis and transformation to acute myeloid leukemia (AML) (2). Life expectancy for patients with Ph-negative MPNs vary from as few as 5–7 years in patients with MF to more than 15 years for patients with PV and ET (3).
Physicians treating patients with MPNs are limited in the choice of drugs for these diseases. Patients with PV and ET are usually treated with cytoreductive agents (e.g. hydroxyurea, anagrelide, busulphan, pipobroman) which can effectively control elevated blood cell counts and decrease the risk of thrombosis, but may also be associated with an increased risk of transformation to AML and/or post-PV/ET MF (4–7). Therefore, apart from hydroxyurea, there are few drugs available for treating these patients without incurring in significant late side effects. For patients with MF, therapy is usually palliative and directed to alleviation of symptoms caused by splenomegaly and/or cytopenias (8). There are no approved drugs for this disease. Hydroxyurea, thalidomide and conventional chemotherapeutic agents are some of the agents used for treating splenomegaly, and response rates are in the order of 20–40% (9–11). Splenectomy can be a therapeutic option in cases of marked splenomegaly, but is associated with a post-operative mortality rate of up to 7% (12). MF is also associated with increased levels of pro-inflammatory cytokines which lead to constitutional symptoms (weight loss, night sweats, fatigue, fever) and can worsen patients’ quality of life (13).
Recently, great advances were made in the understanding of the pathogenesis of these disorders with the discovery of an activating mutation of the JAK2 tyrosine kinase (TK) (JAK2 V617F) in patients with Ph-negative MPNs (14–17). The JAK2 V617F mutation leads to constitutive signalling through the JAK2 TK, leading to increased cellular proliferation and resistance to apoptosis in hematopoietic cells. More importantly, the discovery of JAK2 V617F led to the development of JAK2 inhibitors for therapy of patients with Ph-negative MPNs, following the same rationale used to target BCR-ABL1 in chronic myeloid leukemia with imatinib. At this moment, there are several JAK2 inhibitors in clinical trials for patients with Ph-negative MPNs, and herein we summarize the rationale for developing these drugs and the most relevant clinical data.
The JAK Family of Kinases
a) Discovery and Structure
JAK kinases were first identified in 1989 and were named after the two-faced roman god Janus (Janus kinases) due to their unique structure, characterized by the presence of two tyrosine kinase domains (18). There are four members of the JAK family of TK: JAK1, JAK2, JAK3 and TYK2. Structurally, all members of the JAK family contain seven distinct domains: JH1-7 (JAK homology domains 1–7) (figure 1) (19). The TK domain (JH1) and the pseudokinase domain (JH2) are located in the carboxy-terminal portion of the molecule. The JH1 domain is a “true” TK domain and is responsible for the kinase activity of JAKs (20). The pseudokinase domain has no kinase activity and its function might be to inhibit and regulate the activity of the JH1 domain, as deletion of the JH2 domain leads to increased kinase activity (21). Domains JH3-JH4 are structurally similar to SH2 (Src-homology 2) domains (22). However, unlike classic SH2 domains, domains JH3-JH4 do not bind phosphotyrosine residues in interacting proteins, and their role is still unknown (23). The JH5-JH7 domains are located in the amino-terminal portion of the molecule and contain a FERM (Band 4.1, ezrin, radixin and moesin) motif, which is important for binding of the JAK molecule to the cytokine receptor and in maintaining receptor expression at cell surface (24, 25).
Figure 1. JAK2 structure and mutation sites.
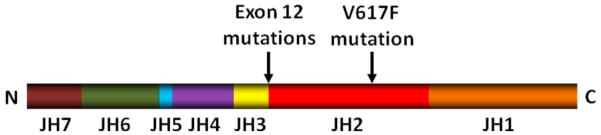
The V617F mutation locates in the pseudokinase domain (JH2 domain) which regulates activity of the TK domain (JH1 domain). Exon 12 mutations of JAK2 (described in patients with JAK2 V617F negative polycythemia vera) cluster in residues F537-E543 and locate between the pseudokinase and SH2-like domain
b) Function
JAK kinases are cytoplasmic TK that associate with the intracellular portion of cytokine and hematopoietic growth factors receptors that do not possess intrinsic TK activity (e.g. interferon receptor [IFNAR, IFNGR], erythropoietin [EPO] receptor [EPOR], thrombopoietin [TPO] receptor [MPL], interleukin-6 receptor [IL6R]) (26). Binding of the ligand to the receptor activates the kinases, leading to transphosphorylation of the receptor and subsequent activation of several distinct intracellular signalling pathways (Figure 2). JAK kinases are known to activate STATs (signal transducers and activators of transcription), forming the JAK-STAT pathway (26). STATs are latent transcription factors, and upon tyrosine phosphorylation they form dimers which translocate to the nucleus, bind to DNA and induce expression of target genes (27). There are seven members of the STAT family (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6); activation of STAT3 and STAT5a/b leads to increased expression of genes related to increased cellular proliferation (CCND1), angiogenesis (VEGF) and resistance to apoptosis (BCL2L1, MCL1, BIRC5) and has been associated with neoplastic transformation (27). Besides the JAK-STAT pathway, there is also cross-talk between JAK kinases and other signalling pathways. JAKs can activate the MAPK (mitogen activated protein kinase) pathway through a Ras-dependent mechanism (28, 29). JAKs can also associate with and stimulate activity of the kinase PI3K (phosphatidylinositol-3-kinase) of the PI3K/Akt/mTOR pathway which inhibits apoptosis and stimulates cellular proliferation (30, 31). Negative regulation of JAK kinases is performed by phosphatases and SOCS proteins (supressors of cytokine signalling). Phosphatases, such as SHP1 (SH2-containing phosphatase 1) and CD45, remove phosphate moieties from tyrosine residues and inactivate JAK TKs (32–34). SOCS proteins are induced by STAT activation and function in a classic negative feedback loop mechanism. The SOCS1 protein binds via its SH2 domain to a tyrosine residue in the activation loop of JAK2, leading to its inactivation (35). Interaction of JAK2 with SOCS1 also leads to JAK2 polyubiquitination and subsequent degradation by the proteasome (36).
Figure 2. Signalling pathways activated by JAK2.

Binding of the putative ligand to cytokine receptors leads to receptor dimerization and approximation of two JAK2 molecules, which trans-phosphorylate and activate each other. Activated JAK2 molecules initiate several distinct signalling pathways. Phosphorylation of STATs (STAT5 depicted in the figure) leads to STAT dimerization, nuclear translocation, binding to DNA and activation of expression of target genes. Other pathways that may be activated by JAKs include the Ras/Raf/MAPKinases and the PI3K/Akt/mTOR pathway.
JAKs play an important role in cytokine intracellular signaling, and studies using knockout mouse models have provided important data on the role of specific members of the JAK family of TKs. JAK1 is essential for signaling by receptors containing the common γc chain (e.g. IL-2 and IL-7 receptors), class II cytokine receptors (including Interferons [IFN]-α,-β and -γ) and cytokine receptors containing gp130 (e.g. IL6R) (37). Deficiency of JAK1 leads to a severe impairment in lymphocyte development from defective IL-7 (interleukin-7) signaling, and JAK1-null mice die at birth from neuronal defects and an inability for suckling (37). JAK2 is essential for pathways that control erythroid, myeloid and megakaryocytic development, but not for lymphopoiesis (38, 39). JAK2 mediates signaling through EPOR, MPL and cytokine receptors that harbor the common β-chain (e.g. IL3 receptor [IL3A]) and IFN-γ receptors (38). Deficiency of JAK2 is lethal to mice embryos due to the absence of definitive erythropoiesis. JAK3 is only activated by cytokines of the γc family (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21), and JAK3 deficiency leads to severe impairment in lymphoid development, with reductions in numbers of T- and B-lymphocytes and NK-cells (40, 41). Mutations in JAK3 have been described in patients with severe combined immunodeficiency (SCID) (42, 43). Mice that are TYK2-null have an increased susceptibility to viral infections and a reduced response to IL-12 (44, 45).
