

Abstract
CodY, a nutritional regulator highly conserved in low G+C Gram-positive bacteria, is essential in Streptococcus pneumoniae (the pneumococcus). A published codY mutant possessed suppressing mutations inactivating the fatC and amiC genes, respectively belonging to iron (Fat/Fec) and oligopeptide (Ami) ABC permease operons, which are directly repressed by CodY. Here we analyzed two additional published codY mutants to further explore the essentiality of CodY. We show that one, in which the regulator of glutamine/glutamate metabolism glnR had been inactivated by design, had only a suppressor in fecE (a gene in the fat/fec operon), while the other possessed both fecE and amiC mutations. Independent isolation of three different fat/fec suppressors thus establishes that reduction of iron import is crucial for survival without CodY. We refer to these as primary suppressors, while inactivation of ami, which is not essential for survival of codY mutants and acquired after initial fat/fec inactivation, can be regarded as a secondary suppressor. The availability of codY - ami + cells allowed us to establish that CodY activates competence for genetic transformation indirectly, presumably by repressing ami which is known to antagonize competence. The glnR codY fecE mutant was then found to be only partially viable on solid medium and hypersensitive to peptidoglycan (PG) targeting agents such as the antibiotic cefotaxime and the muramidase lysozyme. While analysis of PG and teichoic acid composition uncovered no alteration in the glnR codY fecE mutant compared to wildtype, electron microscopy revealed altered ultrastructure of the cell wall in the mutant, establishing that co-inactivation of GlnR and CodY regulators impacts pneumococcal cell wall physiology. In light of rising levels of resistance to PG-targeting antibiotics of natural pneumococcal isolates, GlnR and CodY constitute potential alternative therapeutic targets to combat this debilitating pathogen, as co-inactivation of these regulators renders pneumococci sensitive to iron and PG-targeting agents.
Introduction
The global nutritional regulator CodY is highly conserved in low G+C Gram-positive bacteria [1], and regulates up to 200 genes in Bacillus subtilis [2]. The B. subtilis CodY regulon concerns not only metabolic pathways, but also cellular processes such as sporulation, motility and competence for genetic transformation [1,3,4]. Most of these genes are directly repressed by CodY during exponential growth and induced upon nutrient starvation. In other species, CodY has also been shown to regulate a number of major virulence genes (for reviews, see references [1,3]) by directly binding DNA and repressing the target genes. CodY is activated by branched chain amino acids [5] but also by GTP in certain species, such as B. subtilis [6].
Transcriptome analysis of a codY mutant in the human pathogen Streptococcus pneumoniae showed that CodY mainly regulated amino acid metabolism, biosynthesis and uptake [7]. However, it was recently demonstrated that the codY mutant used in this study had accumulated suppressing mutations allowing tolerance of codY inactivation (collectively called socY for suppressor of c od Y), and that the codY gene could not be readily inactivated by insertion of an antibiotic cassette [8]. A first suppressing mutation was identified in the fatC gene by whole-genome sequencing of the codY mutant [8]. This gene belongs to the fatD–fatC–fecE–fatB operon; this operon (also called piuBCDA or pit1) [9], which is directly repressed by CodY [7], encodes the major ferric iron ABC permease of S. pneumoniae [10], with FatB also shown to bind heme [11]. While the fatC mutation was present in the entire codY - population, a second suppressing, variable mutation was found in the amiC gene [8], encoding a subunit of the Ami oligopeptide ABC permease [12]. It was concluded that the three different amiC mutations identified in the codY - population arose subsequently to fatC in an otherwise codY fatC mutant lineage, presumably providing a selective advantage over ami + cells. Based on these data, CodY was suggested to be an essential protein in S. pneumoniae primarily because repression of the fat/fec operon by CodY was required to avoid uncontrolled iron import resulting in toxicity [8].
Two further pneumococcal codY mutant strains have been published [13,14], including one in which glnR, encoding a regulator of glutamine/glutamate (Glu/Gln) metabolism, was also inactivated by design [13]. Because CodY was previously concluded to be an essential protein in S. pneumoniae [8], we analyzed these mutants to establish whether new suppressing mutations allowed tolerance of codY inactivation in these strains. Here we show that both strains contain mutations truncating the fecE gene, encoding another subunit of the Fat/Fec permease. The independent isolation of three different mutations in the fat/fec operon crucial for tolerance of codY inactivation demonstrates that codY essentiality results from the deregulation of iron import when CodY is absent and unable to repress fat/fec. Furthermore, we provide evidence that the published glnR codY mutant, while possessing no other suppressor than the fecE mutation, is only partially viable on solid medium. We establish that this reduced/poor viability is linked to the co-inactivation of codY and glnR. This mutant was also found to be hypersensitive to the peptidoglycan (PG) targeting antibiotic cefotaxime, as well as to the PG-targeting muramidase enzyme lysozyme, to which the pneumococcus is normally naturally resistant [15]. Although biochemical analysis revealed normal PG composition in the mutant, electron microscopy analysis showed mutant cells to have thicker, irregular and yet less dense cell wall leading us to conclude that co-inactivation of GlnR and CodY regulators impacts pneumococcal cell wall physiology.
Results
Genetic dissection of the glnR codY double mutant
We began our study by investigating whether inactivation of glnR could be responsible for suppressing the essentiality of codY in strain TK108, the published D39 glnR codY::trim double mutant [13]. We initially determined that a glnR::kan 22C cassette, conferring kanamycin resistance (KanR), transformed with normal efficiency into a wildtype recipient strain (Fig 1A). Throughout this study, transformation efficiencies of mutations were always compared to the same reference marker, the rpsL41 point mutation, conferring streptomycin resistance (SmR) [16] and carried on the same donor DNA as the mutation under investigation. An efficiency of 0.25 for the KanR cassette relative to SmR (Fig 1A) is in the range expected for transformation of a heterologous insert compared to a point mutation [17], showing that glnR could readily be inactivated without additional suppressing mutations.
Fig 1. Genetic dissection of codY mutants.
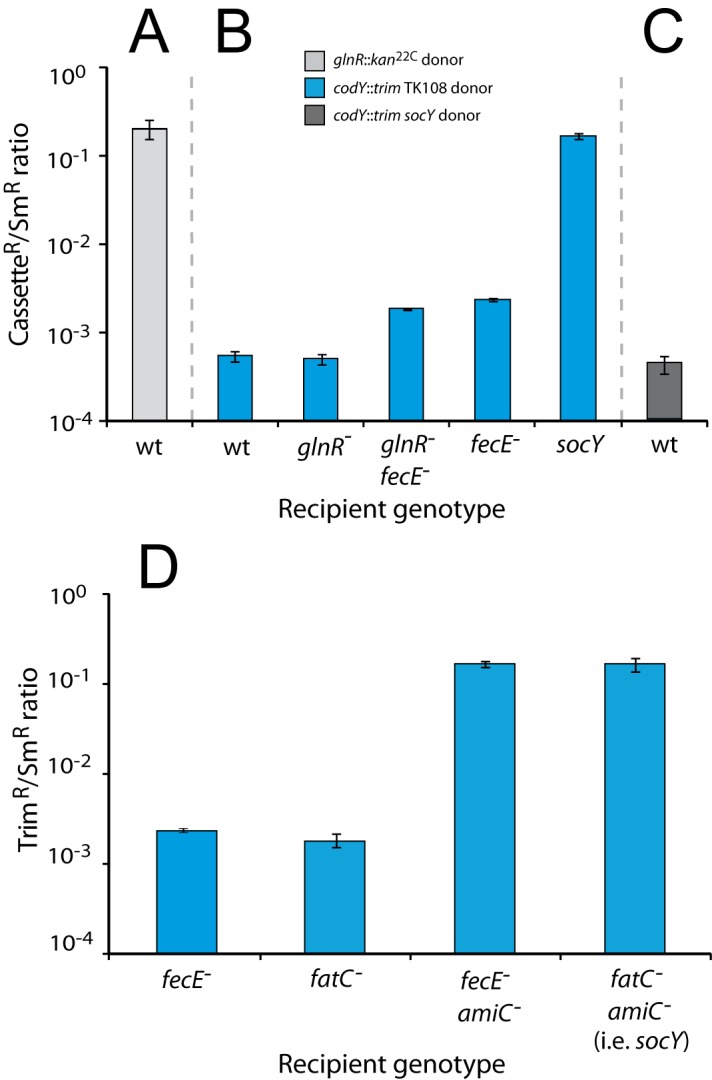
(A) Transformation efficiency of the glnR::kan 22C cassette into wildtype (wt) recipient. Transformation efficiencies calculated from triplicate repeats. Donor strain, R3154; wt recipient strain, TD198. (B) Transformation efficiency of the codY::trim cassette present in the glnR codY (fecE) mutant into wt and mutant recipient strains. Transformation efficiencies calculated as in panel A. Donor strain, TD196 (i.e. TK108 but rpsL41). Recipient strains, wt, TD198; glnR -, TD195; glnR - fecE -, TD227; fecE -, TD230; socY, TD142. (C) Transformation efficiency of the codY::trim cassette from the suppressed codY::trim, socY strain into a wt recipient. Transformation efficiencies calculated as in panel A. Donor strain, TD80; wt recipient as in panel A. (D) Inactivation of fatC and fecE has the same suppressive effect on codY inactivation. Transformation efficiency of the codY::trim cassette present in the TK108 mutant into wt and mutant recipient strains. Transformation efficiencies calculated as in panel A. Donor strain, TD196. Recipient strains, fecE -, TD230; fatC -, TD141; fecE - amiC -, TD228; socY, TD142.
