

Abstract
Excessive activation of the N-Methyl-D-Aspartate (NMDA) receptor and the neurotransmitter dopamine (DA) mediate neurotoxicity and neurodegeneration under many neurological conditions, including Huntington's disease (HD), an autosomal dominant neurodegenerative disease characterized by the preferential loss of medium spiny projection neurons (MSNs) in the striatum. PSD-95 is a major scaffolding protein in the postsynaptic density (PSD) of dendritic spines, where a classical role for PSD-95 is to stabilize glutamate receptors at sites of synaptic transmission. Our recent studies indicate that PSD-95 also interacts with the D1 DA receptor localized in spines and negatively regulates spine D1 signaling. Moreover, PSD-95 forms ternary protein complexes with D1 and NMDA receptors, and plays a role in limiting the reciprocal potentiation between both receptors from being escalated. These studies suggest a neuroprotective role for PSD-95. Here we show that mice lacking PSD-95, resulting from genetic deletion of the GK domain of PSD-95 (PSD-95-ΔGK mice), sporadically develop progressive neurological impairments characterized by hypolocomotion, limb clasping, and loss of DARPP-32-positive MSNs. Electrophysiological experiments indicated that NMDA receptors in mutant MSNs were overactive, suggested by larger, NMDA receptor-mediated miniature excitatory postsynaptic currents (EPSCs) and higher ratios of NMDA- to AMPA-mediated corticostriatal synaptic transmission. In addition, NMDA receptor currents in mutant cortical neurons were more sensitive to potentiation by the D1 receptor agonist SKF81297. Finally, repeated administration of the psychostimulant cocaine at a dose regimen not producing overt toxicity-related phenotypes in normal mice reliably converted asymptomatic mutant mice to clasping symptomatic mice. These results support the hypothesis that deletion of PSD-95 in mutant mice produces concomitant overactivation of both D1 and NMDA receptors that makes neurons more susceptible to NMDA excitotoxicity, causing neuronal damage and neurological impairments. Understanding PSD-95-dependent neuroprotective mechanisms may help elucidate processes underlying neurodegeneration in HD and other neurological disorders.
Introduction
Preferential loss of striatal medium spiny neurons (MSNs) is a hallmark of Huntington's disease (HD), an inherited autosomal dominant neurological disorder characterized by cognitive impairment, psychiatric disturbances, and motor disability irreversibly progressing to death 10-20 years after the onset of symptoms. Although the genetic basis (i.e. the gene encoding huntingtin or Htt) and pathological features (polyglutamine (polyQ) expansion in the N-terminal of Htt) have been at least partially identified (HDCRG, 1993), intensive studies have yet to pinpoint the precise molecular and cellular mechanisms by which MSNs die in HD (Levine et al., 2004; Cha, 2007; Milnerwood and Raymond, 2010). In particular, the ubiquitous expression of Htt does not explain the relatively selective nature of MSN loss, and studies on genetically engineered mice suggest that polyQ extension on Htt is neither necessary nor sufficient for MSN degeneration. It has become evident that while polyQ expansion on Htt may trigger HD, the preferential vulnerability of MSNs may underlie their selective and progressive demise. Thus understanding the mechanisms that regulate MSN vulnerability is fundamentally important.
Several lines of evidence support roles for dopamine (DA)- and N-Methyl-D-Aspartate (NMDA)-mediated toxicity in MSN degeneration. In the striatum, DA and glutamate axon terminals converge on the same dendritic spines on postsynaptic MSNs, forming “synaptic triads” (Freund et al., 1984; Goldman-Rakic et al., 1989; Carr and Sesack, 1996; Yao et al., 2008). Both D1, the predominant D1-class DA receptor, and the NMDA receptor (NMDAR) are concentrated in spine heads and the postsynaptic density (PSD) of MSNs, where most corticostriatal glutamatergic synapses are formed (Hersch et al., 1995). The striatum receives the densest DA innervation of the brain, and HD progresses according to a dorsoventral gradient corresponding to the gradient of DA concentration, suggesting that DA signaling participates in the preferential and progressive vulnerability of MSNs in HD. Indeed, DA can regulate striatal neuron viability via receptor-independent mechanisms involving oxidative stress-induced apoptosis as well as receptor-dependent mechanisms (Bozzi and Borrelli, 2006). Sustained elevation of extracellular DA causes selective degeneration of MSNs (Cyr et al., 2003). Furthermore, the elevated DA tone can also enhance the deleterious effects of polyQ- expanded Htt on striatal function in a mouse model of HD, accompanied by accelerated formation of mutant Htt aggregates in striatal projection neurons (Cyr et al., 2006).
The involvement of NMDAR-mediated excitotoxicity in MSN degeneration has also been well documented (Choi, 1988; Levine et al., 2004; Fan and Raymond, 2007). First, NMDARs are disproportionately lost in the putamen of human HD patients, even in the presymptomatic stage of the disease (Young et al., 1988; Albin et al., 1990). Second, injection of the NMDAR agonist quinolinic acid (QA) into monkey striatum results in behavioral, neurochemical, and neuropathological abnormalities similar to changes seen in HD patients (Hantraye et al., 1990). Third, intrastriatal injection of QA in rodents selectively destroys MSNs but spares interneurons known to be resistant to degeneration in HD (Beal et al., 1986). Finally, transgenic mouse models harboring mutant Htt show increased sensitivity to NMDAR excitotoxicity, mediated primarily by the NR2B-containing receptors (Levine et al., 1999; Zeron et al., 2002; Fan and Raymond, 2007). Thus, mechanisms regulating NMDARs (expression, subunit composition, synaptic targeting and modulation by DA) may dictate MSN vulnerability.
PSD-95 is a prominent member of the MAGUK (membrane-associated guanylate kinases) family synaptic scaffolds in excitatory synapses (Kim and Sheng, 2004). PSD-95 interacts with NMDA NR2 subunits through its first two PDZ domains, which may play a role in functionally localizing these receptors in the synapse (Kim and Sheng, 2004). We and others have shown that PSD-95 also interacts with DA D1 receptor via the C terminus of the receptor and the N terminus of PSD-95 (Zhang et al., 2007; Kruusmagi et al., 2009; Sun et al., 2009; Ha et al. 2012), an interaction that regulates D1 trafficking (Zhang et al., 2007). Consequently, PSD-95, D1, and NMDARs may form a multiprotein complex. Within the complex, PSD-95 may inhibit the physical association between D1 and NMDARs and functionally uncouple D1 receptor trafficking and signaling from modulation by NMDARs (Zhang et al., 2009). These data suggest that PSD-95 plays an important role in survival of neurons, especially MSNs that distinguish themselves from other neuronal populations by their rich DA innervation, via regulating localization, trafficking, and stabilization of both NMDA and DA receptors in the synapse, and by balancing the interplay between DA and glutamate systems in spines and synapses.