The JAK2 V617F mutation in MPNs
a) Description
The first clue that JAKs TKs could be involved in the pathogenesis of MPNs came from studies with the fruit fly Drosophila melanogaster. One report described that a point mutation (E695K) in the JH2 domain of the gene Hopscotch (the JAK equivalent of Drosophila) leads to increased kinase activity and development of a leukemia-like picture with increased numbers of hemocytes (fly blood cells) (46). There was also increased activation of STAT92E (STAT equivalent of Drosophila).
The JAK2 V617F mutation was first described by several groups in 2005. It was identified primarily in patients with PV (90–97%), ET (60%) and PMF (50%) (14–17). The mutation can be detected in rare cases of chronic myelomonocytic leukemia, atypical CML, myelodysplastic syndrome and AML (47). Other reports have described the presence of the JAK2 V617F mutation in blood and endothelial cells from patients with idiopathic splanchnic thrombosis (i.e. Budd-Chiari syndrome, portal vein thrombosis), indicating the presence of a latent MPN in these cases (48, 49). Recent studies have revealed that the presence of a germline JAK2 haplotype (46/1) predisposes to the development of MPNs positive for the JAK2 V617F mutation (50–52).
b) Mechanism of JAK2 activation by the V617F mutation
Intracellularly, the JAK2 V617F mutation leads to constitutive activation of the JAK2 TK with phosphorylation and activation of STAT5 and other downstream targets (Figure 2) (15). The consequence is an increased signaling of cytokine receptors that associate with JAK2 TK, such as EPOR and MPL, with increased proliferation of hematopoietic cells harboring these receptors. The expression of JAK2 V617F in EPOR-positive BaF/3 cells confers EPO-hypersensitivity and EPO-independent growth and survival (17). The mechanism through which the V617F mutation leads to constitutive activity of the TK domain is the subject of much study. As mentioned, the pseudokinase domain inhibits activity of the TK domain (21). Residue V617 is located in a loop connecting two β-strands of the pseudokinase domain which interact with the activation loop of the JH1 domain (22). The JAK2 V617F mutation possibly leads to increased TK activity by disrupting this interaction and the inhibitory activity of the JH2 domain. Recently, one study described that residue F595 is crucial for the constitutive activity of the JAK2 V617F mutant (but not wild-type JAK2) (53). Residue F595 is located in the αC-helix of the JH2 domain, in close proximity to residue V617. In V617F-mutated JAK2, there is an interaction between the two phenylalanine residues (F595 and V617F) which changes conformation of the JH2 αC-helix (53). The conformational change in the JH2 helix is transmitted to the TK domain and possibly leads to its autoactivation (53). The F595 residue is also crucial for the constitutive activity of other JAK2 mutations, such as exon 12 mutations (see below), indicating that changes in the C-helix of the JH2 domain might underlie a common mechanism of JAK2 constitutive activation.
c) Animal Models
Several studies reported on animal models for JAK2 V617F-positive MPNs using retroviral transplantation models and transgenic mice (54–64). Expression of the JAK2 V617F TK in mice HSC and progenitor cells leads to a phenotype similar to PV with increased hematocrit, splenomegaly and BM cellularity. In some studies there was later progression to fibrotic changes in the bone marrow (BM) (54, 55). Recently, one study employing a transgenic mouse model demonstrated that JAK2 V617F exerts differential effects on HSCs and hematopoietic progenitor cells (61). There was a minimal effect of JAK2 V617F on HSCs proliferation, self-renewal and STAT5 activation, and JAK2 V167F-mutated HSCs did not have a selective advantage over wild-type HSCs. The main effect of JAK2 V617F on HSCs was to skew differentiation towards erythroid cells, as assessed by gene expression profiling demonstrating increased expression of genes associated with erythroid differentiation. It is unknown how JAK2 V617F directs differentiation of HSCs to erythroid cells, but it might involve epigenetic modification of histone phosphorylation (65). On the other hand, JAK2 V617F-positive hematopoietic progenitor cells were expanded, skewed towards erythroid lineage and displayed increased phosphorylation of STAT5 (61). These results are compatible with another report which demonstrated that the JAK2 V617F mutation is detected in HSCs from patients with PV and it predisposes these cells to erythroid differentiation (66). Importantly, JAK2 V617F HSCs (but not JAK2 V617F progenitor cells) were able to transmit the disease through transplantation to recipient mice, indicating that they were the disease initiating cells (61). This has implications for therapy of patients with JAK2 inhibitors (see below).
d) Clinical impact of JAK2 V617F mutation
One important question regarding JAK2 V617F is how a single mutation can contribute to diseases with different phenotypes and clinical features. The allelic burden of JAK2 V617F may be related to this. Patients may be heterozygous or homozygous for the JAK2 V617F mutation, and homozygosity is frequently acquired by uniparental disomy of chromosome 9p, where the JAK2 gene locates (67). Patients with JAK2-mutated ET are usually heterozygous (68), and those who are positive for the mutation presents with clinical features similar to PV, such as higher hemoglobin, neutrophil counts, BM erythropoiesis and a higher rate of transformation into PV compared to JAK2 V617F-negative ET (69, 70). Patients with PV are usually homozygous for JAK2 V617F, and those patients with a high (> 75%) allele burden have higher hematocrit, white blood cell (WBC) count and higher incidence of splenomegaly and thrombotic events (68, 71, 72). In one animal model it was demonstrated that the ratio of mutant:wild type JAK2 determined the clinical phenotype: animals presenting with a low ratio had a disease similar to ET, while those with a high ratio developed a clinical picture more reminiscent of PV (59). This suggests that JAK2 V617F-positive PV/ET forms a biological continuum. The allelic burden of the JAK2 V617F mutation also has clinical and prognostic implications in patients with MF, although the reports are conflicting. One study reported that patients with primary MF who had a high allelic burden of JAK2 V617F developed “PV-like” phenotype of the disease with higher hemoglobin and WBC count and a higher risk of leukemic transformation and of developing massive splenomegaly (73). By contrast, 2 other studies reported that MF patients with a low JAK2 V617F allelic burden had lower hemoglobin and low incidence of splenomegaly, but worse survival either due to deaths by systemic infections secondary to cytopenias or due to leukemic transformation (74, 75).
e) JAK2 V617F-negative MPNs
Despite the important discovery of the JAK2 V617F mutation, some patients (particularly ET and MF patients) have a diagnosis of JAK2 V617F-negative MPN. Search for other mutations led to the discovery of JAK2 exon 12 mutations in 3% of PV patients (76) and mutations of MPL (MPL W515K/L) in 8.5% of ET and 10% of MF patients (77, 78). These mutations also lead to increased signaling through cytokine receptors, with increased cellular proliferation and hypersensitivity or independence to hematopoietic growth factors (76, 79). JAK2 exon 12 mutations occur almost exclusively in patients with JAK2 V617F-negative PV (76, 80, 81). Several mutations in exon 12 have been identified, including in-frame deletions, point mutations and duplications, and they cluster in residues F537-E543 (76, 80–82). MPL mutations affect primarily patients with ET and MF, and the most common mutation is MPL W515L (77, 83). The presence of MPL mutation has been linked to a higher incidence of anemia and older age in both patients with ET and MF (84, 85). While the JAK2 V617F and related mutations certainly contribute to the pathogenesis of MPNs, it is probable that they represent a secondary genetic event, and not the disease initiating mutation (86, 87). Support for this hypothesis comes from reports showing that JAK2 V617F-negative erythroid colonies from patients with PV can grow in the absence of EPO (86). Several studies have also reported that patients with JAK2 V617F-positive MPNs can develop JAK2 V617F-negative AML (89, 92). Additionally, more recent studies have revealed a myriad of other mutations in patients with MPNs, such as TET2 mutations which are found in 12% of patients (88). TET2 mutations can either precede or succeed the acquisition of the JAK2 V617F mutation, and in fact appear to occur independently from the latter, giving rise to multiple clones harboring one, the other or both mutations (88–90). Animal models have revealed that the TET2 gene, along with its family members TET1 and TET3, is responsible for DNA demethylation, and it is conceivable that deletions of TET2 are related to the abnormal DNA methylation patterns seen in hematologic malignancies (91). In summary, the molecular biology of Ph-negative MPNs is much more complex than initially thought after the discovery of the JAK2 V617F mutation. Future studies will strive to increase our understanding of the biology of these disorders.
