

Abstract
Although 18F-fluorodeoxyglucose (18F-FDG) is still the most widely used positron emission tomography (PET) radiotracer, there are a few well-known limitations to its use. The last decade has seen the development of new PET probes for in vivo visualization of specific molecular targets, along with important technical advances in the production of positron-emitting radionuclides and their related labeling methods. As such, a broad range of new PET tracers are in preclinical development or have recently entered clinical trials. The topics covered in this review include labeling methods, biological targets, and the most recent preclinical or clinical data of some of the next generation of PET radiopharmaceuticals. This review, which is by no means exhaustive, has been separated into sections related to the PET radionuclide used for radiolabeling: fluorine-18, for the labeling of agents such as FACBC, FDHT, choline, and Galacto-RGD; carbon-11, for the labeling of choline; gallium-68, for the labeling of peptides such as DOTATOC and bombesin analogs; and the long-lived radionuclides iodine-124 and zirconium-89 for the labeling of monoclonal antibodies cG250, and J591 and trastuzumab, respectively.
In the past few years a great deal of interest in the clinical application of positron-emitting radiopharmaceuticals for the molecular imaging of oncology has expanded after the introduction and huge clinical success of 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET). Although 18F-FDG is used extensively and successfully in many cancers,1–3 because of the targeting characteristic of this compound as a marker of glucose metabolism, the sensitivity and specificity of 18F-FDG are not optimal in all cancer types. The shortfalls of imaging with 18F-FDG, such as inadequate differentiations between post-therapy inflammation and tumor, poor imaging in slow-growing tumors, and high uptake in normal cells such as brain and gut, have been shown to be improved with other PET tracers. In recent years, research has been conducted on new PET radiopharmaceuticals that are directed at a wider range of molecular targets, including, for example, DNA, cell surface antigens, cellular receptors, and hypoxia; examples of such radiopharmaceuticals include 18F-3-fluorothymidine (18F-FLT), 18F-1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)thymine (18F-FMAU), 64Cu-labeled diacetyl-bis (N4-methylthiosemicarbazone) (64Cu-ATSM), and fluoromisonidazole (18F-MISO). The clinical translations of these agents are in different stages, and a review of the current status of many of these radiopharmaceuticals has been recently published in The Journal of Nuclear Medicine (Dunphy and Lewis).4
This article will focus on the chemistry, targeting, and clinical implications of a select number of next-generation PET radiopharmaceuticals, including radiolabeled small molecules, peptides, and antibodies that are transitioning into the clinic for early trials in humans. This review can only focus on a small number of the PET tracers that are being researched because of the breadth of the field. The expanding availability of PET nuclides (11C [20.4 minutes]; 68Ga [68 minutes]; 18F [109.8 minutes]; 64Cu [12.7 hours]; 89Zr [18.4 hours]; 124I [4.12 days]) with varying half-lives, matching the kinetics of the compound they are incorporated into, has been instrumental in expanding the repertoire of agents available. As a consequence, the noninvasive interrogation of cancer biology with radiopharmaceuticals will only expand in the future, as the fields of biology and chemistry come together to improve the availability and specificity of the agents used in molecular imaging with PET.
Radio fluorine Labeled Compounds
18F-Choline
Chemistry
Fluoromethyl-dimethyl-2-hydroxyethyl-ammonium, or 18F-choline (18F-FCH; Fig. 1), was developed by DeGrado et al5,6,8 and Hara,7 who reported the synthesis of no-carrier-added 18F-FCH through the intermediate 18F-fluorobromo-methane (18F-FBM). 18F-FBM is prepared from dibromo-methane. 18F-FBM was obtained with a 20%–40% radiochemical yield (RCY) (not decay corrected) by alkylation of dimethylethanolamine (DMEA), with a synthetic time of 40 minutes. 18F-FCH was obtained with excellent radiochemical purity (RCP) after slight modifications to the original synthesis.5
Figure 1.
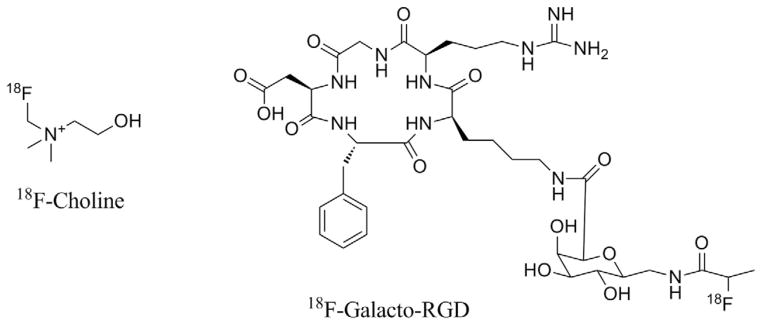
Structures of 18F-choline and 18F-Galacto-RGD.
18F-FCH automation has been reported with varying degrees of success with respect to RCP and RCY. Iwata et al9 reported the use of a remotely operated system for the distillation of 18F-FBM and its conversion to the more reactive 18F-fluoromethyl trifluoromethanesulfonate (triflate) (18F-CH2FOTf). 18F-FCH was successfully prepared via 18F-CH2FOTf alkylation of DMEA on a solid support, by passing 18F-FBM over a silver trifluoromethanesulfonate column at 180°C. 18F-FCH was obtained with a 40% RCY within 30 minutes. Cimitan et al10 cite the convenience of a fully automated module from Argos Zyklotron (Klagenfurt, Austria) for the production of 18F-FCH. Excellent RCP and low DMEA contamination were achieved in the final sample. Recently, Kryza et al11 reported a fully automated 18F-FCH synthesis by using GE TracerLab (GE HealthCare, St Giles, UK) MX FDG modules. The device used in the conversion of 18F-FBM to triflate could not be adapted to the disposable system; therefore, 18F-FBM was used as the methylating alternative to DMEA. 18F-FCH was obtained with a 15%–25% RCY (not decay corrected) within 35 minutes.
Biomedical Targets and Clinical Applications
The advantages of PET imaging with choline were apparent after initial studies with 11C-choline. However, imaging with 11C produces logistical limitations because of its short half-life (20.4 minutes). In 2001, Hara7 wrote a brief review comparing 11C-choline PET with early 18F-FCH studies for various tumor types. These early studies showed 18F-FCH PET most promising for imaging prostate, hepatocellular, and brain tumors.5,12–15
18F-FCH demonstrates higher avidity for prostate cancers than does 18F-FDG and has been shown to provide better localization of lesions on imaging.16,17 Cimitan et al10 studied the potential of early and delayed 18F-FCH PET/computed tomography (CT) scans to monitor the recurrence of prostate cancer after treatment with radical prostatectomy, radiotherapy, or hormonal therapy alone. Early PET scans are required when imaging prostate cancer patients because of bladder uptake and excretion of 18F-FCH, a phenomenon not observed with 11C-choline. Furthermore, the authors noted uptake in the prostate, bone, and abdominal and pelvic lymph nodes. True-positive PET/CT scans were found in patients with serum prostate specific antigen (PSA) levels >4 ng/mL; 46 of the 100 patients with low yet rising PSA levels receiving PET scans; however, they did not show 18F-FCH uptake. Mean PSA of this group was 1.98 ng/mL, and none of these patients were shown to have clinical recurrence. Cimitan et al theorized that 18F-FCH PET/CT will not impact the care of patients with a biochemical recurrence of prostate cancer until the PSA level increases above 4 ng/mL; however, 18F-FCH PET/CT can be used to exclude distant metastatic disease even at an early time point. To investigate clinical management, a comparison of 18F-FCH with conventional imaging of prostate cancer and 18F-FDG was conducted in 16 patients for staging or restaging. 18F-FCH was shown to provide a more accurate and beneficial treatment plan in 88% of patients, compared to other imaging modalities which provided improved planning in only 56% of cases.18
Increasing 18F-FCH accumulation in the skeleton in delayed PET images is highly predictive of bone metastases; however, hormonal therapy can increase the difficulty of interpreting uptake.19 Beheshti et al20 investigated the use of 18F-FCH to uncover bone metastases (BM) in prostate cancer patients, finding sensitivity, specificity, and accuracy of 79%, 97%, and 84%, respectively, in the discovery of BM in 70 patients. Husarik et al21 examined 111 patients (43 patients for staging and 68 patients for restaging), concluding that 18F-FCH PET/CT accurately depicts BM in prostate cancer patients.
18F-FCH PET is superior to 18F-FDG in the detection of brain lesions because choline exhibits low uptake in normal brain tissue versus the brain uptake of glucose. Benign brain lesions also display many of the same features as malignant disease with conventional imaging.22,23 Twenty-one patients with histologically confirmed high-grade glioma or brain metastasis and 9 patients with benign disease were imaged; statistically significant increases in standard uptake values (SUVs) were observed in malignant lesions, with metastatic lesions expressing the highest SUVs.24 18F-FCH PET also permitted detection of recurrent tumors in patients previously treated by intracranial radiation, where radiation therapy usually produces reactive changes that make accurate diagnoses difficult with in vivo imaging.24
18F-Fluoro-5α-dihydrotestosterone
Chemistry
β-18F-fluoro-5α-dihydrotestosterone (18F-FDHT) is a fluorinated analog of dihydrotestosterone (DHT), the native androgen receptor-binding ligand. The synthesis of 18F-FDHT by Liu et al25,26 in 1992 has been reported as the standard method of preparation, with only minor modifications since initial evaluation. According to the procedure of nonradiolabeled FDHT of Liu et al, fluorine is introduced by fluoride displacement of reactive triflate.26 This strategy is suitable for subsequent radiolabeling; consequently, the intermediate triflate, 16α-[[(trifluoromethyl) sulfonyl] oxy]-3,3-(ethylene-dioxy) androstan-17-one, obtained in 3 steps from 5α-androstanedione, can also be used as the precursor for radiosynthesis. In the preparation of 18F-FDHT, the 16α-triflate displaces 18F-fluoride by nucleophilic substitution, with nBu4N18F. Subsequent reduction of the 17-ketone function occurs with LiAlH4 in a mixed Et2O-THF solution, in a stereoselective manner, to yield the hydroxyl group. Finally, deprotection of the ketal, in acidic conditions, leads to 18F-FDHT (Fig. 2). The total synthesis time, including normal-phase high-performance liquid chromatography (HPLC) purification, is 90 minutes. 18F-FDHT was obtained with a decay-corrected yield of 31%–48%25. The final product was formulated in a 5%–10% ethanolic saline solution, and the specific activity was 1151 Ci/mmol. 18F-FDHT is currently in clinical trials.
Figure 2.

Synthesis of 18F-FDHT from the intermediate 16α-triflate.
An automated protocol for 18F-FDHT with a plastic cassette-type 18F-FDG synthesizer was developed by Mori et al.27 By using the triflate precursor previously discussed, 18F-FDHT was obtained with 52.4% ± 8.6% (n = 10) radiochemical yield, which was estimated by analytical HPLC (55-minute synthetic time). The clinical demand and use of 18F-FDHT are thought to increase with radiolabeling developments such as those described by Mori et al.
Biomedical Targets and Clinical Applications
Prostate cancer is an androgen-driven neoplasm. Most prostate cancers initially respond to treatment with androgen depravation, leading to disease regression. However, as the cancer progresses, many lesions lose responsiveness and become castrate resistant. Castrate resistance occurs after biology distortion in the normal cellular pathway of the androgen response, which has been shown through mutations affecting ligand specificity, and increased cellular enzyme activity.28,29
Androgen receptors (ARs) are principal components along the pathway to prostate cancer and are the target of 18F-FDHT.30 The histopathologic AR profile of metastatic lesions is variable; therefore, an in vivo imaging paradigm to accurately and noninvasively identify prostate cancer would improve diagnostic sensitivity and clinical management.31 18F-FDHT binds sex hormone binding globulin within the plasma, thereby slowing the breakdown of endogenous androgens and assisting intracellular transport.32 Beattie et al31 exploited PET to quantitate AR levels in clinical pharmacokinetic modeling analyses of 18F-FDHT. In vitro cell binding via 18F-FDHT, the cold compound (FDHT), DHT, and AR-dependent cells (CWR22-rv1) corroborated 18F-FDHT specific uptake. In vivo dynamic 18F-FDHT PET scans were acquired at the metastatic lesion site (13 patients) and at the aorta (25 patients) to obtain regions of interest (ROIs) and blood time-activity information. Before dynamic 18F-FDHT PET, patients were scanned by whole body 18F-FDG PET/CT for prospective lesion localization. Time-activity curves (derived from aorta ROI) were converted to SUVs. Most of the metastases were in bone, which is the prevailing metastatic site.33 Radiolabeled metabolite assessment and clearance of 18F-FDHT, established from patient blood samples, were used as input in the pharmacokinetic modeling and classified for statistical fitness. The authors concluded that 18F-FDHT PET is a reasonable method for estimating AR expression in metastatic prostate cancer.31
In a separate study, AR targeting by 18F-FDHT PET and binding selectivity to the AR by the antagonist flutamide were measured in patients with recurrent or metastatic prostatic adenocarcinoma.34 Parameters such as tumor-to-muscle ratio (T/M) and ROI evaluated tumor 18F-FDHT uptake and SUVmax, respectively. The sensitivity of 18F-FDHT PET in detecting AR expression was reported as 63% on a patient basis and as 86% on a lesion-specific basis. 18F-FDHT–positive patients had significantly higher PSA values relative to 18F-FDHT–negative patients. In addition, tumor uptake of 18F-FDHT decreased on flutamide administration (mean SUV and T/M decreased), thus validating AR targeting by 18F-FDHT.