We previously described a unique transgenic mouse strain in which a targeted deletion of the GK (Guanylate Kinase-like) domain of PSD-95 completely abolishes the production of the full-length PSD-95 (Yao et al., 2004). Mice lacking PSD-95 display impaired learning (Migaud et al., 1998), enhanced locomotor sensitivity to psychostimulants and DA agonists (Yao et al., 2004; Zhang et al., 2007), and altered behavioral responses to hallucinogen, antipsychotics, and alcohol (Abbas et al., 2009; Camp et al., 2011). Over the years working with our mouse colony, we have observed sporadic and progressive neurological impairments in a subset of mutant mice, including motor defects, limb clasping, and striatal neurodegeneration. In this study, we have characterized these phenotypes at the behavioral, immunohistochemical, and electrophysiological levels. Our results suggest that enhanced DA D1 receptor-mediated signaling and excessive DA-glutamate interaction in the absence of PSD-95 contribute to striatal NMDA receptor hyperfunction, MSN cell death, and the neurological phenotypes observed in mutant mice.
Results
Motor and Neurological Impairments in PSD-95-ΔGK Mice
The PSD-95-ΔGK mice were engineered to carry a stop codon inserted in frame into exon 13 of the PSD-95 gene, intended to remove the GK domain of the protein. Unexpectedly, homozygous mice for this mutation do not produce detectable levels of PSD-95, thus representing a null strain of PSD-95 (Yao et al., 2004). The reason for the complete loss of PSD-95 in mutant mice remains unknown, but is likely due to the premature stop codon and non-sense mediated mRNA decay. The PSD-95-ΔGK homozygote females are sterile. Heterozygous mice were intercrossed and the genetic features of the mice were determined. From 37 litters, 268 mice were genotyped at p11: 89 (33%) were wild type (WT), 145 (54%) were heterozygotes, and 34 (13%) were homozygotes, demonstrating a distortion from the expected Mendelian ratio.
In locomotor activity tests, PSD-95-ΔGK homozygous mice were substantially less active, consistently traveling shorter distances when placed in a novel environment compared with their WT littermates (Figure 1a). The total horizontal and vertical activity was significantly reduced in homozygous mutant mice (Figure 1b). In follow-up locomotor tests in the same chambers, PSD-95-ΔGK homozygotes displayed consistently lower locomotor activity on subsequent days (Figure 1c). Interestingly, heterozygous mice displayed intermediate locomotor activity between WT and homozygous mutant mice, suggesting a potential gene dosage effect. These results support and extend the hypoactivity phenotype previously reported for these mice (Yao et al., 2004; Zhang et al., 2007).
Figure 1.
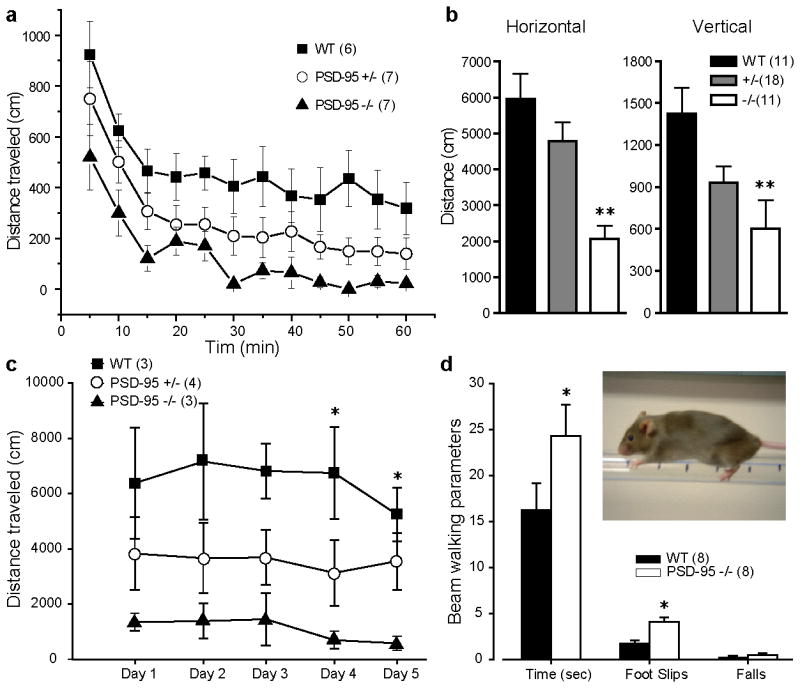
Reduced locomotor activity and impaired motor coordination in PSD-95 deficient mice. (a) Time courses of horizontal activity per 5 min of WT, PSD-95+/-, and PSD-95-/- mice when placed in a novel environment, an open field activity chamber. (b) Summary of total horizontal and vertical activity of WT, PSD-95+/- and PSD-95-/- mice in response to a novel environment. **, p < 0.01, one-way ANOVA with post-hoc Dunnett's test vs. WT. (c) Total horizontal activity of WT, PSD-95+/-, and PSD-95-/-mice during a 5-day test period. Mice were placed in the same locomotor activity monitor on test days for 60 min. Horizontal and vertical activity was measured as distance traveled in blocks of 5 min. *, p < 0.05, one-way ANOVA with post-hoc Dunnett's test vs. corresponding WT controls. (d) Summary of beam walking data. Inset, a WT mouse performing the beam walking test. *, p < 0.05 versus WT, Student's t test. Numbers in parentheses designate numbers of mice used.
To investigate other aspects of motor dysfunction in PSD-95-ΔGK mice, we performed a beam walking assay (Stanley et al., 2005; Allbutt and Henderson, 2007) that measures motor coordination. During beam walking tests, mice walk on an elevated beam and the time a mouse travels on the beam, the number of foot slips, and the number of falls are measured. PSD-95-ΔGK mice took significantly longer time traveling from one end of an elevated beam to the other end and had significantly more foot slips while on the beam (Figure 1d), suggesting motor coordination impairments associated with these mice.
In addition to the hypoactivity and motor coordination deficits, we also observed other neurological phenotypes associated with the mutant mice (Figure 2). These phenotypes include occasional signs of seizure, increased alertness and stress during handling (such as the removal of a cage lid or removal/return of cagemates), and dyskinesia of the limbs (Figure 2a). When suspended by their tail, a subpopulation of mutant mice retracted their limbs toward their trunks in a dystonic fashion, rather than extending them as observed in WT (Figure 2a). The severity of the phenotype varied from short clasping between hindlimbs or forelimbs and between hind and forelimbs to full body clasping that maintained for the duration of the test (Figure 2a). In a controlled study using a cohort of 132 mice, we observed that the majority of homozygous mutants (>70%), as well as a small population of heterozygotes (∼23%), displayed the limb clasping phenotype to various degrees (Figure 2b). The limb clasping phenotype started as early as 4 weeks and was progressive and more severe in older mice (Figure 2c).
Figure 2.