Why target the JAK2 V617F mutation, for what clinical benefit?
With the development of the tyrosine kinase inhibitor (TKI) imatinib for therapy of CML the field of hematology-oncology entered the era of kinase inhibitors, where small molecules target kinases aberrantly activated in cancer cells, with a greater therapeutic index and safety compared to traditional chemotherapeutic agents. As soon as 2 years after the publication of the first reports of the JAK2 V617F mutation in MPNs, several JAK2 inhibitors were entering clinical trials for patients with MF and later PV/ET, with differences in potency and kinase specificity (Table 1). Pre-clinical studies have confirmed activity of these compounds, with induction of apoptosis in both in vitro and in vivo models (93, 94). Most TKI in current clinical development are small molecules that act by competing with ATP (adenosine tri-phosphate) for the ATP-binding catalytic site in the TK domain, since ATP is the source of phosphate groups utilized by TK for phosphorylating protein targets (95). The V617F mutation locates outside the TK domain of JAK2, and current JAK2 inhibitors target both wild-type and mutated JAK2 indiscriminately. This could explain why these drugs are active in patients with both wild-type and mutated JAK2, as described below. Targeting of wild-type JAK2 is expected to lead to myelosuppression, and this probably explains reported therapy-related anemia and thrombocytopenia in clinical trials with these compounds.
Table 1.
Profiles of JAK2 inhibitors in clinical development, with reported clinical results
| Drug | Company | Current Phase | Target(s) | IC50 Value (nM)
|
||
|---|---|---|---|---|---|---|
| JAK2 | JAK1 | JAK3 | ||||
| CEP-701 | Cephalon | I/II | JAK2, FLT3 | 1 | ? | ? |
| INCB018424 | Incyte | III | JAK2, JAK1 | 2.8 | 3.3 | 322 |
| SB1518 | S*Bio | I | JAK2, FLT3 | 23 | 1280 | 520 |
| TG101348 | TargeGen | II | JAK2, FLT3 | 3 | 105 | 996 |
| XL019 | Exelixis | Discontinued | JAK2 | 2 | 132 | 250 |
So far, most clinical studies with JAK2 inhibitors have focused on patients with MF. Clinical responses usually consist of marked reductions in spleen size, improvement in systemic symptoms, weight, and exercise capacity (96). In the majority of studies so far there was no overall significant change in JAK2 V617F allele burden, BM fibrosis and other markers suggestive of clonal eradication. There are several possible reasons for this. First, compared to chronic phase CML, which is a disease driven essentially by a single gene (BCR-ABL1), MF has a more complex biology, characterized by mutations in several different genes (JAK2, MPL, TET2), cytogenetic abnormalities in up to 40% of cases and multiple clones and subclones (97). It might be that targeting just one pathway with a single TKI is not enough for eradication of the diseased cell. Second, it has been hypothesized that the mechanism of action of JAK2 inhibitors is due to inhibition of proinflammatory cytokine signaling in both normal and neoplastic cells (98). MF is an inflammatory disorder characterized by increased levels of pro-fibrotic, -inflammatory and -angiogenic cytokines (i.e. transforming growth factor-beta1 [TGF-β1], bone morphogenetic protein 1 [BMP1], tumor necrosis factor-α [TNF-α], IL-6, IL-8, basic fibroblast growth factor [bFGF], platelet derived growth factor [PDGF], thrombopoietin [TPO] vascular endothelial growth factor [VEGF]) which lead to splenomegaly, BM fibrosis, osteosclerosis, increased angiogenesis and development of systemic symptoms (99–105). JAK2 inhibitors, which may not only inhibit JAK2 but also the closely related JAK1 in some cases, by blocking cytokine receptors signaling lead to significant improvements in spleen size and systemic symptoms for patients with MF (103). Some studies of JAK2 inhibitors have revealed a decrease in the serum levels of proinflammatory cytokines with continued therapy, a finding which has not been confirmed in all reports (96). Cytokines might also be involved in resistance to JAK2 inhibitors, and there is in vitro data showing that elevated concentrations of cytokines reduce growth arrest induced by JAK2 inhibitors in JAK2 V617F-positive cells (106). Finally, there’s recent evidence to suggest that, similar to imatinib in CML, the disease-initiating cell in MPNs is spared from the action of JAK2 inhibitors (61). In a transgenic mouse model of JAK2 V617F MPNs, therapy with the JAK2 inhibitor TG101348 lead to significant improvements in spleen size and a reduction in the number of erythroid precursor cells, but it did not eliminate the disease initiating cells (contained in the HSCs compartment) as JAK2 V617F-mutated HSCs obtained from mice treated with TG101348 were still able to transmit the disease when transplanted into lethally irradiated mice recipients. This suggests that these compounds are only able to eradicate progenitor cells, sparing the disease-initiating cells, which has potential implications for the long-term outcome of patients treated with these drugs.
Results of Clinical Trials with JAK2 Inhibitors in MPNs
a) MF (Table 2)
Table 2.
Preliminary results of clinical studies with JAK2 inhibitors in MF
| Study | Therapy | N | Responses | Toxicity |
|---|---|---|---|---|
| Santos et al.(107) | CEP-701 80mg twice daily (Phase II) | 22 | CI by IWG criteria: 27% (reduction in spleen size and improvement in cytopenias) | GI toxicity, cytopenias |
| Hexner et al.(110) | CEP-701 80–160mg twice daily (Phase I) | 26 | Reduction in splenomegaly (median 5.8 cm) | GI toxicity, cytopenias |
| Verstovsek et al.(96) | INCB018424 10–25mg twice daily (Phase I/II) | 153 | CI by IWG criteria: 44% (reduction in spleen size); response rate 52% with optimized dose schedule; transfusion independency 14%; improvement in exercise ability, systemic symptoms, decrease in cytokines in majority of patients | Thrombocytopenia, anemia |
| Verstovsek et al.(113) | SB1518 100–600mg once daily (Phase I) | 43 | CI by IWG criteria: 28% (reduction in spleen size) | GI toxicity, thrombocytopenia |
| Seymour et al.(114) | SB1518 100–600mg once daily (Phase I) | 20 | Reduction in spleen size in 11/13 evaluable patients (2 patients ≥ 50%); transfusion independency in 2/9 patients | GI toxicity |
| Pardanani et al.(117) | TG101348 30–800mg once daily (Phase I/II) | 59 | Reduction in splenomegaly (CI: 49%), improvement in systemic symptoms, leukocytosis, thrombocytosis; improvement in BM fibrosis in selected cases | Anemia, Thrombocytopenia, GI toxicity |
| Shah et al.(119) | XL019 25–300mg once daily or 25mg thrice a week (Phase I) | 30 | Improvement in splenomegaly, systemic symptoms, anemia, leukocytosis | Neurological toxicity (study closed) |
Abbreviations: MF indicates myelofibrosis; CI, clinical improvement; IWG-MRT, International Working Group-Myelofibrosis Research and Treatment; GI, gastrointestinal
a.1) CEP-701
CEP-701 (also known as lestaurtinib) is a indolocarbazole alkaloid that has activity as a FLT3 and JAK2 inhibitor. In a pre-clinical study, CEP-701 inhibited both wild-type and mutated JAK2, with a half-maximal inhibitory concentration (IC50) of 1nM for wild-type JAK2 (93). CEP-701 also inhibited growth of JAK2 V617F-positive HEL.92 cells xenografted into nude mice.