Larson et al33 contrasted 18F-FDHT and 18F-FDG for tumor discrimination in progressive metastatic prostate cancer patients. The inclusion criterion was castrate patients. The patients underwent a bone scan, an 18F-FDG PET scan, and dynamic and whole body 18F-FDHT PET scans, with blood sampling for metabolite analyses. Pharmacokinetics indicated 18F-FDHT bound tightly to plasma proteins, with uptake occurring within 10 minutes. Lesions were classified individually and per patient. Semiquantitative evaluations of the 2 tracers were made using a 5-point scale to distinguish tumor localization, with only 4-point (probably positive) or 5-point (definitely positive) lesions considered for analyses. ROI tools located 18F-FDG lesions and 18F-FDHT lesions, which were superimposed using dual monitors. 18F-FDG PET detected 97% of lesions (SUVmax = 5.22), whereas 18F-FDHT PET detected 78% of lesions (SUVmax = 5.28). One patient positive for 18F-FDHT underwent biopsy and was also positive for AR overexpression by immunohistochemistry. Three patients received follow-up scans after treatment with pharmaceuticals expected to reduce the AR effect; there were changes in 18F-FDHT uptake, implying competition between 18F-FDHT and DHT for the AR, and 18F-FDHT selectivity in androgen-independent prostate cancer.
In an important examination detailing radiation absorbed dose, Zanzonico et al35 developed dosimetry estimates for 18F-FDHT in the normal tissues of advanced prostate cancer patients. Ten whole body 18F-FDHT distribution PET scans were acquired from 7 progressive prostatic cancer patients; serial blood samples were collected for dosimetric analyses. Calculations were made concerning the maximum radioactivity in the red marrow (MBq/g), the total marrow activity, the mean activities (kBq/mL) in the liver, spleen, and total body, total organ activities, and the 18F residence time. 18F intestinal residence times were calculated by assuming liver elimination via hepatobiliary excretion. All measurements were corrected for radioactive decay from the time of 18F-FDHT injection. Mean organ absorbed doses (cGy/MBq) were calculated on the basis of a 70-kg man. Each tissue received a mean absorbed dose <0.003 cGy/MBq, except the liver (0.00333 cGy/MBq) and the wall of the urinary bladder (0.00868 cGy/MBq), which received the greatest absorbed doses. These values fall below the maximum dose limit per study to normal tissues (5 cGy). The authors maintain the absorbed doses are comparable to other 18F-labeled radiopharmaceuticals.35 This study presumes total liver elimination in the intestines of 18F by the hepatobiliary route, thereby discounting urinary excretion and potentially miscalculating 18F residence time. However, this is a clever survey of whole body 18F-FDHT absorbed radiation dose to normal organs.
18F-Galacto-arginine-glycine Aspartic Acid
Chemistry
18F-Galacto-arginine-glycine aspartic acid (18F-RGD) (Fig. 1) was the first 18F-labeled RGD-containing glycopeptide described for noninvasive imaging of αvβ3 integrin expression with PET; the design, synthesis, and radiolabeling of 18F-Galacto-RGD was reported by Haubner et al.36,37 Haubner et al designed first-generation radioiodinated cyclic RGD peptides to image αvβ3 integrin expression, showing receptor-specific tumor accumulation in various mouse models. Limitations of the tracers, however, included high liver and intestinal retention. Therefore, pharmacokinetic considerations led the authors to investigate conjugating the RGD monomer cyclo (-Arg-Gly-Asp-D-Tyr-Lys-) with a galacto sugar amino acid in an approach known for improving peptide pharmacologic properties.36 Moreover, the galactose derivative, used as a sugar amino acid moiety, permitted radiolabeling through the 4-nitrophenyl-2-18F-fluoropropionate (18F-NFP) prosthetic group; the reaction proceeds through a fast acylation step, with a good yield. The authors chose the 18F-NFP prosthetic group to minimize the negative effects of hydrophilicity on 18F-Galacto-RGD.36 Synthesis of 18F-Ga-lacto-RGD requires building the partially protected cyclo(-Arg-Gly-Asp-D-Tyr-Lys-) on solid support, followed by formulation of the Fmoc-N-protected galacto amino acid derivative from peracetylated galactose. Subsequent glycosylation and deprotection resulted in 18F-Galacto-RGD within 2.5 minutes at a mild temperature, with 76% yield. 18F-NFP is obtained in a time-consuming, 3-step procedure; however, no improvements have been reported in the literature.38
Biomedical Targets and Clinical Applications
Integrins are heterodimers of transmembrane glycoproteins involved in cellular communication, both between cells and between the cell and the surrounding matrix.39 The receptor αvβ3 is an integrin implicated in metastasis, angiogenesis, and proliferation40; faint expression is observed in resting endothelial cells and normal organs.41 Obstruction of receptor uptake on tumor cells retards invasion and metastasis.42 αvβ3 binds matrix proteins at the peptide sequence RGD.43 18F-Galacto-RGD is a glycosylated cyclic pentapeptide derived from the first-generation [125I]-3-iodo-DTyr4-cyclo(-Arg-Gly-Asp-D-Tyr-Val).44 In a recent study, 18F-Galacto-RGD was formulated to target αvβ3 expression and to describe the dosimetry,45 pharmacokinetics, and biodistribution46 of the tracer uptake in many human cancers, including musculoskeletal, melanoma, colon, breast, head or neck, sarcoma, and osseous metastases.45–47 An inclusion criterion was malignancy; radiometabolites were analyzed of blood samples, revealing >95% intact tracer 120 minutes after injection. Dynamic and static whole body PET scans were acquired to accommodate the tumor regions. ROIs were drawn over major organs, images were calibrated to SUVs or Bq/mL, and the residence time of 18F (maximum = 2.64 hours) was determined to estimate organ activities and uptake of 18F-Galacto-RGD. The effective dose was 18.7 ± 2.42 μSv/MBq. The mean effective absorbed radiation dose was higher in women (20.2 ± 2.5 μSv/MBq) than in men (17.1 ± 0.7 μSv/MBq). The urinary bladder wall received the greatest dose (218 μGy/MBq). The dose to the bladder wall was higher in women, as were the radiation absorbed dosages to the kidneys, liver, small intestine, and lungs, which were also comparatively high. The high activity in the bladder wall was implied as αvβ3 overexpression from intestinal smooth muscle cells. Time-activity curves verified rapid renal excretion and low background. Imaging was proposed 60 minutes after injection for optimum contrast. 18F-Galacto-RGD PET detected 79.3% malignant lesions in 89.4% of patients. Tumor accumulations were rapid under 10 minutes. Large variations in intratumor and intertumor uptake were noted in primary tumors and metastases, denoting a range of αvβ3 expression, possibly because of phases along the course of metastatic disease. Image quality in the urinary tract and bladder was poor because of high accumulations. Blood pool (1.8% injected dose [ID]/L ± 0.6, 72 minutes post injection) and muscle (0.7% [ID]/L ± 0.19, 72 minutes post injection) accumulations were low, allowing feasible PET detection of 18F-Galacto-RGD targeted αvβ3 receptors in the thorax, extremities, and abdomen.
Beer et al48 conducted PET imaging with 18F-Galacto-RGD in squamous cell carcinoma of the head and neck in 11 patients and showed visualization of all primary tumors >5 mm; no correlations between tumor size and SUV were observed. Immunohistochemistry associated uptake of 18F-Galacto-RGD with αvβ3 expression in the neovasculature.
Haubner et al49 showed 18F-Galacto-RGD targets αvβ3 receptors and monitors tumor-induced angiogenesis in mouse models of human A431 squamous cell carcinoma and M21 melanoma. Each transplanted xenograft had positive and negative controls, one that transcribed the αvβ3 gene and one without the transgene. Nude mice were inoculated with increasing amounts of the M21 cell line (αvβ3 positive) to determine the level of αvβ3 expression; previous reports exhibited 18F-Galacto-RGD selectivity for M21 versus M21-L (αv-negative).36 Small animal PET (supine and prone, 90 minutes after injection.), immunohistochemistry (human anti-αvβ3 and murine β3), and Western blot analyses (anti-αv of lysed tumor tissue) were performed. Increasing 18F-Galacto-RGD tumor uptake paralleled the increasing percentage of receptor-positive injected cells. The PET imaging results were compared with αv subunit analysis by Western blot, with positive correlation. Tumor-to-background and tumor-to-muscle ratio correlations with αv expression were low (r = 0.76); however, the authors maintain that the ROI also contains normal tissue with low 18F-Galacto-RGD uptake (eg, muscle, lung).49 The A431 tumor cells established induced angiogenesis, because they do not express the αvβ3 receptor (confirmed with anti-human αvβ3 monoclonal antibody). 18F-Galacto-RGD PET revealed αvβ3 receptor expression on activated endothelial cells, suggesting that angiogenesis can be observed by small animal PET. Tumor vessel endothelial cell expression of β3 was substantiated by using a polyclonal antibody against the murine β3 subunit. The tumor was detected with PET, and specificity was confirmed with blocking studies. Clinical studies were also performed, concentrating on patients with melanoma and sarcoma; confirmation was by histopathology and immunohistochemistry of resected specimens. Areas with the highest density of αvβ3 expression were correlated with tracer uptake by mean SUV. Renal elimination of the tracer was observed, with the highest accumulations in the kidneys (SUV = 5.5 ± 3.7, 79 minutes after injection), liver (SUV= 2.4 ± 0.5, 79 minutes after injection), and spleen (SUV 2.5 ± 0.5, 79 minutes after injection). Tumor SUV ranged from 1.2–10.0, with a 60-minute retention time of 18F-Galacto-RGD; the activity concentration decreased with time in other organs. The urinary bladder wall received the highest absorbed dose (0.20 ± 0.04 mGy/MBq), and the tumor-to-blood ratio (T/B) (3.8 ± 2.6, 79 minutes after injection) and T/M (8.8 ± 6.0, 79 minutes after injection) were calculated (again with great variation). In this study, the authors translated findings from the laboratory into the clinic by demonstrating noninvasive imaging of αvβ3 receptor expression with 18F-Galacto-RGD targeting, level of receptor expression, and expression in the tumor vasculature to establish and image angiogenesis.
Anti-18F-Fluoro-cyclobutyl-1-carboxylic Acid
Chemistry
The synthetic L-leucine anti-1-amino-3-18F-fluoro-cyclobutyl-1-carboxylic acid (anti-18F-FACBC) is an analog of 1-aminocyclobutane-1-11C-carboxylic acid (11C-ACBC).50 The synthesis of anti-18F-FACBC was first reported by Shoup et al51,52 in an 11-step formulation (from epichlorohydrin) of the triflate precursor syn-1-(tert-butoxycarbonyl)amino-3-[[(trifluoromethyl) sulfonyl] oxy]-cyclobutane-1-carboxylic acid methyl ester (Fig. 3). Radiosynthesis and deprotection by acid hydrolysis were performed with a self-developed remote manual system. The synthetic route was performed in 60 minutes, with a RCY of 12% (end of bombardment) after purification.
Figure 3.
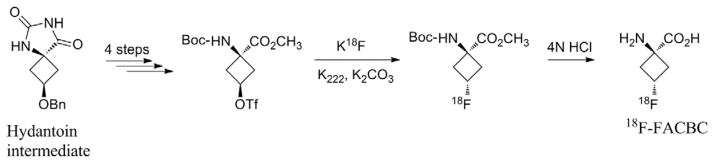
Synthesis of 18F-FACBC from the hydantoin intermediate (major isomer).
McConathy et al53 disclosed several drawbacks to the anti-18F-FACBC synthesis of Shoup et al.51,52 The authors improved the radiolabeling route of the precursor with a fully automated radiosynthesis to facilitate transition to clinical studies.53 Subsequent hydrolysis of the isomeric mixture resulted in a new mix, from which the desired amino acid was isolated by reverse-phase HPLC; this route precludes the gram scale synthesis of the labeling precursor. The synthesis of the labeling precursor involves the formation of a hydantoin intermediate in a 3:1 isomeric ratio and favors the syn diastereoisomer in variable yields. Anti-18F-FACBC was obtained in 70 minutes after the delivery of 18F-fluoride (5.2–7.4 GBq), with a decay-corrected yield (DCY) of 24% ± 4% (n = 8). The triflate group was displaced by 18F-fluoride at 90°C with Kriptofix (K222) and potassium carbonate; radiochemical purity was >99%. The acidic conditions used in deprotection differed slightly from those initially described by Shoup et al51,52 and led to incomplete hydrolysis of the methyl ester. Thus, analogous radiolabeling was performed with the tert-butyl ester analog; hydrolysis was complete, but the DCY was <10%. An exhaustive, fully automated quality control for anti-18F-FACBC is referenced as a standard for anti-18F-FACBC production.54–56 In 2007, Oka et al54 used a slightly modified, nonautomated method (with respect to deprotection) by using a mixture of trifluoroacetic acid in dichloromethane as opposed to 4N hydrochloric acid. The RCY of anti-18F-FACBC was 43.5% ± 8.6% (n = 9) within 90 minutes, and the RCP was 95.3% ± 6.1% (n = 9), which was determined by radio-thin-layer chromatography.