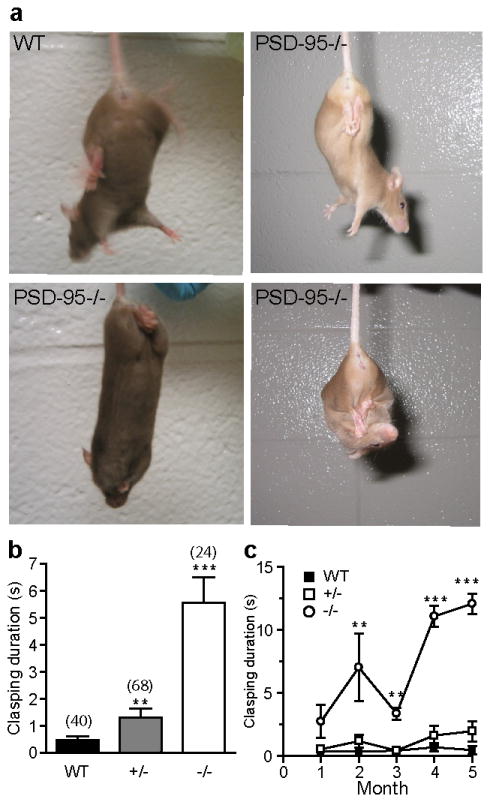
Progressive limb-clasping symptoms in PSD-95 deficient mice. (a) Examples of hind-limb, all-limb and full body clasping in PSD-95-/- mice compared to a WT control. (b) Clasping duration in a 15-s interval. **, p < 0.01; ***, p≪0.001, Student's t-tests. A clasping event is defined by the retraction of limbs into the body and toward the midline. (c) Developmental progression of clasping phenotype. **, p < 0.01; ***, p≪0.001, one-way ANOVA followed by post hoc Newman–Keuls tests. Mice were observed during a 15 s tail suspension test to monitor the progression of the clasping phenotype between 4 and 20 weeks of age. Numbers in parentheses designate numbers of mice used.
Striatal Neurodegeneration in Aged PSD-95-ΔGK Mice
Hindlimb clasping is a manifestation of motor dysfunction often linked to striatal degeneration (Mangiarini et al., 1996; Reddy et al., 1998; Yamamoto et al., 2000; Mantamadiotis et al., 2002; Cyr et al., 2003). To determine whether neurodegenration occurs in the striatum of PSD-95-ΔGK mice, we carried out immunohistochemical and quantitative stereological analysis of the striatal integrity in young (3-month) and aged (>12-month-old) symptomatic PSD-95-ΔGK mice and age-matched WT littermates (Figure 3). We focused on MSNs, the GABAergic projection neurons in the striatum that account for 90-95% of total striatal cell populations. MSNs are particularly prone to damage by excitotoxicity, DA toxicity, or oxidative stress, and are preferentially targeted for degeneration in HD. We identified MSNs by immunohistochemical staining with an antibody against DARPP-32 (Dopamine- and cAMP-regulated phosphoprotein, Mr 32 kDa; Figure 3b,c), a molecular marker of MSNs. DARPP-32-positive cells from serial striatal sections were analyzed by stereology to estimate the total MSN number, striatal volume, and MSN density. There was a significant loss of total DARPP-32-positive cells and cell density in the striatum of aged PSD-95 deficient mice compared to WT controls (Figure 3d,e). Striatal volume was also significantly reduced in aged mutant mice (Figure 3f). In comparison, cell count and cell density were modestly reduced in young PSD-95-ΔGK mice, but the reduction did not reach significance levels (Figure 3d,e). The striatal volume was similar between young mutants and their WT controls (Figure 3f). Some PSD-95-ΔGK mice also displayed an enlarged ventricle (Figure 3b), a hallmark of striatal neurodegeneration (Dragatsis et al., 2000; Yamamoto et al., 2000). Finally, direct comparison of WT striatum at different ages also revealed an age-dependent loss of MSNs, as DARPP-32-positive cells were significantly fewer in >1-yr-old mice compared to 3-month-old mice. Together, these results suggest that a portion of striatal MSNs is lost as mice age and the absence of PSD-95 may speed up the degeneration process.
Figure 3.
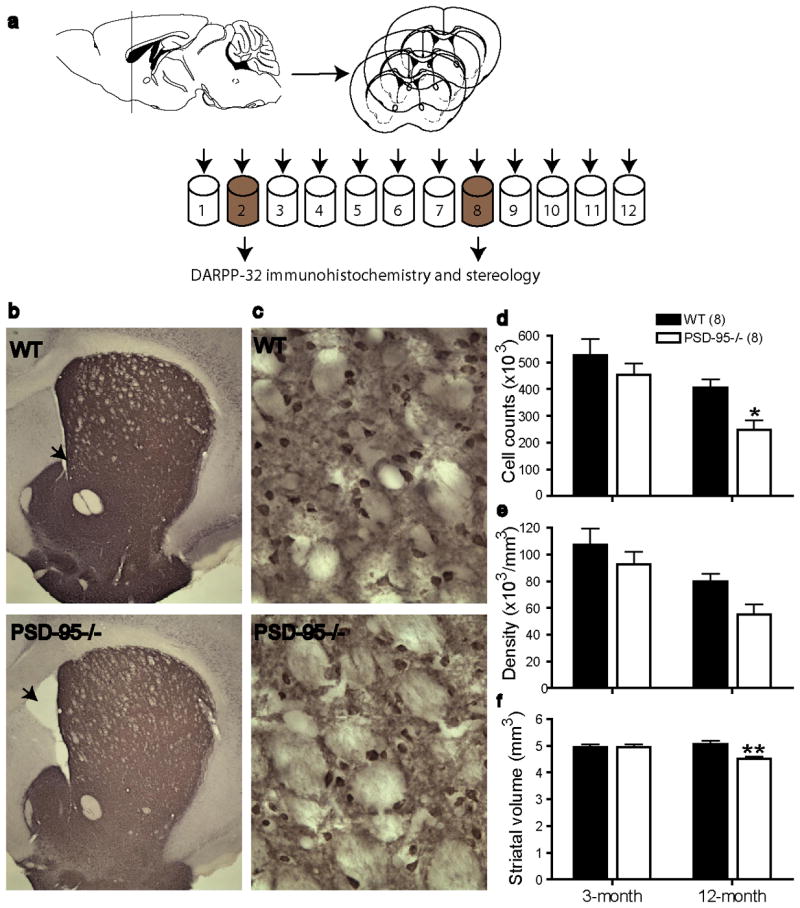
Loss of striatal MSNs in aged symptomatic PSD-95 deficient mice. (a) Sectioning scheme for immunohistochemistry and stereological analysis. Serial coronal sections (240 μm apart) from Tubes #2 and #8 were used in DARPP-32 staining and subsequent cell counting. (b, c) DARPP-32 staining of a WT and a PSD-95-/- mouse striatal section, viewed at 2.5× (b) and 40× (c). Arrows indicate ventricles. (d-f) Summary of total DARPP-32-positive cells (d), cell density (e), and striatal volume (f) in 3-month- and >12-month-old WT and PSD-95-/- symptomatic mice. Cell counting was made at 40×, using Stereo Investigator 7. n = 8 mice each group, *, p < 0.05; **, p < 0.01 vs. WT, two-way ANOVA with Bonferroni post-hoc tests.
