

Abstract
To obtain mechanistic insights into the cross talk between lipolysis and autophagy, two key metabolic responses to starvation, we screened the autophagy-inducing potential of a panel of fatty acids in human cancer cells. Both saturated and unsaturated fatty acids such as palmitate and oleate, respectively, triggered autophagy, but the underlying molecular mechanisms differed. Oleate, but not palmitate, stimulated an autophagic response that required an intact Golgi apparatus. Conversely, autophagy triggered by palmitate, but not oleate, required AMPK, PKR and JNK1 and involved the activation of the BECN1/PIK3C3 lipid kinase complex. Accordingly, the downregulation of BECN1 and PIK3C3 abolished palmitate-induced, but not oleate-induced, autophagy in human cancer cells. Moreover, Becn1+/− mice as well as yeast cells and nematodes lacking the ortholog of human BECN1 mounted an autophagic response to oleate, but not palmitate. Thus, unsaturated fatty acids induce a non-canonical, phylogenetically conserved, autophagic response that in mammalian cells relies on the Golgi apparatus.
Keywords: ATG5, beclin 1, brefeldin A, GFP-LC3, RFP-FYVE
See also: VA Bankaitis (April 2015)
Introduction
Macroautophagy (here referred to as “autophagy”) relies on the sequestration of cytoplasmic structures in double-membraned vesicles that are commonly known as autophagosomes. Upon closure, autophagosomes fuse with lysosomes to generate autolysosomes, resulting in the degradation of the autophagosomal cargo and inner membrane by acidic hydrolases. The nucleation of autophagosomes is often initiated by unc-51-like autophagy-activating kinase 1 (ULK1) and generally requires phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3, also known as VPS34). PIK3C3 encodes the catalytic subunit of a class III phosphatidylinositol-3 kinase that operates within a large macromolecular complex involving beclin 1, autophagy related (BECN1), ATG14 and phosphoinositide-3-kinase, regulatory subunit 4 (PIK3R4, also known as VPS15) (Weidberg et al, 2011). The elongation of autophagosomal membranes relies on two conjugation reactions, that is, the conjugation of ATG12 to ATG5 and that of mammalian orthologs of yeast Atg8 to phosphatidylethanolamine. In this setting, microtubule-associated protein 1 light chain 3 (MAP1LC3, best known as LC3) and GABA(A) receptor-associated protein (GABARAP) family members are conjugated to phosphatidylethanolamine in process that resembles ubiquitination. Such a cascade of reactions involves ATG7 (an E1-like enzyme) and ATG3 (an E2-like enzyme), eventually yielding phosphatidylethanolamine-conjugated and autophagosome-associated LC3 and GABARAP proteins (Rubinsztein et al, 2012). Thus, the formation of autophagosomes can be quantified by following the redistribution of LC3B (the most studied member of the LC3/Atg8 family) to cytoplasmic puncta or by assessing its lipidation (which increases its electrophoretic mobility). In addition, the so-called autophagic flux (i.e. the actual ability of autophagosomes to degrade intracellular components) can be monitored by measuring the turnover of long-lived proteins or by determining the degradation of specific autophagic substrates such as sequestosome 1 (SQSTM1, best known as p62) (Bjorkoy et al, 2009). Of note, autophagic responses are negatively regulated by mechanistic target of rapamycin (MTOR) complex I (MTORCI), a key hub for the control of cell survival, growth and proliferation (Laplante & Sabatini, 2012).
Autophagy plays a major role in the differentiation and function of adipocytes (Singh et al, 2009b; Zhang et al, 2009), in the mobilization of lipid droplets within hepatocytes (a process that has been dubbed “lipophagy”) (Singh et al, 2009a), as well as in the oxidation of fatty acids (FAs) by cancer cells (Guo et al, 2013), underscoring its significant impact on lipid metabolism. Conversely, several saturated FAs (SFAs) and unsaturated FAs (UFAs) appear to modulate autophagy (Brenner et al, 2013). Moreover, neutral lipid droplets have recently been shown to contribute to autophagic responses by providing substrates for the formation of autophagosomes (Dupont et al, 2014a).