Mechanism and Clinical Applications
Uncontrolled cell proliferation of cancer cells results in increased nutritional requirements of tumors. In particular, the requirements of some amino acids increase and lead to up-regulation of amino acid transporters.57 Membrane transport of amino acids is via carrier protein; amino acid transport, rather than protein synthesis, predominates tumor uptake of radiolabeled amino acids.58 Amino acids are fundamental to all biological processes, and they are building blocks of proliferating tumor cells.59–61 18F-FACBC was used by Shoup et al62 as a brain tumor imaging agent alternative to L-methyl-11C-methionine (half-time = 20 minutes) because it has a longer half-life (109.8 minutes). 18F-FACBC, as well as other non-naturally occurring amino acids, is metabolically stable (versus naturally occurring amino acids) and capable of radiolabel by longer-lived isotopes.63 Amino acid measurement in protein synthesis might be able to be used as a sensitive and specific target for malignancy.
Within the brain, CT and magnetic resonance imaging (MRI) are the most commonly used imaging modalities for malignancies. However, these modalities are not optimal for differentiating malignant and benign lesions or to assess post-radiation or surgery burden compared with tissue injury. 18F-FDG is currently the most widely used PET tracer in oncology; however, 18F-FDG exhibits very high uptake in normal brain and inflammatory tissue after therapy. Shoup et al62 present 18F-FACBC as a potential PET brain imaging tracer because of significant tumor-to-brain ratios.
The authors inoculated rats with 9L gliosarcoma cells by intracranial injection before intravenous administration of 18F-FACBC and performed biodistribution and toxicity assays.62 18F-FACBC was shown with low brain uptake and high accumulation in the brain tumor (6.61 tumor-to-brain ratio at 60 minutes). Clearance in other organs (60 minutes) was good relative to the tumor (excluding the liver), and marrow activity did not increase, indicating stability of the tracer. The T/B ratio was 5.4 at 60 minutes, indicating good clearance in the blood (0.32% ID/g, 60 minutes; 1.72% ID/g tumor). The authors recommend healthy doses of 18F-FACBC up to 9.5 mg/kg. PET imaging of a glioblastoma multiforme patient was also performed with 18F-FACBC.62 Rat biodistribution data (%[ID]/g) were converted to human values (%[ID]/organ) and expressed as residence time (18F) to estimate dose. 18F-FACBC PET identified the tumor on dynamic imaging in the frontal region of the left hemisphere, with T/B ratio of 6 at 20 minutes after injection. The absorbed dose (rad/mCi) to the urinary bladder wall was estimated at 0.14. The total estimated body dose for 18F-FACBC was 0.046 rad/mCi. Meanwhile, normal brain uptake was low (29 nCi/mL, 60 minutes), with greater accumulation at the tumor site (146 nCi/mL, 60 minutes). Time-activity curves revealed that in vivo measurements can be performed 10 minutes after injection to illustrate amino acid brain distribution.
Radiation dosimetry of anti-18F-FACBC was done in an examination of healthy volunteers, resulting in a mean radiation-absorbed dose estimate to the total body of 12.8μGy/MBq.64 The liver (52.2 μGy/MBq), pancreas (31.5μGy/MBq), and kidneys (22.1 μGy/MBq) received the highest doses; other organs with high anti-18F-FACBC dose estimates were the wall of the heart (22.3 μGy/MBq), spleen (20.2 μGy/MBq), muscle (14.7μGy/MBq), and the urinary bladder wall (11.9 μGy/MBq). The breasts (4.4 μGy/MBq) received the lowest estimated dose of anti-18F-FACBC.
The analog anti-18F-FACBC has also been reported with increased uptake in the prostate.65,66 A small number of patients (n = 5) with recurrent prostate cancer were imaged with PET scans showing tumor uptake and the identification of confirmed lesions; bladder excretion was limited, which allowed enhanced visualization.67 In a case report, Jani et al showed prostate uptake of 18F-FACBC in prostate cancer (Fig. 4).
Figure 4.

For a representative axial slice, a CT image (A), an FACBC PET image (B), and a registered image (C) are shown; this figure demonstrates how the FACBC PET scan information was registered with the planning CT scan. (Adapted with permission from Jani AB, et al. Case study of anti-1-amino-3 18F-fluorocyclobutane-1-carboxylic acid (anti-18F-FACBC) to guide prostate cancer radiotherapy target design. Clin Nucl Med 34:279–284, 2009.)
Radiocarbon Labeled Compounds
11C-Choline
Biomedical Targets and Clinical Applications
Phosphatidylcholine is the most abundant phospholipid of the eukaryotic cell membrane.68 Choline, a phosphatidylcholine precursor, can be targeted to the cell membrane to image proliferation in many cancer models such as breast,68 lung,69 and prostate.70 Tumor uptake of 11C-choline results in biochemical phosphorylation and entrapment within the membrane.69 PET imaging with 11C-choline was first investigated in the late 1990s by Hara et al69 to image brain tumors as a segregate marker of 18F-FDG, which has poor differentiation of lesions in the brain because of large 18F-FDG uptake by normal brain. Hara et al have also detected primary mediastinal lymph node lesions and metastases originating from non–small-cell lung cancer (NSCLC), with histopathologic follow-up of resections.69 18F-FDG PET images were also acquired in the patients; however, 18F-FDG was not able to resolve small metastases. The sensitivity of 11C-choline PET (100%) was greater than that of CT (19%) or 18F-FDG PET (75%) in localizing mediastinal lymph node metastases in patients with NSCLC, implying superiority of 11C-choline as a PET marker in imaging lung cancer.
11C-choline PET was performed in normal subjects, patients with cerebrovascular disease, and cancer. Imaging was obtained with 11C-choline and H215O on 24 patients diagnosed with malignant brain tumors to determine tumor uptake versus blood flow. 11C-choline tumor uptake was positive in all lesions and independent of the rate of tumor blood flow. Four patients with cerebral infarcts or hemorrhage exhibited no uptake of 11C-choline. In addition, 3 case reports presented 4 patients with brain tumors (glioblastoma multiforme, hemangioblastoma, and metastatic adenocarcinoma originating from lung). In these cancers, tumor uptake of 11C-choline was high, with tumor-to-normal brain ratios of 13.4, 27.5, and 10.7.71
Locally advanced prostate cancer is notoriously difficult to image; current imaging modalities perform poorly in the diagnosis and staging of disease. A patchwork of various imaging modalities is used to obtain information concerning disease burden, including transrectal ultrasound, MRI, CT, and bone scan. These techniques have not been shown to be sensitive or specific enough to detect metastatic or recurrent disease,72–75 defined as a PSA level increase in the blood after curative treatment with surgery or radiation therapy. PET imaging in prostate cancer is infrequent because of the limited success of clinical 18F-FDG PET. 18F-FDG is excreted by the kidneys in the urine, causing an early build-up of activity within the bladder and obscuring the prostate on imaging. The nature of prostate tumor growth kinetics also leads to a suboptimal uptake of 18F-FDG for imaging. In comparison, urinary excretion of 11C-choline is negligible; furthermore, 11C-choline uptake is prostate specific, rapid, and stable.76 Hara found 11C-choline uptake high in primary prostate cancer, as well as in metastatic sites, concluding 11C-choline PET more sensitive than bone scintigraphy in detecting bone metastases. 18F-FDG PET was not as sensitive a diagnostic tool for detecting bone metastases derived from prostate cancer.76
11C-choline has been reported to monitor the response to therapy in prostate cancer treatment. Krause et al70 used androgen-independent, PSA-negative PC3 (human prostate cancer athymic xenograft) mice to assess treatment response to docetaxel (antimitotic chemotherapeutic) at 3 mg/kg body weight (3-week treatment schedule). The therapeutic response was monitored with 11C-choline PET before, throughout, and after treatment, revealing high tumor uptake as well as high accumulations in the kidneys and liver. There was no significant difference in the tumor-to-muscle ratios (T/Mmean) between the control group and the treatment (docetaxel) group during the first week. 11C-choline tumor uptake decreased as early as week 1 and remained low throughout the study period in the treatment group. A significant difference in tumor growth was observed between the control and treated animals (P = 0.025); the tumor volume of the control animals increased, whereas inhibition of tumor volume was observed in the docetaxel-treated animals. This was a carefully designed study predicting 11C-choline as a marker of early treatment response in prostate cancer.
In a survey to analyze 11C-choline as a PET imaging marker of breast cancer metastasis, Zheng et al68 inoculated athymic mice with human MCF-7 (transfected with interleukin-1α) or MDA-MB-435 cells. Biodistribution and small animal PET imaging analyses were performed. The T/Bs were high in both models: 6.9 (T/B, MCF-7), 12.5 (T/B, MDA-MB-435), indicating high tumor uptake of 11C-choline. Small animal PET corresponded with biodistribution data by clearly showing tumor uptake of 11C-choline in both breast cancer models.
Radiogallium Labeled Compounds
Chemistry
68Ga is a 68Ge/68Ga (half-life = 270.8 days) generator produced short-lived radionuclide (half-life = 68 minutes), decaying at 89% through positron emission (maximum energy of 1.92 MeV). Production of 68Ga represents an alternative to costly cyclotron-produced radionuclides. It is an excellent candidate for high specific activity labeling of small peptides with fast blood clearance and target localization. 68Ga-DOT-ATOC is the most widely used 68Ga-based PET radiopharmaceutical77,78 and has been used to optimize 68Ga-labeling methods for DOTA-derivatized peptides.79–81 For example, Asti et al82 described an effective purification of the 68Ge/68Ga eluate (a crucial labeling step81), which was applied to the production of 68Ga-DOTATOC and is suitable for clinical use. Recently, Decristoforo et al80 described a fully automated synthesis for 68Ga-labeled peptides with good, reproducible yields. The clinical applications of 68Ga-labeled ligands are discussed below.
68Ga-DOTATOC
Biomedical Targets and Clinical Applications
Neuroendocrine tumors (NETs) originate from gastroentero-pancreatic tracts, expressing specific amine and peptide receptors (eg, somatostatin [SMS], vasointestinal peptide receptors, bombesin, cholecystokinin, gastrin, and/or substance P). Furthermore, NETs are highly differentiated.83 The peptide DOTATOC ([D]-Phe1-Thy3-octreotide) delineates lesions with up-regulated SMS receptors such as localized NETs84 and well-differentiated thyroid tumors (DTCs).85–87 Hofmann et al84 compared 68Ga-DOTATOC with the single photon emission computed tomography (SPECT) radiotracer 111In-octreotide to associate SMS receptors in primary and metastatic disease. On authentication of metastasis from the histology of surgical specimens, 8 patients were enrolled in the imaging trial. The patients were scanned by whole body 111In-octreotide planar (4 hours and 24 hours after injection) and SPECT (abdominal, 4 hours after injection; thoracic, 24 hours after injection) imaging, CT, and/or MRI. Dynamic and static PET acquisitions were acquired to accommodate the abdominal organs. Quantitative tumor kinetics were completed on lesions with diameters ≥3 cm, revealing fast renal clearance and peak tumor uptake of 68Ga-DOTATOC as early as 38 minutes after injection and tumor accumulations at 75 minutes after injection. At 90 minutes after injection, ROIs were drawn around the kidney, liver, spleen, and tumor to compare (by semiquantitative examination) the mean SUVs; the kidney-to-spleen mean SUV ratios varied from 0.13-0.5. PET disclosed SUVs in the lung of 3 and liver SUVs of 20. 68Ga-DOTATOC detected SMS receptors in the central nervous system with tumor-to-nontumor ratio of 100:1, whereas the metastases:normal tissue ratio was >3:1 in the liver. The authors reported 68Ga-DOTATOC PET discriminated lesions (100%) not recognized on morphologic imaging (CT, MRI) and not discerned by 111In-octreotide planar and SPECT imaging (85%).84 The authors conclude 68Ga-DOTATOC PET is superior to 111In-octreotide SPECT in distinguishing SMS-positive receptors in primary tumor and metastatic disease.84
Middendorp et al85 evaluated recurrent DTCs by targeting SMS receptors with 68Ga-DOTATOC in a retrospective comparison of 18F-FDG PET. However, 18F-FDG does not target SMS. Instead, SMS scintigraphy with111In-labeled analogs is usually combined with analysis by 18F-FDG PET. In this study, SMS targeting was not performed, and tumor imaging was associated by tracer uptake. Seventeen patients were treated by thyroidectomy, 131I (for planar whole body scintigraphy [WBS]), and external beam radiation; inclusion criteria were tumor recurrence. Patients underwent thyroid-stimulating hormone suppressive therapy during imaging; however, before imaging, thyroglobulin (Tg) and Tg-antibody values were assessed after thyroid-stimulating hormone stimulation. Both 68Ga-DOTATOC and 18F-FDG patients were fasted before scanning and divided into radioiodine-positive and -negative groups; lesions were defined as total number detected by at least 1 of the 3 modalities (131IWBS, 18F-FDG PET, or 68Ga-DOTATOC PET). The authors found comparable diagnostic PET performance on radioiodine-positive tumors with 68Ga-DOTATOC and 18F-FDG; however, 18F-FDG was suggested as preferable with radioiodine-negative DTC relapse (to localize lung and bone metastases).85 In DTCs, regional relapses occur at a rate of 20% during disease course history (10-year survival rate >90%).88 Both PET methods identified the same quantity of metastases. Lesion detectability varied by Tg level. For example, below 10 ng/mL, there were no significant differences between the 3 methods; however, above 10 ng/mL Tg, 131I WBS detected 25 lesions, 18F-FDG PET detected 90 lesions, and 68Ga-DOTATOC PET detected 43 lesions. The authors concluded no differences between 18F-FDG and 68Ga-DOTATOC on a patient basis, no differences in radioiodine-positive lesions, and significantly higher labeling of radioiodine-negative lesions with 18F-FDG85; they disclosed that 18F-FDG is usually the tracer of choice when scanning radioiodine-negative lesions with PET in DTCs with increased Tg. The most prominent limitation of this study is that the detection rates of the 2 tracers were not comparable.
