

Abstract
Purpose of review
This review will explore two new aspects of the involvement of viruses in multiple sclerosis pathogenesis. The first aspect is the complex interactions between viruses. The second aspect is the proposal of a mechanism by which autoreactive T cells are able to escape thymic selection and potentially recognize self and a pathogen.
Recent findings
With regard to viruses, recent work has demonstrated that one virus may enhance the replication of another virus, potentially leading to an increase in inflammation and disease progression. Also, interactions between human endogenous retroviruses, which likely do not replicate, and certain herpes viruses, may also play a role in disease pathogenesis. Mechanistically, T cells expressing dual T-cell receptors would be able to recognize self and a foreign antigen specifically. Therefore, human endogenous retroviruses potentially play a role in multiple sclerosis pathogenesis, and both interactions between multiple viruses and autoreactive CD8+ T cells with dual T-cell receptors may play a role in the pathogenesis of the disease.
Summary
The complex interactions between multiple viral infections, either within the central nervous system or in the periphery, and the host immune response to viral infection may be such that a variety of viral specificities result in the activation of T cells that recognize self and induce multiple sclerosis. Therefore, it is unlikely that any one microbe will be determined to be the causative agent of multiple sclerosis as reflected by the number of potential triggering mechanisms of the disease.
Keywords: autoreactive, CD8+ cytotoxic T lymphocytes, dual T-cell receptor
Introduction
Much has been written over the years on the role that viruses are thought to play in multiple sclerosis (MS). Several, recent, well written reviews have covered the topic in depth [1,2▪,3,4]. However, there are new aspects of the involvement of viruses in MS that deserve a closer look. Therefore, this review will first briefly discuss the disease itself. This will be followed by a discussion on how interactions between multiple viruses may play a role in MS pathogenesis. Finally, the involvement of CD8+ T cells and three possible mechanisms for the generation of autoreactive CD8+ T cells upon viral infection will be examined.
The Disease
Autoimmune diseases are characterized by the body's intolerance to self-antigens. The most common autoimmune disease of young adult Caucasians is MS [5]. The cause of MS is unknown, thus making the classification of MS as a strict autoimmune disease debatable [6▪]. However, experimental evidence suggests, and it is widely accepted, that an immune response directed against self-antigens in the central nervous system (CNS) is important in disease progression. Clinically, MS patients can present with disturbances in vision, sensation and motor control. The disease is diagnosed by the presence of focal demyelinated plaques in the brain. The clinical course of MS is variable; it ranges from a majority of patients who have attacks separated by periods of no clinical symptoms (relapsing–remitting) to a more progressive clinical manifestation of the disease (primary progressive or secondary progressive). The heterogeneity of the clinical presentation could be the result of the variety of immune cells, including monocytes/macrophages, activated glial cells and T cells specific for different myelin or CNS antigens/epitopes, associated with disease progression [7–9]. Although MS is believed to be a CD4+ T-cell-mediated autoimmune disease, CD8+ T cells may be involved in MS pathogenesis due to their being the predominate leukocyte in the lesions of MS patients. Lesions are not restricted to white matter in the CNS and abnormalities in the cortical and deep gray matter are observed in the early stages of MS [10–12,13▪▪]. CNS inflammation can lead to oligodendrocyte death and axonal transection (reviewed in [14,15]). Axonal loss inevitably leads to disability in MS patients. Disease-modifying therapeutics for the treatment of MS, such as interferon (IFN)-β, glatimer acetate (copaxone), mitoxantrone and natalizumab, all modulate the immune system [16,17], thereby providing evidence supporting a central role for the immune system in driving disease progression. Unfortunately, the available therapeutics do not prevent axonal loss over time in progressive forms of MS indicating that new therapeutics directed at axonal preservation and a better understanding of what cell types are involved in MS pathogenesis are necessary.
