

Abstract
Proton radiation is touted for improved tumor targeting, over standard gamma radiation, due to the physical advantages of ion beams for radiotherapy. Recent studies from our laboratory demonstrate that in addition to these targeting advantages, proton irradiation can inhibit angiogenic and immune factors critical to “hallmark” processes that impact cancer progression, thereby modulating tumor development. Outside the therapeutic utilization of protons, high-energy protons constitute a principal component of galactic cosmic rays and thus are a consideration in carcinogenesis risk for space flight. Given that proton irradiation modulates fundamental biological processes known to decrease with aging (e.g. angiogenesis and immunogenicity), we investigated how proton irradiation impacts tumor advancement as a function of host age, a question with both therapeutic and carcinogenesis implications. Tumor lag time and growth dynamics were tracked, after injection of murine Lewis lung carcinoma (LLC) cells into syngeneic adolescent (68 day) vs. old (736 day) C57BL/6 mice with or without coincident irradiation. Tumor growth was suppressed in old compared to adolescent mice. These differences were further modulated by proton irradiation (1 GeV), with increased inhibition and a significant radiation-altered molecular fingerprint evident in tumors grown in old mice. Through global transcriptome analysis, TGFβ1 and TGFβ2 were determined to be key players that contributed to the tumor dynamics observed. These findings suggest that old hosts exhibit a reduced capacity to support tumor advancement, which can be further reduced by proton irradiation.
INTRODUCTION
Over the past decade, proton therapy has attracted considerable attention within the radiation oncology community. There are now about 40 proton centers dedicated to treatment of a wide range of cancers (1). Accepted advantages demonstrated for proton therapy over conventional X-ray radiotherapy include decreased dosing of normal tissue with consequent decreased side effects and improved targeting of treatment to tumors within close proximity of vital organs (2). Recently in preclinical models, proton irradiation has been shown to modulate several key processes critical in tumor advancement and progression, including angiogenesis and immunogenicity (2, 3). Girdhani et al. demonstrated that proton irradiation (1 GeV) reduced levels of VEGF, IL6, IL8 and HIF1a, both in vitro and in vivo (3). The findings of inhibited angiogenic factor expression are in line with other recent work showing significant inhibition of in vivo blood vessel formation in a zebrafish model following proton irradiation with 1, 2 and 5 Gy at 35 MeV (4). Grabham et al. also found inhibition of developing vessel formation, using in vitro 3D cultures, subsequent to proton exposure (1 GeV) at doses as low as 0.4 Gy (5). These findings add to the accumulating data suggesting that proton irradiation can inhibit biological processes that are critical to cancer progression, with some of these processes also found to be reduced in older hosts. Given that proton irradiation modulates the same fundamental processes of angiogenesis and immunogenicity that are known to decrease with increasing age, we investigated how proton irradiation modulates tumor advancement as a function of host age, a question with both therapeutic and carcinogenesis implications.
A vast number of studies have been conducted on the impact of age on carcinogenesis. Epidemiological studies show that with the onset of middle age, the increase in incidence starts to decline and in old age tumor incidence actually decreases (6, 7). Literature addressing carcinogenesis as a function of age has primarily focused on the accumulation of DNA damage and mutations in potential cancer cells. Here our focus is not on age-accumulated DNA damage driving carcinogenic cell transformation, but rather on the impact of host age on overall tumor advancement after injection of transformed cells into syngeneic mice. We examine the role of interactions between cancer and host cells, and how tumors develop differently as a function of age and irradiation.
The effects of protons (1 GeV) on 68- and 736-day-old mice implanted with syngeneic cancer cells were investigated. Tumor establishment, advancement, growth rates and their molecular underpinnings were examined. We observed and characterized distinct behaviors that strongly correlated to both age and radiation exposure. A global transcriptome analysis performed on the set of all excised tumors from the different age groups, with and without proton irradiation was undertaken. It revealed a number of the upstream regulators involved, thereby shedding light on the tumor dynamics observed. Two ligands from the TGFβ family (8, 9), TGFβ1 (9, 10) and TGFβ2 (11), were found to be centrally involved. The identification of these factors points to age-dependent processes ongoing during tumor progression that can be differentially modulated by proton irradiation.
MATERIALS AND METHODS
Cell Culture
Murine Lewis lung carcinoma (LLC) cells were obtained from American Type Culture Collection [LL/2 (LLC1), ATCC® CRL1642™, Manassas, VA] and cultured under standard conditions (12) in DMEM, high glucose (Gibco® Invitrogen Cell Culture, Carlsbad, CA) with 10% FBS (Gibco Invitrogen Cell Culture). LLC cells were passaged for a month from frozen stocks expanded for fewer than 2 months from the original stock obtained from ATCC. ATCC characterized the LLC cell line with routine mycoplasma detection, morphology check by microscopy and species verification by isoenzyme analysis.
Irradiation of Mice
Thirty C57BL/6 male mice (68 days old) were purchased from Jackson Laboratory (Bar Harbor, ME) and thirty mice (736 days old) from the National Institute of Aging were used in this study. Scaling of mouse age to human age was accomplished using published criteria (13). Mice 68 and 736 days old, referred to as “adolescent” and “old”, respectively, at tumor injection are considered to correspond to 17- and 75-year-old humans. Ten adolescent and ten old mice were given whole-body irradiation once a day for three days with 0.5 Gy of proton ions (1 GeV; LET 0.24 keV/μm; plateau, non-Bragg peak, region, 0.5 Gy/min dose rate) at Brookhaven National Laboratory (Upton, NY). Mice were restrained and were not anesthetized during irradiation. An additional twenty adolescent and twenty old mice were not irradiated, but otherwise handled identically and used as sham, nonirradiated controls. All animals, control and irradiated, were injected with syngeneic LLC cells.
Cancer Cell Injections
Injections of C57BL/6 mice were made subcutaneously in the middorsal region of the back with 1 × 106 LLC cells, 8 h after the first of three dose fractions. Nonirradiated mice, serving as controls, were similarly injected with 1 × 106 LLC cells. The timing of the dosing relative to cancer cell injection was chosen to maximize the likelihood of the impact of proton irradiation on tumor development and advancement, by combining the situation where the first irradiation fraction acts on the normal cells of the host and later fractions modulate interactions between the host and the cancer cells. In a previous study with 56Fe irradiation, we performed a similar experimental setup and fractionation schedule (7). Mice were anesthetized by inhalation of 4% isoflurane, for injection of 1 × 106 LLC cells suspended in 0.2 ml phosphate-buffered saline (PBS). Nonirradiated control mice were treated identically. Mice were monitored regularly and tumor size was measured with calipers by a single individual with 30 years of experience. Once tumors reached approximately 1.5 cm3, mice were sacrificed, tissues processed and LLC tumor histology characterized and confirmed.