18F-FDG uptake is low in slow-growing tumors such as carcinoid, which show low proliferation rates.89,90 68Ga-DOTATOC PET can be used to monitor the expression of SMS receptors in patients with NETs for radioimmunotherapy. The earliest NET 68Ga-DOTATOC PET studies were conducted by Hoffman et al,84 showing high tumor-to-non-tumor ratios and identifying metastatic lesions not uncovered by SPECT or conventional imaging.91 In another early study by von Falck et al,92 22 metastatic NET patients were evaluated by PET. 68Ga-DOTATOC uptake was increased in 72 of 74 lesions (97%). When compared with 18F-FDG PET, 68Ga-DOTATOC identified a greater, more accurate extension of tumor volume, which consisted of differentiated and undifferentiated tumor masses. In a larger study comparing 68Ga-DOTATOC to 18F-FDG, Koukouraki et al93 acquired dynamic PET images of 15 patients for both radiotracers. Tumor uptake of 18F-FDG was observed in 43 of 63 lesions (68%), whereas 68Ga-DOTATOC showed uptake in 57 of 63 lesions (90%) (Fig. 5).
Figure 5.

(A) Example of patient with resected paraganglioma and recurrent mass in retroperitoneum. Both tracers, 18F-FDG and 68Ga-DOTATOC, showed clearly enhanced uptake in retroperitoneal mass seen on CT scan. (B) Both tracers demonstrated a metastatic lesion in lower lobe of left lung. The small pulmonary lesion (CT and zoom of the outlined red area) was better delineated on 68Ga-DOTATOC study than on 18F-FDG study. (C) Another small lesion in upper left anterior mediastinum (CT overview and zoom of left anterior mediastinum) was clearly delineated on 68Ga-DOTATOC study but not on 18F-FDG study. (With kind permission from Springer Science+Business Media: Koukouraki S, et al. Comparison of the pharmacokinetics of 68Ga-DOTATOC and 18F-FDG in patients with metastatic neuroendocrine tumours scheduled for 90Y-DOTATOC therapy. Eur J Nucl Med Mol Imaging 33:1115–1122, 2006.)
SPECT imaging with the SMS analog 111In-DTPAOC is also available to evaluate patients with NET. Krenning et al94,95 and Kwekkeboom et al96 compared 68Ga-DOTATOC PET and 111In-DTPAOC SPECT in 27 patients. 68Ga-DOTATOC PET was found superior to 111In-DTPAOC SPECT in detecting NET manifestations in the lung and skeleton, while analogous for lesions in the liver and brain.91 The potential to image tumors with 68Ga-bombesin receptors was reported after successful NET 68Ga-DOTADOC PET. An early treatment (imatinib) trial of 17 patients with primary, recurrent, and metastatic disease evaluated by 68Ga-bombesin PET and 18F-FDG PET showed visualization in 25 of 30 lesions with 18F-FDG, whereas tumor uptake of 68Ga-bombesin was reported in 8 of 30 lesions.97
68Ga-Bombesin
Biomedical Targets and Clinical Applications
Bombesin receptors, a subtype of the regulatory gastrin-releasing peptide (GRP),98 are expressed in tumors such as breast,99 prostate,100 and lung,101 and have been speculated to function with mitogenic activity.102 In an effort to target GRPr-positive tumors in mouse models of prostate cancer, Mansi et al103 compared a bombesin PET tracer antagonist (68Ga-RM2) with the analogous SPECT tracer (111In-RM2). The authors questioned affinity of bombesin-derived antagonists to in vivo GRPr over bombesin agonists.104 In addition, LNCaP (androgen dependent, low GRPr expression) and PC3 (androgen independent, high GRPr expression), tumor-bearing nude mice were studied to assess the relationship between prostate cancer and androgen dependence. RM2 was fabricated by solid-phase peptide synthesis and conjugated with DOTA for 68Ga radiolabeling (microwave, 95°C, 30 seconds, 4×). 111In-RM2 labeling was performed at 95°C (30 minutes, 2×). In vitro receptor affinity and selectivity for GRPr were confirmed by using a universal bombesin receptor radioligand (125I-Tyr4-bombesin). In vitro cell saturation and blocking experiments were completed to substantiate binding specificity, and also, internalization studies with a cold ligand were carried out to establish nonspecific internalization. Pharmacokinetics (111In-RM2, PC3) and biodistribution (both tracers and tumor models) were executed to analyze tumor uptake and nonspecific binding. SPECT/CT (n = 2, PC3) and PET/CT (LNCaP and PC3) were exploited to image each group of animals with the appropriate tracer. The authors showed 68Ga-RM2 targets GRPr by binding the surface of PC3 with selective affinity.103 For example, tumor uptake was lower in animals preinjected with the cold compound, signifying a GRPr-mediated response. Tumor uptake in the PC3 xenografts increased with time, whereas 68Ga-RM2 uptake in other GRPr-positive organs decreased. 68Ga-RM2 targeted GRPr expression in LNCaP xenografts, with a lower tumor uptake compared with PC3 xenografts. In addition, 68Ga-RM2 showed rapid blood clearance. The PET images showed high specific uptake with low background; SPECT images were comparable. The authors presume the bombesin antagonist 68Ga-RM2 is suitable for targeting GRPr expression in the prostate, irrespective of androgen dependence.103
Glutamate linkers have been used to design heterodimers of RGD and bombesin (BBN) to target the expression of integrin αvβ3 and GRPr.105 The RGD-BBN was conjugated with 1,4,7-triazacyclononanetriacetic acid (NOTA) for 68Ga radiolabeling and preclinical PET imaging of human PC3 and MDA-MB-435 melanoma xenograft mice. The authors investigated dual tumor targeting because binding affinity might be lower with monomeric peptides106; however, they also compared the radiolabeled heterodimer 68Ga-NOTA-RGD-BBN with the corresponding monomers 68Ga-NOTA-RGD, 68Ga-NOTA-BBN. The competitive radioligands 125I-c[RGDyK] (integrin expressing U87MG glioma cells) and 125I[Tyr4] (PC3 cells) were used to characterize in vitro intregrin αvβ3 and GRPr binding and specificity of RGD-BBN, NOTA-RGD-BBN, BBN, and RGD. Cell uptake studies were done with PC3 cells. In vivo biodistribution of PC3 tumor-bearing mice and immunofluorescence staining for antibodies against GRPr and integrin αvβ3 were also performed. The authors showed RDG-BBN and RGD-NOTA-BBN selectively targeted αvβ3 in the U87MG cell line, whereas RGD-BBN, NOTA-RGD-BBN, and BBN selectively targeted GRPr in PC3 cells.105 PC3 cells experienced high in vitro uptake of 68Ga-NOTA-BBN and low cellular uptake of 68Ga-NOTA-RGD. Protein targeting was demonstrated in PC3 tumor and normal tissue with human αvβ3 and murine β3; positive correlations were shown with immunofluorescence. Dynamic and static PET scans were acquired of PC3 or MDA-MB-435 mice injected with 68Ga-NOTA-RGD-BBN, whereas 68Ga-NOTA-RGD or 68Ga-NOTA-BBN was administered as controls. The 68Ga-NOTA-RGD-BBN and 68Ga-NOTA-BBN heterodimers targeted αvβ3 and GRPr, whereas 68Ga-NOTA-RGD showed low tumor uptake on PET (because of low αvβ3 expression and RGD affinity in PC3). In addition, PC3 mice were coinjected with c(RGDyK) (RGD), Aca-BBN (BBN), or RGD for in vivo blocking experiments that substantiated tumor uptake of 68Ga-NOTA-RGD-BBN. PET tumor quantification was validated by ROI. For example, tumor uptake of 68Ga-NOTA-RGD-BBN (maximum tumor uptake, 6.70% ID/g, at 30 minutes post injection.) was significantly greater than that of 68Ga-NOTA-RGD and 68Ga-NOTA-BBN. Tumor-to-nontumor ratios increased with time for all tracers. In the MBA-MD-435 (αvβ3 expression) melanoma model, in vivo imaging did not reveal significant BBN tumor uptake, whereas 68Ga-NOTA-RGD had good contrast at 1 hour after injection (ie, RGD-αvβ3 recognition). Similarly, 68Ga-NOTA-RGD and 68Ga-NOTA-RGD-BBN targeted αvβ3 at 60 minutes after injection, with good tumor contrast (1.63% ± 0.59% and 3.23% ± 0.86% ID/g), whereas 68Ga-NOTA-BBN targeting of GRPr expression was low, with poor tumor uptake (0.38% ± 0.50% ID/g) at 60 minutes after injection. In summary, PET visualized protein-to-receptor targeting of GRPr and integrin αvβ3 expression, with good results in some tumor-ligand interactions (eg, PC3 cells with 68Ga-NOTA-RGD-BBN and 68Ga-NOTA-BBN) and with minimal results in others (68Ga-NOTA-RGD). However, in vitro binding showed the opposite trend. For example, the PC3 cell binding trend was as follows: 68Ga-NOTA-BBN > 68Ga-NOTA-RGD-BBN > 68Ga-NOTA-RGD. The authors explained the inconsistency by inferring that BBN is an internalized receptor, whereas αvβ3 is recognized on the cell surface105; these characteristics influence tumor uptake and retention, which consequently impact image visualization.
Finally, there is a family of 3 total human bombesin receptors, of which GRPr is a member; the other 2 are neuro-medin-B receptor (NMB-R) and BN receptor subtype-3 (BB3).107 The bombesin analog 68Ga-DOTA-PESIN was synthesized for receptor targeting and in vivo imaging of tumor uptake in all 3 receptor subtypes with the PC3 mouse model.108 Cell internalization, externalization, and binding affinity supplemented biodistribution data to determine specificity and uptake profiles. 68Ga-DOTA-PESIN showed good binding affinity for NMB-R and GRPr, with no significant binding to BB3-R. Blocking doses on in vivo biodistribution verified receptor-mediated uptake in GRPr-positive tissues (eg, tumor, pancreas, pituitary, adrenals, spleen, bowel, stomach). PET images were acquired from the intact, sacrificed mouse 1 hour after injection, and 68Ga-DOTA-PESIN accumulation was confirmed in the tumor, pancreas, and kidney to indicate GRPr targeting.
Radiozirconium Labeled Compounds
Chemistry
89Zr is a promising metallo-radionuclide for developing new immuno-PET agents because of its favorable decay characteristics. The radiochemical half-life of 78.41 hours matches the biological half-life of most antibodies, allowing for optimal tumor-to-nontumor ratios to be achieved during the imaging window (typically 2–4 days).109,110 The principal advantage of 89Zr over other long-lived radionuclides (eg, 124I, half-life = 4 days) is perhaps the retention of the radio-metal within the target cell after internalization.111,112 Effective 89Zr radiolabeling, with regard to demetalation or ligand dissociation from the biomolecule, requires the biomolecule to be conjugated to an appropriate bifunctional chelate.113 To date, the bifunctional chelate desferrioxamine B (DFO) is the most widely used for complexing 89Zr4+ ions.114 This chelate was initially used by Meijs et al,115,116 who investigated the reaction between N-(S-acetyl) mercaptoacetyl-DFO and a lysine-modified monoclonal antibody (mAb) for conjugation via a succinimide ring-thioether unit. However, in 2003, Verel et al109 reported poor in vivo stability with this system and therefore developed a new succinylated DFO derivative, coupled to lysine residues of a mAb via a stable amide bond. They reported a 6-step procedure for efficient 89Zr labeling via this chelate (Fig. 6). Consequently, several mAbs have been radiolabeled (with high specific radioactivity) using this approach, the utility of which has been demonstrated with high-quality PET images obtained from preclinical and clinical studies.117 This is a 6-step procedure; recently Vosjan et al117,118 reported the development of an alternative DFO derivative, p-isothiocyanate-benzyl-DFO, that might enable a more efficient and rapid preparation of 89Zr-labeled mAbs. Supplementary preclinical and clinical data are necessary to assess the efficiency of this new bifunctional chelate; the facile labeling procedure, the stability of the tested conjugates, and the results of comparative biodistribution studies in small animals are promising.117
Figure 6.