The cause of MS is not known. Although a variety of myelin self-antigens recognized by the immune system have been identified in MS patients, no single self-antigen has been definitively shown to be responsible for MS pathogenesis. Environmental factors such as bacterial and viral infections have been implicated in MS pathogenesis. Implicated viruses include some of the herpes viruses, influenza virus, paramyxoviruses and picornaviruses (recently reviewed in [1,2▪,3,4]). There is evidence that peripheral (outside of the CNS) viral infections, such as with picornaviruses, can trigger exacerbations of MS [18]. Although the triggering factor or event is unknown, epidemiological and experimental evidence shows that certain individuals are genetically susceptible to MS in combination with environmental factors. Genome-wide association studies found that the most common alleles associated with MS are human leukocyte antigen (HLA)-DR1501 and HLA-DQ0601, which encode for molecules that present antigen to CD4+ T cells [19,20], and the interleukin (IL)-2 receptor α and IL-7 receptor α (CD127) genes [20].
Several mechanisms could explain how viral infections lead to MS. The first mechanism involves direct infection of the brain by a virus. Viral infection of the CNS can potentially cause inflammation and damage to cells that produce myelin. This damage releases fragments of myelin that are recognized by autoreactive T cells which then are activated within the inflammatory milieu. This results in epitope spreading in which T cells that recognize epitopes of myelin proteins then trigger a series of events that result in more inflammation in the CNS and more myelin destruction. Another mechanism, that need not include epitope spreading, involves a persistent viral infection within the CNS resulting in inflammatory demyelinating disease due to the immune response attempting to eliminate infected cells within the CNS. An appealing alternate mechanism is that peripheral infections (outside of the CNS) can activate cross-reactive T cells such that the immune response to the virus cross-reacts with CNS myelin or ‘self’. In this way, the activated T cells have the ability to recognize both the virus as well as myelin. These T cells then enter the CNS and cause inflammatory demyelinating lesions and MS. These mechanisms are not mutually exclusive and in different individuals could potentially lead to MS. A largely unanswered question is how viral infection in the periphery can generate cross-reactive autoreactive T cells that induce inflammation and demyelination within the CNS.
Interactions between Viruses
Most studies trying to determine if there is a single causative virus for MS have not been successful; however, interactions between viruses have not been fully characterized as a potential mechanism involved in MS pathogenesis. Matullo et al. [21] demonstrated, using a transgenic mouse model in which measles virus replication was restricted to the CNS, that peripheral infection could potentially trigger CNS pathology. It was found that peripheral infection of these mice with lymphocytic choriomeningitis virus (LCMV) resulted in peripheral LCMV-specific CD8+ T cells being recruited to the CNS where they caused neurological damage. These CD8+ T cells caused damage despite no detectable LCMV in the CNS. These authors proposed that human CNS diseases with an unknown cause, such as MS, need not be associated with just one infectious agent. Any inflammation leading to the breakdown of the blood–brain barrier could leave the CNS vulnerable to pathogen-independent immune attack.
In support of the hypothesis that one virus may enhance another virus's replication, Borkosky et al. [22▪▪] used torque teno virus (TTV) isolates obtained from the brains of MS patients to infect both lymphoblastoid and Burkitt's lymphoma cell lines. Next, infecting the TTV-infected cell lines with Epstein–Barr virus (EBV) led to enhanced TTV replication, suggesting that interactions between two viruses in the CNS could synergistically cause and promote disease progression.