Real-Time Quantitative PCR
RNA was isolated from tumor tissue in in TRIzol (Invitrogen, Carlsbad, CA) using standard methods and homogenized using a Tissue Lyser II (Qiagen, Valencia, CA). Tissue with TRIzol was extracted according to the manufacturer’s instruction. The quality and quantity of the RNA was measured by an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA). Synthesis of cDNAs from 1 μg of total RNA was performed using a High-Capacity cDNA Reverse Transcription kit (ABI, Applied Biosystems, Carlsbad, CA) and stored at −20°C until use. Expression levels of RNA transcripts were quantified by real-time PCR (7300 Real Time PCR System, Applied Biosystems®). The cDNAs were mixed with TaqMan Universal PCR Master Mix (Applied Biosystems) and appropriate primer probes (TGFβ1 and TGFβ2) before performing real-time PCR. The PCR protocol was denaturation at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 1 min. Samples were profiled both for the target sequence and 18S (ribosomal RNA) expression as endogenous control. For each amplification reaction, a threshold cycle was observed in exponential phase and quantification of relative expression levels achieved using standard curves for both target and controls. Assays were performed with technical duplicates and data was analyzed using the method described by Schmittgen and Livak (14).
Genome-Wide Expression Analysis
Genome-wide expression profiling of tumor tissue was done using MouseWG-6 bead array chips (Illumina Inc., San Diego, CA). Total RNA was amplified with Ambion Illumina TotalPrep Amplification Kit (Ambion®, Austin, TX) and labeled from all replicate biological samples for each condition. For tumor replicates, 30 tumor samples from adolescent mice and 30 tumor samples from old mice, for a total of 60 tumor samples, were used. All replicate samples were run individually. For each age group 10 samples of tumors from animals that had received proton irradiation were used, while 20 samples of tumors from mice that were nonirradiated mice. Total RNA was isolated, purified using TRIzol (Invitrogen) and quantified using an Agilent Bioanalyzer. Samples were deemed suitable for amplification and hybridization if they had 28s/18s = 2:1, RIN > 7. A total RNA of 500 ng per sample was amplified using Ambion TotalPrep and 1.5 μg of the product was loaded onto the chips. After hybridization at 55°C, chips were washed and scanned using the Illumina iScan System. Data was checked with GenomeStudio (Illumina) (background subtracted and rank invariant normalization applied) for quality control, then imported into MultiExperiment Viewer (15) for statistical analysis. Statistically significant genes were determined using MultiExperiment Viewer by applying a one-way ANOVA analysis with standard Bonferroni correction with a FDR < 0.05 that resulted in a list of significant genes. Average gene expression signals less than 10 were filtered out due to signal being close to background. The resulting list of significant genes was used for the remaining analysis.
Further pathway analysis was performed by using a log2-fold change comparing all samples to each other and observing pathway relations using Ingenuity® Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA).
Statistical Analysis
Student’s t tests were used for statistical analysis as appropriate. All P values were calculated using two-tailed tests. OriginPro (Origin-Lab®, Northampton, MA) was used for graphs. Differences were considered statistically significant if P < 0.05. Error bars in the graphs represent standard errors.
To quantify tumor growth, we used nonlinear regression to fit the tumor growth phase data with curves of the form V = bect, where V is tumor volume and t is time as described in Beheshti et al. (7). Here, c is interpreted as the tumor growth rate once the tumor starts to grow more or less exponentially and b is interpreted as the extrapolated “effective” volume at the implant time, t = 0. “Effective” means the volume is inferred by backward extrapolation to earlier time points from the fit to later time points.
Tumor progression data in each group was calculated by two different methods also described in Beheshti et al. (7). In the first method, the “areas under the curves” [denoted “Atotal” (Table 1)] were calculated on days 7, 9, 11, 14 and 15. In the second method the final measured average volumes at the last day for which all tumor size data were available (day 11) were utilized [denoted “Vlast” (Table 1)] in each group.
TABLE 1.
Growth Rate Calculations
| Group | b (mm3) | c (day−1) | Atotal (mm3 days) | Vlast (mm3) |
|---|---|---|---|---|
| Adolescent (0 Gy) | 202.0 ± 23.8 | 0.154 ± 0.008 | 8112.11 | 927.1 ± 98.8 |
| Old (0 Gy) | 236.8 ± 29.9 | 0.122 ± 0.005 | 6414.08 | 733.4 ± 63.2 |
| Adolescent (0.5 Gy × 3 proton) | 211.1 ± 33.3 | 0.157 ± 0.010 | 9441.59 | 1091.6 ± 84.8 |
| Old (0.5 Gy × 3 proton) | 148.7 ± 32.8 | 0.138 ± 0.010 | 4674.56 | 513.8 ± 63.8 |
Notes. Growth rate calculations were done by an exponential fit V = bect to individual tumors after the approximate start of exponential growth starts with V = tumor volume (mm3) and t = time (days). The parameters in the table are defined as: b = “effective” initial tumor volume; c = growth rate; Atotal = total area under the data points shown in Fig. 2; Vlast = tumor size at day 11 that corresponds to the final measured tumor volume at the last day all tumor size data existed in all groups. The Materials and Methods section describes how average growth rate c and effective initial tumor volume b were calculated for each group and how they are interpreted biologically.
RESULTS
Tumor Progression in Old and Adolescent Mice with and without Proton Irradiation
For these studies, we utilized an in vivo model of tumor establishment and advancement, which mimics aspects of tumor progression, that involved wild-type C57BL/6 (adolescent or old) mice injected subcutaneously with syngeneic Lewis lung carcinoma. To investigate the effect of both host age and irradiation, 10 adolescent and 10 old irradiated mice were treated with three 0.5 Gy fractions of whole-body proton irradiation, with cancer cells injected subcutaneously after the first of three daily radiation dose fractions. Twenty age-matched, sham-irradiated control mice were otherwise treated identically.
Measurements of tumor volume as a function of time for nonirradiated mice revealed that tumor growth was substantially inhibited in the old mice compared to the adolescent mice (Fig. 1A), consistent with analogous studies from our laboratory looking at middle aged vs. young mice (7). Comparisons of tumor volumes for all time points taken at greater than 11 days post-cancer cell injection reveal that the differences in volumes were statistically significant [indicated by asterisks (Fig. 1A)]. Strikingly, proton irradiation of old animals had a significant inhibitory effect on tumor advancement in the older hosts (Fig. 1D) compared to age-matched nonirradiated controls. While, in contrast, proton irradiation did not significantly modulate tumor progression in the adolescent animals (Fig. 1B). Thus, tumors in the proton-irradiated aged mice exhibited a very marked reduction in overall advancement, (resulting from the combined inhibition of both age itself and the proton-induced response of the aged host) compared to that of tumors developing in the irradiated adolescent mice (Fig. 1C).
FIG. 1.