There are two studies which evaluated CEP-701 for therapy of patients with JAK2 V617F-mutated MF. The first study was a single center phase II trial that enrolled 22 patients with primary or post-PV/ET MF (107). All patients were positive for the JAK2 V617F mutation and 36% were transfusion-dependent. Median spleen size was 19 cm from left costal margin. Patients received CEP-701 as a liquid formulation at an initial dose of 80 mg twice daily, based on previous studies conducted in patients with FLT3-positive AML (108). Responses were evaluated by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria (109). Overall, responses were seen in six patients (27%) and consisted of clinical improvements (CI) in all cases: reduction in spleen size (N=3), transfusion independency (N=2) and reduction in spleen size with improvement in cytopenias (N=1). Responders had significant lower levels of phosphorylated STAT3 than non-responders, suggesting that responses were obtained by inhibition of JAK2 activity. Median response duration was 14+ months. There was no change in JAK2 V617F allele burden, BM fibrosis and cytokine levels among responders. Toxicity was considerate and included myelosuppression (grade 3–4 anemia 14%; grade 3–4 thrombocytopenia 23%) and GI toxicity (diarrhea, any grade 72%, grade 3–4 9%; nausea, grade 1–2 only, 50%; vomiting grade 1–2 only, 27%).
A phase I/II trial conducted by the MPD Research Consortium is exploring the hypothesis that higher dosages and a different formulation of CEP-701 might achieve a better response rate in patients with JAK2 V617F-positive MF (110). In this study, only reported in abstract form, patients receive escalated doses of CEP-701 in both liquid (80–100 mg twice daily) and capsule (100–160 mg twice daily) formulations. Twenty-six patients (PMF=16, Post-PV=7, Post-ET=3) have been recruited for this multicenter trial. Fifty-percent were transfusion-dependent, median JAK2 V617F allele burden was 78.4% and mean spleen size was 16.7 cm. Six patients had a decrease in spleen size by median of 5.8 cm. There was no change in transfusion requirements and/or reduction in WBC count. Among 6 patients with paired baseline/week 12 JAK2 V617F samples, three patients had a decrease in allele burden of 4.3–12.9%. Similar to the previous trial, GI toxicity was the most common side effect observed. In patients receiving the liquid formulation, dose limiting toxicity was diarrhea (grade 3: 1 case), 71% had grade 1–2 diarrhea and 29% had grade 1–2 nausea and/or vomiting. Patients who received the capsule formulation had a better tolerability, with 37% experiencing grade 1–2 diarrhea (one patient had grade 3 diarrhea) and 37% reported grade 1–2 nausea. Late toxicity (beyond 28 days of therapy) included diarrhea (all grades: 7 patients; grade 3–4: 1 patient), thrombocytopenia (grade 3–4: 2 patients) and neutropenia (grade 3–4: 1 patient). Overall, both trials seem to demonstrate that CEP-701 has at best modest efficacy in patients with JAK2 V617F-positive MF. GI toxicity is considerate. Further results from the second study are awaited in the near future.
a.2) INCB018424
INCB018424 is a potent, orally available inhibitor of both JAK1 (IC50=3.3 nM) and JAK2 (IC50=2.8 nM). It is more selective against JAK3 (IC50=428 nM) and TYK2 (IC50=1.9 nM). In a pre-clinical study, INCB018424 inhibited activation and phosphorylation of JAK2 V617F and downstream targets ERK, STAT5 and STAT3 in HEL cells and Ba/F3 cells expressing JAK2 V617F (94). Inhibition of JAK2 induced apoptosis of JAK2 V617F-positive cells (IC50=126 nM for Ba/F3 cells expressing JAK2 V617F). INCB018424 inhibited clonogenic growth of erythroid progenitor cells from patients with JAK2 V617F-mutated PV, and mutated cells were more sensitive to JAK1/2 inhibition than healthy cells. In a mouse model of MPN, treatment with INCB018424 reduced levels of pro-inflammatory cytokines (IL-6, TNF-α), improved spleen size and increased survival of treated mice (94).
A phase I/II clinical trial with INCB018424 was conducted in patients with primary or post-PV/post-ET MF independent of JAK2 mutational status (96). Patients were eligible if they required therapy and were refractory, relapsed or intolerant to previous therapy or, if newly diagnosed, with intermediate or high-risk by Lille score criteria. Patients with neutropenia (ANC ≤ 1.5×109/L) or thrombocytopenia (platelet count ≤ 100×109/L) were excluded from the trial. A total of 153 patients were enrolled in this study, and 115 (75%) are still on therapy, with a median follow-up of 14.7 months. Median age was 65 years, 82% were JAK2 V617F-positive and 92% had splenomegaly at beginning of therapy. Median spleen size was 19 cm below left costal margin. During the phase I part of the trial, several once daily and twice daily schedules were evaluated in a classic 3+3 cohort design. MTDs were defined as 25 mg twice daily and 100 mg once daily, and thrombocytopenia was the DLT. During the phase II portion of the trial, the cohorts of 25 mg twice daily and 50 mg once daily were expanded, and additional dose-schedules regimens were evaluated to determine the most effective regimen with a reduced incidence of thrombocytopenia. Based on response and toxicity, an optimized schedule of 15 mg twice daily (10 mg twice daily if platelets were 100–200×109/L) with monthly dose escalations up to 25 mg twice daily (if no response and no toxicity was observed) was chosen as the best regimen. By IWG-MRT criteria, 61 patients (44%) achieved a CI due to a ≥ 50% reduction in spleen size during the first 3 months of treatment. Response rate was 52% and 49% in the cohorts receiving 15 mg twice daily and 25 mg twice daily. Response was maintained at 12 months in 73% and 78% of responders of the 15 mg and 25 mg cohorts, respectively. There was no difference in response rate by JAK2 V617F mutational status (51% [mutated] vs. 45% [wild-type]) and type of MF (49% [primary] vs. 45% [post-PV] vs. 62% [post-ET]). Magnetic resonance imaging confirmed reductions in both spleen and liver size and volume.
Therapy with INCB018424 led to improvement in hemoglobin and achievement of transfusion independence in 14% of patients who were transfusion dependent at baseline. There was also decrease in WBC count and platelet count in selected cases. Significant improvements in constitutional symptoms and exercise ability were observed. Majority of patients who received 10–25 mg twice daily had ≥ 50% improvement in total and/or individual symptoms scores as assessed by the Myelofibrosis Symptom Assessment Form (MFSAF) (111). Study patients (N=27) had an improvement from baseline in the 6-minute walking test of 34, 57 and 71 meters after 1, 3 and 6 months of therapy. Patients also gained weight and had an overall increase in performance status while on therapy. Alleviation of symptoms and increases in exercise ability correlated with reduction in pro-inflammatory cytokines (including IL-6, TNF-α, IL-1Ra). There was no correlation between JAK2 mutational status and improvement in systemic symptoms or reduction in cytokine levels. There were 111 patients with data on allele burden after 3 months of treatment. At baseline, median allele burden was 70%, and INCB018424 led to an average decrease in the allele burden of 11% at 1 year and 18% at 2 years. A greater than 25% reduction was seen in 19 patients and a greater than 50% reduction was seen in 8 patients only.
Side effect profile revealed that INCB018424 was well tolerated. Non-hematological toxicity was rare (< 10%) and usually grade 1–2. Most common side effects were related to myelosuppression and included anemia and thrombocytopenia. Importantly, the regimen of 15 mg twice daily with adapted dose escalation led to lower rates of myelosuppression than the regimen of 25 mg twice daily (grade 3–4 anemia: 8.3% vs. 26.7%; new-onset transfusion dependent anemia: 4.7% vs. 20%; grade 3–4 thrombocytopenia: 2.9% vs. 30%). Only 3 patients have transformed to AML (rate of 0.016 patients/year), less than expected based on historical cohorts (73). INCB018424 is an effective and well tolerated drug for controlling splenomegaly and systemic symptoms in patients with MF. Mechanism of action may be related to blocking of cytokine signalling through inhibition of JAK1 and JAK2 activation. Recently, two phase III randomized trials have been conducted evaluating INCB018424 in patients with MF.
a.3) SB1518
SB1518 is a TKI active against wild type JAK2 (IC50=22 nM), JAK2 (IC50=19 nM) and FLT3 (IC50=22 nM) (112). Pre-clinical evaluation confirmed that SB1518 inhibits proliferation of Ba/F3 cells expressing both EPOR and JAK2 V617F, and this is associated with reduced phosphorylation of JAK2 and STAT5. SB1518 also demonstrated in vivo activity when administered to nude mice xenografted with JAK2 V617F-positive cells. There are two phase I/II trials that evaluated SB1518 in the therapy of patients with MF.