Schematic representation of 89Zr-N-SucDFO-mAb conjugate. (Color version of figure is available online.)
89Zr-DFO-J591
Biomedical Targets and Clinical Applications
The antibody J591 was found to target the expression of the transmembrane glycoprotein prostate-specific membrane antigen (PSMA) and has been the basis of several SPECT and radiotherapeutic agents.114,119 To take advantage of the quantitative aspect of PET, 89Zr was conjugated to the J591 via DFO and examined in preclinical models of prostate cancer.120,121 LNCaP and PC3 mice were assayed by in vivo biodistribution and small animal PET to assess tumor and organ uptake of 89Zr-DFO-J591. The specificity of 89Zr-DFO-J591 for PSMA was verified in vivo by showing limited binding to PC3 cells and an average immunoreactive fraction of 0.95 ± 0.03 of 89Zr-DFO-J591 in the LNCaP cell line. The LNCaP mice had high tumor uptake at 24 hours after injection (34.4% ± 3.2% ID/g), which steadily increased (38.0% ± 6.2% ID/g, 48 hours; 40.4% ± 4.8% ID/g, 96 hours). The tumor uptake in the PSMA-negative mice was predictably not as high (15.6% ± 2.1% ID/g, 24.0% ± 2.6% ID/g, 96 hours). The visualization of PSMA-positive and -negative tumors is shown in Fig. 3. The authors report relatively high bone uptake of 89Zr in the LNCaP mice and low bone accumulation of 89Zr in the PC3 mice, while speculating possible recirculation of labeled metabolites after slow intratumoral metabolism.114 However, in vivo competitive inhibition studies corroborated in vitro data, signifying 89Zr-DFO-J591 specificity for the PSMA. Small animal PET recorded 89Zr-DFO-J591 uptake in LNCaP mice <24 hours after injection. The maximum % ID/g (from PET) was 38.2 ± 4.9, and the mean T/M ratio at 48 hours was 15.85, which increased by 120 hours (22.49) and 144 hours (25.89) (Fig. 7). This study verifies 89Zr-DFO-J591 PET imaging of tumor specificity for PSMA expression in prostate cancer; tumor uptake was high, with good tumor-to-background ratios.
Figure 7.

Temporal immuno-PET images of 89Zr-DFO-J591 (10.9–11.3 MBq [295–305 μCi], 60–62 μg of mAb, in 200 μL of sterile saline) recorded in LNCaP tumor-bearing (PSMA-positive, left shoulder) (A) and PC3 tumor-bearing (PSMA-negative, right shoulder) (B) mice between 3 and 144 hours after injection. Transverse and coronal planar images intersect center of tumors, and mean T/M ratios derived from volume-of-interest analysis of immuno-PET images are given. Upper thresholds of immuno-PET have been adjusted for visual clarity, as indicated by scale bars. (Reprinted by permission of the Society of Nuclear Medicine from: Holland JP, et al. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med 51:1293–1300, 2010, Figure 4.)
Ruggiero et al122 correlated Cerenkov luminescence imaging (CLI) data with small animal PET images of LNCaP mice retro-orbitally injected with 89Zr-DFO-J591. The Cerenkov effect describes the continuous emission of ultraviolet and visible light from decayed radionuclides in the condensed phase123; radiation results when charged particles travel faster than the speed of light through optically transparent material, emitting visible light as polarized molecules relax to equilibrium.122 89Zr-DFO-J591 continued to provide excellent tumor-to-background ratios and clearly distinguished tumor volume (both flanks were injected, resulting in small and large LNCaP tumors). 89Zr-DFO-J591 uptake was 72.3% ± 4.6% ID/g (large tumors) and 32.0% ± 5.4% ID/g (small tumors). Tumor uptake steadily increased throughout the experimental time course. Ex vivo optical imaging of excised organs was done in addition to in vivo optical imaging (after the PET experiment). Tumor uptake of 89Zr-DFO-J591 was reported ex vivo biodistribution and with in vivo CLI; likewise, background tissues were consistent, showing 89Zr clearance (96 hours after injection). This work provides the first example of dual CLI/PET imaging of tumor uptake of a 89Zr labeled mAb.
89Zr-DFO-Trastuzumab
Biomedical Targets and Clinical Applications
The mAb trastuzumab (Herceptin; Genentech, Inc, South San Francisco, CA) targets the human epidermal growth factor receptor kinase (ERBB or HER) signaling network, and has a history as a therapeutic for metastatic or adjuvant treatment of oncological disease.124 Up-regulation of the ERB2 (HER2/neu) receptor has been associated with metastasis and poor prognosis in many cancers, including breast (incidence, 20%–30%), ovarian, lung, colorectal, and prostate.125,126 Specifically, HER receptors stimulate growth and regulate survival and differentiation.127 The most widely used methods of determining HER2/neu status are invasive and include immunohistochemistry or fluorescence in situ hybridization at the time of diagnosis of the primary tumor; however, HER2/neu tumor status can show a time-dependent change after therapy and can be different across tumor lesions in a single patient.128–130 Because of variable HER2/neu receptor expression over time, noninvasive and dynamic measurement methods would be ideal for monitoring potential treatment and disease prognoses.
Holland et al131 radiolabeled trastuzumab with 89Zr (conjugated to DFO) to investigate HER2/neu expression in human breast cancer xenografts (BT-474 cells, HER2/neu positive; MDA-MB-468 cells, HER2/neu negative) by immuno-PET. For immuno-PET studies, the mice were induced with tumors in each flank. In addition, the pharmacodynamic effects of a novel anti-HER2/neu therapeutic (HSP90 inhibitor PU-H71), were measured. The immunoreactive fraction of 89Zr-DFO-trastuzumab (0.87 ± 0.07) was determined by cellular binding assays and competitive inhibition with both cell lines (BT-474, MDA-MB-468) and non-labeled trastuzumab. BT-474 tumor uptake of 89Zr-DFO-trastuzumab and an intraperitoneal injection of PU-H71 four hours before 89Zr-DFO-trastuzumab administration were evaluated by acute biodistribution. Furthermore, in vivo competitive inhibition studies were performed, with biodistribution at the 24-hour time point. To analyze the pharmacodynamic profile of PU-H71 on HER2/neu expression, a subset of mice were implanted with sustained release 17β-estradiol before intraperitoneal PU-H71 administration. Tumors were collected at fixed time points and homogenized for Western blot analysis. The authors verified PU-H71 decreased the expression of HSP90 client oncoproteins such as HER2/neu, Akt, and RAF1.131 At 24 hours post injection, BT-474 tumor uptake of 89Zr-DFO-trastuzumab was 64.68% ± 13.06% ID/g, which denotes that 89Zr-DFO-trastuzumab targets the expression of the HER2/neu receptor. Tumor uptake at 120 hours was 75.54 ± 13.47. In vivo competitive inhibition studies confirmed specificity of 89Zr-DFO-trastuzumab for the HER2/neu antigen; pharmacodynamics also revealed PU-H71 specificity for HER2/neu. The immuno-PET images disclosed high maximum T/M ratios in the BT-474 mice of 11.90 (5 hours after injection), compared with lower maximum T/M ratios in the MDA-MB-468 mice (4.15; 5 hours after injection). There were no reported differences in the T/M in the PU-H71–treated mice; however, there were differences in the vehicle-treated mice (BT-474 T/M = 7.63; MDA-MB468 T/M = 2.82), consistent with decreases in HER2/neu expression observed on in vivo Western blot. This study validated the efficacy of immuno-PET to target and localize HER2/neu positive tumors, and to monitor the therapeutic effects of anti-HER2/neu theraupeutics, such as PU-H71.
Dijkers et al132 performed the first animal and clinical PET scans for 89Zr-trastuzumab. In vivo biodistribution data performed in animals revealed significantly higher uptake of radioactivity in the HER2/neu-positive tumor than in the HER2/neu-negative tumor, with 19.3% ± 2.0% ID/g uptake after 1 day that increased to 33.4% ± 7.6% ID/g at day 6.111 Fourteen patients were enrolled in the clinical trial and received PET scans throughout 7 days post injection. The optimal imaging time for delineation of the presence of tumors was 4–5 days. The PET images produced with 89Zr-trastuzumab revealed high spatial resolution and a good signal-to-noise ratio (SNR), which was superior to 111In-trastuzumab SPECT.133,134 Excellent tumor uptake and visualization of HER2-positive metastatic liver, lung, bone, and brain tumor lesions were obtained (Fig. 8). 89Zr-trastuzumab PET visualized bony metastatic disease in 7 of 9 patients. 89Zr-trastuzumab PET detected known lesions in 6 of 12 patients.132 These early studies show great promise for the potential of 89Zr-trastuzumab in immuno-PET.
Figure 8.

Examples of 89Zr-trastuzumab uptake 5 days after injection. (A) Patient with liver and bone metastases; (B and C) 2 patients with multiple bone metastases. Several lesions have been specifically indicated by arrows. (Reprinted by permission from Macmillan Publishers Ltd: Dijkers EC., et al. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin Pharmacol Ther 87:586–592, 2010.)
Radioiodine Labeled Compounds
124I-cG250
Chemistry
124I is a promising positron emitter for PET that has been extensively reviewed by Koehler et al.135 As with 89Zr, the radioactive half-life (4.2 days) of 124I permits PET imaging of long biological processes such as in vivo pharmacokinetics with radiolabeled mAbs. It has been recognized for nearly 20 years that antibodies can be directly iodinated with 124I without losing immunobiological activity; however, efficient production schemes of 124I have only recently been established. Although expensive to produce, 124I has become more readily available as its clinical use has increased for protein and small molecule radiolabeling.135,136
Radiolabeling of mAbs can be achieved either by direct electrophilic radioiodination or with various prosthetic groups.137 For direct labeling, radioiodination occurs at tyrosyl residues with oxidizing agents such as Iodogen (Thermo Scientific Pierce Protein Research Products, Rock-ford, IL) or chloramine-T. Recently, the mAb 124I-cG250 was used to investigate the expression of carbonic anhydrase IX (CA IX), a protein stimulated by hypoxia and involved in angiogenesis. 124I-cG250 targeted PET imaging of CA IX expression, the basis of a recent phase III clinical trial in the United States, is discussed below.
Biomedical Targets and Clinical Applications
CA IX is a transmembrane glycoprotein. It closely associates overexpression of the hypoxia inducible factor-1 (HIF-1) transcription factor,138 regulation of pH,139 and cellular proliferation,140 with regard to the malignant transformation.141 The mAb G250 (cG250, less immunogenic chimeric derivative) targets CA IX,142 and binds clear cell renal carcinomas (RCC) in humans143 and animals.144 RCC is the most common primary neoplasm of the kidney and is one of the most aggressive tumors with respect to metastasis. Because of the large proportion of patients presenting with stage IV disease, a systemic imaging agent or directed treatment is desirable. Because CA IX is expressed in >94% of human RCC145 and is absent in most normal tissues,146 radiolabeled cG250 is a functional imaging agent for tumor localization. Steffens et al147 conducted a phase I trial with 131I-labeled cG250 in 16 patients with clinically verified RCC. All 13 patients with G250-positive primary tumor had uptake and visualization of the lesion after 24 hours; 100% of known metastatic disease in these patients was also visualized and verified by conventional imaging.146 Likewise, a fragmented portion of the cG250 antibody, F(ab′)2, has also been studied, showing similar biodistribution patterns to the full cG250 antibody when both were labeled with 131I.148
In a clinical trial by Divgi et al,149 PET antibody imaging accurately detected RCC with 124I-cG250. Patients selected for kidney resection (classified by CT, MRI, or ultrasound) were imaged with 124I PET before surgery to assess tumor antibody uptake; upon resection, immunohistochemical verification of CA IX correlated positive tumor uptake with PET. With patients administered 124I-cG250 and having a diagnosis of pathologically proven RCC, PET accurately identified 15 of 16 cases (94% sensitivity). The specificity of immuno-PET to correctly determine patients without RCC was 100%.149 The high sensitivity and specificity of this paradigm, with confirmed histopathology, supports the authors’ hypothesis that 124I-cG250 differentiates RCC from other kidney masses.149
Furthermore, Strong et al150 localized 124I-cG250 in surgically extracted RCC specimens with handheld simultaneous detection beta and gamma PET probes. The dual probes (1-to 2-mm detection range from probe tip) intraoperatively evaluated suspected tumor regions before surgery with real-time count per second (cps) readouts and automatic background subtraction. The authors suggest that the beta probe is more reliable than the gamma probe for tumor detection versus normal renal parenchyma.141
Lawrentschuk et al144 evaluated the relationships between hypoxia, oxygen tension, and CA IX expression in human RCC xenograft murine models. PET/CT imaging, in vivo bio-distribution, and differential tumor volume oxygenation were established after tumor initiation. Tumor PO2 was resolved by timed optical 2-channel sensing probes, then calibrated against muscle PO2, and compared with whole body 124I-cG250 PET, biodistribution data, and CA IX immunohistochemistry. Pharmacokinetics and autoradiography were also performed, followed by serum stability assays at days 0 and 7 (97% and 81% respectively), which indicated protein binding capacity. Although intratumoral hypoxia was noted (35.08 ± 7.48 mm Hg vs 5.02 ± 2.41 mm Hg), it was not correlated with CA IX expression; the authors suggest the lack of CA IX expression might have been due to a small sample size or to the cell line (SKRC-52).144 As such, 124I-cG250 PET localized renal tumors. The localization paralleled in vivo biodistribution, autoradiography, histology, and immunohistochemistry data. The limitation of this study is that CA IX expression was not correlated with tumor hypoxia. The authors maintain that the degree of expression might modulate hypoxia, and that CA IX levels are variable.144
This review has discussed the chemistry, targeting, and initial clinical investigations of a select number of new PET radiopharmaceuticals in oncology. The efficient production and isolation of additional PET nuclides for clinical investigations and research into stable chelators between these and biologically significant molecules are revolutionizing the field of molecular imaging with PET. A wider variety of molecular targets are now being investigated to image cancer. In the current paradigm of oncological PET imaging, 18F-FDG is still a valuable tool in the clinical evaluation of cancer; however, many of these novel radiopharmaceuticals in PET will provide clinicians greater insight into the molecular and cellular backgrounds of various forms of cancer to improve patient outcomes and change disease management. The field of PET in oncology will continue to grow and enhance the discovery and treatment of cancer.