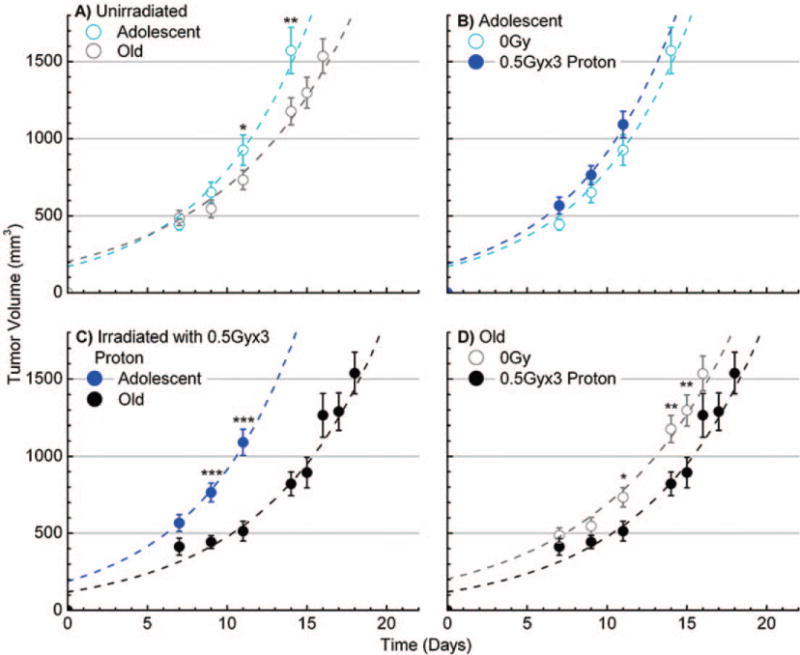
Tumor growth in adolescent and old mice, with or without proton irradiation. Panels A–D: The data showed average measured tumor volumes, after injection at time 0, of murine Lewis lung carcinoma (LLC) in C57BL/6 mice with and without 3 × 0.5 Gy of proton irradiation (1 GeV). All 0 Gy conditions have n = 20 mice and all proton-irradiated conditions have n = 10 mice. Panels A and C: Compare data from adolescent vs. old mice without and with irradiation, respectively. Panel B: Depicts data for proton-irradiated adolescent vs. nonirradiated and panel D: proton-irradiated old vs. nonirradiated mice groups. The time post-cell-injection is shown on the x axis. Error bars show ±SE and asterisks indicate time points where there is a statistically significant differential even when ignoring information from the other time points; *P < 0.05, **P < 0.01 and ***P < 0.001. The dashed lines indicate a generated best fits in terms of two quantities, tumor growth rates after approximately exponential growth starts and “effective” initial tumor volumes as explained in the Materials and Methods section.
Tumor Growth Rates and Effective Initial Tumor Volumes
Two quantitative aspects of tumor development were taken to be the growth rate c and the “effective initial tumor volume” b, obtained by extrapolating the fitted exponential curve backwards to time zero and interpreted as a measure of how supportive the host microenvironment was for tumor organization and establishment prior to the near exponential growth phase (see Materials and Methods). The curves in Fig. 1 show results for the fits V = b*exp(ct), with coefficients determined as described in the Materials and Methods and shown in Table 1.
Overall, the data show that tumor growth rates in nonirradiated mice are indeed dependent on host age and that there was a statistically significant decrease in average growth rates c for tumors developing in the nonirradiated old hosts compared to the nonirradiated adolescent hosts (P ≤ 0.001). Regarding perturbation by proton irradiation, the following results were found: (A) the effective initial volume b in old mice was significantly decreased by irradiation (P ≤ 0.05), while in adolescent mice no difference in initial volume with irradiation was exhibited; and (B) the overall tumor progression decrease observed with irradiation for the old mice was due exclusively to a dramatic decrease in effective initial volume of these tumors with no significant difference in c. In sum, these studies demonstrate that for this syngeneic system, older hosts provide a systemic environment that inhibits tumor growth. Further, a novel finding of this investigation is that proton irradiation only impacted and inhibited tumor establishment and advancement in the older hosts, even though tumor growth in these animals was already significantly reduced compared to that of adolescent animals.
Molecular Differences in Tumors as a Function of Age and Proton Irradiation
Transcriptome analysis of the tumors demonstrate that clear gene expression differences arise between the different age and radiation status groups that parallel the observed tumor dynamics. Euclidean cluster analysis of the global gene expression data was conducted to compare tumor samples from mice as a function of age and irradiation status. Interestingly, overall gene expression data for the tumor samples from the old, proton-irradiated mice significantly deviated from that for all the other groups, i.e. for the old, nonirradiated mice and for both the adolescent irradiated and nonirradiated mice (Fig. 2A). Moreover, differences were also detected in distinct regions of the global gene expression patterns of the proton-irradiated old group compared to the nonirradiated old group (Fig. 2B). It was found that 1,537 genes were significantly differentially expressed between these groups, with an FDR < 0.05. Logarithmic scatter plots of gene expression for comparison between each of the pairs of groups revealed distinct differential patterns in expression levels (Fig. 2C). Expanding on Fig. 2C, Table 2 shows the number of genes that are 1.2-fold up- and down-regulated. Surprisingly, overall there were more genes up-regulated in the tumors growing in the old hosts than there were in those tumors from the younger, adolescent hosts. Proton irradiation was seen to further enhance this differential. When comparing tumor samples from irradiated old mice to those from nonirradiated old mice, a significant number of genes were up-regulated beyond 1.2-fold, while only a few 1.2-fold down-regulated genes were found. When making these same comparisons for irradiation of adolescent hosts, for tumor samples from proton irradiated vs. those from nonirradiated adolescent hosts, more genes with a greater than 1.2-fold change were found to be down-regulated than up-regulated (Table 2). These findings of opposite gene regulation clearly indicate that proton modulation of the overall tumor transcriptome is highly dependent on the host’s age.
FIG. 2.
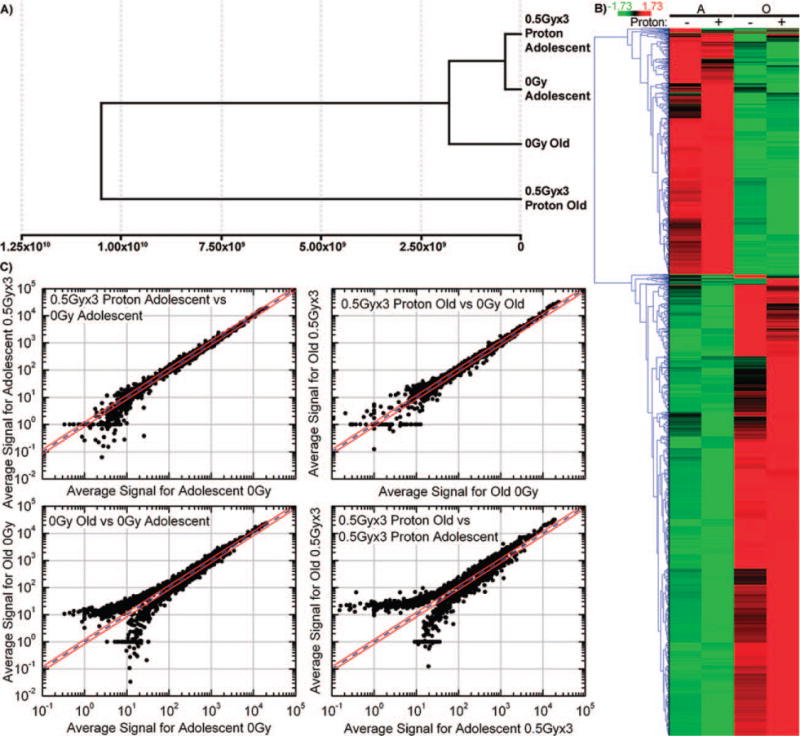
Gene regulation changes for tumors as a function of host age and proton irradiation status. Panel A: Cluster analysis of expression data, done by Euclidean distance calculation displayed as a dendrogram, for LLC tumors growing in adolescent and old mice, with or without 3 × 0.5 Gy whole-body proton irradiation. Data expression patterns from tumors in the irradiated and nonirradiated adolescent groups cluster together, with that for tumors in nonirradiated old mice being similar. The overall expression pattern of tumors growing in the old mice is observed to be considerably perturbed by exposure to protons. Panel B: Hierarchical clustering of genes by average linkage (UPGMA) and Euclidean distance calculation between age groups adolescent (A) and old (O) and irradiation groups (− for 0 Gy and + for 3 × 0.5 Gy proton). Panel C: Logarithmic scatter plots comparing gene expression in: adolescent mice (0 Gy) vs. adolescent mice (3 × 0.5 Gy proton); old mice (0 Gy) vs. old mice (3 × 0.5 Gy proton); adolescent mice (0 Gy) vs. old mice (0 Gy); and adolescent mice (3 × 0.5 Gy proton) vs. old mice (3 × 0.5 Gy proton). The blue line is referred to as the identity line, while the two outer red lines represent the window of genes that are up- or down-regulated 1.2-fold from the identity line. Points that fall outside the 1.2-fold window represent genes that were found to be more than 1.2-fold up- or down-regulated between the samples compared.