The first clinical trial reported recruited 43 patients (MF=36, AML=7) to receive SB1518 at doses ranging from 100–600 mg orally once daily continuously over 28-days cycles (113). Seventy-eight percent of patients were positive for the JAK2 V617F mutation, and 65% had splenomegaly, with a median spleen size of 13 cm. The MTD on this trial was 500 mg and DLT included abdominal pain and diarrhea. Side effects included diarrhea (all grades: 33%; grade 3: 4%), nausea (grade 1–2 only: 13%), and thrombocytopenia (grades 3–4: 4%). Among the 25 patients with MF and splenomegaly who had a follow-up physical examination, 7 (28%) had a CI by IWG-MRT criteria, manifested by a ≥ 50% reduction in spleen size. Seven additional patients had reduction in spleen size of 35–50%. Similar to INCB018424, responses to SB1518 were observed both in patients with (6/23) and without (1/2) the JAK2 V617F mutation. Three patients with AML had evidence of benefit (≥ 50% reduction in spleen size=1; stable disease=2). Correlative studies demonstrated reduction in JAK2 and STAT5 phosphorylation after patients received SB1518.
Data from the second phase I study was recently presented (114). A total of 20 patients with MF who required therapy were recruited. Splenomegaly was present in 85% (median spleen size 17 cm) and 85% were positive for JAK2 V617F. Similar to the other trial, DLTs were observed at 600 mg once daily (nausea, fatigue, diarrhea, dehydration), and MTD was determined to be 500 mg once daily. Median time on study so far is 4.8 months. Most common side effect was diarrhea (all grades: 85%; grade 3–4: 10%), nausea (all grades: 35%) and vomiting (all grades: 35%). Myelosuppression was minimal, as no dose dependent neutropenia or thrombocytopenia was observed, and 25% developed anemia (grade 3–4: 5%). Responses were observed with reduction in splenomegaly (11 of 13 with post-baseline assessment, including 2 patients with ≥ 50% reduction), transfusion independence in 2 of 9 patients with transfusion dependent anemia and improvement in WBC and platelet counts in selected patients. Even though 500 mg once daily was the MTD, pharmacokinetic analysis demonstrated no increase in drug levels with doses higher than 400 mg once daily. Based on this, the recommended dose of SB1518 for phase II trials is 400 mg once daily.
a.4) TG101348
TG101348 is an orally available, potent JAK2 inhibitor (IC50= 3 nM) with greater selectivity against JAK1 (IC50= 105 nM) and JAK3 (IC50= 996 nM). TG101348 inhibits proliferation of JAK2 V617F-positive cells both in vitro and in vivo (115). In a mouse model of PV, administration of TG101348 led to improvement in hematocrit, spleen size and spleen architecture, reduction in extramedullary hematopoiesis and in some cases reduction of bone marrow fibrosis (116).
TG101348 was investigated in a phase I clinical trial in patients with MF who had symptomatic splenomegaly and intermediate/high-risk disease (117). Patients received TG101348 at doses from 30–800 mg once daily continuously during 28-days cycles. MTD was 680 mg daily and DLTs included grade 3–4 hyperamylasemia (6%) and hyperlipasemia (13%). At the time of last report, 59 patients had been enrolled, including 40 patients in the expanded cohort at MTD. Eighty-six percent of patients were positive for the JAK2 V617F mutation, 98% had splenomegaly (median size 18 cm below left costal margin) and 37% were transfusion dependent. TG101348 produced marked reduction in spleen size, with 49% of patients experiencing CI based on reduction of splenomegaly. Responses were durable, and at the time of last report, no patient had relapsed after responding to TG101348. Elevated WBC counts (> 11×109/L) normalized in 73% of evaluable patients. Platelet count also normalized in the majority of patients who presented with thrombocytosis (> 450×109/L). There was improvement in constitutional symptoms, including fatigue, pruritus, night sweats and early satiety. Improvement in symptoms did not correlate with a reduction in pro-inflammatory cytokines. JAK2 V617F allele burden reduced in 59% of 22 evaluable patients, with a median decrease of 60%. In selected cases there was also improvement in BM cellularity and fibrosis. Side effects were mainly GI in nature, including diarrhea (all grades: 76%; grade 3: 13%), nausea (all grades: 70%; grade 3: 5%) and vomiting (all grades: 69%; grade 3: 3%). Grade 3–4 hematological toxicities at MTD included neutropenia (grade 3: 15%), thrombocytopenia (grade 3–4: 33%) and new-onset transfusion dependent anemia (67%). These preliminary results demonstrate that TG101348 is able to induce responses in splenomegaly in the majority of patients with MF, and this may be due to a direct effect on inhibition of JAK2 V617F. Future studies will confirm these observations.
a.5) XL019
The TKI XL019 is a potent, reversible and selective inhibitor of JAK2 (IC50=2 nM) (118). XL019 inhibits both wild-type and mutated JAK2 V617F. In a xenograft tumor model of HEL 92.1 JAK2 V617F-positive cells, administration of XL019 to xenografted mice suppressed STAT5 phosphorylation, inhibited tumor cell growth and induced tumor cell apoptosis.
A phase I clinical trial with XL019 in patients with primary or post-PV/ET MF was conducted (119). Thirty patients were enrolled and received XL019 at doses from 25–300 mg using different schedules of administration. Initial dose escalation started at 100 mg for three weeks every 28 days. However, patients who received ≥ 100 mg/day of XL019 developed reversible peripheral neuropathy. The dose schedule was amended, and patients received 25–50 mg once daily or 25 mg thrice weekly. Among the 21 patients who received ≤ 50 mg of XL019, there was spleen reduction all 12 patients with splenomegaly who were JAK2 V617F-mutated, with a ≥ 50% reduction in 5 patients. There was reduction in WBC counts in 7 of 9 patients who presented with WBC count ≥ 15×109/L. Improvements in anemia and constitutional symptoms (pruritus, asthenia) were seen in 4 and 6 patients, respectively. Four patients presented with pre-leukemic MF, with blast counts between 10–19%. XL019 led to reduction of circulating blasts in 3 of these patients, with normalization of BM blasts in 2 cases. Even though XL019 had great selectivity for JAK2, minimal hematological toxicity was observed. Toxicity was mainly neurological, and included peripheral neuropathy, weakness, paresthesia, formication and unsteady gait. Due to the high incidence of these symptoms, even at low doses, the trial of XL019 was closed and no further development of this compound is planned.
b) PV and ET (Table 3)
Table 3.