Increased Striatal NMDAR Activity in PSD-95-ΔGK Mice
Synaptic dysfunction and pathophysiology often precede neurodegeneration and the onset of symptoms and are considered to be a mechanism that renders neurons and synapses more prone to damage. In particular, abnormal enhancement of NMDAR activity is thought to mediate the increased sensitivity of MSNs to excitotoxicity in mouse models of HD (Milnerwood and Raymond, 2010). Thus, we characterized striatal NMDARs in young (p30-p50) PSD-95-ΔGK mice; mutant mice at these ages are generally pre-symptomatic (Figure 2) which allows us to determine whether NMDAR function is already disturbed that may contribute to the neurological phenotypes observed in the symptomatic mice at older ages. We first measured the ratio (NMDA/AMPA ratio) of excitatory postsynaptic currents (EPSCs) mediated by NMDA and AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors. We employed a kinetics-based method, which allowed AMPAR- and NMDAR-mediated components to be distinguished based on their different activation and inactivation kinetics (Figure 4). The NMDA/AMPA ratio was significantly increased in the MSNs of PSD-95-ΔGK mice compared to age-matched WT mice. To directly monitor AMPAR-mediated corticostriatal transmission in PSD-95-ΔGK mice, we recorded miniature EPSCs (mEPSCs) at -60 mV in the presence of extracellular Mg2+ (Figure 5). No difference was found in the amplitude of AMPA-mEPSCs between WT and mutant mice (Figure 5a, b), suggesting that postsynaptic AMPARs are normal in PSD-95-ΔGK mice. Thus, the increased NMDA/AMPA ratio in the mutant synapse was likely due to an enhancement in NMDAR number and/or function. The frequency of AMPA-mEPSCs was significantly reduced in mutant mice (Figure 5c), suggesting a decrease in the probability of neurotransmitter release. This presynaptic impairment is consistent with the recognized role of PSD-95 in presynaptic terminal development and maturation (El-Husseini et al., 2000; Futai et al., 2007).
Figure 4.
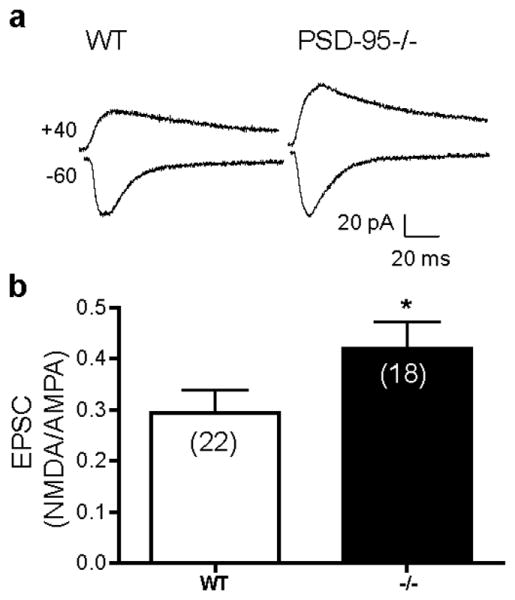
Increased striatal NMDA/AMPA ratio in PSD-95-ΔGK mice. (a) Representative EPSCs recorded from WT and PSD-95-/- MSNs at holding potentials of -60 (to record AMPA-EPSCs) and +40 mV (to record both AMPA-EPSCs and NMDAR-EPSCs). (b) Summary of mean NMDA/AMPA ratios, defined as the amplitude of the NMDAR component 80 ms after stimulation at -40 mV divided by the peak AMPAR component at -60 mV. Numbers in parentheses indicate numbers of cells analyzed. *, p < 0.05 vs. WT; Student's t tests.
Figure 5.
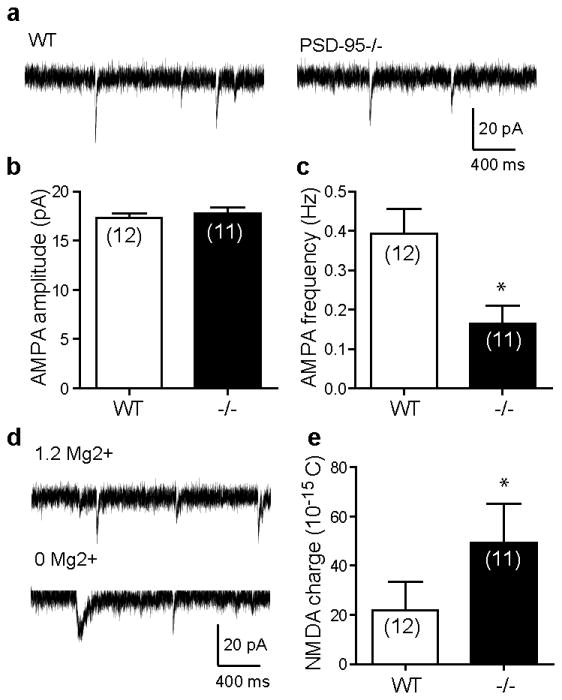
Increased striatal NMDAR activity in PSD-95-ΔGK mice. (a) Representative AMPAR-mediated mEPSCs recorded from WT and mutant MSNs. (b,c) Summaries of mean AMPA-mEPSC amplitudes (b) and frequencies (c). (d) Representative mEPSCs in the absence (to record total mEPSCs mediated by both NMDA and AMPA receptors) followed by the presence (to record AMPA-mEPSCs) of Mg2+. (e) Summary of charge transfer mediated through NMDA-mEPSCs. The NMDAR component of mEPSCs was derived by subtracting the average AMPA-mEPSC from the average total mEPSC, and the area under the resultant NMDA-mEPSC was measured. Numbers in parentheses indicate numbers of cells analyzed. *, p < 0.05 vs. WT, Student's t tests.
To directly test the hypothesis that postsynaptic NMDAR number and/or function was enhanced in PSD-95-ΔGK mice, we analyzed NMDAR-mediated mEPSCs (NMDA-mEPSCs) at -60 mV. Following the recording of AMPA-mEPSCs, extracellular Mg2+ was removed to record the total mEPSCs mediated by both AMPARs and NMDARs (Figure 5d). NMDA-mEPSC was then calculated by subtracting the average AMPA-mEPSC from the average total mEPSC and measured as the charge transfer. This analysis showed a significantly increased NMDA-mEPSC in PSD-95-ΔGK mice (Figure 5e). Together, striatal NMDAR activity is enhanced in PSD-95-ΔGK mice.
Enhanced Modulation of NMDAR Currents by D1 Activation in PSD-95-ΔGK Cortical Neurons
We hypothesized that the enhanced NMDAR activity in PSD-95-ΔGK mice was due, at least in part, to increased D1 signaling and D1-dependent modulation in these mice (Zhang et al., 2007; Zhang et al., 2009). We tested this hypothesis by measuring the potentiation of NMDAR-EPSCs in prefrontal cortex neurons by SKF81297, a D1/D5-selective agonist (Figure 6). We used cortical pyramidal neurons in this experiment because D1 and D2 DA receptors are segregated into different populations of neurons in the striatum (Gerfen, 1992), and without live fluorescence labeling it was not feasible to identify D1-expressing MSNs. NMDA-EPSCs were recorded from layer V pyramidal cells at -60 mV in the presence of the AMPAR antagonist CNQX (6-cyano-7-nitroquinoxaline-2,3-dione; 20 μM) and in the absence of Mg2+. In WT mice, a brief bath application of SKF81297 at a low dose (1 μM) significantly potentiated NMDA-EPSCs (152.2 ± 2.7% vs. baseline (101.3 ± 1.0%), measured at 50 min, p < 0.05). The delayed and long-lasting potentiation (often irreversible after agonist washout) is consistent with the literature and is mediated by modulation of postsynaptic D1 receptors (Seamans et al., 2001). This lasting potentiating response was significantly increased in PSD-95-ΔGK mice (WT, 152.2 ± 2.7%; ΔGK, 181.3 ± 6.4%; measured at 50 min; p < 0.05), suggesting that NMDARs in the mutant mice are more sensitive to D1 activation.