TABLE 2.
The Number of Genes with 1.2-fold Up- or Down-Regulation for each Comparison Group
| Old vs. adolescent
|
0.5 Gy × 3 proton vs. 0 Gy
|
|||
|---|---|---|---|---|
| 0 Gy | 0.5 Gy × 3 | Adolescent | Old | |
| Number of 1.2-fold up-regulated genes | 886 | 986 | 220 | 446 |
| Number of 1.2-fold down-regulated genes | 552 | 503 | 464 | 186 |
Further analysis was performed using IPA software to determine key molecular differences occurring between each group, which could shed insight on the tumor dynamics observed. Through pathway and upstream regulator analysis, TGFβ1 and TGFβ2 were indicated to be key players involved in regulating the observed dynamics. The transforming growth factor-β (TGFβ) family contains three ligands, TGFβ1, TGFβ2 and TGFβ3, which have demonstrated activity and involvement in cell growth, differentiation and migration of cancer cells (16). Paradoxically, TGFβ expression has been shown to facilitate tumor suppression at early stages of tumorigenesis, but enhance tumor growth for late stage tumors (10, 11, 16). The samples used under this analysis can be considered late-stage tumors, as these were collected at the time of sacrifice of the mice. From pathway analysis conducted with MeV (15) and IPA, it was predicted that both TGFβ1 and TGFβ2 are down-regulated in tumors from proton-irradiated old mice compared to tumors from nonirradiated old mice (Fig. 3). In contrast, tumors from proton-irradiated adolescent mice showed an up-regulation of TGFβ2, with no alteration in regulation of TGFβ1, compared to tumors from the similarly nonirradiated aged controls.
FIG. 3.
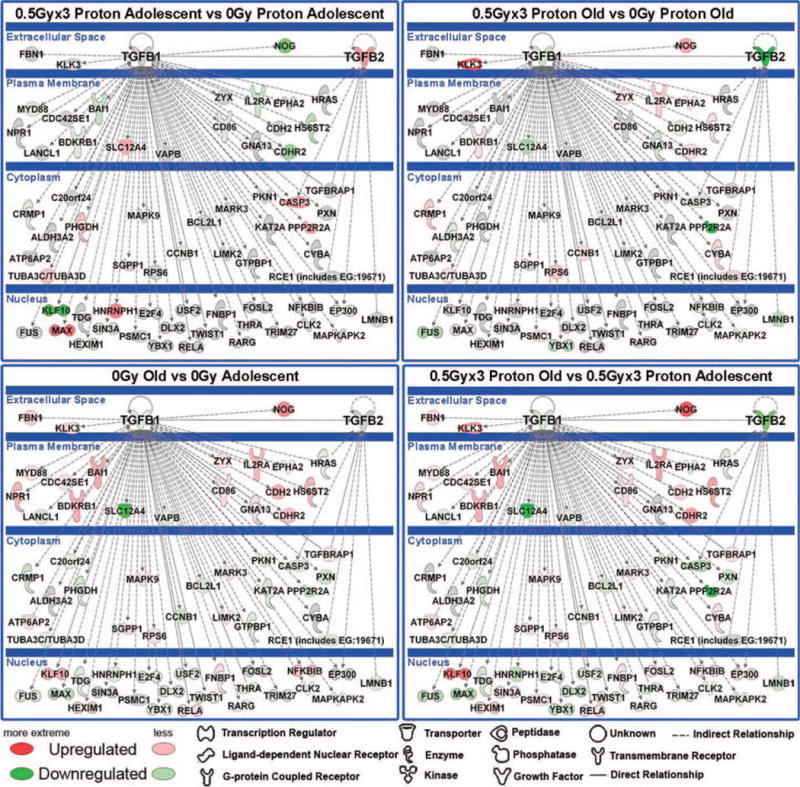
Gene network analysis for key genes involved in age-dependent tumor progression as a function of proton irradiation. Pathway analysis was done with Ingenuity Pathway Analysis (IPA) software. The network depicted contains central nodes from TGFβ1 and TGFβ2 with direct (solid lines) and indirect (dashed lines) relationships to these molecules. Log2-fold changes to the gene expression were used to obtain different shades of green for regulation levels for 1.2-fold change in down-regulated genes, while different shades of red depict regulation levels for 1.2-fold change in up-regulated genes. Grey depicts genes that exist in the network without a significant 1.2-fold change under the perturbation investigated. The darker the shade of green or red, the greater the fold change.
To validate these findings on gene regulation, quantitative real-time PCR (RT-PCR) was performed investigating TGFβ1 and TGFβ2 expression within the four groups (Fig. 4A and B). For each group RT-PCR results were compared with the 0 Gy adolescent samples, which were normalized to one. In agreement with the array data, it was determined that there was a significant decrease in both TGFβ1 and TGFβ2 mRNA expression in tumor samples from proton-irradiated old mice compared to those for nonirradiated old mice (~twofold decreases in expression for TGFβ1 with P = 0.0002, and ~fourfold decrease in expression for TGFβ2 with P = 0.0055). While no significant differences in TGFβ1 were observed between the adolescent, proton-irradiated mice and the nonirradiated mice (considering the standard errors for the irradiated samples), a significant increase in TGFβ2 mRNA expression was noted for tumors from the irradiated mice. The decrease observed with TGFβ1 and TGFβ2 expression varies in accordance with the decrease observed in the tumor growth (Fig. 1). As stated, the literature shows that for late stage tumors, decreases in TGFβ1 and TGFβ2 have been shown to decrease tumor growth (15).
FIG. 4.