The UFAs oleate (C18:1) and linoleate (C18:2) stimulate the autophagic flux in both mammary epithelial cells (Pauloin et al, 2010) and hepatocytes (Mei et al, 2011). Along similar lines, polyunsaturated FAs including di-homo-γ-linoleic acid (C20:3), arachidonic acid (C20:4) and eicosapentaenoic acid (C20:5) induce autophagy in multiple cell types (Fukui et al, 2013; O'Rourke et al, 2013). Conversely, the autophagy-modulatory potential of the SFA palmitate (C16:0) exhibits a significant degree of variation. Indeed, palmitate has been shown to inhibit (Las et al, 2011; Kim et al, 2013) as well as to activate (Khan et al, 2012; Martino et al, 2012) autophagy in pancreatic islet and vascular endothelial cells. Moreover, palmitate reportedly limits autophagy in hepatocytes (Mei et al, 2011), yet promotes it in mouse embryonic fibroblasts (Tan et al, 2012). At least in some cell types, the autophagic response to palmitate involves protein kinase C (PKC) (Tan et al, 2012), eukaryotic translation initiation factor 2, subunit 1α, 35 kDa (EIF2S1, best known as eIF2α) and its substrate eIF2α kinase 2 (EIF2AK2, best known as PKR), and mitogen-activated protein kinase 8 (MAPK8, best known as JNK1) (Shen et al, 2012). In addition, palmitate has been shown to stimulate or inhibit AMP-activated protein kinase (AMPK), an upstream regulator of autophagy impinging on MTORCI signaling, depending on experimental variables including cell type, concentration and exposure time (Fediuc et al, 2006; Sun et al, 2008). Although these discrepancies have not been investigated in detail, they may reflect the ability of palmitate to modulate autophagy in a manner that changes with time, initially promoting it and then inhibiting it. In support of this notion, feeding mice with a high-fat diet for a prolonged period has been shown to promote alterations in intracellular lipids that are accompanied by defects in the fusion between autophagosomes and lysosomes (Koga et al, 2010).
Driven by the consideration that distinct FAs influence the composition of cellular membranes and affect long-term health in a different manner (Wijendran & Hayes, 2004; Cascio et al, 2012), we performed a systematic analysis of FAs for their capacity to induce autophagy in short-term experiments. Here, we report the unexpected finding that SFAs and UFAs promote autophagy by activating different molecular mechanisms. Thus, while SFAs stimulate canonical autophagy, UFAs trigger a BECN1- and PIK3C3-independent autophagic response that is accompanied by the redistribution of LC3 to Golgi-associated vesicles.
Results
Palmitate and oleate induce autophagy in vitro and in vivo
To identify FAs that trigger autophagy, we exposed human osteosarcoma U2OS cells that stably express a green fluorescent protein (GFP)-LC3 chimera (Shen et al, 2011) to a panel of 26 FAs differing in the length of the carbon chain as well as in saturation status, followed by assessment of GFP-LC3+ puncta by automated fluorescence microscopy. SFAs with 15–18 carbon atoms such as pentadecanoic, hexadecanoic, heptadecanoic and octadecanoic acid (stearate), but neither smaller nor larger SFAs, promoted the formation of GFP-LC3+ dots in U2OS cells (Fig1A). Similarly, several UFAs with 14–20 carbon atoms, including myristoleic, palmitoleic, oleic, linoleic and arachidonic acid, efficiently triggered the aggregation of GFP-LC3 in cytoplasmic dots (Fig1A). Based on solubility considerations and natural abundance in human cells (as well as in commonly employed oils, i.e. palm and olive oil), we chose to focus our study on one UFA, that is, oleic acid (oleate) and one SFA, that is, hexadecanoic acid (palmitate).
Figure 1.

- A Screening for autophagy-inducing fatty acids. U2OS cells expressing GFP-LC3 were cultured in control conditions (Co) or treated with the indicated saturated (in black) or unsaturated (in blue) fatty acids for 4 h, followed by the quantification of the number of cytoplasmic GFP-LC3+ dots per cell by automated fluorescence microscopy. Data are means ± SEM of at least three independent experiments (*P < 0.05, **P < 0.01 versus untreated cells).
- B–D Autophagic flux induced by palmitate, stearate and oleate. Wild-type (WT, C) and GFP-LC3-expressing (B) U2OS cells as well as HeLa cells (D) were maintained in control conditions, exposed to nutrient-free (NF) conditions or treated with 500 μM oleate (OL), 500 μM stearate (ST) or 500 μM palmitate (PA), alone or in combination with 10 μg/ml E-64d and 10 μg/ml pepstatin (Pep) for 4 (D) or 6 (B, C) h. Thereafter, the number of cytoplasmic GFP-LC3+ dots per cell (B), LC3 lipidation and p62 degradation (C) or the degradation of labeled long-lived proteins (D) was quantified. In (B) and (D), data are means ± SEM (B) or normalized means ± SD (D) of at least three independent experiments (B), or four replicate assessments from one representative (D) experiment(s) out of three performed (*P < 0.05, **P < 0.01 versus untreated cells; ##P < 0.01 versus cells treated with PA or OL only; n.s., not significant versus cells treated with E-64d plus Pep). In (C), β-actin levels were monitored to ensure equal loading of lanes, and densitometry was employed to quantify the abundance of lipidated LC3 (LC3-II) and p62 (both normalized to β-actin levels).