Preliminary results of clinical studies with JAK2 inhibitors in PV and ET
| Study | Therapy | N | Responses | Toxicity |
|---|---|---|---|---|
| Moliterno et al.(120) | CEP-701 80mg twice daily (Phase II) | 39 | Reduction in spleen size: 83%; reduction in phlebotomy requirements: 3/5 (60%) | GI toxicity, 5 thrombotic episodes |
| Verstovsek et al.(121) | INCB018424 10–25mg twice daily (Phase II) | 73 | LeukemiaNet criteria-PV: 97% (CR 45%; PR 52%); ET: 90% (CR: 13%; PR: 77%) | Anemia, Thrombocytopenia |
Abbreviations: PV indicates polycythemia vera; ET, essential thrombocythemia; GI, gastrointestinal; CR, complete response; PR, partial response
b.1) CEP-701
A multicenter phase II trial is evaluating CEP-701 in patients with high-risk JAK2 V617F-positive PV/ET (120). Inclusion criteria include absolute neutrophil count > 7×109/L (PV) or use of hydroxyurea (PV or ET). Initial dose is 80 mg twice daily with optional escalation to 120 mg twice daily, with treatment up to 18 months. At the time of the last report, 39 patients (PV=27, ET=12) have been enrolled. Responses observed consisted of reduction in spleen size (> 5 cm or to non-palpable) in 15/18 patients and improvement in pruritus in 5/5 patients evaluable. Reduction in phlebotomies requirements were observed in 3/5 patients, which only occurred after 6 months of therapy. There was no reduction in platelet count and WBC counts, and some patients even had an increase in these parameters during treatment. A greater than 15% reduction in JAK2 V617F allelic burden was observed in 20% of evaluable patients. Most common adverse events were related to GI toxicity, similar to clinical trials with this compound in MF. More worrisome, 5 patients experienced serious thrombotic events (3 venous and 2 arterial), a side effect which has not been reported in other clinical trials with CEP-701. This suggests that this compound might not prevent thrombosis in high-risk patients with PV/ET.
b.2) INCB018424
A clinical trial is currently evaluating INCB018424 in patients with PV/ET who are refractory/intolerant to hydroxyurea (121). Additional inclusion criteria include: hematocrit (Ht) ≥ 45% and/or phlebotomy dependence (PV); platelet count ≥ 650×109/L (ET). Responses are assessed by the European LeukemiaNet criteria (122). The first part of the trial determined the best dose cohort (PV: 10 mg twice daily; ET: 25 mg twice daily) which was then expanded in the second part of the study. Seventy-three patients have been enrolled (PV=34, ET=39), and median follow-up is 10.4 months. Among patients with PV, 74% were refractory to hydroxyurea, 76% were dependent on phlebotomies and 100% were positive for the JAK2 V617F mutation. Therapy with INCB018424 achieved normalization of the Ht in the absence of phlebotomy, with a mean Ht of 39% at 6 months. After starting treatment, only 2 patients needed phlebotomies in the first 2 weeks, and none ever since. There was also sustained improvement in WBC counts, platelet counts, spleen size, pruritus, bone pain and night sweats. CR rate by LeukemiaNet criteria was 45%, and 52% had a partial response (PR) (overall response rate [ORR]=97%). Sixty-eight percent achieved complete hematological response. Among patients with ET, 87% were refractory to hydroxyurea and 67% were positive for JAK2 V617F. In patients with ET, INCB018424 led to rapid and sustained normalization of platelet counts (baseline: 884×109/L; 6-months: 558×109/L) and 85% achieved a platelet count < 600×109/L. ORR was 90% (CR=13%, PR=77%).
Overall, the drug was well tolerated, and most common grade 3 toxicities included thrombocytopenia (PV: 3%) and neutropenia (ET: 5%). Grade 2 anemia was observed in 12% of PV patients and 18% of ET patients. No grade 4 toxicity has been recorded so far. Six patients have discontinued drug therapy at the time of last report, and 3 of these were due to adverse side effects (atrial flutter, renal insufficiency and diarrhea/nausea/vomiting in one each). INCB018424 appears to be highly effective for therapy of patients with PV/ET who are refractory/intolerant to hydroxyurea.
Conclusion
The development of JAK2 inhibitors has started the era of targeted therapies for patients with Ph-negative MPNs. Even though these drugs do not seem to eradicate the malignant clone, there is still great benefit to be gained from the use of these compounds. Improvements in systemic symptoms and splenomegaly can significantly impact on the quality of life of patients with MF, which is significant clinical benefit. For patients with PV and ET, initial studies suggest that these drugs represent a safe alternative for patients who fail therapy with hydroxyurea, the most commonly employed first line agent. Despite these initial advances, there is still a lot to be learned from these compounds. We still do not fully understand their mechanism of action, and if their benefit is derived from inhibition of JAK2 in neoplastic cells and/or due to a more global down-regulation of cytokine signalling. Response does not seem to be dependent on JAK2 mutation status, and determining biomarkers for response could permit better selection of patients to be treated with these drugs. In conclusion, JAK2 inhibitors are a novel class of agents with promising results for treating patients with MF, PV and ET. Further studies are needed to better understand and define their role in the treatment of Ph-negative MPNs.
Figure 3.

Biochemical structures of selected JAK2 inhibitors in clinical trials.
Practice Points.
JAK2 inhibitors can lead to improvement in splenomegaly, systemic symptoms, exercise capacity and quality of life in patients with MF
No improvement in bone marrow fibrosis or reduction of JAK2 mutated allelic burden is usually seen
In patients with PV and ET, JAK2 inhibitors lead to sustained improvement in peripheral blood counts and freedom from phlebotomies
Clinical benefits are seen both in patients with and without the JAK2 V617F mutation
Common side effects include anemia, thrombocytopenia and gastrointestinal disturbances
Research Agenda.
Determine the mechanism of action of these drugs and which cells they are targeting – neoplastic cells, normal cells, or both
Discover biomarkers predictive of response to JAK2 inhibitors
Evaluate the impact of JAK2 inhibitors on survival and leukemic transformation of Ph-negative MPNs
Footnotes
Conflicts of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tefferi A, Gilliland G. Classification of chronic myeloid disorders: from Dameshek towards a semi-molecular system. Best Pract Res Clin Haematol. 2006;19(3):365–85. doi: 10.1016/j.beha.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000 Apr 27;342(17):1255–65. doi: 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- 3.Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976–1995. Am J Hematol. 1999 May;61(1):10–5. doi: 10.1002/(sici)1096-8652(199905)61:1<10::aid-ajh3>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 4.Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008 Aug;22(8):1494–502. doi: 10.1038/leu.2008.177. [DOI] [PubMed] [Google Scholar]
- 5.Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005 Apr 1;105(7):2664–70. doi: 10.1182/blood-2004-09-3426. [DOI] [PubMed] [Google Scholar]
- 6.Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005 Jul 7;353(1):33–45. doi: 10.1056/NEJMoa043800. [DOI] [PubMed] [Google Scholar]
- 7.Thoennissen NH, Krug UO, Lee DH, Kawamata N, Iwanski GB, Lasho T, et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood. 2010 Apr 8;115(14):2882–90. doi: 10.1182/blood-2009-07-235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mesa RA, Barosi G, Cervantes F, Reilly JT, Tefferi A. Myelofibrosis with myeloid metaplasia: disease overview and non-transplant treatment options. Best Pract Res Clin Haematol. 2006;19(3):495–517. doi: 10.1016/j.beha.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Trillos A, Gaya A, Maffioli M, Arellano-Rodrigo E, Calvo X, Diaz-Beya M, et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol. 2010 Jun 22; doi: 10.1007/s00277-010-1019-9. [DOI] [PubMed] [Google Scholar]
- 10.Mesa RA, Steensma DP, Pardanani A, Li CY, Elliott M, Kaufmann SH, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. 2003 Apr 1;101(7):2534–41. doi: 10.1182/blood-2002-09-2928. [DOI] [PubMed] [Google Scholar]
- 11.Siragusa S, Vaidya R, Tefferi A. Hydroxyurea Effect On Marked Splenomegaly Associated with Primary Myelofibrosis: Response Rates and Correlation with JAK2V617F Allele Burden [abstract] Blood. 2009 Nov 20;114(22):Abstract 4971. [Google Scholar]
- 12.Mesa RA, Nagorney DS, Schwager S, Allred J, Tefferi A. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer. 2006 Jul 15;107(2):361–70. doi: 10.1002/cncr.22021. [DOI] [PubMed] [Google Scholar]
- 13.Mesa RA, Niblack J, Wadleigh M, Verstovsek S, Camoriano J, Barnes S, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007 Jan 1;109(1):68–76. doi: 10.1002/cncr.22365. [DOI] [PubMed] [Google Scholar]