In addition, there is circumstantial evidence for involvement of human endogenous retroviruses (HERVs) in MS pathogenesis based on the detection of HERV viral RNA and/or protein in MS patients in comparison to healthy donors [23,24,25▪]. HERVs are RNA viruses and are able to express reverse transcriptase. Approximately 8% of every human genome is HERVs [26]. Although HERVs are most likely not able to replicate, evidence suggests that HERVs could potentially be involved in MS pathogenesis by being expressed as proteins [27], by possessing immunogenic properties [28] and by being activated and responding to immune stimuli [29]. Thus, HERVs are hypothesized to be ‘retroelement’ genes that are potentially able to impact cellular functions [30]. Recently, Perron et al. [25▪] used quantitative PCR and antibody staining to detect and quantify HERV-W multiple sclerosis retrovirus (MSRV) expression in peripheral blood mononuclear cells and tissues obtained from MS patients, thereby demonstrating an association between MS disease and MSRV in comparison to healthy controls. Similar work by Laska et al. [31▪] provided further evidence that the RNA titer of HERV-Fc1 was markedly higher in MS patients when compared with healthy controls. A potential mechanism by which HERVs could be involved in MS pathogenesis was proposed by Mameli et al. [32▪] in that EBV-activated HERV-W/MSRV/synctin-1 in cells derived from peripheral blood and astrocytes, thereby suggesting that EBV could be an initial trigger for MS and that EBV and HERV-W/MSRV/synctin-1 are the main contributors to MS pathogenesis [32▪]. MS has been associated with increased reactivity to certain HERVs either individually or in association with herpes viruses [33]. Petersen et al. [34▪▪] monitored the seroreactivity to EBV, Epstein-Barr nuclear antigen 1 and varicella zoster virus, and compared antibody levels to these viruses in MS patients that either did or did not respond to IFN-β therapy. This longitudinal study [34▪▪] demonstrated that disease activity was related to HERVs and certain herpes viruses, suggesting that disease progression and efficacy of IFN-β treatment in MS patients were directly linked to the immune response to these viruses, thereby implicating HERVs as potentially having a role in MS pathogenesis.
The Involvement of CD8+ T Cells
In MS, although both CD4+ and CD8+ T cells can be found in lesions, activated CD8+ T cells outnumber CD4+ T cells by approximately 10-fold [35,36]. Furthermore, these large numbers of CD8+ T cells are found to be clonally expanded in sites of active demyelination [37,38]. Various groups have shown that class I genes such as HLA-A3 increase the risk of developing MS [39–43]. This is consistent with the observations that CD8+ T cells are clonally expanded in the cerebrospinal fluid and in the circulation of MS patients [44]. In addition, cloned CD8+ T cell lines have been generated from MS patients that are HLA-A2-restricted and specific for myelin basic protein (MBP), myelin proteolipid protein (PLP) and myelin-associated glycoprotein [45]. These CD8+ cytotoxic T lymphocyte (CTL) clones produced tumor necrosis factor (TNF)-α and some also produced IFN-γ. Interestingly, some HLA-A3 restricted CD8+ T cells cloned from MS patients have been found to lyse targets coated with PLP peptide as well as targets coated with peptide derived from Saccharomyces cerevisiae, the chromosome region maintenance 1770–778 peptide [46]. These reports indicate that within the autoreactive CD8+ T-cell population, cross-reactive T cells exist that have the potential to react with microbes/viruses and self, suggesting a potential role for either molecular mimicry or for CD8+ T cells having more than one T-cell receptor (TCR). For human and mouse T cells in the periphery, the frequency of T cells expressing more than one TCR β chain is approximately 1% [47,48]. About 30% of human T cells in the periphery have two functional TCR α chains [49] and approximately 8–15% of murine T cells in the periphery express more than one TCR α chain on the cell surface [50,51]. Interestingly, while studying TCR expression on myelin-specific T cells, Alli et al. [52▪,53] identified T cells that expressed dual TCRs: one TCR combination was specific for myelin and one reacted against hair follicles. Almost all transgenic mice expressing one of the TCR combinations developed alopecia by 4 months, and CD8+ T cells from these mice could adoptively transfer alopecia, demonstrating a potential autoimmune role for dual TCRs.
Potential Mechanisms for The Generation of Autoreactive CD8+ T Cells
A major question to be addressed is how are such autoreactive CD8+ T cells generated and how do they initiate CNS inflammatory disease. We suspect that there are multiple mechanisms for how viral infections can lead to the activation of autoreactive T cells that then go to the target organ causing inflammation and pathology. We hypothesize that the autoreactive CD8+ T cells generated following viral infection are novel in that they express more than one TCR and can induce inflammatory demyelinating autoimmune disease in naïve mice. We do not know, however, if one of the TCRs recognizes both a viral and a self-peptide (molecular mimicry) (Fig. 1, #1) [54▪], if the two separate TCRs are functioning where one TCR recognizes the viral peptide and the other TCR recognizes the self-peptide (Fig. 1, #2), or if the TCR is a chimera having either 2 α chains and 1 β chain or 2 β chains and 1 α chain, which, when present in different combinations, results in one combination recognizing viral peptide and the other combination recognizing self-peptide (Fig. 1, #3).