To establish whether the inactivation of glnR could suppress codY essentiality, we transformed a glnR mutant with genomic DNA from TD196 (i.e. TK108 but rpsL41; S1 Table). The transformation efficiency of codY::trim (conferring trimethoprim resistance, TrimR) was over 350-fold lower than normal efficiency and was comparable to that observed in a wildtype recipient (Fig 1B). This result established that mutation of glnR does not allow cells to tolerate codY inactivation.
Interestingly, the wildtype recipient was transformed with similar efficiency using as donor TD196 or the previously characterized codY::trim socY strain (Fig 1C). Since in the latter case survival of codY - transformants required the simultaneous co-transfer of two independent suppressors (the amiC and fatC mutations to be referred to as socY hereafter) [8], explaining the very low frequency of TrimR clones recovered, these results suggested that yet uncharacterized suppressing mutations could also be present in the glnR codY mutant to allow tolerance of codY inactivation.
A fecE mutation suppresses inviability of codY mutants
Sequence analysis of targeted PCRs showed that the glnR codY mutant possessed wildtype fatC and amiC genes, suggesting the presence of alternative suppressor(s) than the previously detailed socY mutations. Whole-genome sequencing of TK108 was then used (Materials and Methods), identifying only 6 single nucleotide polymorphisms (SNPs) compared to D39 [18], and no mutation in the entire ami operon. Localized PCR and sequencing of TK102, the glnR - parent of TK108, showed that 5 of the 6 SNPs were already present prior to codY::trim transformation and thus unlikely to be involved in suppression of codY essentiality. Interestingly, the only remaining mutation was a C→T mutation introducing a stop codon in the fecE gene (referred to as fecE - hereafter), resulting in the truncation of the FecE protein after 13 of 250 amino acids. This gene is part of the fat/fec iron permease operon, as is fatC. It seemed therefore likely that the fecE mutation was responsible for tolerance of codY inactivation.
To determine whether fecE - suppressed codY essentiality, the point mutation was introduced into the glnR - parent without selection (Materials and Methods) to create a glnR fecE mutant (TD227). The codY::trim cassette was transformed into both fecE - and glnR - fecE - recipient strains, and results showed that this cassette transferred with ∼10-fold higher efficiency into both strains than into the glnR - parent or the wild type (Fig 1B). However, this efficiency remained lower than into the fully suppressed socY recipient strain, showing that fecE - only partially suppressed codY::trim lethality. To confirm the equivalence of fecE and fatC mutations as suppressors of codY - inviability, the fecE - mutation was inserted into a strain mutant for amiC, thus recreating a socY-like combination, and the transformation of codY::trim repeated. Unlike both fatC and fecE single mutants, the fecE amiC and fatC amiC double mutants readily accepted the codY::trim cassette, showing that both mutations of the fat/fec operon are equally suppressive of codY::trim lethality, resulting in full suppression in conjunction with amiC mutation (Fig 1D).
Interestingly, a third pneumococcal codY mutant was recently published [14]. These authors showed that this codY mutant harbored a mutation in the amiC gene [14] but possessed a wildtype fatC gene (Sven Hammerschmidt, personal communication). However, we discovered by sequencing that a deletion of base 409 (G) resulted in truncation of the fecE gene in this codY mutant. Thus, all three independently constructed codY mutants of S. pneumoniae possess an inactive fat/fec encoded iron permease, providing strong evidence that this inactivation is necessary to allow pneumococci to tolerate codY inactivation. Since it is impossible to create a pneumococcal strain with only glnR and codY inactivated, we will hereafter refer to the mutant under study as glnR - codY - (fecE -) to highlight the presence of an accompanying necessary suppressing mutation in fecE.
The glnR codY (fecE) mutant displays a severe colony-forming defect
Comparison of growth of glnR - codY - (fecE -), codY - socY and wildtype strains in liquid media (C+Y, CAT, TSB and THY) showed that codY mutants grew slower than wildtype cells in all instances (Fig 2A and S1 Fig). However, the codY socY mutant generally grew even slower and/or displayed a longer lag than its glnR codY (fecE) counterpart (particularly in C+Y medium, Fig 2A). This supported our conclusion that glnR - codY - (fecE -) cells were ami + as inferred from whole-genome sequencing data, since ami - cells have been repeatedly observed over years in our laboratory to grow significantly slower than wildtype cells.
Fig 2. The glnR codY fecE triple mutant is only partially viable on solid medium.

(A) Growth curves of strains in C+Y medium. After initial growth to identical cellular densities (OD 0.2) in C+Y medium, cells were diluted 1/100 in 300 μL final volume of C+Y medium in microtitre plate, and OD492 readings taken every 10 min for 600 min. Strain identities, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75. (B) Time-lapse microscopy of wildtype and codY mutant cells growing in CAT-agarose. Strain identities as in panel A. (C) Loss of viability is maintained in newly generated glnR fecE codY triple mutants. Viable counts performed on newly created codY::trim transformants with varying parental genotypes. Colony-forming units per ml (cfu mL-1) counted after growth to OD550 0.2 in C+Y and plating on CAT agar + 5% horse blood. From left to right, columns 1 and 2 represent viable (TD195) and non-viable (TK108) controls; columns 3–5 represent parent strains (TD212 black, TD227 dark grey and TD230 light grey respectively); three TrimR transformant clones (appropriately coloured panels numbered 1–3) from each parent transformed by codY::trim PCR fragment. Average cfu mL-1 calculated from triplicate repeats.
We then analyzed glnR codY (fecE) mutant cells by fixing them on microscope slides using polylysin (Materials and Methods), and exploring their morphology (S1 Text and S2 and S3 Figs). Mutant cells appeared to form short chains, with an average number of cells per chain of 4.6 compared to 1.7 for the wild type (S2G Fig), where one chain should give rise to one cfu after plating. Notably, we observed a ~3-fold reduction in individual cell numbers (counted irrespective of whether these were part of chains or not) compared to wild type for aliquots from cultures of the same initial optical density, suggesting that a significant proportion (>66%) of the glnR codY (fecE) mutant cells lysed in these conditions (S2F Fig). To account for such a deficit in cell numbers, which is unlikely to come from a loss of viability in liquid culture since cells were grown to the same OD and we do not see a substantial difference in doubling time in liquid culture (Fig 2A), we suggest that fixing the cells on microscope slides using polylysin may have caused lysis of a fraction of the population. In contrast to glnR codY (fecE) mutant cells, the codY - socY mutant while also forming short chains (average number of cells per cfu of 5.7; Fig 2G) showed only a modest deficit (∼25%) in individual cell numbers compared to wild type (as well as to a glnR fecE mutant and a codY + complemented glnR codY (fecE) mutant; S3E Fig), suggesting more limited cell lysis (S2F Fig). On the other hand, a glnR fecE mutant displayed characteristics similar to wildtype (S3 Fig).
Comparing viable counts of the glnR codY (fecE) mutant with wild type and the codY socY mutant then revealed that the former displayed a ~20-fold loss of colony-forming units (cfu) compared to wild type when plated on CAT-agar (a deficit also observed in THY-agar, and using either Select Agar from Invitrogen or Ultrapure Agarose from Sigma) (data not shown). Viable glnR - codY - (fecE -) clones picked and regrown in liquid medium displayed the same loss of viability upon plating, indicating that these clones had not accumulated suppressing mutations but represented random low frequency survivors. In contrast, the codY - socY strain was fully viable, establishing that mortality is specific to the glnR codY (fecE) mutant. glnR - codY - (fecE -) colonies were nevertheless similar in size to wild type, in contrast to codY - socY, where ami inactivation results in very small colonies, further confirming that the former were ami+. Altogether, these observations suggested that the loss of viability was unrelated to overall growth rate but could be associated with plating on solid medium.
To further document the loss of viability, time-lapse microscopy was used to visualize the morphology and growth of glnR codY (fecE) mutant cells on CAT-agarose (Materials and Methods). Firstly, the rather low cell density of these cultures compared to control culture revealed that the majority of cells lysed shortly (i.e. within less than 1 hour) after transition from liquid to solid medium, leaving only cell debris. Secondly, cells that did not lyse immediately displayed aberrant cell morphology, generally being significantly larger than wildtype cells, and started lysing after 75–100 min growth on solid medium (Fig 2B). Clearly some of these cells must survive, as colonies are formed on solid medium, but these results confirm that the majority of cells are rapidly lost.
To confirm that the colony-forming defect observed was really due to the co-inactivation of codY and glnR (and accompanying necessary suppressor mutation in fecE or fatC), we recreated strains by transforming a codY::trim PCR fragment into glnR fatC, glnR fecE and fecE mutants. Three TrimR transformants were recovered from each transformation, and grown in identical conditions to determine viable counts. Results showed that unlike the generated fecE codY double mutants, the resulting glnR fecE codY and glnR fatC codY triple mutants all displayed a loss of viability upon plating (Fig 2C). These observations established that it is the simultaneous inactivation of glnR and codY which results in lysis and associated loss of viability in solid medium.
Restoration of normal colony-forming ability of the glnR codY (fecE) mutant
We then checked whether reintroduction of an ectopic, wildtype copy of codY into the glnR codY (fecE) mutant restored full viability. This experiment involved transformation with DNA from two strains in parallel, one possessing a CEPM -codY + cassette introducing wildtype codY at an ectopic chromosomal location [19] and the other possessing the same cassette but with codY inactivated by insertion of a mariner minitransposon (CEPM -codY::spc 3A). Transformation efficiency of CEPM-codY + was ~100-fold higher than CEPM -codY::spc, establishing that introduction of ectopic codY + restored normal viability on solid medium (Fig 3A). The abnormally high codY +/SmR ratio observed (almost 90-fold excess codY + cassette transformants over the expected number) providing strong support for this conclusion is fully explained by the fact that the SmR but not the codY + transformants display the glnR - codY - (fecE -) specific colony-forming defect. A large proportion of SmR transformants are thus lost, skewing the resulting codY +/SmR ratio.