- 14.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005 Mar 19–25;365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 15.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005 Apr 28;434(7037):1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 16.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005 Apr 28;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 17.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005 Apr;7(4):387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 18.Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989 Mar;86(5):1603–7. doi: 10.1073/pnas.86.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng. 2002 Sep;15(9):727–37. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 20.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991 Apr;11(4):2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002 Dec 6;277(49):47954–63. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 22.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001 Jan;14(1):27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 23.Radtke S, Haan S, Jorissen A, Hermanns HM, Diefenbach S, Smyczek T, et al. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J Biol Chem. 2005 Jul 8;280(27):25760–8. doi: 10.1074/jbc.M500822200. [DOI] [PubMed] [Google Scholar]
- 24.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001 Dec;8(6):1327–38. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 25.Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. 2005 Jul 22;280(29):27251–61. doi: 10.1074/jbc.M501376200. [DOI] [PubMed] [Google Scholar]
- 26.Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 27.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004 Feb;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 28.Winston LA, Hunter T. JAK2, Ras, and Raf are required for activation of extracellular signal-regulated kinase/mitogen-activated protein kinase by growth hormone. J Biol Chem. 1995 Dec 29;270(52):30837–40. doi: 10.1074/jbc.270.52.30837. [DOI] [PubMed] [Google Scholar]
- 29.Mizuguchi R, Noto S, Yamada M, Ashizawa S, Higashi H, Hatakeyama M. Ras and signal transducer and activator of transcription (STAT) are essential and sufficient downstream components of Janus kinases in cell proliferation. Jpn J Cancer Res. 2000 May;91(5):527–33. doi: 10.1111/j.1349-7006.2000.tb00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Shami A, Naccache PH. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. Involvement of Jak2 in the stimulation of phosphatidylinositol 3-kinase. J Biol Chem. 1999 Feb 26;274(9):5333–8. doi: 10.1074/jbc.274.9.5333. [DOI] [PubMed] [Google Scholar]
- 31.Bouscary D, Pene F, Claessens YE, Muller O, Chretien S, Fontenay-Roupie M, et al. Critical role for PI 3-kinase in the control of erythropoietin-induced erythroid progenitor proliferation. Blood. 2003 May 1;101(9):3436–43. doi: 10.1182/blood-2002-07-2332. [DOI] [PubMed] [Google Scholar]
- 32.Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995 Mar 10;80(5):729–38. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 33.Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci U S A. 1998 Mar 31;95(7):3845–50. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001 Jan 18;409(6818):349–54. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- 35.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997 Jun 26;387(6636):921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 36.Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol. 2002 May;22(10):3316–26. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998 May 1;93(3):373–83. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 38.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998 May 1;93(3):385–95. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 39.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998 May 1;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 40.Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995 Nov 3;270(5237):800–2. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 41.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995 Nov 3;270(5237):794–7. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 42.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995 Sep 7;377(6544):65–8. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 43.Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995 Nov 3;270(5237):797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 44.Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000 Oct;13(4):549–60. doi: 10.1016/s1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- 45.Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000 Oct;13(4):561–71. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 46.Luo H, Rose P, Barber D, Hanratty WP, Lee S, Roberts TM, et al. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. 1997 Mar;17(3):1562–71. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jelinek J, Oki Y, Gharibyan V, Bueso-Ramos C, Prchal JT, Verstovsek S, et al. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005 Nov 15;106(10):3370–3. doi: 10.1182/blood-2005-05-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiladjian JJ, Cervantes F, Leebeek FW, Marzac C, Cassinat B, Chevret S, et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008 May 15;111(10):4922–9. doi: 10.1182/blood-2007-11-125328. [DOI] [PubMed] [Google Scholar]
- 49.Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009 May 21;113(21):5246–9. doi: 10.1182/blood-2008-11-191544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olcaydu D, Harutyunyan A, Jager R, Berg T, Gisslinger B, Pabinger I, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009 Apr;41(4):450–4. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- 51.Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009 Apr;41(4):455–9. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009 Apr;41(4):446–9. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5(6):e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006 Sep 1;108(5):1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 55.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006 Jun 1;107(11):4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res. 2006 Dec 1;66(23):11156–65. doi: 10.1158/0008-5472.CAN-06-2210. [DOI] [PubMed] [Google Scholar]
- 57.Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X, et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood. 2008 May 15;111(10):5109–17. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008 Apr 15;111(8):3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 60.Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008 Jan;22(1):87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 61.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010 Jun 15;17(6):584–96. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010 Apr 29;115(17):3589–97. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010 May 20; doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC, et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood. 2010 May 14; doi: 10.1182/blood-2009-12-257063. [DOI] [PubMed] [Google Scholar]
- 65.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009 Oct 8;461(7265):819–22. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006 Apr 18;103(16):6224–9. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005 Sep 15;106(6):2162–8. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 68.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006 Oct 1;108(7):2435–7. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 69.Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005 Dec 3;366(9501):1945–53. doi: 10.1016/S0140-6736(05)67785-9. [DOI] [PubMed] [Google Scholar]
- 70.Wolanskyj AP, Lasho TL, Schwager SM, McClure RF, Wadleigh M, Lee SJ, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol. 2005 Oct;131(2):208–13. doi: 10.1111/j.1365-2141.2005.05764.x. [DOI] [PubMed] [Google Scholar]
- 71.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007 Aug 1;110(3):840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 72.Tefferi A, Lasho TL, Schwager SM, Strand JS, Elliott M, Mesa R, et al. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer. 2006 Feb 1;106(3):631–5. doi: 10.1002/cncr.21645. [DOI] [PubMed] [Google Scholar]
- 73.Barosi G, Bergamaschi G, Marchetti M, Vannucchi AM, Guglielmelli P, Antonioli E, et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007 Dec 1;110(12):4030–6. doi: 10.1182/blood-2007-07-099184. [DOI] [PubMed] [Google Scholar]
- 74.Guglielmelli P, Barosi G, Specchia G, Rambaldi A, Lo Coco F, Antonioli E, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009 Aug 20;114(8):1477–83. doi: 10.1182/blood-2009-04-216044. [DOI] [PubMed] [Google Scholar]
- 75.Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008 Apr;22(4):756–61. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 76.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007 Feb 1;356(5):459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006 Jul;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008 Jul 1;112(1):141–9. doi: 10.1182/blood-2008-01-131664. [DOI] [PubMed] [Google Scholar]