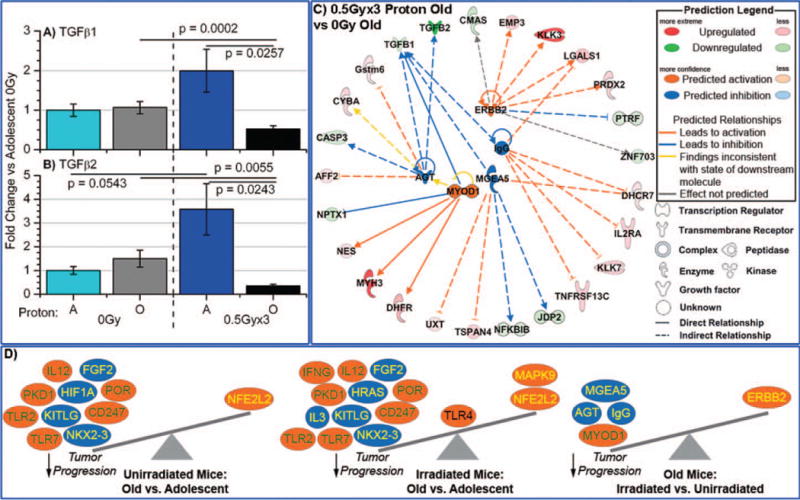
Characterization of TGFβ1 and TGFβ2 in tumors from proton-irradiated and nonirradiated adolescent and old mice. The mRNA expression for panel A: TGFβ1 and for panel B: TGFβ2, was determined by real-time PCR (RTPCR). Fold changes for mRNA expression were determined for each group, compared to 0 Gy adolescent mice. Significance is indicated by the P values within the plots. Panel C: Gene network depiction of upstream regulators predicted to be either activated (orange) or inhibited (blue) in tumors from irradiated old mice vs. from nonirradiated old mice, determined by IPA software. Specific up-regulated (red) and down-regulated (green) genes from the experimental data set involved in determining the activation state of the upstream regulator are shown with direct (solid lines) and indirect (dashed lines) relationships to the upstream regulators. The predicted relationships are color coded to indicate whether it leads to activation (orange) or inhibition (blue). Relationships that are inconsistent with the prediction (yellow) or have an undetermined effect (gray) are also shown. The darker the shade of green or red, the greater the fold change. Panel D: A schematic of the activation states of the upstream regulators from Table 3 shows the balance between the tumor promoters (text in yellow) and tumor suppressors (text in green) with a predicted activation (orange oval) or predicted inhibition (blue oval).
Further insight into the mechanisms involved in the tumor dynamics at play under this investigation can be gained by studying differentials between the various upstream regulators involved in the irradiated and nonirradiated groups, for both old and adolescent mice. Upstream regulators were determined using IPA and were defined as any molecule that affected the expression or function of another molecule. This includes transcription regulators, growth factors, cytokines, enzymes, transmembrane receptors and kinases. The IPA upstream regulator analysis uses expected causal effects calculated from the empirical gene expression data compiled in this study, and then correlates this with a comprehensive database of known upstream regulators compiled from the literature. The activation state of each upstream regulator culled from the experimental data set is determined by calculating the activation z score. For those z scores ≥2, the activation state of the upstream regulator is predicted to be activated and for those with z scores ≤2, the activation state is predicted to be inhibited.
From our array analysis, we determined statistically significant upstream regulators, with predicted activated or inhibited action. This was done for comparisons between all groups (with the exception of adolescent irradiated vs. nonirradiated mice) as shown in Table 3. By displaying a network of the upstream regulators and the related up- and down-regulated genes involved, it was revealed that TGFβ1 and TGFβ2 are also implicated in the network linking most of the determined upstream regulators for irradiated old mice vs. nonirradiated mice (Fig. 4C and Table 3). Interestingly, an examination of the individual upstream regulators involved for the different groups, in conjunction with the reported impact on tumor growth from the literature (Table 3 and Fig. 4D), revealed correlations consistent with the observed tumor dynamics. For each of the upstream regulators, information from the literature was compiled regarding the impact of that specific regulator on tumor growth, i.e. inhibition, promotion or both. The literature indicates that for all groups, those upstream regulators that were predicted to have an inhibited activation state are associated with enhancement of tumor growth (Fig. 4D), suggesting that the inhibition of these regulators in the specific networks under consideration may impede progression. Likewise, the majority of the upstream regulators in all groups that were predicted to be in an activated state are documented to inhibit tumor growth (Table 3 and Fig. 4D). This suggests that overall the net action of the activated upstream regulators may also be facilitating inhibition of tumor advancement. Inhibition of tumor progression is indeed borne out in the data from tumors from irradiated old mice vs. nonirradiated mice (Fig 1D). For the irradiated old mice vs. nonirradiated mice the upstream regulators all point to inhibition of tumor growth with the exception of one, ERBB2 (also known as HER2/neu), which promotes tumor growth (17) (Table 3 and Fig. 4C). We also note, ERBB2 is the only upstream regulator that does not include TGFβ1 in the network compiled from the existing gene expression data (Fig. 4C). Although ERBB2 action may be in opposition, the action of the majority of the indicated upstream regulators (4/5) align with the observed overall inhibition of tumor advancement observed in proton-irradiated mice compared to nonirradiated old mice (Fig. 4D).
TABLE 3.
Activation States of Upstream Regulators
| Group | Molecule type | Upstream regulator | Predicted activation state | Regulation z score | Effect on tumors | Ref. |
|---|---|---|---|---|---|---|
| 0.5 Gy × 3 proton old vs. 0 Gy old | transcription regulator | MYOD1 | Activated | 2.215 | Inhibits | (35, 36) |
| kinase | ERBB2 | Activated | 2.186 | Promotes | (17) | |
| enzyme | MGEA5 | Inhibited | −2.449 | Promotes | (38) | |
| complex | IgG | Inhibited | −2.401 | Promotes | (39) | |
| growth factor | AGT | Inhibited | −2.008 | Promotes | (37) | |
| Old vs. adolescent | ion channel | PKD1 | Activated | 2.611 | Inhibits | (42) |
| transcription regulator | NFE2L2 | Activated | 2.599 | Promotes | (43) | |
| transmembrane receptor | TLR2 | Activated | 2.392 | Inhibits | (28) | |
| transmembrane receptor | TLR7 | Activated | 2.377 | Inhibits | (29) | |
| complex | IL12 | Activated | 2.240 | Inhibits | (30) | |
| enzyme | POR | Activated | 2.236 | Inhibits | (44) | |
| transmembrane receptor | CD247 | Activated | 2.000 | Inhibits | (31) | |
| growth factor | FGF2 | Inhibited | −2.600 | Promotes | (26) | |
| transcription regulator | NKX2-3 | Inhibited | −2.121 | Promotes | (25) | |
| transcription regulator | HIF1A | Inhibited | −2.020 | Promotes | (22) | |
| growth factor | KITLG | Inhibited | −2.000 | Promotes | (45) | |
| 0.5 Gy × 3 proton old vs. 0.5 Gy × 3 proton adolescent | transcription regulator | NFE2L2 | Activated | 2.809 | Promotes | (43) |
| ion channel | PKD1 | Activated | 2.611 | Inhibits | (42) | |
| transmembrane receptor | TLR2 | Activated | 2.577 | Inhibits | (28) | |
| transmembrane receptor | TLR7 | Activated | 2.377 | Inhibits | (29) | |
| complex | IL12 | Activated | 2.240 | Inhibits | (30) | |
| transmembrane receptor | CD247 | Activated | 2.236 | Inhibits | (31) | |
| enzyme | POR | Activated | 2.236 | Inhibits | (44) | |
| cytokine | IFNG | Activated | 2.224 | Inhibits | (46) | |
| transmembrane receptor | TLR4 | Activated | 2.166 | Both | (47) | |
| kinase | MAPK9 (JNK2) | Activated | 2.000 | Promotes | (48) | |
| growth factor | FGF2 | Inhibited | −2.600 | Promotes | (26) | |
| enzyme | HRAS | Inhibited | −2.200 | Promotes | (49) | |
| cytokine | IL3 | Inhibited | −2.186 | Promotes | (50) | |
| transcription regulator | NKX2-3 | Inhibited | −2.121 | Promotes | (25) | |
| growth factor | KITLG | Inhibited | −2.000 | Promotes | (45) |
Notes. Upstream regulators predicted to be activated or inhibited (bold) over the course of tumor growth by increased host age (comparisons between tumors from nonirradiated old vs. nonirradiated adolescent mice) or by proton irradiation (comparisons between tumors from irradiated old mice vs. nonirradiated old mice), or by the combination of age and proton irradiation (comparisons of irradiated old vs. irradiated adolescent), obtained through the use of Ingenuity Pathway Analysis software. Regulation z score indicates the degree of significance. The last two columns denote the effects the upstream regulators have on tumor progression as based on the literature.