Figure 1.
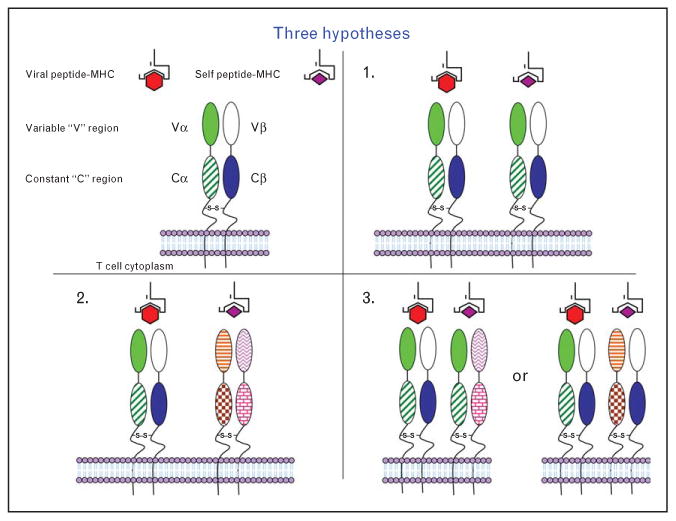
Model for dual TCR reactivity. Three possible mechanisms exist for dual reactivity of the TCR in autoimmune disease: (1) molecular mimicry, where one TCR recognizes virus and self; (2) dual TCRs, where one TCR is specific for virus and the other TCR is specific for self; and (3) a chimeric TCR, where different Vα and Vβ combinations on a single T cell are responsible for recognizing both virus and self. Source: Previously published as Fig. 8 in Ref. [54▪].
Two important observations support our hypothesis that dual TCR expression allows self-reactive T cells to escape thymic selection and be activated by a virus. Recently, the Goverman lab [55] described a MBP transgenic TCR model where the MBP-specific CD8+ T cells encoded multiple TCRs. Experimental autoimmune encephalomyelitis could be induced in these mice by vaccinia virus infection, suggesting that dual-reactive T cells participate in CNS demyelinating disease: one TCR recognizing MBP and the other TCR recognizing a vaccinia virus peptide. However, they did not determine whether one or more than one TCR was involved in the demyelinating disease, nor did they experimentally demonstrate a direct pathogenic effect of these dual expressing T cells. Also, these studies were performed with transgenic mice and using a virus for which the natural host is unknown.
Recently, we have shown that Theiler's murine encephalomyelitis virus (TMEV) infection generates autoreactive T cells [56]. Spleen mononuclear cells (MNCs) isolated from SJL/J mice, 5 weeks after infection, proliferated when stimulated with irradiated TMEV-infected syngeneic antigen-presenting cells (APCs), as well as when stimulated with irradiated uninfected syngeneic APCs [56]. In addition, we also demonstrated that in-vitro stimulated bulk MNCs could kill uninfected syngeneic target cells [57]. Upon further characterization, we ascertained that the autoreactive cells were CD8+ T cells [57]. Using recombinant vaccinia virus encoding TMEV capsid proteins to infect SJL/J mice, we showed that CD8+ T cells from these infected mice could also kill uninfected syngeneic target cells [58]. Infection of mice with a recombinant vaccinia virus encoding β-galactosidase did not result in CD8+ T cells capable of killing uninfected target cells, thus providing evidence that a viral epitope encoded in the viral capsid is sufficient to generate these autoreactive T cells [58]. These data indicate that following virus infection, a population of T cells were generated that could recognize virus and self. Importantly, we have demonstrated that hybridomas generated from cloned TMEV-induced dual-reactive CD8+ T cells were able to induce clinical signs of disease (flaccid hind limb paralysis) upon adoptive transfer into naive mice [54▪]. Dual TCRs were detected on the surface of these hybridomas by flow cytometry, and multiple α chain mRNAs were detected by PCR screening. Although individual T cells having more than one TCR have been previously reported [47,59–66], many of these studies used transgenic systems. Therefore, T cells having more than one TCR are not unique, but their production following peripheral viral infection of the natural host is unique, and this is the first demonstration of antiviral CD8+ T cells having more than one TCR initiating an autoimmune disease in vivo. We hypothesize that this is a potential mechanism for virus-induced autoimmune disease initiated by CD8+ T cells.