Fig 3. Restoration of normal colony-forming ability of the glnR codY (fecE) mutant.
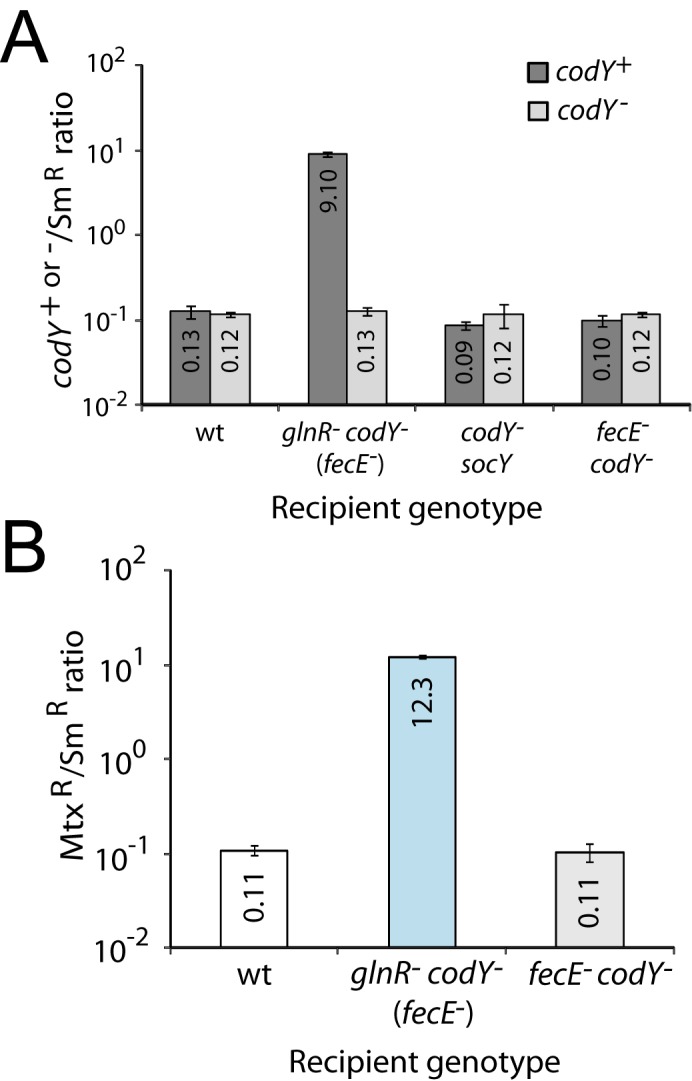
(A) Restoration by reintroduction of an ectopic, wildtype copy of codY. Ratios of KanR transformants obtained from transformation with either CEPM-codY + (dark grey bars, R2437 donor; indicated as codY +) or CEPM-codY::spc 3A (light grey bars, R3701; indicated as codY -). Transformation efficiency of CEPM-kan cassettes normalized as in Fig 1A. Recipient strain identities, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75. fecE - codY -, TD268. Transformation efficiencies calculated from triplicate repeats. Recipient strains in this panel were hexA +. (B) Inactivation of ami restores the viability of glnR codY (fecE) mutant cells. The amiC9 mutation which inactivates the amiC gene renders the Ami permease inactive and confers resistance to methotrexate (MtxR). Ratios of MtxR transformants normalized as in Fig 1A with transformation efficiencies calculated from triplicate repeats. Recipient strain identities, wt, D39; glnR - codY - (fecE -), TK108. Recipient strains in this panel were hexA +.
Secondly, since codY can be readily inactivated in a fecE amiC mutant (Fig 1D), we hypothesized that inactivation of ami would rescue glnR codY (fecE) mutants from plating mortality. To check this, we compared the efficiency of transformation of amiC9, a point mutation inactivating the Ami permease, into wildtype and glnR - codY - (fecE -) recipients. Whilst amiC9 transformed into wild type with expected efficiency compared to SmR, a ~110-fold excess in amiC9 transformants over the expectation was observed in the triple mutant (Fig 3B), establishing that inactivation of ami restored colony-forming ability of glnR codY - (fecE -) cells to the same extent as a second, ectopic copy of codY. In parallel, introduction of ectopic codY + and of amiC9 into fecE - codY - cells occurred with efficiencies similar to wild type (Fig 3) providing further support to the conclusion that the mortality observed is linked to the co-inactivation of codY and glnR.
CodY and competence for genetic transformation
The finding that the fecE codY double mutant was fully viable (Fig 2C) offered us the opportunity to reinvestigate the effect of codY inactivation on pneumococcal competence for genetic transformation [4,20], in the presence of a functional ami operon. CodY actively represses competence in B. subtilis [4,21], but a previous study in an S. pneumoniae codY mutant did not provide a clear answer as to whether CodY played a regulatory role on pneumococcal competence [8]. This is because at the time, the only characterized codY mutant was a codY fatC amiC mutant and ami mutations strongly upregulate competence [22], masking any potential role of CodY.
Competence profiles of codY fecE mutant cells were therefore analyzed throughout incubation at 37°C in C+Y medium with initial pH values between 6.6 and 7.6, since spontaneous competence induction is known to be strongly dependent on the initial pH [8,23] (Materials and Methods). The codY fecE mutant displayed a competence-down phenotype compared with wildtype and fecE mutant strains, as illustrated by the failure to develop competence at pH 6.9 and the strongly reduced competence at pH 7.1 (S4A—S4C Fig), suggesting that CodY may in fact activate pneumococcal competence.
Since CodY represses ami [7], and ami itself antagonizes spontaneous competence development [22], ‘activation’ of competence by CodY could rely primarily on repression of the ami operon. We therefore mutated the CodY binding site (CYbs) in the ami promoter, creating the ami CYbs0 mutation to abolish repression of ami despite the presence of CodY (Materials and Methods). Monitoring competence of ami CYbs0 and fecE ami CYbs0 mutants revealed that ami derepression by the ami CYbs0 mutation decreased competence in a similar manner to the inactivation of codY (S4D and S4E Fig). These results show that unlike in B. subtilis, CodY activates competence in S. pneumoniae but this activation is probably indirect, resulting from CodY-mediated repression of ami.
Sensitivity of glnR codY (fecE) mutant cells to lytic enzymes and cefotaxime
We then wished to establish whether the rapid lysis of glnR - codY - (fecE -) cells in solid medium was dependent on the major pneumococcal autolysin LytA. LytA is an amidase enzyme which cleaves the stem peptide of PG, separating it from the glycan strands at its base thereby disrupting the integrity of the cell wall and causing lysis [24,25]. A lytA::cat cassette [26] was transformed into the glnR codY (fecE) mutant and wild type, comparing its efficiency of transformation to SmR. A ratio of ~0.1 was observed for both recipient strains (data not shown), indicating that in contrast to ami inactivation (Fig 3B), inactivation of lytA did not restore colony-forming ability of glnR codY - (fecE -) cells. Moreover, three glnR codY (fecE) lytA transformants picked at random still displayed reduced colony-forming ability (data not shown). Both results established that LytA is neither responsible nor required for the lysis and resulting loss of viability of the triple mutant in solid medium.
In fact, a puzzling observation was made when comparing the sensitivity to deoxycholate (DOC), a well-known inducer of LytA-dependent autolysis, of the lytA - derivative and its parent (Fig 4A). The glnR - codY - (fecE -) parent itself appeared more resistant to DOC than the wild type. On the other hand, a glnR mutant behaved as wild type, while a codY - socY strain and a glnR codY fecE amiC mutant were also resistant to DOC (Fig 4A). Altogether, these results suggested that the inactivation of codY was responsible for this phenotype, a conclusion fully supported by the restoration of normal DOC sensitivity through ectopic expression of codY in otherwise codY - cells (Fig 4A).
Fig 4. Sensitivity to DOC and lysozyme of various codY derivatives.
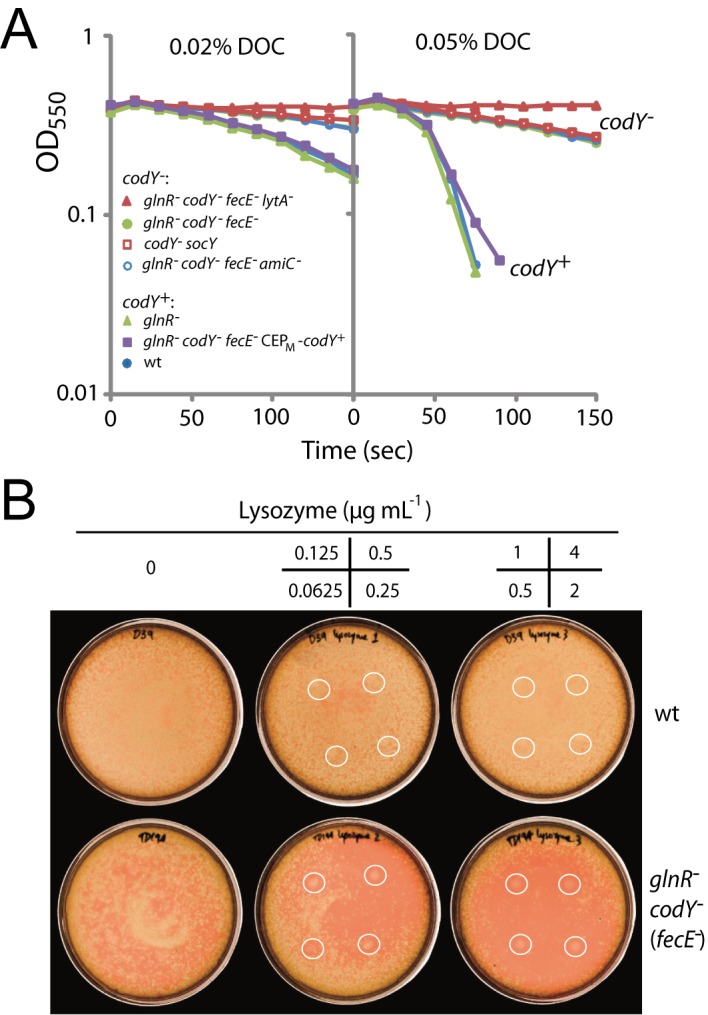
(A) Sensitivity of various strains to 0.02% or 0.05% DOC. Strain identities: wt, D39; glnR -, TD195; codY - socY, TD75; glnR - codY - (fecE -), TK108; glnR - codY - (fecE -) lytA -, TD272; glnR - codY - (fecE -) CEPM -codY +, TD273; glnR - codY - (fecE -) amiC -, TD247. (B) Sensitivity of strains to lysozyme. Lysozyme spotted onto plates in 5 μL volumes at the concentrations shown in the 4-panel grid. Spotting positions represented as white circles on the plates. Strain identities, wt; D39, glnR - codY - (fecE -); TK108.