Figure 6.
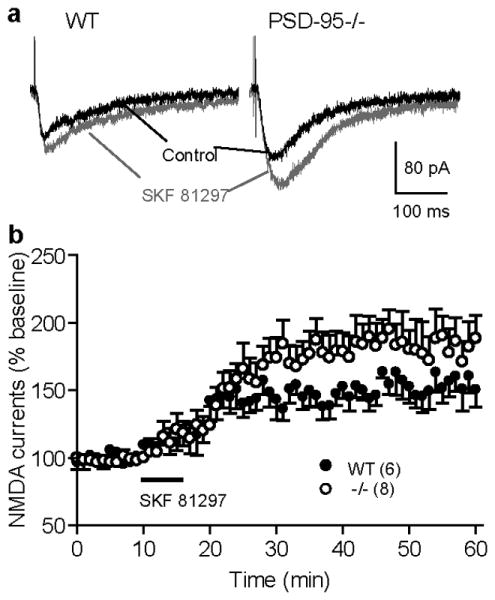
Enhanced SKF81297 modulation of synaptic NMDAR currents in PSD-95-ΔGK mice. (a) Representative NMDA-EPSCs recorded from WT and mutant PFC pyramidal neurons before and (30-40 min) after bath application of SKF81297 (1 μM). (b) Summary of SKF81297 effect on NMDA-EPSCs in WT and PSD-95-/- mice. NMDA-EPSCs were pharmacologically isolated in Mg2+-free ACSF containing CNQX (20 μM), glycine (1 μM), and picrotoxin (100 μM). Neurons were voltage clamped at −60 mV. Following a 10-min baseline recording, a brief (5-10 min) application of SKF81297 elicited a long-lasting potentiation of NMDA-EPSCs in WT neurons, which was further enhanced in PSD-95-ΔGK neurons. Numbers in parentheses indicate numbers of cells analyzed.
Repeated Cocaine Treatments Exacerbate Limb Clasping in PSD-95-ΔGK Mice
The results presented above are consistent with elevated D1 signaling and stronger D1-NMDA receptor interaction in PSD-95-ΔGK mice, as we have shown previously (Yao et al., 2004; Zhang et al., 2007; Zhang et al., 2009). To test if this enhanced D1 signaling may contribute to development of the dyskinesia phenotype in PSD-95-ΔGK mice, we examined whether repeated treatment of asymptomatic mutant mice with cocaine, a psychostimulant and indirect DA agonist, could induce clasping behavior (Figure 7). We found that repeated cocaine administrations (20mg/kg/day for 7 days, i.p.) reliably converted asymptomatic mutant mice to symptomatic clasping mice, whereas the same dose regimen did not produce overt dyskinesia phenotypes in normal mice (Figure 7a). The limb-clasping behavior in mutants emerged rapidly (within 3-7 injections) and persisted for over 2 weeks after the last injection (Figure 7b). Interestingly, the cocaine-induced dyskinesia phenotype gradually disappeared and mice returned to their asymptomatic state after 4-6 weeks. These results support the hypothesis that increased D1 signaling in mutant mice may contribute to the dyskinesia behaviors in these mice. However, it is unlikely that MSN degeneration is involved in the development of this cocaine-elicited dyskinesia phenotype; rather, alterations of function and/or modulation of the basal ganglion circuit may play a more direct role.
Figure 7.
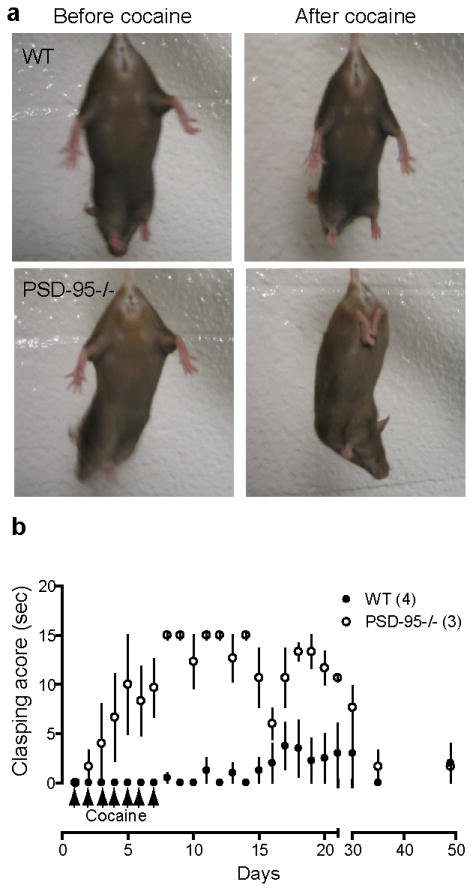
Exacerbating effect of cocaine exposure on limb-clasping behavior. (a) Repeated cocaine administration (7 days, 20 mg/kg, i.p.) induced limb clasping in asymptomatic PSD-95-/- mice, but not in WT mice. The same WT and mutant mice before and after cocaine treatments are shown. (b) Development and recovery time course of cocaine-elicited limb clasping behavior. Clasping score designates clasping duration in a 15-s interval. Numbers in parentheses indicate numbers of mice analyzed. Age = 7 months.
Discussion
Here we report that mice lacking the postsynaptic scaffold PSD-95 develop dyskinesia phenotypes including reduced locomotor activity, impaired motor coordination, and limb clasping. Quantitative stereological analysis reveals significant loss of DARPP-32-positive MSNs in the striatum as well as smaller striatal volume in aged, but not young, mutant mice. Activity of striatal NMDARs is enhanced significantly in young mutant mice. Cortical NMDARs are substantially more sensitive to D1 stimulation, suggesting that the NMDAR hyperfunction could be due, at least in part, to an increased D1 signaling in mutant mice. Repeated stimulation of DA signaling by cocaine injections reliably converted asymptomatic KO mice to clasping symptomatic mice. Combined with our previous studies (Yao et al., 2004; Zhang et al., 2007; Zhang et al., 2009), these results support a working model (Figure 8) where PSD-95 limits D1 activity in dendritic spines and acts as a molecular brake that negatively regulates D1-NMDAR cross-talk. Absence of PSD-95 may thus lead to concomitant overactivation of both D1 and NMDA receptors and make neurons more susceptible to excitotoxicity, damaging synaptic or neuronal function and even causing cell death. Our study identifies PSD-95 as a neuroprotector in the striatum, and provides insights into the molecular mechanisms underlying striatal neurodegeneration in HD, and has broader implications for understanding the neurotoxicity involved in neurological conditions associated with DA and glutamate dysfunction.
Figure 8.