DISCUSSION
Further insight into the biological responses elicited by proton radiation is critical for the optimization of proton therapy and to gain a better understanding of the risk of space travel, since a large component of space radiation derives from protons (18). Recent studies have revealed quite unexpected radiobiological responses to proton irradiation in the laboratory setting (3–5, 19). Our focus was to examine the integrated impact of the modulation of host age and proton irradiation on the dynamics and molecular underpinnings of tumor progression. To date, there remain limited investigations examining tumor growth as a function of host age, and essentially no work exploring how host age impacts the radiobiology of proton irradiation.
We previously reported (7) on a parallel study examining the impact of host age and 56Fe irradiation (1 GeV/amu) on tumor progression. In that study 56Fe irradiation inhibited tumor progression and tumor growth rates in young hosts, while only the growth rates were inhibited by the irradiation in older hosts. Here, as in the previous study, we demonstrated that without exposure to radiation, tumors growing in old mice experience significant inhibition of tumor progression and exhibit slower tumor growth rates. Additionally, we demonstrated here that whole-body exposure to fractionated proton irradiation, with three doses of 0.5 Gy at 1 GeV (LET = 0.24 keV/μm; plateau, non-Bragg peak, region) dramatically inhibited tumor advancement in old mice with little change in growth rates, yet this same dosing had little effect on tumor advancement in the adolescent mice. Proton beams in the clinical setting typically have energies that range from 60–250 MeV, with LET values from ~0.4–1.0 keV/μm, and are delivered in the Spread Out Bragg Peak (SOBP) region as opposed to the plateau region used in this study (2). The results presented here suggest that a potential wealth of information on proton radiobiology is available outside the classic clinical and preclinical arena, which under proper interpretation could be informative and exploited in the clinical setting.
As previously reported (7) and reconfirmed in this study with adolescent and old animals, we found older hosts had a reduced capacity to support tumor establishment and advancement, as well as growth. Consistent with the literature, a number of factors may contribute to this phenomenon, including a generalized inability of older hosts to mount an angiogenic response (20–24), and a loss of potency of hematopoietic stem cells (HSC) and other adult stem cells (5, 8). Building on the global transcriptome analysis, we determined sets of upstream regulators that were predicted to be activated or inhibited in the modulation of tumor growth dynamics under radiation or aging. Like all tumors, the tumor samples analyzed comprise a mix of cell types; along with cancer cells there are various stromal and endothelial components. The upstream regulators that are predicted to be activated or inhibited for tumors of mice from different age and irradiation states are shown in Table 3. Figure 4D not only shows the predicted activation state of these regulators and the documented effect of each on cancer progression, but it also schematically depicts how the net balance of the regulators indicated for each condition as tipping the scale to stimulate or inhibit tumor progression. As the middle panel of Fig. 4D illustrates, the net balance of factors predicts a reduced tumor progression in older mice. One factor predicted to be inhibited in the tumors of the old vs. adolescent hosts is HIF1α. Inhibition of HIF1α has been previously implicated as contributing to slower tumor growth of human breast cancer cells (MDA-MB-231) in aged animals (22). In addition, our analysis also indicated the angiogenic factors FGF2 and NKX2-3, which act as tumor promoters (25, 26), to be inhibited in the LLC tumors growing in the old hosts. Although it has been reported that the immune system in general is suppressed in older hosts, some immune factors are nevertheless maintained despite age (27). In this study, we implicate involvement of a group of upstream regulators related to the immune system that are actually activated in the aged host and are documented in the literature to inhibit or slow tumor progression. This group includes TLR2 (28), TLR7 (29), IL12 (30) and CD247 (31). When activated, Toll-like receptor 2 (TLR2) has even been reported to induce tumor regression (28). These findings support the involvement of two systems in the slower tumor growth in our aged animals: a reduction of angiogenic signaling; and the activation of immune factors that have not declined with age.
In addition to those factors that act to slow tumor growth with increased age, we found that, under certain conditions, proton irradiation can additionally perturb the system to further diminish tumor progression. Global genome analysis showed striking differences in the tumors from the proton-irradiated old hosts compared to those tumors from each of the other groups. Euclidean cluster analysis for all samples demonstrated that proton-irradiated tumor samples from the aged mice showed the greatest transcriptome cluster differential compared to those of the other groups. Interestingly, based on the transcriptome data, tumor samples from the nonirradiated old mice were found to be closer to those of the irradiated and nonirradiated adolescent groups, than to those from the irradiated old mice (Fig. 2A). Through network analysis two key factors, TGFβ1 and TGFβ2, were revealed to contribute to the slower tumor advancement observed in the proton-irradiated old mice compared to that in the nonirradiated old mice. Down-regulation of both TGFβ1 and TGFβ2 was detected in the samples from irradiated old mice compared to the samples from nonirradiated old mice, while TGFβ2 was up-regulated and there was no significant fold change for TGFβ1 when comparing tumors from adolescent irradiated with nonirradiated mice (Figs. 3 and 4). The TGFβ family has been shown to act as both a tumor suppressor and a tumor promoter (9), depending on context. In many early-stage tumors, TGFβ functions as a tumor suppressor, but in late-stage solid tumors as a tumor promoter (10). The tumors in our studies are considered to be late-stage solid tumors, where TGFβ action would be expected to be tumor promoting. The fact that both TGFβ1 and TGFβ2 are down-regulated in tumors from the old proton-irradiated compared to the nonirradiated hosts (Figs. 3, 4A and B), appears consistent with the inhibition in tumor progression we observed in these irradiated hosts (Fig. 1). Several studies have shown, with ionizing gamma radiation, that TGFβ1 can act as a radioprotector (9, 32, 33). More specifically, the work of Bouquet et al. (34) shows that with ionizing radiation, a reduction or inhibition of TGFβ1 increases radiosensitivity of breast cancer cells and promotes radiation-induced tumor latency. There is little information on the impact of proton irradiation in vivo on TGFβ. In vivo studies done using proton ions have typically been carried out on murine models equivalent in age to the adolescent mice in our studies (2, 8). Kajioka et al. (8) compared the effects of proton irradiation to those of photon irradiation using whole-body exposures at 3 Gy (0.4 Gy/min) protons at the SOBP in adolescent-equivalent mice. Although they observed increased levels of TGFβ1 in the plasma of animals exposed to photon radiation, they detected no changes in mice receiving proton irradiation. Correspondingly, the tumors from adolescent mice in our study showed no change in TGFβ1 expression levels between the irradiated and the nonirradiated adolescent group, while we find a clear down-regulation of TGFβ1 and TGFβ2 in tumors growing in aged hosts as a consequence of proton irradiation.