- E,F Involvement of ATG5 and ATG7 in fatty acid-induced autophagy. WT (F) or GFP-LC3-expressing (E) U2OS cells were transfected with a control siRNA (siUNR) or with siRNAs targeting ATG5 (siATG5) or ATG7 (siATG7) for 48 h and either maintained in control conditions or treated with 500 μM PA or 500 μM OL. Six hours later, cells were processed for the quantification of the number of cytoplasmic GFP-LC3+ dots per cell by automated fluorescence microscopy (E) or for the assessment of LC3 lipidation and p62 degradation by immunoblotting (F). In (E), data are means ± SEM of at least three independent experiments (*P < 0.05, **P < 0.01 versus untreated siUNR-transfected cells; #P < 0.05 versus siUNR-transfected cells treated with PA or OL only). In (F), β-actin levels were monitored to ensure equal loading of lanes.
- G Induction of autophagy by fatty acids in mice. C57BL/6 mice were injected i.p. with vehicle only, 100 mg/kg PA or 100 mg/kg OL. One or two hours later, animals were euthanatized and LC3 lipidation, p62 degradation and AMPK phosphorylation were assessed by immunoblotting in the indicated tissues. GAPDH levels were monitored to ensure equal loading of lanes. Densitometry was employed to quantify the abundance of lipidated LC3 (LC3-II) and p62 (both normalized to GAPDH levels) and AMPK phosphorylation (normalized to total AMPK levels). Results are means ± SD of three mice (*P < 0.05, **P < 0.01, ***P < 0.001, versus vehicle-treated mice).
The increase in GFP-LC3+ puncta, LC3 lipidation and p62 degradation induced by palmitate and oleate was exacerbated by the addition of E-64d and pepstatin (Fig1B and C), two well-known inhibitors of lysosomal proteases. This indicates that palmitate and oleate do not stimulate the mere accumulation of LC3 at cytoplasmic structures, but truly increase the autophagic flux. Of note, the same results were obtained with stearate (C18:0) (Fig1B and C), indicating that palmitate constitutes an appropriate saturated control for oleate despite bearing a slightly smaller carbon chain. Along similar lines, both palmitate and oleate accelerated the degradation of long-lived proteins, which is one of the hallmarks of functional autophagy (Klionsky et al, 2012), in thus far resembling nutrient deprivation (Fig1D). The autophagy-stimulatory activity of palmitate and oleate was equally suppressed by the small interfering RNA (siRNA)-mediated knockdown of two proteins critically involved in the late steps of autophagosomal elongation, that is, ATG5 and ATG7 (Fig1E). Moreover, the depletion of ATG5 and ATG7 virtually abolished the ability of palmitate and oleate to promote the lipidation of endogenous LC3 and the degradation of p62 in wild-type U2OS cells (Fig1F). Palmitate and oleate administered i.p. caused a rapid autophagic response in the heart, liver and skeletal muscle of wild-type (WT) C57BL/6 mice, which was characterized by LC3 lipidation, p62 degradation and phosphorylation of AMPK (Fig1G).
Altogether, these findings indicate that saturated as well as unsaturated FAs stimulate a rapid autophagic response, both in vitro and in vivo.
Requirement for the Golgi apparatus in the autophagic response to oleate
To monitor the implication of different subcellular compartments in the pro-autophagic activity of palmitate and oleate, we assessed the colocalization of LC3 aggregates with organelle-specific markers. Oleate, but not palmitate, promoted the accumulation of GFP- or red fluorescence protein (RFP)-tagged variants of LC3 in dots that colocalized with several proteins of the trans-Golgi network, including trans-Golgi network protein 2 (TGOLN2) and galactose-1-phosphate uridylyltransferase (GALT) (Fig2A and B). As a control, no such colocalization was induced by brefeldin A (BFA), which disrupts the Golgi apparatus (Fig2A and B). Along similar lines, oleate-induced GFP-LC3+ puncta, but not their palmitate-induced counterparts, colocalized with lectin, mannose-binding, 1 (LMAN1) (Fig2C), a marker of the endoplasmic reticulum–Golgi intermediate compartment. In addition, oleate was more efficient than palmitate at inducing the colocalization of GFP-LC3+ dots with RAB7A, a protein residing in late endosomes, while BFA completely failed to do so (Fig2D). In order to test whether this observation also applied to other FAs, we exposed U2OS cells stably co-expressing RFP-LC3 and a GFP-tagged variant of GALT to the 26 FAs initially screened for their ability to promote autophagy (Fig1A). We found that several UFAs other than oleate, but none of the SFAs tested here, promote the accumulation of RFP-LC3+ dots that colocalize with the trans-Golgi apparatus (Supplementary Fig S1A).
Figure 2.