Acknowledgments
This work was funded in part by the Office of Science (BER), U. S. Department of Energy (Award DE-SC0002456), and the National Institutes of Health (R25T CA046945).
References
- 1.Delbeke D. Oncological applications of FDG PET imaging: brain tumors, colorectal cancer, lymphoma and melanoma. J Nucl Med. 1999;40:591–603. [PubMed] [Google Scholar]
- 2.Weber WA, Avril N, Schwaiger M. Relevance of positron emission tomography (PET) in oncology. Strahlenther Onkol. 1999;175:356–373. doi: 10.1007/s000660050022. [DOI] [PubMed] [Google Scholar]
- 3.Glaser M, Luthra SK, Brady F. Applications of positron-emitting halogens in PET oncology. Int J Oncol. 2003;22:253–267. [PubMed] [Google Scholar]
- 4.Dunphy MP, Lewis JS. Radiopharmaceuticals in preclinical and clinical development for monitoring of therapy with PET. J Nucl Med. 2009;50(Suppl 1):106S–121S. doi: 10.2967/jnumed.108.057281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeGrado TR, Baldwin SW, Wang S, et al. Synthesis and evaluation of (18)F-labeled choline analogs as oncologic PET tracers. J Nucl Med. 2001;42:1805–1814. [PubMed] [Google Scholar]
- 6.DeGrado TR, Coleman RE, Wang S, et al. Synthesis and evaluation of 18F-labeled choline as an oncologic tracer for positron emission tomography: initial findings in prostate cancer. Cancer Res. 2001;61:110–117. [PubMed] [Google Scholar]
- 7.Hara T. 18F-fluorocholine: a new oncologic PET tracer. J Nucl Med. 2001;42:1815–1817. [PubMed] [Google Scholar]
- 8.DeGrado TR, Reiman RE, Price DT, et al. Pharmacokinetics and radiation dosimetry of 18F-fluorocholine. J Nucl Med. 2002;43:92–96. [PubMed] [Google Scholar]
- 9.Iwata E, Wakabayashi Y, Matsuse S, et al. Induction of primer pheromone production by dihydrotestosterone in the male goat. J Vet Med Sci. 2001;63:347–348. doi: 10.1292/jvms.63.347. [DOI] [PubMed] [Google Scholar]
- 10.Cimitan M, Bortolus R, Morassut S, et al. [18F]fluorocholine PET/CT imaging for the detection of recurrent prostate cancer at PSA relapse: experience in 100 consecutive patients. Eur J Nucl Med Mol Imaging. 2006;33:1387–1398. doi: 10.1007/s00259-006-0150-2. [DOI] [PubMed] [Google Scholar]
- 11.Kryza D, Tadino V, Filannino MA, et al. Fully automated [18F]fluorocholine synthesis in the TracerLab MX FDG Coincidence synthesizer. Nucl Med Biol. 2008;35:255–260. doi: 10.1016/j.nucmedbio.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 12.Talbot JN, Fartoux L, Balogova S, et al. Detection of hepatocellular carcinoma with PET/CT: a prospective comparison of 18F-fluorocholine and 18F-FDG in patients with cirrhosis or chronic liver disease. J Nucl Med. 2010;51:1699. doi: 10.2967/jnumed.110.075507. [DOI] [PubMed] [Google Scholar]
- 13.Kwee SA, Wei H, Sesterhenn I, et al. Localization of primary prostate cancer with dual-phase 18F-fluorocholine PET. J Nucl Med. 2006;47:262–269. [PubMed] [Google Scholar]
- 14.Heinisch M, Dirisamer A, Loidl W, et al. Positron emission tomography/computed tomography with F-18-fluorocholine for restaging of prostate cancer patients: meaningful at PSA < 5 ng/mL? Mol Imaging Biol. 2006;8:43–48. doi: 10.1007/s11307-005-0023-2. [DOI] [PubMed] [Google Scholar]
- 15.Schmid DT, John H, Zweifel R, et al. Fluorocholine PET/CT in patients with prostate cancer: initial experience. Radiology. 2005;235:623–628. doi: 10.1148/radiol.2352040494. [DOI] [PubMed] [Google Scholar]
- 16.Price DT, Coleman RE, Liao RP, et al. Comparison of [18 F]fluorocholine and [18 F]fluorodeoxyglucose for positron emission tomography of androgen dependent and androgen independent prostate cancer. J Urol. 2002;168:273–280. [PubMed] [Google Scholar]
- 17.Kwee SA, Coel MN, Lim J, et al. Prostate cancer localization with 18fluorine fluorocholine positron emission tomography. J Urol. 2005;173:252–255. doi: 10.1097/01.ju.0000142099.80156.85. [DOI] [PubMed] [Google Scholar]
- 18.Beauregard JM, Williams SG, Degrado TR, et al. Pilot comparison of F-fluorocholine and F-fluorodeoxyglucose PET/CT with conventional imaging in prostate cancer. J Med Imaging Radiat Oncol. 2010;54:325–332. doi: 10.1111/j.1754-9485.2010.02178.x. [DOI] [PubMed] [Google Scholar]
- 19.Eschmann SM, Pfannenberg AC, Rieger A, et al. Comparison of 11C-choline-PET/CT and whole body-MRI for staging of prostate cancer. Nuklearmedizin. 2007;46:161–168. doi: 10.1160/nukmed-0075. quiz N147–168. [DOI] [PubMed] [Google Scholar]
- 20.Beheshti M, Langsteger W, Fogelman I. Prostate cancer: role of SPECT and PET in imaging bone metastases. Semin Nucl Med. 2009;39:396–407. doi: 10.1053/j.semnuclmed.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Husarik DB, Miralbell R, Dubs M, et al. Evaluation of [(18)F]-choline PET/CT for staging and restaging of prostate cancer. Eur J Nucl Med Mol Imaging. 2008;35:253–263. doi: 10.1007/s00259-007-0552-9. [DOI] [PubMed] [Google Scholar]
- 22.Enzinger C, Strasser-Fuchs S, Ropele S, et al. Tumefactive demyelinating lesions: conventional and advanced magnetic resonance imaging. Mult Scler. 2005;11:135–139. doi: 10.1191/1352458505ms1145oa. [DOI] [PubMed] [Google Scholar]
- 23.Law M, Meltzer DE, Cha S. Spectroscopic magnetic resonance imaging of a tumefactive demyelinating lesion. Neuroradiology. 2002;44:986–989. doi: 10.1007/s00234-002-0872-1. [DOI] [PubMed] [Google Scholar]
- 24.Kwee SA, Ko JP, Jiang CS, et al. Solitary brain lesions enhancing at MR imaging: evaluation with fluorine 18 fluorocholine PET. Radiology. 2007;244:557–565. doi: 10.1148/radiol.2442060898. [DOI] [PubMed] [Google Scholar]
- 25.Liu A, Dence CS, Welch MJ, et al. Fluorine-18-labeled androgens: radiochemical synthesis and tissue distribution studies on six fluorine-substituted androgens, potential imaging agents for prostatic cancer. J Nucl Med. 1992;33:724–734. [PubMed] [Google Scholar]
- 26.Liu A, Carlson KE, Katzenellenbogen JA. Synthesis of high affinity fluorine-substituted ligands for the androgen receptor: potential agents for imaging prostatic cancer by positron emission tomography. J Med Chem. 1992;35:2113–2129. doi: 10.1021/jm00089a024. [DOI] [PubMed] [Google Scholar]
- 27.Mori T, Kiyono Y, Asai T, et al. Automated synthesis of 16{beta}-[18F]fluoro-5{alpha}-dihydrotestosterone using a plastic cassette-type FDG synthesizer. J Nucl Med Meeting Abstracts. 2010;51:1525. [Google Scholar]
- 28.Agus DB, Cordon-Cardo C, Fox W, et al. Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999;91:1869–1876. doi: 10.1093/jnci/91.21.1869. [DOI] [PubMed] [Google Scholar]
- 29.Agus DB, Golde DW, Sgouros G, et al. Positron emission tomography of a human prostate cancer xenograft: association of changes in deoxyglucose accumulation with other measures of outcome following androgen withdrawal. Cancer Res. 1998;58:3009–3014. [PubMed] [Google Scholar]
- 30.Choe YS, Lidstrom PJ, Chi DY, et al. Synthesis of 11 beta-[18F]fluoro-5 alpha-dihydrotestosterone and 11 beta-[18F]fluoro-19-nor-5 alpha-dihydrotestosterone: preparation via halofluorination-reduction, receptor binding, and tissue distribution. J Med Chem. 1995;38:816–825. doi: 10.1021/jm00005a009. [DOI] [PubMed] [Google Scholar]
- 31.Beattie BJ, Smith-Jones PM, Jhanwar YS, et al. Pharmacokinetic assessment of the uptake of 16beta-18F-fluoro-5alpha-dihydrotestosterone (FDHT) in prostate tumors as measured by PET. J Nucl Med. 2010;51:183–192. doi: 10.2967/jnumed.109.066159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonasera TA, O’Neil JP, Xu M, et al. Preclinical evaluation of fluorine-18-labeled androgen receptor ligands in baboons. J Nucl Med. 1996;37:1009–1015. [PubMed] [Google Scholar]
- 33.Larson SM, Morris M, Gunther I, et al. Tumor localization of 16beta-18F-fluoro-5alpha-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J Nucl Med. 2004;45:366–373. [PubMed] [Google Scholar]
- 34.Dehdashti F, Picus J, Michalski JM, et al. Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur J Nucl Med Mol Imaging. 2005;32:344–350. doi: 10.1007/s00259-005-1764-5. [DOI] [PubMed] [Google Scholar]
- 35.Zanzonico PB, Finn R, Pentlow KS, et al. PET-based radiation dosimetry in man of 18F-fluorodihydrotestosterone, a new radiotracer for imaging prostate cancer. J Nucl Med. 2004;45:1966–1971. [PubMed] [Google Scholar]
- 36.Haubner R, Wester HJ, Weber WA, et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–1785. [PubMed] [Google Scholar]
- 37.Haubner R, Kuhnast B, Mang C, et al. [F-18]Galacto-RGD: synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug Chem. 2004;15:61–69. doi: 10.1021/bc034170n. [DOI] [PubMed] [Google Scholar]
- 38.Li Z, Conti PS. Radiopharmaceutical chemistry for positron emission tomography. Adv Drug Deliv Rev. 2010;62:1031–1051. doi: 10.1016/j.addr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Cox D, Aoki T, Seki J, et al. The pharmacology of the integrins. Med Res Rev. 1994;14:195–228. doi: 10.1002/med.2610140203. [DOI] [PubMed] [Google Scholar]
- 40.Brooks PC, Montgomery AM, Rosenfeld M, et al. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 41.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 42.Teti A, Migliaccio S, Baron R. The role of the alphaVbeta3 integrin in the development of osteolytic bone metastases: a pharmacological target for alternative therapy? Calcif Tissue Int. 2002;71:293–299. doi: 10.1007/s00223-001-2071-1. [DOI] [PubMed] [Google Scholar]
- 43.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 44.Haubner R, Wester HJ, Reuning U, et al. Radiolabeled alpha(v)beta3 integrin antagonists: a new class of tracers for tumor targeting. J Nucl Med. 1999;40:1061–1071. [PubMed] [Google Scholar]
- 45.Beer AJ, Haubner R, Wolf I, et al. PET-based human dosimetry of 18F-galacto-RGD, a new radiotracer for imaging alpha v beta3 expression. J Nucl Med. 2006;47:763–769. [PubMed] [Google Scholar]
- 46.Beer AJ, Haubner R, Goebel M, et al. Biodistribution and pharmacokinetics of the alphavbeta3-selective tracer 18F-galacto-RGD in cancer patients. J Nucl Med. 2005;46:1333–1341. [PubMed] [Google Scholar]
- 47.Vonlaufen A, Wiedle G, Borisch B, et al. Integrin alpha(v)beta(3) expression in colon carcinoma correlates with survival. Mod Pathol. 2001;14:1126–1132. doi: 10.1038/modpathol.3880447. [DOI] [PubMed] [Google Scholar]
- 48.Beer AJ, Grosu AL, Carlsen J, et al. [18F]galacto-RGD positron emission tomography for imaging of alphavbeta3 expression on the neovasculature in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6610–6616. doi: 10.1158/1078-0432.CCR-07-0528. [DOI] [PubMed] [Google Scholar]
- 49.Haubner R, Weber WA, Beer AJ, et al. Noninvasive visualization of the activated alphavbeta3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005;2:e70. doi: 10.1371/journal.pmed.0020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Washburn LC, Sun TT, Byrd B, et al. 1-aminocyclobutane[11C]-carboxylic acid, a potential tumor-seeking agent. J Nucl Med. 1979;20:1055–1061. [PubMed] [Google Scholar]