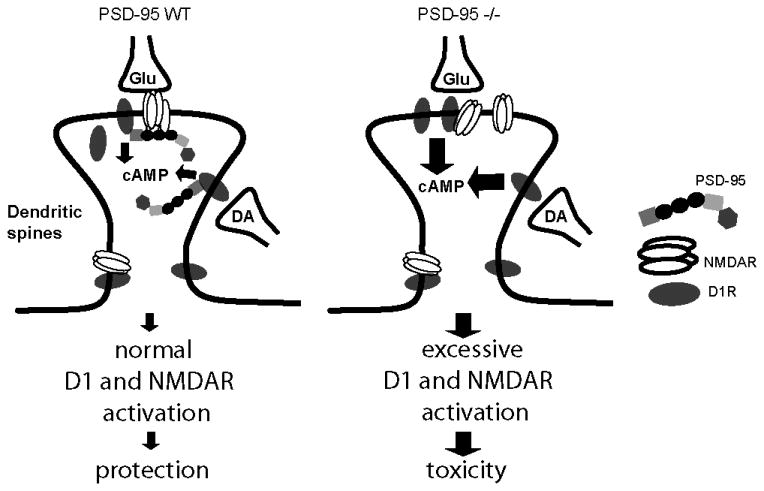
Proposed model by which PSD-95 may regulate neuronal integrity by regulating NMDAR function, DA D1 signaling, and DA-glutamate interaction at synapses. In dendritic spines PSD-95 regulates D1 trafficking and inhibits D1 signaling, and dampens the positive coupling between D1 and NMDAR. Deletion of PSD-95 in mutant mice produces enhanced D1 signaling, measured as increased cAMP production, which in turn further enhances NMDAR activity. The concomitant overactivation of both D1 and NMDAR make the neurons more susceptible to NMDAR-mediated excitotoxicity, causing cell death and the neurological phenotypes observed in mutant mice.
The dyskinetic phenotypes associated with PSD-95-ΔGK mice are consistent with a central role of PSD-95 as a structural and signaling scaffold in excitatory synapses in the mammalian brain (Kim and Sheng, 2004; Gardoni, 2008). PSD-95 is believed to organize ionotropic glutamate receptors and their associated signaling proteins as well as DA receptors in the PSD, thus regulating the strength, plasticity, and dopaminergic modulation of corticostriatal circuitry (Kim and Sheng, 2004; Yao et al., 2008). Dysregulation of PSD-95-NMDAR-D1 receptor complexes is linked to experimental Parkinson's disease (PD) and L-DOPA–induced dyskinesia (LID), a debilitating motor complication of DA replacement therapy for PD (Nash et al., 2005; Fiorentini et al., 2006; Gardoni et al., 2006; Gardoni, 2008; Porras et al., 2012). Interestingly, striatal PSD-95 level is markedly elevated in rat and macaque models of LID (Nash et al., 2005; Porras et al., 2012) and reducing PSD-95 levels or disrupting PSD-95-D1 interaction have been reported to alleviate LID (Porras et al., 2012). Therefore, either too much or too little PSD-95 in the striatum can cause motor function impairments. Future studies are needed to elucidate how PSD-95-NMDAR-D1 receptor complexes are differentially altered, leading to motor deficits associated with different DA disease conditions.
Our results reveal a 39% loss of MSNs and 11 % reduction of striatal volume in aged, but not young PSD-95-ΔGK mice. Because motor deficits and dyskinesia occurred well before MSN loss (Figure 2) and 3-month old symptomatic mutants did not have a significant loss of MSNs (Figure 3), it is unlikely that the behavioral phenotypes are directly correlated with striatal degeneration, although cell loss at late stages should contribute to the behavioral phenotypes observed in mutant mice. In addition, cocaine-induced dyskinesia in PSD-95 deficient mice mostly likely resulted from disturbance of the basal ganglia synapses and circuit function in mutant mice, rather than a rapid loss of striatal MSNs following cocaine exposure, because the cocaine-induced phenotype was reversible over time (Figure 7). It remains to be determined whether cell loss is limited to striatal GABAergic MSNs, as well as the underlying degeneration process, i.e. apoptosis, necrosis, or neurotoxicity.
The mechanisms underlying the preferential degeneration of striatal MSNs continue to be elusive, but may involve excessive DA signaling and NMDA excitotoxicity. Our study provides support for the idea that concomitant over-activation of both D1 and NMDARs, resulting from a positive feedback loop between these receptors, can cause neurotoxicity and cell death (Cepeda and Levine, 2006; Zhang et al., 2009). Our results also directly support that PSD-95 is a neuroprotection molecule that dampens D1 signaling and D1-NMDA receptor interaction and can prevent runaway potentiation of NMDARs and excitotoxicity during sustained activation of DA signaling. PSD-95-mediated regulation of D1 and D1-NMDAR interaction offers a novel neuroprotective mechanism, because except for the classical D2 receptor-dependent neuroprotection against glutamate excitotoxicity in cortical and hippocampal neurons (Bozzi and Borrelli, 2006; Kihara et al., 2002), no other DA receptor-dependent neuroprotective mechanisms have been described.
However, PSD-95 may regulate neuronal integrity through other mechanisms. First, when coexpressed in oocytes with NR1/NR2B, PSD-95 decreases glutamate sensitivity of the receptor channels and inhibits the PKC-mediated potentiation of the channels (Yamada et al., 1999). Thus PSD-95 may protect certain cells, e.g. MSNs and hippocampal neurons where NR2B is abundantly expressed, against NMDAR-mediated neurotoxicity. Consistently, antisense knockdown of PSD-95 has been shown to drive neuronal death in cultured hippocampal neurons (Gardoni et al., 2002). Second, PSD-95 may regulate the proper localization of NMDAR subunits, particularly different NR2 subunits, in the synapse (Kim and Sheng, 2004). Synaptic and extrasynaptic receptors differ in their subunit compositions (Rumbaugh and Vicini, 1999), are associated with different intracellular signaling pathways, and play different roles in neuronal survival (Riccio and Ginty, 2002). Particularly, activation of synaptic NR2A-containing NMDARs is neuroprotective whereas stimulation of extrasynaptic NR2B-containing NMDARs is neurotoxic (Hardingham and Bading, 2002). However, it should be noted that NMDAR-mediated currents are generally unaffected in hippocampal neurons derived from PSD-95-deficient mice (Migaud et al., 1998; Beique et al., 2006; Elias et al., 2006). Finally, in contrast to the neuroprotective roles, PSD-95 may also confer neurotoxic effects to neurons. PSD-95 interacts with both NMDARs and neuronal nitric oxide synthase (nNOS), coupling NMDAR activity to the production of nitric oxide (NO), a molecule that triggers NMDAR-dependent excitotoxicity (Brenman et al., 1996; Sattler et al., 1999). Transient disruption of NMDAR-PSD-95 interactions in cortical neurons has been shown to be neuroprotective against ischemic brain damage (Aarts et al., 2002). This mechanism may be less dominant in MSNs, as nNOS expression is low in these neurons (Zucker et al., 2005). We suggest that the net effects of PSD-95 in neuronal integrity may depend on the competition between neuroprotective and neurotoxic mechanisms, likely in a cell type-dependent manner.