The resistance of codY mutant cells to DOC appeared surprising and quite antagonistic with the rapid lysis of glnR codY (fecE) mutant cells upon plating. This apparent contradiction prompted us to examine other phenotypes, such as sensitivity to β-lactam antibiotics, which is frequently modified in pneumococcal isolates harboring peptidoglycan (PG) modifications [27–29]. A comparison of the effect of cefotaxime on various codY mutants and the wild type revealed that the glnR codY (fecE) mutant was much more sensitive to this antibiotic than the wild type, while glnR and fecE single mutants, a fecE codY double mutant and a codY - socY strain all behaved similarly to wild type (Table 1). Similarly, the glnR codY (fecE) mutant was hypersensitive to ampicillin and ceftriaxone, suggesting that sensitivity may extend to all β–lactam antibiotics (S2 Table).
Table 1. Sensitivity to cefotaxime of S. pneumoniae strain D39 and its glnR, codY and/or fat-fec mutant derivatives used in this study, measured as size of inhibition zone (mm).
| Genotype a | Cefotaxime (μg mL-1) | |||||
|---|---|---|---|---|---|---|
| 4 | 1 | 0.25 | 0.062 | 0.016 | 0.0039 | |
| wt | 22.0 ± 1.4 | 10.8 ± 0.4 | 1.0 ± 1.4 | 0 | 0 | 0 |
| fecE - | 21.3 ± 0.4 | 10.5 ± 0.7 | 0 | 0 | 0 | 0 |
| glnR - | 22.5 ± 1.4 | 15.0 ± 2.8 | 2.5 ± 3.5 | 0 | 0 | 0 |
| glnR - fecE - | 20.8 ± 1.1 | 11.0 ± 0.7 | 0 | 0 | 0 | 0 |
| codY - fecE - | 23.5 ± 1.4 | 15.8 ± 2.8 | 4.0 ± 0.7 | 0 | 0 | 0 |
| codY - fatC - amiC - | 23.0 ± 1.4 | 13.5 ± 0.7 | 3.0 ± 1.4 | 0 | 0 | 0 |
| glnR - codY - (fecE -) | 31.7 ± 0.4 | 27.3 ± 0.4 | 18.0 ± 1.4 | 15.3 ± 1.8 | 8.0 ± 0.7 | 0 |
| glnR - codY - fecE - amiC - | 25.5 ± 0.7 | 17.8 ± 1.8 | 10.0 ± 2.8 | 3.0 ± 1.4 | 0 | 0 |
a Strains used from top to bottom: TD249, TD230, TD195, TD227, TD268, TD75, TK108 and TD247.
The β-lactam sensitivity of the glnR - codY - (fecE -) mutant together with its colony-forming defect suggested some alteration of the cell wall. This led us to investigate its sensitivity to lysozyme, a muramidase enzyme also targeting PG (Fig 5A). While S. pneumoniae is normally resistant to lysozyme [15], the triple mutant turned out to be hypersensitive (Fig 4B). Altogether, these observations suggested that the simultaneous absence of CodY and GlnR affected cell wall physiology.
Fig 5. Pneumococcal cell wall composition, documented alterations and potential connections with GlnR-CodY through Glu/Gln metabolism.

(A) Diagram of pneumococcal cell-wall composition, with LytA, LytB and lysozyme cleavage sites, as well as cell-wall alterations known to affect hydrolytic activities. The glycan chains in pneumococcal PG are made of alternating MurNAc (M) and GlcNAc (G) residues. WTA is composed of repeating units containing two N-acetylgalactosamine residues (Gn), ribitol phosphate (R), glucose (Gc) and 2-acetamido-4-amino-2,4,6-trideoxygalactose (Ga). WTA chains are linked to PG via an unknown linkage (dotted line). Each R moiety can further be alanylated, although an initially proposed a-GalpNAc substitution [48] could not be confirmed in a later work [32]. The amino acids in the stem peptide are designated in single-letter code, with an asterisk indicating d-configuration. Modifications in the cell wall are indicated by numbers: (1) decoration of WTA by phosphocholine (P-Cho); (2) deacetylation of G by PgdA; (3) O-acetylation of M by Adr; (4) Branching between muropeptides by MurMN; (5) Amidation at 2nd stem peptide residue by MurT/GatD. (B) GlnR and CodY in the regulation of Gln/Glu metabolism of S. pneumoniae. Both regulators directly repress gdhA, involved in Glu synthesis. GlnR also represses glnA, involved in Gln synthesis, with DNA binding of GlnR to DNA shown to depend on GlnA [13]. Gln acts as a donor for amidation of the 2nd stem peptide residue (see panel A) catalyzed by the MurT/GatD amidotransferase.
Simultaneous inactivation of glnR and codY impacts cell wall physiology
Inactivation of both glnR and codY is expected to strongly affect Gln metabolism, as a result of full derepression of Gln synthetase and Glu dehydrogenase (Fig 5B). Since D-Gln is a key component of the PG stem peptide (Fig 5A), we hypothesized that imbalance in Gln/Glu could interfere with PG synthesis, resulting in the sensitivity phenotypes of the glnR codY (fecE) mutant. To establish whether the glnR codY (fecE) mutant displayed cell wall modifications, the composition of its PG was determined and compared with that of the wild type and other mutants used in this study. No difference was observed in the PG composition, including the percentage of unamidated peptides, the degree of stem peptide cross-linkage, and the deacetylation of muropeptides of the glnR codY (fecE) mutant compared to other strains (S2 Table and S5 Fig). In addition, we analyzed lipoteichoic acid (LTA; Materials and Methods) as a representative for pneumococcal teichoic acids, since both LTA and wall teichoic acid (WTA) have the same structure within their repeating units [30–32] and share the same biosynthesis pathway [33]. This analysis revealed no significant difference between the wild type and the glnR codY (fecE) mutant (S6 Fig).
Despite their normal PG and LTA composition, it was possible that mutant cells had alterations in their cell wall architecture. To assess this, glnR - codY - (fecE -) cells were visualized by transmission electron microscopy (TEM). Interestingly, these cells displayed a number of altered morphological features compared to wildtype and CEPM-codY + complemented glnR - codY - (fecE -) cells (Fig 6A). Firstly, cell shape was altered, consistent with observations in S2 Fig. Secondly, the cell wall of mutant cells appeared altered and less homogenous than that of wildtype cells. Indentations into the PG were frequently observed (green arrows), as well as areas where the PG was detached from the membrane (red arrows). Thirdly, we observed oddly shaped mutant cells with small blebs on the cell surface (yellow arrows). Fourthly, septa were frequently wrongly positioned and sometimes thicker than normal (blue arrow) suggesting that the mutations affect the cell division process. In addition, the cell wall of the glnR - codY - (fecE -) cells was less electron-dense and defined than the cell wall of wildtype or CEPM-codY + complemented cells (Fig 6B). Finally, we observed electrolucent foci within the mutant cells (white arrows) which were much less frequent in wildtype cells (Fig 6A). Their origin remains elusive but they do not appear to be surrounded by a membrane and may reflect the accumulation of some unknown metabolite, like fatty acids, that are extracted during the sample preparation and treatment for TEM analysis. We suggest that the morphological changes observed by TEM are caused by alterations in cell wall architecture and/or composition, explaining also the sensitivity to PG-targeting agents as a consequence of co-inactivation of glnR and codY.
Fig 6. Altered cell morphology of the glnR codY (fecE) mutant visualized by TEM.