Conclusion
Investigators have reported the occurrence of T cells with more than one TCR [47,59–66] and have hypothesized that such cells could initiate autoimmune disease. We hypothesize that autoreactive CD8+ T cells are generated during viral infection. T cells having more than one TCR could explain why no one microbe has been consistently associated with MS. We hypothesize that a variety of viral specificities/peptides could activate T cells which can also recognize self, thereby initiating disease. The induction of MS by viruses could potentially be through multiple mechanisms (Fig. 1) and future work is needed to determine the relevant mechanism(S).
Key Points.
The immune response to viral infection could mediate multiple sclerosis.
A causative agent for multiple sclerosis could be the interactions between viruses.
Autoreactive CD8+ T cells generated following viral infection that express more than one TCR could be involved in MS pathogenesis.
Acknowledgments
We are grateful to Mr. Daniel J. Harper for preparation of the manuscript.
This work was supported by NIH grants T32AI055434 (MFC) and 5R01NS065714–03 and the Emma Mary Deland Foundation.
Footnotes
Conflicts of interest: There are no conflicts of interest.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 557).
- 1.Libbey JE, Fujinami RS. Potential triggers of MS. Results Probl Cell Differ. 2010;51:21–42. doi: 10.1007/400_2008_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2▪.Virtanen JO, Jacobson S. Viruses and multiple sclerosis. CNS Neurol Disord Drug Targets. 2012;11:528–544. doi: 10.2174/187152712801661220. This is an in-depth review on the role of viruses in MS pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owens GP, Gilden D, Burgoon MP, et al. Viruses and multiple sclerosis. Neuroscientist. 2011;17:659–676. doi: 10.1177/1073858411386615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kakalacheva K, Munz C, Lunemann JD. Viral triggers of multiple sclerosis. Biochim Biophys Acta. 2011;1812:132–140. doi: 10.1016/j.bbadis.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 6▪.Wootla B, Eriguchi M, Rodriguez M. Is multiple sclerosis an autoimmune disease? Autoimmune Dis. 2012;2012:969657. doi: 10.1155/2012/969657. This study provides evidence for and against MS as a classical autoimmune disease and proposes that viruses can be a potential trigger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown A, McFarlin DE, Raine CS. Chronologic neuropathology of relapsing experimental allergic encephalomyelitis in the mouse. Lab Invest. 1982;46:171–185. [PubMed] [Google Scholar]
- 8.Lassmann H. Axonal and neuronal pathology in multiple sclerosis: what have we learnt from animal models. Exp Neurol. 2010;225:2–8. doi: 10.1016/j.expneurol.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kidd D, Barkhof F, McConnell R, et al. Cortical lesions in multiple sclerosis. Brain. 1999;122:17–26. doi: 10.1093/brain/122.1.17. [DOI] [PubMed] [Google Scholar]
- 11.De Stefano N, Matthews PM, Filippi M, et al. Evidence of early cortical atrophy in MS: relevance to white matter changes and disability. Neurology. 2003;60:1157–1162. doi: 10.1212/01.wnl.0000055926.69643.03. [DOI] [PubMed] [Google Scholar]
- 12.Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7:841–851. doi: 10.1016/S1474-4422(08)70191-1. [DOI] [PubMed] [Google Scholar]
- 13▪▪.Magliozzi R, Serafini B, Rosicarelli B, et al. B-cell enrichment and epstein-barr virus infection in inflammatory cortical lesions in secondary progressive multiple sclerosis. J Neuropathol Exp Neurol. 2013;72:29–41. doi: 10.1097/NEN.0b013e31827bfc62. This study shows that lesions found in MS patients are not restricted to white matter in the CNS and proposes that a potential mechanism of intracortical inflammation could be sustained by an EBV-driven immunopathologic response. [DOI] [PubMed] [Google Scholar]