Our study identifies PSD-95 as a risk factor associated with the pathogenesis of HD. Because of its ubiquitous expression, the disease-causing protein Htt alone cannot explain the relatively selective nature of MSN loss. It is possible that PolyQ-expanded Htt may couple to MSN-specific susceptibility mechanisms to confer the preferential vulnerability of MSNs. Consistent with this idea, in vitro studies suggest that normal Htt binds PSD-95 at the SH3 domain and PolyQ expansion interferes with the ability of Htt to interact with PSD-95 (Sun et al., 2001). This interaction may link many of the cellular functions proposed for Htt (e.g. transcriptional regulation, anti-apoptosis, and dendritic morphogenesis) to the PSD-95-NMDAR and other signaling complexes in the PSD. Striatal PSD-95 is also decreased in animal models of HD (Jarabek et al., 2004), although the functional significance of this decrease is unclear. In addition to HD and PD, PSD-95 and other MAGUKs are also implicated in Alzheimer's disease, ischemia, schizophrenia, and neuropathic pain (Gardoni, 2008). The underlying mechanisms are not fully understood but likely involve abnormal interactions between these synaptic scaffolds with glutamate and DA receptors, associated signaling complexes, and the pathogenic proteins that alter synapse and neural circuit function. A better understanding of MAGUK regulation of synaptic organization, function, signaling, and plasticity under normal and pathological conditions could lead to the identification of new targets for pharmaceutical intervention of these neurological and psychiatric disorders.
Methods
Mice
Wild-type (WT) and PSD-95-ΔGK mice (Yao et al., 2004) were housed under standard laboratory conditions (12 h light/dark cycle) with food and water provided ad libitum. All experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and with an approved IACUC protocol from the Harvard Medical Area Standing Committee on Animals.
Behavioral Assessments
Locomotion activity was measured in an automated ENV510 activity monitor (Med Associates Inc.) (Zhang et al., 2007). Mice were placed in the monitor and their horizontal and vertical activities were recorded at 5-min intervals for up to 60 min. Horizontal and vertical activities were measured as the distance traveled.
For the beam walking assay, mice were first trained to walk on a 60-cm long, 3-cm diameter plastic rod elevated 26 cm above a bench. A ruler in the middle of the rod was used to measure foot slips. A soft cushion was placed under the beam to provide a protective surface for falling mice. Mice were subjected to the beam test following three days of training. Mice were placed on the beam at one end with inked feet and allowed to walk to the other end of the rod. Mice that fell were returned to the position where they fell, with a maximum time of 60 s allowed on the beam. The time spent on the beam, the number of foot slips (foot slipped down lower than the ruler), and the number of falls were measured. Mice were returned to their home cages after the test.
For limb clasping quantification, mice were observed during a 15 s tail suspension test. A clasping event is defined by the retraction of limbs into the body and toward the midline. The clasping duration was measured and 15 s was set as a cut-off.
Perfusion and Brain Section Preparation
Mice were anesthetized and perfused transcardially with ice-cold PBS, followed by ice-cold 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer, pH 7.4. Brains were post fixed overnight in 4% PFA and cryoprotected with 10% (wt/vol) then 30% sucrose. Brains were sectioned (40 μm) on a freezing microtome, collected in antifreeze solution containing 30% glycerol, 30% ethoxyethanol, 40% 0.1M PBS, and stored at -20°C until the time of use. For each brain, serial sections from bregma 1.5 to -0.95 mm, encompassing nearly the entire striatum, were sequentially collected into 12 tubes, labeled No. 1 through 12. Thus sections in each tube were 480 μm apart along the rostrocaudal axis of the striatum.
Immunohistochemistry
Free-floating brain sections from Tubes Nos. 2 and 8 of each mouse were rinsed in PBS and preincubated in 4% normal donkey serum (Jackson ImmunoResearch) for 60 min. Endogenous peroxidase was quenched with 3% H2O2 for 7-15 min. Sections were incubated with a primary antibody against rabbit DARPP-32 (Chemicon), and followed by secondary antibody diluted in PBS containing 2% normal donkey serum and 0.1% Triton X-100. Bound antibody was visualized using the ABC system (Vectastain ABC Kit, Vector Laboratories). Sections were mounted, air dried, alcohol dehydrated, rinsed with Xylene, and coverslipped.
Stereological Cell Counting
The total number of DARPP-32-positive cells in the striatum was estimated using Stereo Investigator software (MBF Bioscience) and according to stereologic principles (West and Gundersen, 1990). The anatomical boundaries of the striatum were determined according to fiduciary neuroanatomical landmarks and the use of a mouse brain atlas (Paxinos and Franklin, 2001). Design-based stereology was performed using a Zeiss Axiovert microscope (Zeiss) coupled to an Optronics Microfire digital camera (Goleta) for visualization of tissue sections. The total number of immunoreactive cells, from tissue sections separated by 240 μm, was estimated from coded slides using the optical fractionator method. For each tissue section analyzed, section thickness was assessed empirically and guard zones of ∼2 μm thickness were used at the top and bottom of each section. The striatum was outlined under low magnification (2.5×) and the outlined region was analyzed by a systematic random sampling design. Cells were counted under 40× magnification. The coefficient of error was calculated according to the procedure of Gundersen and colleagues, values < 0.10 were considered acceptable (West and Gundersen, 1990; West, 1993). The volume of the region was estimated according to Cavalieri's principle.
Brain Slice Preparation
Mice were sacrificed by decapitation and brains were rapidly removed. Cortical slices (300 μm) containing the striatum or medial prefrontal cortex were cut using a vibratome. Slices were perfused with an ice-cold artificial CSF (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 25 D-glucose, saturated with 95% O2 and 5% CO2. Slices were incubated in ACSF for > 1 hr at room temperature before transferring to a recording chamber.
Electrophysiology
Whole-cell voltage-clamp experiments were performed on individual dorsal medium spiny neurons in the striatum or layer V pyramidal neurons of the prefrontal cortex using an Axoclamp 2B amplifier. Cells were visualized with an upright microscope under infrared illumination and identified by their morphologies. In some cases, the identities of MSNs and pyramidal neurons were confirmed by the high resting membrane potential (-80 to -90 mV) and adaptive firing patterns in response to constant current injections, respectively, under current clamp. Recording pipettes (4.5 - 5.5MΩ) were filled with (in mM): 142 Cs-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 2.5 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25 (with CsOH). Neurons were voltage clamped at -60 or +40 mV unless indicated otherwise. Picrotoxin (50 or 100 μM) was included in the bath to block GABAA receptor-mediated synaptic responses. Tetrodotoxin (1 μM) was added during recordings of mEPSCs. For evoked EPSCs, stimuli were delivered with a concentric bipolar electrode (FHC) placed at layer II/III in the PFC or over the corpus callosum 200 μm dorsal to the recording site in the striatum. Series resistance was monitored throughout whole-cell recordings, and data were discarded if the resistance changed by 15%. All recordings were made at 32°C with a temperature controller (Warner Instruments). Drugs were delivered to the bath with a gravity-driven perfusion system (Harvard Apparatus). Data acquisition and analysis were performed using a pClamp 9.2 software suite (Molecular Devices). mEPSCs were analyzed by Mini Analysis 6 (Synaptosoft).
Statistical Analysis
All data are expressed as mean ± s.e.m. Statistical tests are noted in figure legends (significance was set a priori at 0.05).
Acknowledgments
We thank members of the laboratory for comments and discussions and Ms. Donna Reed for editorial assistance. We thank Drs. Oliver Cooper and Angel Vinuela for helping with striatal stereological experiments. This study was supported by National Institutes of Health grants DA021420 and NS057311 (W.-D.Y.), a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (W.-D.Y.), the William F. Milton Fund of Harvard University (W.-D.Y.), and National Center for Research Resources grant RR000168 (currently OD011103 from the Office of Research Infrastructure Programs, Office of the Director).