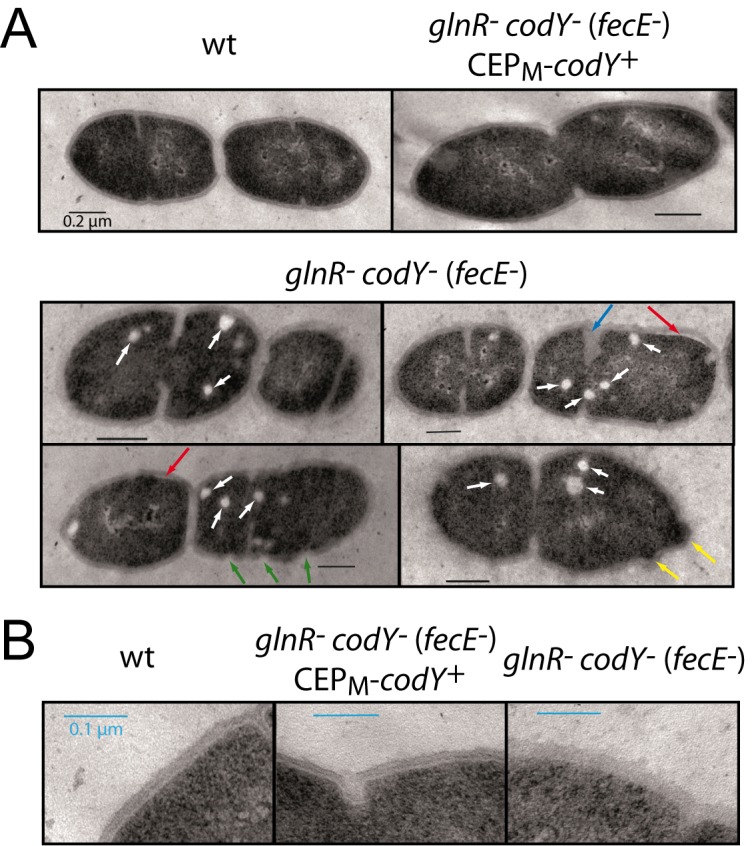
(A) Visualization of wt, glnR - codY - (fecE -) CEPM -codY + and glnR - codY - (fecE -) cells by TEM. Strains used, wt, D39; glnR - codY - (fecE -) CEPM -codY +, TD273; glnR - codY - (fecE -), TK108. Red arrows, detached cell wall; green arrows, gaps in cell wall; white arrows, circular clear bodies present within cells; blue arrow, aberrant septum formation. Black scale bars represent 0.2 μm. (B) Visualization of zoomed images of the strains in panel A focusing on cell wall. Blue scale bars represent 0.1 μm.
Discussion
CodY is essential for control of iron import in pneumococci via the Fat/Fec permease
We previously showed that the global nutritional regulator CodY was essential in the human pathogen S. pneumoniae [8] and that a published codY::trim mutant [7] had a suppressing mutation inactivating the fatC gene [8], belonging to the fat/fec operon which encodes the major ferric iron ABC permease of S. pneumoniae [10]. In this study, we analyzed two further published codY mutants [13,14], showing that both possessed point mutations truncating the fecE gene, which also suppressed CodY essentiality. The detection of fat/fec suppressors in all three published pneumococcal codY mutants implies that co-inactivation of the fat/fec operon is crucial for survival of pneumococci lacking CodY. These data fully support the view that repression of iron transport is primarily responsible for the essentiality of CodY and that mutation of codY can only be tolerated if fat/fec is simultaneously inactivated to prevent iron toxicity, as previously suggested [8]. As a result, all pneumococcal codY mutants studied always have an obligate associated mutation inactivating the fat/fec operon. We refer to this mutation as the primary suppressor of CodY essentiality, crucial for survival of pneumococcal cells lacking CodY.
Control of the Ami permease by CodY is also important though not essential
Suppressing mutations in the amiC gene were first identified through careful analysis of a previously published codY mutant [8]. amiC is part of the ami operon, which is also directly repressed by CodY and encodes the Ami oligopeptide permease. This previous study showed the fatC mutation in 100% of codY - cells but three different amiC mutations in the codY mutant population [8], consistent with a scenario in which the fatC mutation arose first, justifying application of the moniker ‘primary’ suppressor, and the various amiC mutations appeared subsequently, making them secondary suppressors. This suggested that inactivation of the Ami permease aids in tolerance of codY inactivation. The report that another codY mutant also had an inactivated amiC gene [14] (resulting from the very same GGA→TTGC mutation we discovered in our original analysis of codY essentiality [8]; Sven Hammerschmidt, personal communication) provided further support for this interpretation. However, we show that ami inactivation is not compulsory, as indicated by the observation that the glnR codY (fecE) mutant, which harbors no ami mutation, is fully viable in liquid medium (Fig 2A). The ami mutations can thus be viewed as accessory, aiding in but not essential for tolerance of codY inactivation. Nevertheless, the frequent occurrence of ami mutations in pneumococcal codY - strains suggests some level of toxicity when the ami operon is derepressed due to the absence of CodY.
CodY indirectly activates pneumococcal competence by repressing ami
In B. subtilis, CodY plays a regulatory role on competence for genetic transformation, directly repressing the comK gene, encoding the transcriptional activator of competence, as well as sfrA, an operon important for comK activation [4,20,21]. Interestingly, mutation of opp, which encodes the Ami orthologue, resulted in loss of CodY-dependent repression of comK and sfrA, essentially activating competence [21]. Presumably, inactivation of oligopeptide import impacts CodY co-factor (i.e. branched chain amino acids) metabolism in B. subtilis.
In contrast, CodY is not known to directly repress any com gene in S. pneumoniae. Instead, we showed here that CodY activates competence by direct repression of ami (S4 Fig). As Ami antagonizes pneumococcal competence [22] through a mechanism which remains unknown, CodY thus contributes indirectly to the activation of pneumococcal competence. The opposite effects of CodY mutation on competence induction in B. subtilis and S. pneumoniae suggest different ways of responding to nutritional changes in these species. The reason for this difference could lie in the growth phases during which competence is induced in each species. In B. subtilis, competence is induced post-exponentially [34], in conditions of likely nutrient limitation. In contrast, competence in S. pneumoniae is transiently induced during exponential growth phase [35], when nutrients are generally abundant. It therefore makes sense for CodY to regulate competence induction differently in these two bacteria, which develop competence in conditions of widely different nutritional availability. Since it appears beneficial for B. subtilis to induce competence upon nutrient starvation, direct repression of competence by CodY, followed by derepression upon nutrient starvation, regulates the competence circuit to permit this. In the pneumococcus, direct repression by CodY would antagonize competence development during exponential growth, and thus no such direct regulation has been selected during evolution.
CodY also impacts LytA-dependent autolysis
All codY mutants tested, including in combination with ami and fec/fat mutations, displayed increased resistance to DOC, an inducer of LytA-mediated autolysis, compared to wildtype cells (Fig 4A). The activity of the autolysin LytA is dependent on the decoration of WTA by choline (Cho; Fig 5A). LytA activity could therefore be affected as a consequence of altered Cho decoration. However, the putative N-acetylglucosaminidase LytB (Fig 5A), involved in daughter cell separation, is also dependent on Cho-decorated WTA for activity [36]. Although short chains can be observed in the glnR codY (fecE) mutant (S2A Fig), none of the codY mutants formed long chains (hundreds of cells), a hallmark of LytB inhibition [36], which suggests that LytB is at least partly active. However, it is interesting that both tested codY mutants had a propensity to form short chains (S2A Fig). Together with the increased resistance to DOC of all tested codY mutants, this observation suggests that cells lacking CodY may have reduced activity of both LytA and LytB resulting respectively in reduced lysis and increased chaining. It is possible that this is due to an alteration in the Cho-decoration of WTA, which could perturb the activity of these enzymes. However, this phenotype would be specific to all codY mutants, and thus remain separate from the phenotypes observed specifically in the glnR codY (fecE) mutant. The observed loss of cell viability upon plating (Fig 2C) is specific to the latter, and most probably linked to the altered sensitivity to PG-targeting agents observed.
Co-inactivation of glnR and codY severely affects pneumococcal cell wall physiology
glnR - codY - (fecE -) cells display a unique combination of phenotypic traits not observed with other codY mutants. These cells were viable in liquid medium (Fig 2A), but showed a large loss of viability when plated on solid medium (Fig 2B and 2C). Visualization of glnR - codY - (fecE -) cells on CAT-agarose showed cells which lysed rapidly (Fig 2B), confirming the conclusion based on examination of cells fixed on microscopy slides (S2F Fig). On the other hand codY - fecE - cells appeared fully viable (Fig 2C). Inviability is therefore presumably due to derepression of both GlnR and CodY regulons. The major autolysin LytA was not responsible for the colony-forming defect observed in these cells. A specific hypersensitivity to other PG-targeting agents cefotaxime (Table 1), ampicillin, ceftriaxone (S2 Table) and lysozyme (Fig 4B) was also observed for glnR - codY - (fecE -) cells grown on solid medium.
A number of cell wall alterations have been reported to alter pneumococcal sensitivity to certain agents targeting PG (S1 Text and Fig 5A). Potentially connected to our observations, amidation of the second residue of the stem peptide was required for efficient PG cross-linking via transpeptidase activity [31,37]. This amidation, which alters the D-iso-Glu to D-iso-Gln (Fig 5A), is dependent on the amidotransferase enzyme MurT/GatD using L-Gln as donor (Fig 5B) [37–39]. It has also been reported that inhibition of amidation reduced resistance to μ-lactam antibiotics and increased sensitivity to lysozyme in Staphylococcus aureus [38]. Apart from their decreased sensitivity to DOC which can be attributed to codY inactivation (see above), the glnR codY (fecE) mutant shared a phenotype of sensitivity to lysozyme and β-lactams with cells depleted for MurT/GatD. It was therefore tempting to speculate on the alteration in stem peptide synthesis as a cause for phenotype of the glnR codY (fecE) mutant (S1 Text). However, HPLC analysis of muropeptide composition revealed no difference in unamidated peptides as well as in the degree of PG cross-linkage between wildtype and glnR - codY - (fecE -) cells (S2 Table), providing no support to this hypothesis. In addition, while our analysis could not detect O-acetylation, a potential mechanism for lysozyme resistance (Fig 5A, number 3), as (most of) the O-acetyl groups are lost during routine sample preparation, we were able to detect deacetylated muropeptides (another mechanism for lysozyme resistance; Fig 5A, number 2). However, there was no dramatic difference in levels of deacetylated muropeptides in the glnR codY (fecE) mutant that could explain its lysozyme sensitivity.