- 14.Bjartmar C, Kidd G, Mork S, et al. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 15.Dutta R, McDonough J, Chang A, et al. Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of multiple sclerosis patients. Brain. 2007;130:2566–2576. doi: 10.1093/brain/awm206. [DOI] [PubMed] [Google Scholar]
- 16.Libbey JE, Fujinami RS. Experimental autoimmune encephalomyelitis as a testing paradigm for adjuvants and vaccines. Vaccine. 2012;29:3356–3362. doi: 10.1016/j.vaccine.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 18.Kriesel JD, White A, Hayden FG, et al. Multiple sclerosis attacks are associated with Picornavirus infections. Mult Scler. 2004;10:145–148. doi: 10.1191/1352458504ms1005oa. [DOI] [PubMed] [Google Scholar]
- 19.Olerup O, Hillert J. HLA class ll-associated genetic susceptibility in multiple sclerosis: a critical evaluation. Tissue Antigens. 1991;38:1–15. doi: 10.1111/j.1399-0039.1991.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 20.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. The International Multiple Sclerosis Genetics Consortium. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 21.Matullo CM, O'Regan KJ, Curtis M, Rall GF. CNS recruitment of CD8+ T lymphocytes specific for a peripheral virus infection triggers neuropathogenesis during polymicrobial challenge. PLoS Pathog. 2011;7:e1002462. doi: 10.1371/journal.ppat.1002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22▪▪.Borkosky SS, Whitley C, Kopp-Schneider A, et al. Epstein-Barr virus stimulates torque teno virus replication: a possible relationship to multiple sclerosis. PLoS One. 2012;7:e32160. doi: 10.1371/journal.pone.0032160. In this study TTV isolates originally isolated from brains of MS patients were used to infect B cell lines and EBV infection of TTV-infected cells enhanced TTV replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perron H, Garson JA, Bedin F, et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci USA. 1997;94:7583–7588. doi: 10.1073/pnas.94.14.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brudek T, Christensen T, Aagaard L, et al. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology. 2009;6:104. doi: 10.1186/1742-4690-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25▪.Perron H, Germi R, Bernard C, et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult Scler. 2012;18:1721–1736. doi: 10.1177/1352458512441381. This is a comprehensive review on the role of HERVs in MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 27.Blond JL, Lavillette D, Cheynet V, et al. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J Virol. 2000;74:3321–3329. doi: 10.1128/jvi.74.7.3321-3329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patience C, Takeuchi Y, Cosset FL, Weiss RA. Packaging of endogenous retroviral sequences in retroviral vectors produced by murine and human packaging cells. J Virol. 1998;72:2671–2676. doi: 10.1128/jvi.72.4.2671-2676.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnston JB, Silva C, Holden J, et al. Monocyte activation and differentiation augment human endogenous retrovirus expression: implications for inflammatory brain diseases. Ann Neurol. 2001;50:434–442. doi: 10.1002/ana.1131. [DOI] [PubMed] [Google Scholar]
- 30.Antony JM, Deslauriers AM, Bhat RK, et al. Human endogenous retroviruses and multiple sclerosis: innocent bystanders or disease determinants? Biochim Biophys Acta. 2011;1812:162–176. doi: 10.1016/j.bbadis.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31▪.Laska MJ, Brudek T, Nissen KK, et al. Expression of HERV-Fc1, a human endogenous retrovirus, is increased in patients with active multiple sclerosis. J Virol. 2012;86:3713–3722. doi: 10.1128/JVI.06723-11. This study demonstrated a higher RNA titer of HERV-Fc1 in MS patients in comparison to healthy controls. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32▪.Mameli G, Poddighe L, Mei A, et al. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: inference for multiple sclerosis. PLoS One. 2012;7:e44991. doi: 10.1371/journal.pone.0044991. This study provides evidence that EBV could be an initial trigger for MS and that EBV and HERVs are the main contributors to MS pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nexo BA, Christensen T, Frederiksen J, et al. The etiology of multiple sclerosis: genetic evidence for the involvement of the human endogenous retrovirus HERV-Fc1. PLoS One. 2011;6:e16652. doi: 10.1371/journal.pone.0016652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34▪▪.Petersen T, Moller-Larsen A, Ellermann-Eriksen S, et al. Effects of interferon-beta therapy on elements in the antiviral immune response towards the human herpesviruses EBV, HSV, and VZV, and to the human endogenous retroviruses HERV-H and HERV-W in multiple sclerosis. J Neuroimmunol. 2012;249:105–108. doi: 10.1016/j.jneuroim.2012.04.013. This is a longitudinal study of MS patients treated with IFN-β therapy linking HERVS and certain herpes viruses to disease outcome. [DOI] [PubMed] [Google Scholar]
- 35.Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- 36.Gay FW, Drye TJ, Dick GW, Esiri MM. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain. 1997;120:1461–1483. doi: 10.1093/brain/120.8.1461. [DOI] [PubMed] [Google Scholar]
- 37.Babbé H, Roers A, Waisman A, et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bitsch A, Schuchardt J, Bunkowski S, et al. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- 39.McMahon RM, Friis L, Siebold C, et al. Structure of HLA-A*0301 in complex with a peptide of proteolipid protein: insights into the role of HLA-A alleles in susceptibility to multiple sclerosis. Acta Crystallogr D Biol Crystallogr. 2011;67:447–454. doi: 10.1107/S0907444911007888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friese MA, Jakobsen KB, Friis L, et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med. 2008;14:1227–1235. doi: 10.1038/nm.1881. [DOI] [PubMed] [Google Scholar]
- 41.Fogdell-Hahn A, Ligers A, Gronning M, et al. Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens. 2000;55:140–148. doi: 10.1034/j.1399-0039.2000.550205.x. [DOI] [PubMed] [Google Scholar]
- 42.Bertrams HJ, Kuwert EK. Association of histocompatibility haplotype HLA-A3-B7 with multiple sclerosis. J Immunol. 1976;117:1906–1912. [PubMed] [Google Scholar]
- 43.Vandiedonck C, Taylor MS, Lockstone HE, et al. Pervasive haplotypic variation in the spliceo-transcriptome of the human major histocompatibility complex. Genome Res. 2011;21:1042–1054. doi: 10.1101/gr.116681.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mars LT, Saikali P, Liblau RS, Arbour N. Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim Biophys Acta. 2011;1812:151–161. doi: 10.1016/j.bbadis.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuchida T, Parker KC, Turner RV, et al. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci USA. 1994;91:10859–10863. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Honma K, Parker KC, Becker KG, et al. Identification of an epitope derived from human proteolipid protein that can induce autoreactive CD8+ cytotoxic T lymphocytes restricted by HLA-A3: evidence for cross-reactivity with an environmental microorganism. J Neuroimmunol. 1997;73:7–14. doi: 10.1016/s0165-5728(96)00161-0. [DOI] [PubMed] [Google Scholar]
- 47.Padovan E, Giachino C, Cella M, et al. Normal T lymphocytes can express two different T cell receptor β chains: implications for the mechanism of allelic exclusion. J Exp Med. 1995;181:1587–1591. doi: 10.1084/jem.181.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davodeau F, Peyrat MA, Romagné F, et al. Dual T cell receptor β chain expression on human T lymphocytes. J Exp Med. 1995;181:1391–1398. doi: 10.1084/jem.181.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Padovan E, Casorati G, Dellabona P, et al. Expression of two T cell receptor a chains: dual receptor T cells. Science. 1993;262:422–424. doi: 10.1126/science.8211163. [DOI] [PubMed] [Google Scholar]