Through upstream regulator analysis done with IPA, we identified several factors that are likely to contribute to the tumor dynamics observed for the irradiated old mice compared to nonirradiated mice (Table 3, Fig. 4C and D). A known tumor suppressor, transcription regulator MYOD1 (35, 36) was predicted to be activated in tumors growing in the irradiated vs. nonirradiated old mice. We also highlight several factors that are predicted to be inhibited including: an angiogenic factor, AGT or angiotensin II (37); an enzyme, MGEA5, found to aid potentiation of polyploidy through defective cytokinesis (38); and immunoglobulin G (IgG), which has been shown to promote tumor growth and survival of cancer cells by regulating cell migration and tumor MMP9 activity (39). Interestingly, these factors were also shown to be linked to TGFβ1 signaling (Fig. 4C). Overall, proton irradiation was here seen to decrease involvement of TGFβ1 and TGFβ2 in tumors growing in older hosts, which likely is a central contributor to the decrease in tumor progression observed.
In conclusion, further insight into the complexity of molecular mechanisms governing biological response to proton irradiation, at various energies and LET values and in different tissues, will not only prove advantageous in cancer therapy, but will provide an improved basis for cancer risk assessment for space travel. Syngeneic tumors growing in adolescent hosts demonstrated no modification in tumor dynamics and little change in intratumor molecular signaling after whole-body fractionated doses of proton radiation. However, tumors in old hosts, under the same proton irradiation scenario, exhibited significantly suppressed tumor progression, seemingly due in large part to changes driven by TGFβ1 and TGFβ2 (Fig. 4D). When considering the carcinogenesis risk of long-term space missions, astronaut age is an important in factor. In previous studies investigating 56Fe whole-body irradiation, using these same tumor models, we demonstrated a radiation-induced reduction in tumor progression for both young and middle-aged hosts, with a greater impact evident in the young hosts (7). In contrast, in these studies, proton irradiation induced a reduction in tumor progression only in the aged hosts. For cancer therapy it is clear that patient age is a contributing factor in the determination of response and an important consideration in making inroads to more individualized medicine. Preclinical in vivo studies on proton therapy are usually performed using young animals (2, 4). Since proton therapy is utilized for tumor treatment of children (due to the superior targeting advantages over gamma radiation) as well as middle-age to older patients (40, 41), it is important to investigate the efficacy of such therapy, along with the radiobiology of protons in general, as a function of host age, gender and genetics. We have demonstrated proton-induced tumor signaling to be modulated with age and found suppression of TGFβ to play a key role in inhibiting tumor growth in proton-irradiated old mice. Future studies unraveling the biological responses of proton irradiation as a function of host age are needed to further optimize the efficacy of proton cancer therapy and improve cancer risk assessments for space travel.
Acknowledgments
We thank Adam Rusek, Michael Sivertz, and the staff at Brookhaven National Laboratories and NASA Space Radiation Laboratories for their assistance with irradiations. We thank Janusz Weremowicz for assistance with animals, Caitlin Coelho for technical assistance, and Swati Girdhani and J. Tyson McDonald for reading and editing of the manuscript. This material is based upon work supported by the National Aeronautics and Space Administration under NSCOR grants NNJ06HA28G and NNX11AK26G issued through the Human Research Program and by award number U54CA149233 from the National Cancer Institute to LH.
References
- 1.Timmermann B. Proton beam therapy for childhood malignancies: status report. Klinische Padiatrie. 2010;222:127–33. doi: 10.1055/s-0030-1249657. [DOI] [PubMed] [Google Scholar]
- 2.Girdhani S, Sachs R, Hlatky L. Biological effects of proton radiation: what we know and don’t know. Radiat Res. 2013;179:257–72. doi: 10.1667/RR2839.1. [DOI] [PubMed] [Google Scholar]
- 3.Girdhani S, Lamont C, Hahnfeldt P, Abdollahi A, Hlatky L. Proton irradiation suppresses angiogenic genes and impairs cell invasion and tumor growth. Radiat Res. 2012;178:33–45. doi: 10.1667/rr2724.1. [DOI] [PubMed] [Google Scholar]
- 4.Jang GH, Ha JH, Huh TL, Lee YM. Effect of proton beam on blood vessel formation in early developing zebrafish (Danio rerio) embryos. Arch Pharm Res. 2008;31:779–85. doi: 10.1007/s12272-001-1226-1. [DOI] [PubMed] [Google Scholar]
- 5.Grabham P, Hu B, Sharma P, Geard C. Effects of ionizing radiation on three-dimensional human vessel models: differential effects according to radiation quality and cellular development. Radiat Res. 2011;175:21–8. doi: 10.1667/RR2289.1. [DOI] [PubMed] [Google Scholar]
- 6.Ries LAG, Melbert D, Krapcho M, Stinchcomb DG, Howlader N, Horner MJ, et al. SEER Cancer Statistics Review 1975–2005. 1. Bethesda, MD: National Cancer Institute; 2008. http://seer.cancer.gov/csr/1975_2005/, based on November 2007 SEER data submission. [Google Scholar]
- 7.Beheshti A, Sachs RK, Peluso M, Rietman E, Hahnfeldt P, Hlatky L. Age and space irradiation modulate tumor progression: implications for carcinogenesis risk. Radiat Res. 2013;179:208–20. doi: 10.1667/RR3100.1. [DOI] [PubMed] [Google Scholar]
- 8.Kajioka EH, Andres ML, Mao XW, Moyers MF, Nelson GA, Gridley DS. Hematological and TGF-beta variations after whole-body proton irradiation. In Vivo. 2000;14:703–8. [PubMed] [Google Scholar]
- 9.Nguyen DH, Martinez-Ruiz H, Barcellos-Hoff MH. Consequences of epithelial or stromal TGFbeta1 depletion in the mammary gland. J Mammary Gland Biol Neoplasia. 2011;16:147–55. doi: 10.1007/s10911-011-9218-0. [DOI] [PubMed] [Google Scholar]
- 10.Perera M, Tsang CS, Distel RJ, Lacy JN, Ohno-Machado L, Ricchiuti V, et al. TGF-beta1 interactome: metastasis and beyond. Cancer Genomics Proteomics. 2010;7:217–29. [PubMed] [Google Scholar]
- 11.Hau P, Jachimczak P, Schlaier J, Bogdahn U. TGF-beta2 signaling in high-grade gliomas. Curr Pharm Biotechnol. 2011;12:2150–7. doi: 10.2174/138920111798808347. [DOI] [PubMed] [Google Scholar]
- 12.Bertram JS, Janik P. Establishment of a cloned line of Lewis Lung Carcinoma cells adapted to cell culture. Cancer Lett. 1980;11:63–73. doi: 10.1016/0304-3835(80)90130-5. [DOI] [PubMed] [Google Scholar]
- 13.Carnes BA, Grahn D, Hoel D. Mortality of atomic bomb survivors predicted from laboratory animals. Radiat Res. 2003;160:159–67. doi: 10.1667/rr3029. [DOI] [PubMed] [Google Scholar]
- 14.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 15.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–93. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 16.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Rafn B, Nielsen CF, Andersen SH, Szyniarowski P, Corcelle-Termeau E, Valo E, et al. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol Cell. 2012;45:764–76. doi: 10.1016/j.molcel.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 18.Durante M, Cucinotta FA. Heavy ion carcinogenesis and human space exploration. Nat Rev Cancer. 2008;8:465–72. doi: 10.1038/nrc2391. [DOI] [PubMed] [Google Scholar]