- 79.Chaligne R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, et al. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia. 2008 Aug;22(8):1557–66. doi: 10.1038/leu.2008.137. [DOI] [PubMed] [Google Scholar]
- 80.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008 Feb 1;111(3):1686–9. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 81.Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007 Sep;21(9):1960–3. doi: 10.1038/sj.leu.2404810. [DOI] [PubMed] [Google Scholar]
- 82.Butcher CM, Hahn U, To LB, Gecz J, Wilkins EJ, Scott HS, et al. Two novel JAK2 exon 12 mutations in JAK2V617F-negative polycythaemia vera patients. Leukemia. 2008 Apr;22(4):870–3. doi: 10.1038/sj.leu.2404971. [DOI] [PubMed] [Google Scholar]
- 83.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006 Nov 15;108(10):3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 84.Guglielmelli P, Pancrazzi A, Bergamaschi G, Rosti V, Villani L, Antonioli E, et al. Anaemia characterises patients with myelofibrosis harbouring Mpl mutation. Br J Haematol. 2007 May;137(3):244–7. doi: 10.1111/j.1365-2141.2007.06565.x. [DOI] [PubMed] [Google Scholar]
- 85.Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, Barosi G, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008 Aug 1;112(3):844–7. doi: 10.1182/blood-2008-01-135897. [DOI] [PubMed] [Google Scholar]
- 86.Nussenzveig RH, Swierczek I, Jelinek J, Gaikwad A, Liu E, Verstovsek S, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007 Jan;35(1):32–8. doi: 10.1016/j.exphem.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 87.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010 Jun;24(6):1128–38. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009 May 28;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 89.Beer PA, Delhommeau F, LeCouedic JP, Dawson MA, Chen E, Bareford D, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010 Apr 8;115(14):2891–900. doi: 10.1182/blood-2009-08-236596. [DOI] [PubMed] [Google Scholar]
- 90.Schaub FX, Looser R, Li S, Hao-Shen H, Lehmann T, Tichelli A, et al. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood. 2010 Mar 11;115(10):2003–7. doi: 10.1182/blood-2009-09-245381. [DOI] [PubMed] [Google Scholar]
- 91.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010 Jul 18; doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tam CS, Nussenzveig RM, Popat U, Bueso-Ramos CE, Thomas DA, Cortes JA, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood. 2008 Sep 1;112(5):1628–37. doi: 10.1182/blood-2008-02-138230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008 Jun 15;111(12):5663–71. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010 Apr 15;115(15):3109–17. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005 Jul 14;353(2):172–87. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 96.Verstovsek S, Kantarjian H, Mesa R, Pardanani A, Cortes J, Thomas D, et al. Safety and Efficacy of JAK1 & JAK2 Inhibitor, INCB018424, in Myelofibrosis. N Engl J Med. 2010 doi: 10.1056/NEJMoa1002028. accepted, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kralovics R. Genetic complexity of myeloproliferative neoplasms. Leukemia. 2008 Oct;22(10):1841–8. doi: 10.1038/leu.2008.233. [DOI] [PubMed] [Google Scholar]
- 98.Vannucchi AM. How do JAK2-inhibitors work in myelofibrosis: an alternative hypothesis. Leuk Res. 2009 Dec;33(12):1581–3. doi: 10.1016/j.leukres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 99.Schmitt A, Jouault H, Guichard J, Wendling F, Drouin A, Cramer EM. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000 Aug 15;96(4):1342–7. [PubMed] [Google Scholar]
- 100.Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007 Aug 1;110(3):986–93. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bock O, Hoftmann J, Theophile K, Hussein K, Wiese B, Schlue J, et al. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am J Pathol. 2008 Apr;172(4):951–60. doi: 10.2353/ajpath.2008.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. 2000 Jul;157(1):15–9. doi: 10.1016/S0002-9440(10)64511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tefferi A, Kantarjian HM, Pardanani AD, Mesa RA, Newton RC, Scherle PA, et al. The Clinical Phenotype of Myelofibrosis Encompasses a Chronic Inflammatory State That Is Favorably Altered by INCB018424, a Selective Inhibitor of JAK1/2 [abstract] Blood. 2008 Nov 16;112(11):Abstract 2804. [Google Scholar]
- 104.Emadi S, Clay D, Desterke C, Guerton B, Maquarre E, Charpentier A, et al. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood. 2005 Jan 15;105(2):464–73. doi: 10.1182/blood-2003-12-4415. [DOI] [PubMed] [Google Scholar]
- 105.Wang JC, Chen C, Lou LH, Mora M. Blood thrombopoietin, IL-6 and IL-11 levels in patients with agnogenic myeloid metaplasia. Leukemia. 1997 Nov;11(11):1827–32. doi: 10.1038/sj.leu.2400846. [DOI] [PubMed] [Google Scholar]
- 106.Jedidi A, Marty C, Oligo C, Jeanson-Leh L, Ribeil JA, Casadevall N, et al. Selective reduction of JAK2V617F-dependent cell growth by siRNA/shRNA and its reversal by cytokines. Blood. 2009 Aug 27;114(9):1842–51. doi: 10.1182/blood-2008-09-176875. [DOI] [PubMed] [Google Scholar]
- 107.Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010 Feb 11;115(6):1131–6. doi: 10.1182/blood-2009-10-246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004 May 15;103(10):3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 109.Tefferi A, Barosi G, Mesa RA, Cervantes F, Deeg HJ, Reilly JT, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT) Blood. 2006 Sep 1;108(5):1497–503. doi: 10.1182/blood-2006-03-009746. [DOI] [PubMed] [Google Scholar]
- 110.Hexner E, Goldberg JD, Prchal JT, Demakos EP, Swierczek S, Weinberg RS, et al. A Multicenter, Open Label Phase I/II Study of CEP701 (Lestaurtinib) in Adults with Myelofibrosis; a Report On Phase I: A Study of the Myeloproliferative Disorders Research Consortium (MPD-RC) [abstract] Blood. 2009 Nov 20;114(22):Abstract 754. [Google Scholar]
- 111.Mesa RA, Schwager S, Radia D, Cheville A, Hussein K, Niblack J, et al. The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009 Sep;33(9):1199–203. doi: 10.1016/j.leukres.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goh KC, Ong WC, Hu C, Hentze H, Liang AL, Stunkel W, et al. SB1518: A Potent and Orally Active JAK2 Inhibitor for the Treatment of Myeloproliferative Disorders [abstract] Blood. 2007 Nov 16;110(11):Abstract 538. [Google Scholar]
- 113.Verstovsek S, Odenike O, Scott B, Estrov Z, Cortes J, Thomas DA, et al. Phase I Dose-Escalation Trial of SB1518, a Novel JAK2/FLT3 Inhibitor, in Acute and Chronic Myeloid Diseases, Including Primary or Post-Essential Thrombocythemia/Polycythemia Vera Myelofibrosis [abstract] Blood. 2009 Nov 20;114(22):Abstract 3905. [Google Scholar]
- 114.Seymour JF, To B, Goh A, Meadows S, Ethirajulu K, Wood J, et al. First Report of the Phase I Study of the novel oral JAK2 inhibitor SB1518 in patients with Myelofibrosis [abstract] Haematologica. 2010 Jun 10–13;95(suppl 2):Abstract 1444. [Google Scholar]
- 115.Geron I, Abrahamsson AE, Barroga CF, Kavalerchik E, Gotlib J, Hood JD, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008 Apr;13(4):321–30. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 116.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008 Apr;13(4):311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 117.Pardanani AD, Gotlib JR, Jamieson C, Cortes J, Talpaz M, Stone R, et al. A Phase I Evaluation of TG101348, a Selective JAK2 Inhibitor, in Myelofibrosis: Clinical Response Is Accompanied by Significant Reduction in JAK2V617F Allele Burden [abstract] Blood. 2009 Nov 20;114(22):Abstract 755. [Google Scholar]
- 118.Verstovsek S, Pardanani AD, Shah NP, Sokol L, Wadleigh M, Gilliland DG, et al. A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Primary Myelofibrosis and Post-Polycythemia Vera/Essential Thrombocythemia Myelofibrosis [abstract] Blood. 2007 Nov 16;110(11):Abstract 553. [Google Scholar]
- 119.Shah NP, Olszynski P, Sokol L, Verstovsek S, Hoffman R, List AF, et al. A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Primary Myelofibrosis, Post-Polycythemia Vera, or Post-Essential Thrombocythemia Myelofibrosis [abstract] Blood. 2008 Nov 16;112(11):Abstract 98. [Google Scholar]
- 120.Moliterno AR, Hexner E, Roboz GJ, Carroll M, Luger S, Mascarenhas J, et al. An Open-Label Study of CEP-701 in Patients with JAK2 V617F-Positive PV and ET: Update of 39 Enrolled Patients [abstract] Blood. 2009 Nov 20;114(22):Abstract 753. [Google Scholar]
- 121.Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen P, Levy R, et al. A Phase 2 Study of INCB018424, An Oral, Selective JAK1/JAK2 Inhibitor, in Patients with Advanced Polycythemia Vera (PV) and Essential Thrombocythemia (ET) Refractory to Hydroxyurea [abstract] Blood. 2009 Nov 20;114(22):Abstract 311. [Google Scholar]
- 122.Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009 May 14;113(20):4829–33. doi: 10.1182/blood-2008-09-176818. [DOI] [PubMed] [Google Scholar]