- 51.Shoup TM, Goodman MM. Synthesis of [F-18]-1-amino-3-fluorocy-clobutane-1-carboxylic acid (FACBC): a PET tracer for tumor delineation. Journal of Labelled Compounds & Radiopharmaceuticals. 1999;42:215–225. [Google Scholar]
- 52.Shoup TM, Olson J, Hoffman JM, et al. Synthesis and evaluation of [F-18]1-amino-3-fluorocyclobutane-1-carboxylic acid to image brain tumors. J Nucl Med. 1999;40:331–338. [PubMed] [Google Scholar]
- 53.McConathy J, Voll RJ, Yu W, et al. Improved synthesis of anti-[18F]FACBC: improved preparation of labeling precursor and automated radiosynthesis. Appl Radiat Isot. 2003;58:657–666. doi: 10.1016/s0969-8043(03)00029-0. [DOI] [PubMed] [Google Scholar]
- 54.Oka S, Hattori R, Kurosaki F, et al. A preliminary study of anti-1-amino-3-F-18-fluorocyclobutyl-1-carboxylic acid for the detection of prostate cancer. J Nucl Med. 2007;48:46–55. [PubMed] [Google Scholar]
- 55.Schuster DM, Votaw JR, Nieh PT, et al. Initial experience with the radio-tracer anti-1-amino-3-F-18-fluorocyclobutane-1-carboxylic acid with PET/CT in prostate carcinoma. J Nucl Med. 2007;48:56–63. [PubMed] [Google Scholar]
- 56.Schuster DM, Nye JA, Nieh PT, et al. Initial experience with the radiotracer anti-1-amino-3-[F-18]fluorocyclobutane-1-carboxylic acid (anti-[F-18] FACBC) with PET in renal carcinoma. Mol Imaging Biol. 2009;11:434–438. doi: 10.1007/s11307-009-0220-5. [DOI] [PubMed] [Google Scholar]
- 57.Holley RW. A unifying hypothesis concerning the nature of malignant growth. Proc Natl Acad Sci U S A. 1972;69:2840–2841. doi: 10.1073/pnas.69.10.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laverman P, Boerman OC, Corstens FH, et al. Fluorinated amino acids for tumour imaging with positron emission tomography. Eur J Nucl Med Mol Imaging. 2002;29:681–690. doi: 10.1007/s00259-001-0716-y. [DOI] [PubMed] [Google Scholar]
- 59.Grosu AL, Weber WA, Franz M, et al. Reirradiation of recurrent high-grade gliomas using amino acid PET (SPECT)/CT/MRI image fusion to determine gross tumor volume for stereotactic fractionated radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63:511–519. doi: 10.1016/j.ijrobp.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 60.Jager PL. Improving amino acid imaging: hungry or stuffed? J Nucl Med. 2002;43:1207–1209. [PubMed] [Google Scholar]
- 61.McConathy J, Goodman MM. Non-natural amino acids for tumor imaging using positron emission tomography and single photon emission computed tomography. Cancer Metastasis Rev. 2008;27:555–573. doi: 10.1007/s10555-008-9154-7. [DOI] [PubMed] [Google Scholar]
- 62.Shoup TM, Olson J, Hoffman JM, et al. Synthesis and evaluation of [18F]1-amino-3-fluorocyclobutane-1-carboxylic acid to image brain tumors. J Nucl Med. 1999;40:331–338. [PubMed] [Google Scholar]
- 63.Yu W, Williams L, Camp VM, et al. Synthesis and biological evaluation of anti-1-amino-2-[18F]fluoro-cyclobutyl-1-carboxylic acid (anti-2-[18F]FACBC) in rat 9L gliosarcoma. Bioorg Med Chem Lett. 2010;20:2140–2143. doi: 10.1016/j.bmcl.2010.02.048. [DOI] [PubMed] [Google Scholar]
- 64.Nye JA, Schuster DM, Yu W, et al. Biodistribution and radiation dosimetry of the synthetic nonmetabolized amino acid analogue anti-18F-FACBC in humans. J Nucl Med. 2007;48:1017–1020. doi: 10.2967/jnumed.107.040097. [DOI] [PubMed] [Google Scholar]
- 65.Oka S, Hattori R, Kurosaki F, et al. A preliminary study of anti-1-amino-3-18F-fluorocyclobutyl-1-carboxylic acid for the detection of prostate cancer. J Nucl Med. 2007;48:46–55. [PubMed] [Google Scholar]
- 66.Schuster DM, Votaw JR, Nieh PT, et al. Initial experience with the radiotracer anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid with PET/CT in prostate carcinoma. J Nucl Med. 2007;48:56–63. [PubMed] [Google Scholar]
- 67.Savir-Baruch B, Schuster DM, Jarkas N, et al. Pilot evaluation of anti-1-amino-2-[(18)F] fluorocyclopentane-1-carboxylic acid (anti-2-[(18)F] FACPC) PET-CT in recurrent prostate carcinoma. Mol Imaging Biol. 2010 doi: 10.1007/s11307-010-0445-3. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng QH, Stone KL, Mock BH, et al. [11C]Choline as a potential PET marker for imaging of breast cancer athymic mice. Nucl Med Biol. 2002;29:803–807. doi: 10.1016/s0969-8051(02)00339-6. [DOI] [PubMed] [Google Scholar]
- 69.Hara T, Inagaki K, Kosaka N, et al. Sensitive detection of mediastinal lymph node metastasis of lung cancer with 11C-choline PET. J Nucl Med. 2000;41:1507–1513. [PubMed] [Google Scholar]
- 70.Krause BJ, Souvatzoglou M, Herrmann K, et al. [11C]Choline as pharmacodynamic marker for therapy response assessment in a prostate cancer xenograft model. Eur J Nucl Med Mol Imaging. 2010;37:1861–1868. doi: 10.1007/s00259-010-1493-2. [DOI] [PubMed] [Google Scholar]
- 71.Hara T, Kosaka N, Shinoura N, et al. PET imaging of brain tumor with [methyl-11C]choline. J Nucl Med. 1997;38:842–847. [PubMed] [Google Scholar]
- 72.Parivar F, Hricak H, Shinohara K, et al. Detection of locally recurrent prostate cancer after cryosurgery: evaluation by transrectal ultrasound, magnetic resonance imaging, and three-dimensional proton magnetic resonance spectroscopy. Urology. 1996;48:594–599. doi: 10.1016/S0090-4295(96)00250-6. [DOI] [PubMed] [Google Scholar]
- 73.Yu KK, Hricak H. Imaging prostate cancer. Radiol Clin North Am. 2000;38:59–85. viii. doi: 10.1016/s0033-8389(05)70150-0. [DOI] [PubMed] [Google Scholar]
- 74.Kurhanewicz J, Vigneron DB, Males RG, et al. The prostate: MR imaging and spectroscopy: present and future. Radiol Clin North Am. 2000;38:115–138. viii–ix. doi: 10.1016/s0033-8389(05)70152-4. [DOI] [PubMed] [Google Scholar]
- 75.Nudell DM, Wefer AE, Hricak H, et al. Imaging for recurrent prostate cancer. Radiol Clin North Am. 2000;38:213–229. doi: 10.1016/s0033-8389(05)70157-3. [DOI] [PubMed] [Google Scholar]
- 76.Hara T, Kosaka N, Kishi H. PET imaging of prostate cancer using carbon-11-choline. J Nucl Med. 1998;39:990–995. [PubMed] [Google Scholar]
- 77.Maecke HR, Hofmann M, Haberkorn U. (68)Ga-labeled peptides in tumor imaging. J Nucl Med. 2005;46(Suppl 1):172S–178S. [PubMed] [Google Scholar]
- 78.Bauwens M, Chekol R, Vanbilloen H, et al. Optimal buffer choice of the radiosynthesis of (68)Ga-Dotatoc for clinical application. Nucl Med Commun. 2010;31:753–758. doi: 10.1097/MNM.0b013e32833acb99. [DOI] [PubMed] [Google Scholar]
- 79.Breeman WA, de Jong M, de Blois E, et al. Radiolabelling DOTA-peptides with 68Ga. Eur J Nucl Med Mol Imaging. 2005;32:478–485. doi: 10.1007/s00259-004-1702-y. [DOI] [PubMed] [Google Scholar]
- 80.Decristoforo C, Knopp R, von Guggenberg E, et al. A fully automated synthesis for the preparation of 68Ga-labelled peptides. Nucl Med Commun. 2007;28:870–875. doi: 10.1097/MNM.0b013e3282f1753d. [DOI] [PubMed] [Google Scholar]
- 81.Zhernosekov KP, Filosofov DV, Baum RP, et al. Processing of generator-produced 68Ga for medical application. J Nucl Med. 2007;48:1741–1748. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- 82.Asti M, De Pietri G, Fraternali A, et al. Validation of (68)Ge/(68)Ga generator processing by chemical purification for routine clinical application of (68)Ga-DOTATOC. Nucl Med Biol. 2008;35:721–724. doi: 10.1016/j.nucmedbio.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 83.Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25:458–511. doi: 10.1210/er.2003-0014. [DOI] [PubMed] [Google Scholar]
- 84.Hofmann M, Maecke H, Borner R, et al. Biokinetics and imaging with the somatostatin receptor PET radioligand (68)Ga-DOTATOC: preliminary data. Eur J Nucl Med. 2001;28:1751–1757. doi: 10.1007/s002590100639. [DOI] [PubMed] [Google Scholar]
- 85.Middendorp M, Selkinski I, Happel C, et al. Comparison of positron emission tomography with [(18)F]FDG and [(68)Ga]DOTATOC in recurrent differentiated thyroid cancer: preliminary data. Q J Nucl Med Mol Imaging. 2010;54:76–83. [PubMed] [Google Scholar]
- 86.Ain KB, Taylor KD, Tofiq S, et al. Somatostatin receptor subtype expression in human thyroid and thyroid carcinoma cell lines. J Clin Endocrinol Metab. 1997;82:1857–1862. doi: 10.1210/jcem.82.6.4013. [DOI] [PubMed] [Google Scholar]
- 87.Rodrigues M, Traub-Weidinger T, Leimer M, et al. Value of 111In-DOTA-lanreotide and 111In-DOTA-DPhe1-Tyr3-octreotide in differentiated thyroid cancer: results of in vitro binding studies and in vivo comparison with 18F-FDG PET. Eur J Nucl Med Mol Imaging. 2005;32:1144–1151. doi: 10.1007/s00259-005-1820-1. [DOI] [PubMed] [Google Scholar]
- 88.Stokkel MP, Duchateau CS, Dragoiescu C. The value of FDG-PET in the follow-up of differentiated thyroid cancer: a review of the literature. Q J Nucl Med Mol Imaging. 2006;50:78–87. [PubMed] [Google Scholar]
- 89.Strauss LG, Conti PS. The applications of PET in clinical oncology. J Nucl Med. 1991;32:623–648. discussion 649–650. [PubMed] [Google Scholar]
- 90.Adams S, Baum R, Rink T, et al. Limited value of fluorine-18 fluorodeoxyglucose positron emission tomography for the imaging of neuroendocrine tumours. Eur J Nucl Med. 1998;25:79–83. doi: 10.1007/s002590050197. [DOI] [PubMed] [Google Scholar]
- 91.Buchmann I, Henze M, Engelbrecht S, et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2007;34:1617–1626. doi: 10.1007/s00259-007-0450-1. [DOI] [PubMed] [Google Scholar]
- 92.von Falck C, Boerner AR, Galanski M, et al. Neuroendocrine tumour of the mediastinum: fusion of 18F-FDG and 68Ga-DOTATOC PET/CT datasets demonstrates different degrees of differentiation. Eur J Nucl Med Mol Imaging. 2007;34:812. doi: 10.1007/s00259-006-0350-9. [DOI] [PubMed] [Google Scholar]
- 93.Koukouraki S, Strauss LG, Georgoulias V, et al. Comparison of the pharmacokinetics of 68Ga-DOTATOC and [18F]FDG in patients with metastatic neuroendocrine tumours scheduled for 90Y-DOT-ATOC therapy. Eur J Nucl Med Mol Imaging. 2006;33:1115–1122. doi: 10.1007/s00259-006-0110-x. [DOI] [PubMed] [Google Scholar]
- 94.Krenning EP, Bakker WH, Kooij PP, et al. Somatostatin receptor scintigraphy with indium-111-DTPA-D-Phe-1-octreotide in man: metabolism, dosimetry and comparison with iodine-123-Tyr-3-octreotide. J Nucl Med. 1992;33:652–658. [PubMed] [Google Scholar]
- 95.Krenning EP, Kwekkeboom DJ, Bakker WH, et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716–731. doi: 10.1007/BF00181765. [DOI] [PubMed] [Google Scholar]
- 96.Kwekkeboom DJ, Krenning EP, Bakker WH, et al. Somatostatin analogue scintigraphy in carcinoid tumours. Eur J Nucl Med. 1993;20:283–292. doi: 10.1007/BF00169802. [DOI] [PubMed] [Google Scholar]
- 97.Dimitrakopoulou-Strauss A, Hohenberger P, Haberkorn U, et al. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: comparison with 18F-FDG. J Nucl Med. 2007;48:1245–1250. doi: 10.2967/jnumed.106.038091. [DOI] [PubMed] [Google Scholar]