Despite this, visualization of cells by TEM uncovered stark differences in cell wall morpohology between wildtype and glnR codY (fecE) mutant cells, with less dense cell wall observed in the mutant (Fig 6B), along with regions where the cell wall appears disrupted or detached from the cell (Fig 6A). These results strongly suggest that the glnR codY (fecE) mutant has altered PG, which we propose results in the increased sensitivity to PG-targeting agents. However, both the exact physical nature of the alterations observed and the underlying mechanism(s) which can be indirect, owing to alterations in central metabolism in the absence of both CodY and GlnR regulators, remain to be established.
Connections between iron toxicity, cell wall physiology and Ami
While glnR - codY - (fecE -) cells display a unique phenotype among codY mutants, the observation that inactivation of ami restored viability of these cells (Fig 3B) indicates that the very same combination of suppressing mutations allowing survival of codY mutant cells (i.e. ami and fat/fec) can compensate for the absence of both GlnR and CodY. The nature of the compound(s) imported via the Ami permease which is ‘toxic’ for the cell remains unknown. In view of the observation that an Ami homologue allowed use of heme as an iron source by Escherichia coli [40], we previously suggested that Ami could similarly contribute to iron toxicity in S. pneumoniae [8]. However, no direct support for this hypothesis could be obtained since we observed that plating with or without horse blood (i.e. in the presence of widely different heme concentrations) did not alter survival of glnR - codY - (fecE -) cells (data not shown). In addition, even if iron toxicity proved to be responsible for inviability of codY and codY glnR mutants, the link between cell wall physiology and iron toxicity would remain unclear. Alternatively, Ami toxicity may not be connected with iron toxicity but somehow linked to the recycling of peptidoglycan during growth. In Escherichia coli, peptidoglycan recycling is known to rely primarily on the AmpG transporter, which is specific for uptake of anhydromuropeptides, whilst the oligopeptide permease Opp (the orthologue of Ami) has only a minor role in uptake of cell wall derived peptides. In contrast, with Gram-positives lacking AmpG, Ami-like permeases may constitute the major cell wall recycling pathway [41]. It is thus possible that Ami is responsible for peptidoglycan recycling in S. pneumoniae, and altered recycling in the absence of codY is toxic on solid medium. Alternatively, the effect of ami inactivation could be indirect, resulting from the reduced growth rate conferred by ami mutations. A reduction in growth rate could for example compensate for metabolism jamming by allowing more time to the cell to build the envelope.
Concluding remarks
The genetic dissection of previously published codY mutants allowed us to demonstrate here that the primary suppressing mutation necessary to tolerate absence of CodY is inactivation of the fat/fec iron permease operon. This strongly suggests that unregulated iron import is toxic to codY - pneumococci. Furthermore, we showed that a glnR codY (fecE) mutant was only partly viable on solid medium and displayed a unique phenotype among codY mutants, suggestive of altered cell wall physiology, a conclusion fully supported by TEM analysis of mutant cells. While increasing resistance to PG-targeting antibiotics such as β-lactams is problematic for treatment of pneumococcal infections, hypersensitivity of this mutant to PG-targeting agents suggests GlnR and CodY as novel therapeutic targets. Simultaneous targeting of these important nutritional regulators would render pneumococci hypersensitive to both iron and classic PG-targeting antibiotics, and could thus be useful in the constant fight against this debilitating pathogen.
Materials and Methods
Bacterial strains, growth and transformation conditions
S. pneumoniae strain growth and transformation were carried out as described [8]. Strain and primer information can be found in S1 Table. Unless stated, recipient strains were rendered hex - by insertion of the hexA::ermAM cassette as previously described [42], to negate any effect of the mismatch repair system on transformation efficiencies via rejection of mismatched DNA in transformation heteroduplexes [43]. Antibiotics were used at the following concentrations; trimethoprim 200 μg mL-1, erythromycin 2 μg mL-1, kanamycin 250 μg mL-1, streptomycin 200 μg mL-1, methotrexate μg mL-1. Previously described cassettes used for transfer were glnR::kan22 C [17] and ΔcodY::trim [13]. Growth curves were followed in a 96-well Spectrophotometer (ThermoScientific) with OD492 readings every 10 min after 1/100 dilution of cultures grown to OD 0.2 into a final volume of 300 μL. Doubling times were calculated as the time required to observe a doubling of OD during the fastest phase of exponential growth. As a result, this figure does not take into account any lag phase prior to active growth that can be seen in certain conditions with different mutants.
Competence was monitored as previously described [44] in pH gradients in C+Y medium on strains containing the ssbB-luc transcriptional fusion between the promoter of the single-stranded DNA-binding protein SsbB, which is induced during competence [4], and the firefly luciferase gene luc, the fusion thus reporting on competence through light emission by luciferase [45]. Briefly, cells were grown in conditions where competence does not spontaneously develop (2 mL C+Y pH 6.6) to OD550 0.11, pelleted and resuspended in 1 mL C+Y. 100 μL aliquots were diluted 1/10 in C+Y, and 30 μL added to 250 μL C+Y culture of desired pH and 20 μL luciferin (3 mg mL-1) in microtitre plate wells. Emission of light and OD492 values were followed throughout growth.
Genome sequencing
Genome sequencing of the TK108 strain was carried out by LGC genomics. The resulting sequence was aligned to the D39 genome present in the NCBI database (NCTC 7466), and point mutations, insertions and deletions were identified by LGC genomics.
Transfer of fecE - point mutation without selection
Transfer of the fecE - point mutation of TK108 into a D39 recipient strain was achieved without selection. The mutation was amplified from TK108 by PCR with 500 bp flanking DNA on either side. The PCR product (~1,000 bp) was purified using a purification kit (QIAGEN), and transformed into the TD195 strain to create strain TD227. Briefly, 100 μL pre-competent TD195 cells were resuspended in 900 μL C+Y medium with 100 ng μL-1 CSP. Cells were incubated at 37°C for 10 min to induce competence, and 1 μg of fecE - PCR product was added to 100 μL competent culture. This transformation mixture was incubated at 30°C for 20 min, before addition of 1.4 mL C+Y medium, and further incubation at 37°C for 4 h to allow correct integration of the mutation and cell separation. Cells were then plated on CAT agarose plates + 5% defibrinated horse blood (Biomérieux), and incubated overnight at 37°C. Colonies are then picked and analyzed by PCR and sequencing to identify those that had acquired the fecE - point mutation, before sub-cloning to ensure a pure fecE mutant culture.
Mutation of CodY binding site in ami promoter
In order to mutate the CYbs in the ami promoter region, a fragment of DNA of 1,000 bp, with the CYbs (AATTTTCAGAATATT) replaced by a sequence containing an NcoI restriction site (GCTAGGGATCCGCTA) and flanked on either side by 500 bp of DNA was synthesized (Genscript). This fragment was transformed into S. pneumoniae strains, as described for fecE -, in the absence of antibiotic selection, and transformant clones were identified by PCR of the ami promoter and restriction by NcoI.
Microscopy
For static images of cells analyzed in S2 and S3 Figs, cells were grown in C+Y medium to an OD492 of 0.2, and 2 μL of these cultures was spotted onto a microscope slide. A polylysin-coated coverslip was placed on top to fix the cells in position. Images were captured and processed using the Nis-Elements AR software (Nikon). Analysis of cell dimensions was carried out using the MATLAB-based open source software MicrobeTracker [46]. Cell contours were obtained using the alg4 spneumoniae3 algorithm implemented in MicrobeTracker, a derivative of alg4 ecoli2 with parameters spliltTreshold, joindist and joinangle refined to fit the shape of S. pneumoniae.
Time-lapse microscopy was carried out as previously described [47], with modifications. Briefly, pneumococcal precultures were grown in liquid CAT medium at 37°C to an OD492 of 0.2, and 2 μL of these cultures was spotted onto a microscope slide containing a slab of 1.2% CAT agarose before imaging. Images were captured at 5 min intervals for 6 hours and processed using the Nis-Elements AR software (Nikon).
Sensitivity tests
Sensitivity to cefotaxime, ampicillin, ceftriaxone and lysozyme was measured by growing strains to OD550 0.2 in C+Y medium, followed by plating ~400–600 cells on CAT-agarose supplemented with 5% horse blood (Biomérieux). After drying, the inhibitory agent was spotted onto these plates in 5 μL volumes (4 spots per plate) at 4-fold dilutions starting at 4 μg mL-1 (see Table 1). Plates were incubated at 37°C overnight and inhibition zones were measured. Sensitivity to DOC was measured by growing strains to OD550 ~0.4 in C+Y medium, followed by addition of 0.02% or 0.05% DOC to 1 mL of culture. OD550 readings were taken every 15 sec for 150 sec.
Isolation and analysis of peptidoglycan
Peptidoglycan was isolated as previously published [31]. Digestion of peptidoglycan with cellosyl (kindly provided by Hoechst, Germany) yielded muropeptides, which were reduced with sodium borohydride and analyzed by high-pressure liquid chromatograhy as published [31].
Isolation and analysis of LTA
LTA from S. pneumoniae D39 and TK108 (glnR - codY - (fecE - )) was isolated as previously described [32]. Pneumococci were grown in 5 L cultures THY medium. Samples were dissolved in a water/propan-2-ol/7 M triethylamine/acetic acid mixture (50:50:0.06:0.02, v/v/v/v) and analysed using a APEX Qe Fourier-Transform Ion Cyclotron Resonance Mass Spectrometer (Bruker Daltonics, USA) equipped with the Triversa Nanomate (Advion, USA) as ion source applying a spray voltage of -1.1 kV. Mass spectra were recorded in negative-ion mode in the broadband acquisition mode, the mass scale was externally calibrated with glycolipids of known structure, and spectra were smoothed, baseline corrected, and charge deconvoluted.