- 19.Wang M, Hada M, Saha J, Sridharan DM, Pluth JM, Cucinotta FA. Protons sensitize epithelial cells to mesenchymal transition. PLoS One. 2012;7:e41249. doi: 10.1371/journal.pone.0041249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahluwalia A, Narula J, Jones MK, Deng X, Tarnawski AS. Impaired angiogenesis in aging myocardial microvascular endothelial cells is associated with reduced importin alpha and decreased nuclear transport of HIF1 alpha: mechanistic implications. J Physiol Pharmacol. 2010;61:133–39. [PubMed] [Google Scholar]
- 21.Bojovic B, Crowe DL. Chronologic aging decreases tumor angiogenesis and metastasis in a mouse model of head and neck cancer. Int J Oncol. 2010;36:715–23. doi: 10.3892/ijo_00000547. [DOI] [PubMed] [Google Scholar]
- 22.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan W, Gao L, Wu X, Hade EM, Gao JX, Ding H, et al. Expression of a mutant p53 results in an age-related demographic shift in spontaneous lung tumor formation in transgenic mice. PLoS One. 2009;4:e5563. doi: 10.1371/journal.pone.0005563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed MJ, Karres N, Eyman D, Cruz A, Brekken RA, Plymate S. The effects of aging on tumor growth and angiogenesis are tumor-cell dependent. Int J Cancer. 2007;120:753–60. doi: 10.1002/ijc.22351. [DOI] [PubMed] [Google Scholar]
- 25.Li S, Lu X, Chi P, Pan J. Identification of Nkx2-3 and TGFB1I1 expression levels as potential biomarkers to predict the effects of FOLFOX4 chemotherapy. Cancer Biol Ther. 2012;13:443–49. doi: 10.4161/cbt.19298. [DOI] [PubMed] [Google Scholar]
- 26.Zaravinos A, Volanis D, Lambrou GI, Delakas D, Spandidos DA. Role of the angiogenic components, VEGFA, FGF2, OPN and RHOC, in urothelial cell carcinoma of the urinary bladder. Oncol Rep. 2012;28:1159–66. doi: 10.3892/or.2012.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tesar BM, Walker WE, Unternaehrer J, Joshi NS, Chandele A, Haynes L, et al. Murine [corrected] myeloid dendritic cell-dependent toll-like receptor immunity is preserved with aging. Aging Cell. 2006;5:473–86. doi: 10.1111/j.1474-9726.2006.00245.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, et al. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J Immunol. 2011;186:1963–9. doi: 10.4049/jimmunol.1002320. [DOI] [PubMed] [Google Scholar]
- 29.Koga-Yamakawa E, Dovedi SJ, Murata M, Matsui H, Leishman AJ, Bell J, et al. Intratracheal and oral administration of SM-276001: a selective TLR7 agonist, leads to antitumor efficacy in primary and metastatic models of cancer. Int J Cancer. 2013;132:580–90. doi: 10.1002/ijc.27691. [DOI] [PubMed] [Google Scholar]
- 30.Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol. 2010;11:1030–8. doi: 10.1038/ni.1947. [DOI] [PubMed] [Google Scholar]
- 31.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen DH, Oketch-Rabah HA, Illa-Bochaca I, Geyer FC, Reis-Filho JS, Mao JH, et al. Radiation acts on the microenvironment to affect breast carcinogenesis by distinct mechanisms that decrease cancer latency and affect tumor type. Cancer Cell. 2011;19:640–51. doi: 10.1016/j.ccr.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012;72:4119–29. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–65. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, et al. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008;3:e2656. doi: 10.1371/journal.pone.0002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lirk P, Berger R, Hollmann MW, Fiegl H. Lidocaine time- and dose-dependently demethylates deoxyribonucleic acid in breast cancer cell lines in vitro. Br J Anaesth. 2012;109:200–7. doi: 10.1093/bja/aes128. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues-Ferreira S, Abdelkarim M, Dillenburg-Pilla P, Luissint AC, di-Tommaso A, Deshayes F, et al. Angiotensin II facilitates breast cancer cell migration and metastasis. PLoS One. 2012;7:e35667. doi: 10.1371/journal.pone.0035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krzeslak A, Forma E, Bernaciak M, Romanowicz H, Brys M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin Exp Med. 2012;12:61–5. doi: 10.1007/s10238-011-0138-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelegrina LT, Lombardi MG, Fiszman GL, Azar ME, Morgado CC, Sales ME. Immunoglobulin g from breast cancer patients regulates MCF-7 cells migration and MMP-9 activity by stimulating muscarinic acetylcholine receptors. J Clin Immunol. 2013;33:427–35. doi: 10.1007/s10875-012-9804-y. [DOI] [PubMed] [Google Scholar]
- 40.Green LM, Tran DT, Murray DK, Rightnar SS, Todd S, Nelson GA. Response of thyroid follicular cells to gamma irradiation compared to proton irradiation: II. The role of connexin 32. Radiat Res. 2002;158:475–85. doi: 10.1667/0033-7587(2002)158[0475:rotfct]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 41.Di Pietro C, Piro S, Tabbi G, Ragusa M, Di Pietro V, Zimmitti V, et al. Cellular and molecular effects of protons: apoptosis induction and potential implications for cancer therapy. Apoptosis. 2006;11:57–66. doi: 10.1007/s10495-005-3346-1. [DOI] [PubMed] [Google Scholar]
- 42.Zhang K, Ye C, Zhou Q, Zheng R, Lv X, Chen Y, et al. PKD1 inhibits cancer cells migration and invasion via Wnt signaling pathway in vitro. Cell Biochem Funct. 2007;25:767–74. doi: 10.1002/cbf.1417. [DOI] [PubMed] [Google Scholar]
- 43.Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287:9873–86. doi: 10.1074/jbc.M111.312694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutherland M, Gill JH, Loadman PM, Laye JP, Sheldrake HM, Illingworth NA, et al. Antitumor activity of a duocarmycin analogue rationalized to be metabolically activated by cytochrome P450 1A1 in human transitional cell carcinoma of the bladder. Mol Cancer Ther. 2013;12:27–37. doi: 10.1158/1535-7163.MCT-12-0405. [DOI] [PubMed] [Google Scholar]
- 45.Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811–15. doi: 10.1038/ng.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lange F, Rateitschak K, Fitzner B, Pohland R, Wolkenhauer O, Jaster R. Studies on mechanisms of interferon-gamma action in pancreatic cancer using a data-driven and model-based approach. Mol Cancer. 2011;10:13. doi: 10.1186/1476-4598-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oblak A, Jerala R. Toll-like receptor 4 activation in cancer progression and therapy. Clin Dev Immunol. 2011;2011:609579. doi: 10.1155/2011/609579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cellurale C, Girnius N, Jiang F, Cavanagh-Kyros J, Lu S, Garlick DS, et al. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2011;72:472–81. doi: 10.1158/0008-5472.CAN-11-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.To MD, Rosario RD, Westcott PM, Banta KL, Balmain A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene. 2012;32:4028–33. doi: 10.1038/onc.2012.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dentelli P, Rosso A, Olgasi C, Camussi G, Brizzi MF. IL-3 is a novel target to interfere with tumor vasculature. Oncogene. 2011;30:4930–40. doi: 10.1038/onc.2011.204. [DOI] [PubMed] [Google Scholar]