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Abbas AI, Yadav PN, Yao WD, Arbuckle MI, Grant SG, Caron MG, Roth BL. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J Neurosci. 2009;29:7124–7136. doi: 10.1523/JNEUROSCI.1090-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;322:1293–1298. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- Allbutt HN, Henderson JM. Use of the narrow beam test in the rat, 6-hydroxydopamine model of Parkinson's disease. J Neurosci Methods. 2007;159:195–202. doi: 10.1016/j.jneumeth.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Camp MC, Feyder M, Ihne J, Palachick B, Hurd B, Karlsson RM, Noronha B, Chen YC, Coba MP, Grant SG, Holmes A. A novel role for PSD-95 in mediating ethanol intoxication, drinking and place preference. Addict Biol. 2011;16:428–439. doi: 10.1111/j.1369-1600.2010.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;2006:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- Cha JH. Transcriptional signatures in Huntington's disease. Prog Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Cyr M, Beaulieu JM, Laakso A, Sotnikova TD, Yao WD, Bohn LM, Gainetdinov RR, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci U S A. 2003;100:11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Sotnikova TD, Gainetdinov RR, Caron MG. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 2006;20:2541–2543. doi: 10.1096/fj.06-6533fje. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Rizzetti MC, Busi C, Bontempi S, Collo G, Spano P, Missale C. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol Pharmacol. 2006;69:805–812. doi: 10.1124/mol.105.016667. [DOI] [PubMed] [Google Scholar]
- Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F. MAGUK proteins: new targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol. 2008;585:147–152. doi: 10.1016/j.ejphar.2008.01.048. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Viviani B, Marinovich M, Meli E, Pellegrini-Giampietro DE, Cattabeni F, Di Luca M. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. Eur J Neurosci. 2002;16:777–786. doi: 10.1046/j.1460-9568.2002.02141.x. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J Neurosci. 2006;26:2914–2922. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:9015–9019. doi: 10.1073/pnas.86.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CM, Park D, Han JK, Jang JI, Park JY, Hwang EM, Seok H, Chang S. Calcyon forms a novel ternary complex with dopamine D1 receptor through PSD-95 protein and plays a role in dopamine receptor internalization. J Biol Chem. 2012;287:31813–31822. doi: 10.1074/jbc.M112.370601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantraye P, Riche D, Maziere M, Isacson O. A primate model of Huntington's disease: behavioral and anatomical studies of unilateral excitotoxic lesions of the caudate-putamen in the baboon. Exp Neurol. 1990;108:91–104. doi: 10.1016/0014-4886(90)90014-j. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta. 2002;1600:148–153. doi: 10.1016/s1570-9639(02)00455-7. [DOI] [PubMed] [Google Scholar]
- HDCRG (The Huntington's Disease Collaborative Research Group) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarabek BR, Yasuda RP, Wolfe BB. Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington's mouse model. Brain. 2004;127:505–516. doi: 10.1093/brain/awh058. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Kanki R, Yamashita H, Akaike A. Protective effect of dopamine D2 agonists in cortical neurons via the phosphatidylinositol 3 kinase cascade. J Neurosci Res. 2002;70:274–282. doi: 10.1002/jnr.10426. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kruusmagi M, Kumar S, Zelenin S, Brismar H, Aperia A, Scott L. Functional differences between D(1) and D(5) revealed by high resolution imaging on live neurons. Neuroscience. 2009;164:463–469. doi: 10.1016/j.neuroscience.2009.08.052. [DOI] [PubMed] [Google Scholar]
- Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Genetic mouse models of Huntington's and Parkinson's diseases: illuminating but imperfect. Trends Neurosci. 2004;27:691–697. doi: 10.1016/j.tins.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nash JE, Johnston TH, Collingridge GL, Garner CC, Brotchie JM. Subcellular redistribution of the synapse-associated proteins PSD-95 and SAP97 in animal models of Parkinson's disease and L-DOPA-induced dyskinesia. FASEB J. 2005;19:583–585. doi: 10.1096/fj.04-1854fje. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 4. San Diego: Academic; 2001. [Google Scholar]
- Porras G, Berthet A, Dehay B, Li Q, Ladepeche L, Normand E, Dovero S, Martinez A, Doudnikoff E, Martin-Negrier ML, Chuan Q, Bloch B, Choquet D, Boue-Grabot E, Groc L, Bezard E. PSD-95 expression controls L-DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest. 2012;122:3977–3989. doi: 10.1172/JCI59426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ginty DD. What a privilege to reside at the synapse: NMDA receptor signaling to CREB. Nat Neurosci. 2002;5:389–390. doi: 10.1038/nn0502-389. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S. Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci. 1999;19:10603–10610. doi: 10.1523/JNEUROSCI.19-24-10603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley JL, Lincoln RJ, Brown TA, McDonald LM, Dawson GR, Reynolds DS. The mouse beam walking assay offers improved sensitivity over the mouse rotarod in determining motor coordination deficits induced by benzodiazepines. J Psychopharmacol. 2005;19:221–227. doi: 10.1177/0269881105051524. [DOI] [PubMed] [Google Scholar]
- Sun P, Wang J, Gu W, Cheng W, Jin GZ, Friedman E, Zheng J, Zhen X. PSD-95 regulates D1 dopamine receptor resensitization, but not receptor-mediated Gs-protein activation. Cell Res. 2009;19:612–624. doi: 10.1038/cr.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276:24713–24718. doi: 10.1074/jbc.M103501200. [DOI] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Chochi Y, Ko JA, Sobue K, Inui M. Activation of channel activity of the NMDA receptor-PSD-95 complex by guanylate kinase-associated protein (GKAP) FEBS Lett. 1999;458:295–298. doi: 10.1016/s0014-5793(99)01171-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yao WD, Spealman RD, Zhang J. Dopaminergic signaling in dendritic spines. Biochem Pharmacol. 2008;75:2055–2069. doi: 10.1016/j.bcp.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- Young AB, Greenamyre JT, Hollingsworth Z, Albin R, D'Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with Huntington's disease. Science. 1988;241:981–983. doi: 10.1126/science.2841762. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu TX, Hallett PJ, Watanabe M, Grant SG, Isacson O, Yao WD. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J Neurosci. 2009;29:2948–2960. doi: 10.1523/JNEUROSCI.4424-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Vinuela A, Neely MH, Hallett PJ, Grant SG, Miller GM, Isacson O, Caron MG, Yao WD. Inhibition of the dopamine D1 receptor signaling by PSD-95. J Biol Chem. 2007;282:15778–15789. doi: 10.1074/jbc.M611485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker B, Luthi-Carter R, Kama JA, Dunah AW, Stern EA, Fox JH, Standaert DG, Young AB, Augood SJ. Transcriptional dysregulation in striatal projection- and interneurons in a mouse model of Huntington's disease: neuronal selectivity and potential neuroprotective role of HAP1. Hum Mol Genet. 2005;14:179–189. doi: 10.1093/hmg/ddi014. [DOI] [PubMed] [Google Scholar]