TEM
Wild type and glnR - codY - (fecE -) cells were exponentially grown at 37°C in C+Y medium. Samples were then collected, centrifuged and fixed overnight with 5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.5) at 4°C. Postfixation with 1% osmium tetroxide in cacodylate buffer was carried out for 1 h at room temperature. These fixed cells were dehydrated using a graded series of ethanol and embedded in LR White at 60°C for 48 h. Ultrathin sections (50 nm) were obtained using a Leica UC7 microtome and were counterstained with uranyl acetate and lead citrate (Reichert Ultrostainer, Leica, Germany). Samples were examined with a Philips CM120 transmission electron microscope equipped with a Gatan Orius SC200 CCD camera.
Supporting Information
(A-C) Growth curves of strains in CAT, TSB and THY media respectively. After initial growth to identical cellular densities (OD 0.2) in medium, cells were diluted 1/100 in 300 μL final volume of appropriate medium in microtitre plate and OD492 readings taken every 10 min for 600 min. Strain identities, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75.
(TIF)
(A) Wildtype, glnR - codY - (fecE -) and codY - socY cells observed on polylysin slides. Strains used, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75. (B) Length of cells observed on polylysin slides. Data represented as percentage of cell population fitting into different length fractions. It is of note that every cell in a chain (panel G) was nevertheless treated as a single cell; therefore chaining should have had no influence on the calculation of the cell length parameter. The vertical dashed red line represents the average value (indicated above the line) of wildtype cells. Strains used as in panel A. (C) Width of cells observed on polylysin slides. Strains and analysis as in panel B. (D) Area of cells observed on polylysin slides. Strains and analysis as in panel B. (E) Hypothetical volume of cells observed on polylysin slides. Hypothetical value calculated from other values of cell dimension. Strains and analysis as in panel B. (F) Mean number of individual cells counted per image. Strains as in panel A. (G) Percentage of cells per image which form part of chains. Strains as in panel A.
(TIF)
(A) Length of cells observed on polylysin slides. Data represented as percentage of cell population fitting into different length fractions. Strains used, wt, D39; glnR - fecE -, TD227; glnR - codY - (fecE -) CEPM-codY +, TD273. (B) Width of cells observed on polylysin slides. Strains, cells and analysis as in panel A. (C) Area of cells observed on polylysin slides. Strains, cells and analysis as in panel A. (D) Hypothetical volume of cells observed on polylysin slides. Strains, cells and analysis as in panel A. Hypothetical value calculated from other values of cell dimension. (E) Mean number of cells counted per image. Strains as in panel A. (F) Percentage of cells per image which form part of chains. Strains as in panel A.
(TIF)
(A) Development of competence in wt background monitored by following expression of ssbB-luc transcriptional fusion. Competence development (RLU/OD) was plotted against growth (OD492) in a gradient of varying starting pHs. Strain used, TD259. (B) Development of competence in a fecE mutant. Strain used TD263. Experimental information and figure layout as in panel A. (C) Development of competence in a fecE codY double mutant. Strain used TD265. Experimental information and figure layout as in panel A. (D) Derepression of ami antagonizes competence revealed by monitoring competence development in an ami CYbs0 mutant. Strain used TD260. Experimental information and figure layout as in panel A. (E) Development of competence in a fecE ami CYbs0 double mutant. Strain used TD264. Experimental information and figure layout as in panel A.
(TIF)
Peptidoglycan was digested with the muramidase cellosyl and the resulting muropeptides were reduced with sodium borohydride and analysed by high-pressure liquid chromatography. Strains used (from top to bottom: R6, TD249, TD227, TK108, TD247 and TD75) are indicated on the right side.
(TIF)
(A) Section (5500–10500 Da) of the charge deconvoluted ESI-FT-ICR-MS spectrum of pnLTA isolated from strain D39 (wt, black) and TK108 (glnR - codY - (fecE -), red). (B) Enlarged image of the ion cluster with highest intensity (8450–8750 Da). Differences in the signal pattern of the two strains are caused by varying percentage of sodium adduct ion cluster, as indicated. For a clear visualization of mass differences, the most intensive peak of an ion cluster has been chosen instead of the monoisotopic peak.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We thank Bernard Martin for helpful discussions and critical reading of the manuscript. We gratefully acknowledge Simone Thomsen and Brigitte Kunz for their technical assistance during LTA analysis. We thank Sven Hammerschmidt for communication of unpublished data and for providing genomic DNA of the codY mutant and associated parent strain published previously [14].
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by the European Community’s Seventh Framework Programme FP7/2007-2013 under grant agreement number HEALTH-F3-2009-222983 (Pneumopath project) and the CNRS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sonenshein AL (2005) CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr Opin Microbiol 8: 203–207. [DOI] [PubMed] [Google Scholar]
- 2. Molle V, Nakaura Y, Shivers RP, Yamaguchi H, Losick R, Fujuta Y et al. (2003) Additional targets of the Bacillus subtilis global regulator CodY identified by chromatin immunoprecipitation and genome-wide transcript analysis. J Bacteriol 185: 1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sonenshein AL (2007) Control of key metabolic intersections in Bacillus subtilis . Nat Rev Microbiol 5: 917–927. [DOI] [PubMed] [Google Scholar]
- 4. Claverys JP, Prudhomme M, Martin B (2006) Induction of competence regulons as general stress responses in Gram-positive bacteria. Annu Rev Microbiol 60: 451–475. [DOI] [PubMed] [Google Scholar]
- 5. Guédon E, Serror P, Ehrlich SD, Renault P, Delorme C (2001) Pleitropic transcriptional regulator CodY senses the intracellular pool of branched-chain amino acides in Lactococcus lactis . Mol Microbiol 40: 1227–1239. [DOI] [PubMed] [Google Scholar]
- 6. Handke LD, Shivers RP, Sonenshein AL (2008) Interaction of Bacillus subtilis CodY with GTP. J Bacteriol 190: 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hendriksen WT, Bootsma HJ, Estevão S, Hoogenboezem T, De Jong A, De Groot R et al. (2008) CodY of Streptococcus pneumoniae: link between nutritional gene regulation and virulence. J Bacteriol 190: 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caymaris S, Bootsma HJ, Martin B, Hermans PWM, Prudhomme M and Claverys JP (2010) The global nutritional regulator CodY is an essential protein in the human pathogen Streptococcus pneumoniae . Mol Microbiol 78: 344–360. [DOI] [PubMed] [Google Scholar]
- 9. Brown JS, Gilliland SM, Ruiz-Albert J, Holden DW (2002) Characterization of pit, a Streptococcus pneumoniae iron uptake ABC transporter. Infect Immun 70: 4389–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ulijasz AT, Andes DR, Glasner JD, Weisblum B (2004) Regulation of iron transport in Streptococcus pneumoniae by RitR, an orphan response regulator. J Bacteriol 186: 8123–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tai SS, Yu C, Lee JK (2003) A solute binding protein of Streptococcus pneumoniae iron transport. FEMS Microbiol Lett 220: 303–308. [DOI] [PubMed] [Google Scholar]
- 12. Alloing G, de Philip P, Claverys JP (1994) Three highly homologous membrane-bound lipoproteins participate in oligopeptide transport by the Ami system of the Gram-positive Streptococcus pneumoniae . J Mol Biol 241: 44–58. [DOI] [PubMed] [Google Scholar]
- 13. Kloosterman TG, Hendriksen WT, Bijlsma JJ, Bootsma HJ, van Hijum SA, Kok J et al. (2006) Regulation of glutamine and glutamate metabolism by GlnR and GlnA in Streptococcus pneumoniae . J Biol Chem 281: 25097–25109. [DOI] [PubMed] [Google Scholar]
- 14. Hartel T, Eylert E, Schulz C, Petruschka L, Gierok P et al. (2012) Characterization of central carbon metabolism of Streptococcus pneumoniae by isotopologue profiling. J Biol Chem 287: 4260–4274. 10.1074/jbc.M111.304311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis KM, Akinbi HT, Standish AJ, Weiser JN (2008) Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae . PLoS Pathog 4: e1000241 10.1371/journal.ppat.1000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salles C, Créancier L, Claverys JP, Méjean V (1992) The high level streptomycin resistance gene from Streptococcus pneumoniae is a homologue of the ribosomal protein S12 gene from Escherichia coli . Nucl Acids Res 20: 6103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnston C, Martin B, Granadel C, Polard P, Claverys JP (2013) Programmed protection of foreign DNA from restriction allows pathogenicity island exchange during pneumococcal transformation. PLoS Pathogens 9: e1003178 10.1371/journal.ppat.1003178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ et al. (2007) Genome Sequence of Avery's Virulent Serotype 2 Strain D39 of Streptococcus pneumoniae and Comparison with That of Unencapsulated Laboratory Strain R6. J Bacteriol 189: 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guiral S, Hénard V, Laaberki M-H, Granadel C, Prudhomme M, Martin B et al. (2006) Construction and evaluation of a chromosomal expression platform (CEP) for ectopic, maltose-driven gene expression in Streptococcus pneumoniae . Microbiology (Special Issue on Pneumococcus) 152: 343–349. [DOI] [PubMed] [Google Scholar]
- 20. Johnston C, Martin B, Fichant G, Polard P, Claverys JP (2014) Bacterial transformation: distribution, shared mechanisms and divergent control. Nature Rev Microbiol 12: 181–196. 10.1038/nrmicro3199 [DOI] [PubMed] [Google Scholar]