- 50.Heath WR, Miller JFAP. Expression of two a chains on the surface of T cells in T cell receptor transgenic mice. J Exp Med. 1993;178:1807–1811. doi: 10.1084/jem.178.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corthay A, Nandakumar KS, Holmdahl R. Evaluation of the percentage of peripheral T cells with two different T cell receptor α-chains and of their potential role in autoimmunity. J Autoimmun. 2001;16:423–429. doi: 10.1006/jaut.2001.0504. [DOI] [PubMed] [Google Scholar]
- 52▪.Alii R, Nguyen P, Boyd K. A mouse model of clonal CD8+ T lymphocyte-mediated alopecia areata progressing to alopecia universalis. J Immunol. 2012;188:477–486. doi: 10.4049/jimmunol.1100657. This study provides evidence that dual expressing CD8+ T cells can lead to an autoimmune disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J Immunol. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54▪.Libbey JE, Cusick MF, Tsunoda I, Fujinami RS. Antiviral CD8+ T cells cause an experimental autoimmune encephalomyelitis-like disease in naive mice. J NeuroVirol. 2012;18:45–54. doi: 10.1007/s13365-012-0077-2. This is the first demonstration of dual TCR-expressing T cells being derived from a natural infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol. 2010;11:628–634. doi: 10.1038/ni.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsunoda I, Lane TE, Blackett J, Fujinami RS. Distinct roles for IP-10/CXCL10 in three animal models, Theiler's virus infection, EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP-10. Mult Scler. 2004;10:26–34. doi: 10.1191/1352458504ms982oa. [DOI] [PubMed] [Google Scholar]
- 57.Tsunoda I, Kuang LQ, Fujinami RS. Induction of autoreactive CD8+ cytotoxic cells during Theiler's murine encephalomyelitis virus infection: implications for autoimmunity. J Virol. 2002;76:12834–12844. doi: 10.1128/JVI.76.24.12834-12844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsunoda I, Libbey JE, Kobayashi-Warren M, Fujinami RS. IFN-γ production and astrocyte recognition by autoreactive T cells induced by Theiler's virus infection: role of viral strains and capsid proteins. J Neuroimmunol. 2006;172:85–93. doi: 10.1016/j.jneuroim.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Elliott JI, Altmann DM. Dual T cell receptor a chain T cells in autoimmunity. J Exp Med. 1995;182:953–959. doi: 10.1084/jem.182.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He X, Janeway CA, Jr, Levine M, et al. Dual receptor T cells extend the immune repertoire for foreign antigens. Nat Immunol. 2002;3:127–134. doi: 10.1038/ni751. [DOI] [PubMed] [Google Scholar]
- 61.Sharma R, Ju ACY, Kung JT, et al. Rapid and selective expansion of nonclonotypic T cells in regulatory T cell-deficient, foreign antigen-specific TCR-transgenic scurfy mice: antigen-dependent expansion and TCR analysis. J Immunol. 2008;181:6934–6941. doi: 10.4049/jimmunol.181.10.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou P, Borojevic R, Streutker C, et al. Expression of dual TCR on DO11.10 T cells allows for ovalbumin-induced oral tolerance to prevent T cell-mediated colitis directed against unrelated enteric bacterial antigens. J Immunol. 2004;172:1515–1523. doi: 10.4049/jimmunol.172.3.1515. [DOI] [PubMed] [Google Scholar]
- 63.Gavin MA, Rudensky AY. Dual TCR T cells: gaining entry into the periphery. Nat Immunol. 2002;3:109–110. doi: 10.1038/ni0202-109. [DOI] [PubMed] [Google Scholar]
- 64.Legrand N, Freitas AA. CD8+ T lymphocytes in double αβ TCR transgenic mice. II. Competitive fitness of dual αβ TCR CD8+ T lymphocytes in the peripheral pools. J Immunol. 2001;167:6158–6164. doi: 10.4049/jimmunol.167.11.6158. [DOI] [PubMed] [Google Scholar]
- 65.Paterson RK, Bluethmann H, Tseng P, et al. Development and function of autospecific dual TCR+ T lymphocytes. Int Immunol. 1999;11:113–119. doi: 10.1093/intimm/11.1.113. [DOI] [PubMed] [Google Scholar]
- 66.Munthe LA, Sollien A, Dembic Z, Bogen B. Preferential positive selection of T lymphocytes which express two different TCR alpha chains, an endogenous and a transgenic. Scand J Immunol. 1995;42:651–661. doi: 10.1111/j.1365-3083.1995.tb03708.x. [DOI] [PubMed] [Google Scholar]