- 98.Gugger M, Reubi JC. Gastrin-releasing peptide receptors in non-neoplastic and neoplastic human breast. Am J Pathol. 1999;155:2067–2076. doi: 10.1016/S0002-9440(10)65525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Halmos G, Wittliff JL, Schally AV. Characterization of bombesin/gastrin-releasing peptide receptors in human breast cancer and their relationship to steroid receptor expression. Cancer Res. 1995;55:280–287. [PubMed] [Google Scholar]
- 100.Markwalder R, Reubi JC. Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res. 1999;59:1152–1159. [PubMed] [Google Scholar]
- 101.Toi-Scott M, Jones CL, Kane MA. Clinical correlates of bombesin-like peptide receptor subtype expression in human lung cancer cells. Lung Cancer. 1996;15:341–354. doi: 10.1016/0169-5002(95)00597-8. [DOI] [PubMed] [Google Scholar]
- 102.Cuttitta F, Carney DN, Mulshine J, et al. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature. 1985;316:823–826. doi: 10.1038/316823a0. [DOI] [PubMed] [Google Scholar]
- 103.Mansi R, Wang X, Forrer F, et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging. 2010;38:97–107. doi: 10.1007/s00259-010-1596-9. [DOI] [PubMed] [Google Scholar]
- 104.Mansi R, Wang X, Forrer F, et al. Evaluation of a 1,4,7,10-tetraazacy-clododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin Cancer Res. 2009;15:5240–5249. doi: 10.1158/1078-0432.CCR-08-3145. [DOI] [PubMed] [Google Scholar]
- 105.Liu Z, Niu G, Wang F, et al. (68)Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging. 2009;36:1483–1494. doi: 10.1007/s00259-009-1123-z. [DOI] [PubMed] [Google Scholar]
- 106.Jia B, Liu Z, Shi J, et al. Linker effects on biological properties of 111In-labeled DTPA conjugates of a cyclic RGDfK dimer. Bioconjug Chem. 2008;19:201–210. doi: 10.1021/bc7002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang H, Chen J, Waldherr C, et al. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004;64:6707–6715. doi: 10.1158/0008-5472.CAN-03-3845. [DOI] [PubMed] [Google Scholar]
- 108.Zhang H, Schuhmacher J, Waser B, et al. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2007;34:1198–1208. doi: 10.1007/s00259-006-0347-4. [DOI] [PubMed] [Google Scholar]
- 109.Verel I, Visser GW, Boellaard R, et al. 89Zr immuno-PET: comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J Nucl Med. 2003;44:1271–1281. [PubMed] [Google Scholar]
- 110.Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dijkers EC, Kosterink JG, Rademaker AP, et al. Development and characterization of clinical-grade 89Zr-trastuzumab for HER2/neu immunoPET imaging. J Nucl Med. 2009;50:974–981. doi: 10.2967/jnumed.108.060392. [DOI] [PubMed] [Google Scholar]
- 112.Tinianow JN, Gill HS, Ogasawara A, et al. Site-specifically Zr-89-labeled monoclonal antibodies for ImmunoPET. Nucl Med Biol. 2010;37:289–297. doi: 10.1016/j.nucmedbio.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 113.Wadas TJ, Wong EH, Weisman GR, et al. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110:2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holland JP, Divilov V, Bander NH, et al. 89ZrDFO-J591 for immuno-PET of prostate-specific membrane antigen expression in vivo. J Nucl Med. 2010;51:1293–1300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meijs WE, Haisma HJ, Klok RP, et al. Zirconium-labeled monoclonal antibodies and their distribution in tumor-bearing nude mice. J Nucl Med. 1997;38:112–118. [PubMed] [Google Scholar]
- 116.Meijs WE, Haisma HJ, VanderSchors R, et al. A facile method for the labeling of proteins with zirconium isotopes. Nucl Med Biol. 1996;23:439–448. doi: 10.1016/0969-8051(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 117.Perk LR, Vosjan MJWD, Visser GWM, et al. p-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur J Nucl Med Mol Imaging. 2010;37:250–259. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vosjan MJWD, Perk LR, Visser GWM, et al. Conjugation and radio-labeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat Protoc. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 119.Li Y, Tian Z, Rizvi SM, et al. In vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J591 antibody against the prostate specific membrane antigen. Prostate Cancer Prostatic Dis. 2002;5:36–46. doi: 10.1038/sj.pcan.4500543. [DOI] [PubMed] [Google Scholar]
- 120.Liu H, Moy P, Kim S, et al. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997;57:3629–3634. [PubMed] [Google Scholar]
- 121.Liu H, Rajasekaran AK, Moy P, et al. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–4060. [PubMed] [Google Scholar]
- 122.Ruggiero A, Holland JP, Lewis JS, et al. Cerenkov luminescence imaging of medical isotopes. J Nucl Med. 2010;51:1123–1130. doi: 10.2967/jnumed.110.076521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Berger SL. The use of Cerenkov radiation for monitoring reactions performed in minute volumes: examples from recombinant DNA technology. Anal Biochem. 1984;136:515–519. doi: 10.1016/0003-2697(84)90254-9. [DOI] [PubMed] [Google Scholar]
- 124.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 125.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 126.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 127.Gross ME, Shazer RL, Agus DB. Targeting the HER-kinase axis in cancer. Semin Oncol. 2004;31:9–20. doi: 10.1053/j.seminoncol.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 128.Solomayer EF, Becker S, Pergola-Becker G, et al. Comparison of HER2 status between primary tumor and disseminated tumor cells in primary breast cancer patients. Breast Cancer Res Treat. 2006;98:179–184. doi: 10.1007/s10549-005-9147-y. [DOI] [PubMed] [Google Scholar]
- 129.Wulfing P, Borchard J, Buerger H, et al. HER2-positive circulating tumor cells indicate poor clinical outcome in stage I to III breast cancer patients. Clin Cancer Res. 2006;12:1715–1720. doi: 10.1158/1078-0432.CCR-05-2087. [DOI] [PubMed] [Google Scholar]
- 130.Dijkers EC, de Vries EG, Kosterink JG, et al. Immunoscintigraphy as potential tool in the clinical evaluation of HER2/neu targeted therapy. Curr Pharm Des. 2008;14:3348–3362. doi: 10.2174/138161208786549425. [DOI] [PubMed] [Google Scholar]
- 131.Holland JP, Caldas-Lopes E, Divilov V, et al. Measuring the pharmacodynamic effects of a novel Hsp90 inhibitor on HER2/neu expression in mice using Zr–DFO-trastuzumab. PLoS One. 2010;5:e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dijkers EC, Oude-Munnink TH, Kosterink JG, et al. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin Pharmacol Ther. 2010;87:586–592. doi: 10.1038/clpt.2010.12. [DOI] [PubMed] [Google Scholar]
- 133.Perik PJ, Lub-De Hooge MN, Gietema JA, et al. Indium-111-labeled trastuzumab scintigraphy in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2006;24:2276–2282. doi: 10.1200/JCO.2005.03.8448. [DOI] [PubMed] [Google Scholar]
- 134.de Korte MA, de Vries EG, Lub-de Hooge MN, et al. 111Indium-trastuzumab visualises myocardial human epidermal growth factor receptor 2 expression shortly after anthracycline treatment but not during heart failure: a clue to uncover the mechanisms of trastuzumab-related cardiotoxicity. Eur J Cancer. 2007;43:2046–2051. doi: 10.1016/j.ejca.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 135.Koehler L, Gagnon K, McQuarrie S, et al. Iodine-124: a promising positron emitter for organic PET chemistry. Molecules. 2010;15:2686–2718. doi: 10.3390/molecules15042686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Divgi CR, Pandit-Taskar N, Jungbluth AA, et al. Preoperative characterisation of clear-cell renal carcinoma using iodine-124-labelled antibody chimeric G250 (I-124-cG250) and PET in patients with renal masses: a phase I trial. Lancet Oncol. 2007;8:304–310. doi: 10.1016/S1470-2045(07)70044-X. [DOI] [PubMed] [Google Scholar]
- 137.Welch MJ, Redvanly CS. Handbook of radiopharmaceuticals: radiochemistry and applications. New York: J. Wiley; 2003. [Google Scholar]
- 138.Wykoff CC, Beasley NJ, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- 139.Loncaster JA, Harris AL, Davidson SE, et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res. 2001;61:6394–6399. [PubMed] [Google Scholar]
- 140.Saarnio J, Parkkila S, Parkkila AK, et al. Immunohistochemical study of colorectal tumors for expression of a novel transmembrane carbonic anhydrase, MN/CA IX, with potential value as a marker of cell proliferation. Am J Pathol. 1998;153:279–285. doi: 10.1016/S0002-9440(10)65569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vermylen P, Roufosse C, Burny A, et al. Carbonic anhydrase IX antigen differentiates between preneoplastic malignant lesions in non-small cell lung carcinoma. Eur Respir J. 1999;14:806–811. doi: 10.1034/j.1399-3003.1999.14d14.x. [DOI] [PubMed] [Google Scholar]
- 142.Uemura H, Nakagawa Y, Yoshida K, et al. MN/CA IX/G250 as a potential target for immunotherapy of renal cell carcinomas. Br J Cancer. 1999;81:741–746. doi: 10.1038/sj.bjc.6690757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oosterwijk E, Divgi CR, Brouwers A, et al. Monoclonal antibody-based therapy for renal cell carcinoma. Urol Clin North Am. 2003;30:623–631. doi: 10.1016/s0094-0143(03)00028-4. [DOI] [PubMed] [Google Scholar]
- 144.Lawrentschuk N, Lee FT, Jones G, et al. Investigation of hypoxia and carbonic anhydrase IX expression in a renal cell carcinoma xenograft model with oxygen tension measurements and (124)I-cG250 PET/CT. Urol Oncol. 2009 doi: 10.1016/j.urolonc.2009.03.028. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 145.Bui MH, Seligson D, Han KR, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res. 2003;9:802–811. [PubMed] [Google Scholar]
- 146.Lam JS, Pantuck AJ, Belldegrun AS, et al. G250: a carbonic anhydrase IX monoclonal antibody. CurrOncol Rep. 2005;7:109–115. doi: 10.1007/s11912-005-0036-7. [DOI] [PubMed] [Google Scholar]
- 147.Steffens MG, Boerman OC, de Mulder PH, et al. Phase I radioimmunotherapy of metastatic renal cell carcinoma with 131I-labeled chimeric monoclonal antibody G250. Clin Cancer Res. 1999;5:3268s–3274s. [PubMed] [Google Scholar]
- 148.Brouwers A, Mulders P, Oosterwijk E, et al. Pharmacokinetics and tumor targeting of 131I-labeled F(ab′)2 fragments of the chimeric monoclonal antibody G250: preclinical and clinical pilot studies. Cancer Biother Radiopharm. 2004;19:466–477. doi: 10.1089/cbr.2004.19.466. [DOI] [PubMed] [Google Scholar]
- 149.Divgi CR, Pandit-Taskar N, Jungbluth AA, et al. Preoperative characterisation of clear-cell renal carcinoma using iodine-124-labelled antibody chimeric G250 (124I-cG250) and PET in patients with renal masses: a phase I trial. Lancet Oncol. 2007;8:304–310. doi: 10.1016/S1470-2045(07)70044-X. [DOI] [PubMed] [Google Scholar]
- 150.Strong VE, Humm J, Russo P, et al. A novel method to localize antibody-targeted cancer deposits intraoperatively using handheld PET beta and gamma probes. Surg Endosc. 2008;22:386–391. doi: 10.1007/s00464-007-9611-3. [DOI] [PubMed] [Google Scholar]