- 21. Serror P, Sonenshein AL (1996) CodY is required for nutritional repression of Bacillus subtilis genetic competence. J Bacteriol 178: 5910–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alloing G, Martin B, Granadel C, Claverys JP (1998) Development of competence in Streptococcus pneumoniae: pheromone auto-induction and control of quorum-sensing by the oligopeptide permease. Mol Microbiol 29: 75–84. [DOI] [PubMed] [Google Scholar]
- 23. Chen JD, Morrison DA (1987) Modulation of competence for genetic transformation in Streptococcus pneumoniae . J Gen Microbiol 133: 1959–1967. [DOI] [PubMed] [Google Scholar]
- 24. Holtje JV, Tomasz A (1976) Purification of the pneumococcal N-acetylmuramyl-L-alanine amidase to biochemical homogeneity. J Biol Chem 251: 4199–4207. [PubMed] [Google Scholar]
- 25. García P, García JL, García E, López R (1986) Nucleotide sequence and expression of the pneumococcal autolysin gene from its own promoter in Escherichia coli . Gene 43: 265–272. [DOI] [PubMed] [Google Scholar]
- 26. Guiral S, Mitchell TJ, Martin B, Claverys JP (2005) Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: genetic requirements. Proc Natl Acad Sci USA 102: 8710–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith AM, Klugman KP (2001) Alterations in MurM, a cell wall muropeptide branching enzyme, increase high-level penicillin and cephalosporin resistance in Streptococcus pneumoniae . Antimicrob Agents Chemother 45: 2393–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Filipe SR, Severina E, Tomasz A (2002) The murMN operon: a functional link between antibiotic resistance and antibiotic tolerance in Streptococcuspneumoniae . Proc Natl Acad Sci U S A 99: 1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garcia-Bustos JF, Chait BT, Tomasz A (1988) Altered peptidoglycan structure in a pneumococcal transformant resistant to penicillin. J Bacteriol 170: 2143–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fischer W, Behr T, Hartmann R, Peter-Katalinic J, Egge H (1993) Teichoic and lipoteichoic acid of Streptococcus pneumoniae possess identical chain structures. A reinvestigation of teichoic acid (C polysaccharide). Eur J Biochem 215: 851–857. [DOI] [PubMed] [Google Scholar]
- 31. Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A et al. (2012) Isolation and analysis of cell wall components from Streptococcus pneumoniae . Anal Biochem 421: 657–666. 10.1016/j.ab.2011.11.026 [DOI] [PubMed] [Google Scholar]
- 32. Gisch N, Kohler T, Ulmer AJ, Muthing J, Pribyl T, Fischer K et al. (2013) Structural reevaluation of Streptococcus pneumoniae Lipoteichoic acid and new insights into its immunostimulatory potency. J Biol Chem 288: 15654–15667. 10.1074/jbc.M112.446963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Denapaite D, Bruckner R, Hakenbeck R, Vollmer W (2012) Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist 18: 344–358. 10.1089/mdr.2012.0026 [DOI] [PubMed] [Google Scholar]
- 34. Hahn J, Albano M, Dubnau D (1987) Isolation and characterization of competence mutants in Bacillus subtilis . J Bacteriol 169: 3104–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tomasz A (1966) Model for the mechanism controlling the expression of competence state in pneumococcus cultures. J Bacteriol 91: 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. García P, González MP, García E, López R, García JL (1999) LytB, a novel pneumococcal murein hydrolase essential for cell separation. Mol Microbiol 31: 1275–1277. [DOI] [PubMed] [Google Scholar]
- 37. Zapun A, Philippe J, Abrahams KA, Signor L, Roper DI, Breukink E et al. (2013) In vitro Reconstitution of Peptidoglycan Assembly from the Gram-Positive Pathogen Streptococcus pneumoniae . ACS Chem Biol 8: 2688–2696. 10.1021/cb400575t [DOI] [PubMed] [Google Scholar]
- 38. Figueiredo TA, Sobral RG, Ludovice AM, Almeida JM, Bui NK, Vollmer W et al. (2012) Identification of genetic determinants and enzymes involved with the amidation of glutamic acid residues in the peptidoglycan of Staphylococcus aureus . PLoS Pathog 8: e1002508 10.1371/journal.ppat.1002508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Munch D, Roemer T, Lee SH, Engeser M, Sahl HG and Schneider T (2012) Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus . PLoS Pathog 8: e1002509 10.1371/journal.ppat.1002509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Létoffé S, Delepelaire P, Wandersman C (2006) The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc Natl Acad Sci U S A 103: 12891–12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reith J, Mayer C (2011) Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl Microbiol Biotechnol 92: 1–11. 10.1007/s00253-011-3486-x [DOI] [PubMed] [Google Scholar]
- 42. Mortier-Barrière I, de Saizieu A, Claverys JP, Martin B (1998) Competence-specific induction of recA is required for full recombination proficiency during transformation in Streptococcus pneumoniae . Mol Microbiol 27: 159–170. [DOI] [PubMed] [Google Scholar]
- 43. Claverys JP, Lacks SA (1986) Heteroduplex deoxyribonucleic acid base mismatch repair in bacteria. Microbiol Rev 50: 133–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prudhomme M, Claverys JP (2007) in: The Molecular Biology of Streptococci, Hakenbeck R, Chhatwal GS, editors. Horizon Scientific Press, Norfolk, UK, pp. 519–524. [Google Scholar]
- 45. Chastanet A, Prudhomme M, Claverys JP, Msadek T (2001) Regulation of Streptococcus pneumoniae clp genes and their role in competence development and stress survival. J Bacteriol 183: 7295–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C (2011) High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol Microbiol 80: 612–627. 10.1111/j.1365-2958.2011.07579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Jong IG, Beilharz K, Kuipers OP, Veening JW (2011) Live Cell Imaging of Bacillus subtilis and Streptococcus pneumoniae using Automated Time-lapse Microscopy. J Vis Exp 53: e3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Draing C, Pfitzenmaier M, Zummo S, Mancuso G, Geyer A, Hartung T et al. (2006) Comparison of lipoteichoic acid from different serotypes of Streptococcus pneumoniae . J Biol Chem 281: 33849–33859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A-C) Growth curves of strains in CAT, TSB and THY media respectively. After initial growth to identical cellular densities (OD 0.2) in medium, cells were diluted 1/100 in 300 μL final volume of appropriate medium in microtitre plate and OD492 readings taken every 10 min for 600 min. Strain identities, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75.
(TIF)
(A) Wildtype, glnR - codY - (fecE -) and codY - socY cells observed on polylysin slides. Strains used, wt, D39; glnR - codY - (fecE -), TK108; codY - socY, TD75. (B) Length of cells observed on polylysin slides. Data represented as percentage of cell population fitting into different length fractions. It is of note that every cell in a chain (panel G) was nevertheless treated as a single cell; therefore chaining should have had no influence on the calculation of the cell length parameter. The vertical dashed red line represents the average value (indicated above the line) of wildtype cells. Strains used as in panel A. (C) Width of cells observed on polylysin slides. Strains and analysis as in panel B. (D) Area of cells observed on polylysin slides. Strains and analysis as in panel B. (E) Hypothetical volume of cells observed on polylysin slides. Hypothetical value calculated from other values of cell dimension. Strains and analysis as in panel B. (F) Mean number of individual cells counted per image. Strains as in panel A. (G) Percentage of cells per image which form part of chains. Strains as in panel A.
(TIF)
(A) Length of cells observed on polylysin slides. Data represented as percentage of cell population fitting into different length fractions. Strains used, wt, D39; glnR - fecE -, TD227; glnR - codY - (fecE -) CEPM-codY +, TD273. (B) Width of cells observed on polylysin slides. Strains, cells and analysis as in panel A. (C) Area of cells observed on polylysin slides. Strains, cells and analysis as in panel A. (D) Hypothetical volume of cells observed on polylysin slides. Strains, cells and analysis as in panel A. Hypothetical value calculated from other values of cell dimension. (E) Mean number of cells counted per image. Strains as in panel A. (F) Percentage of cells per image which form part of chains. Strains as in panel A.
(TIF)
(A) Development of competence in wt background monitored by following expression of ssbB-luc transcriptional fusion. Competence development (RLU/OD) was plotted against growth (OD492) in a gradient of varying starting pHs. Strain used, TD259. (B) Development of competence in a fecE mutant. Strain used TD263. Experimental information and figure layout as in panel A. (C) Development of competence in a fecE codY double mutant. Strain used TD265. Experimental information and figure layout as in panel A. (D) Derepression of ami antagonizes competence revealed by monitoring competence development in an ami CYbs0 mutant. Strain used TD260. Experimental information and figure layout as in panel A. (E) Development of competence in a fecE ami CYbs0 double mutant. Strain used TD264. Experimental information and figure layout as in panel A.
(TIF)
Peptidoglycan was digested with the muramidase cellosyl and the resulting muropeptides were reduced with sodium borohydride and analysed by high-pressure liquid chromatography. Strains used (from top to bottom: R6, TD249, TD227, TK108, TD247 and TD75) are indicated on the right side.
(TIF)
(A) Section (5500–10500 Da) of the charge deconvoluted ESI-FT-ICR-MS spectrum of pnLTA isolated from strain D39 (wt, black) and TK108 (glnR - codY - (fecE -), red). (B) Enlarged image of the ion cluster with highest intensity (8450–8750 Da). Differences in the signal pattern of the two strains are caused by varying percentage of sodium adduct ion cluster, as indicated. For a clear visualization of mass differences, the most intensive peak of an ion cluster has been chosen instead of the monoisotopic peak.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.