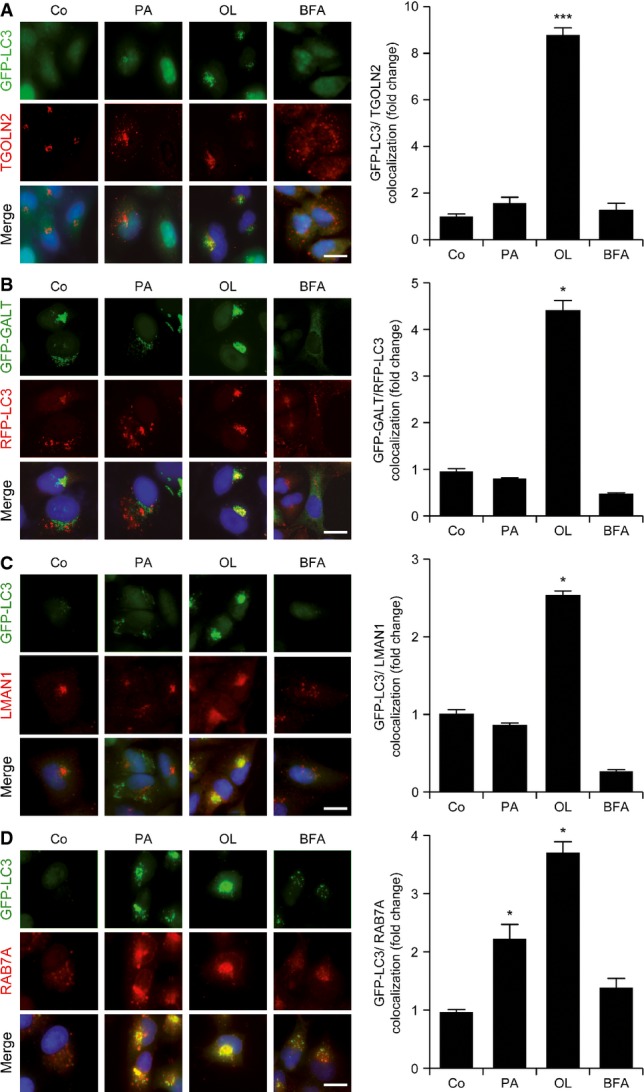
- A–D Colocalization of GFP-LC3+ dots with the Golgi apparatus in cells exposed to oleate. GFP-LC3- or GFP-GALT/RFP-LC3-expressing U2OS cells were maintained in control conditions (Co) or treated with 500 μM palmitate (PA), 500 μM oleate (OL) or 10 μg/ml brefeldin A (BFA) for 6 h. Thereafter, cells were processed to determine the colocalization between GFP-LC3+ or RFP-LC3+ dots and TGOLN2 (A), GALT-GFP (B), LMAN1 (C) and RAB7A (D). Data are normalized means ± SEM of at least three independent experiments (*P < 0.05, ***P < 0.001 versus untreated cells). Scale bars, 10 μm.
To obtain further insights into the involvement of the Golgi apparatus in oleate-triggered autophagy, we performed transmission electron microscopy assessments. Dual immunogold staining revealed that palmitate and oleate reduce the mean distance between GFP-LC3 and p62 (Fig3A), corroborating the notion that both these FAs activate an autophagic response. In addition, while palmitate-treated U2OS cells frequently exhibited fracture-like structures, perhaps reflecting its ability to promote an expansion of the endoplasmic reticulum (Peng et al, 2011), oleate induced vacuolar structures that colocalized with the Golgi apparatus to the nuclear periphery (Fig3B). Interestingly, immunogold labeling allowed us to localize GFP-LC3 to the boundaries of palmitate-induced fractures as well as to the perinuclear vesicles promoted by oleate (Fig3C).
Figure 3.

- A–C Colocalization of autophagic markers with the Golgi apparatus in cells exposed to oleate. GFP-LC3-expressing U2OS cells were maintained in control conditions (Co) or treated with 500 μM palmitate (PA), 500 μM oleate (OL) for 6 h, optionally processed for the differential detection of GFP-LC3 and p62 by immunogold labeling and analyzed by transmission electron microscopy. Solid and empty arrowheads indicate gold particles conjugated to anti-p62 (Ø = 10 nm) and anti-GFP (Ø = 15 nm) antibodies. Data are means ± SD of at least three independent experiments (*P < 0.05, ***P < 0.001 versus untreated cells).
The LC3+ structures accumulating in response to oleate colocalized not only with GALT+ Golgi vesicles (Fig4A), but also with the autophagic substrate p62 (Fig4B and C). Importantly, BFA turned out to abolish the capacity of oleate to promote the accumulation of subcellular structures co-staining positively for autophagosomal markers, proteins of the Golgi apparatus and autophagic substrates (Fig4A–C). Along similar lines, BFA limited the ability of oleate (but not palmitate) to accelerate the degradation of p62 and the lipidation of LC3 in U2OS cells, as well as the catabolism of long-lived proteins in human cervical carcinoma HeLa cells (Fig4D and E). This is remarkable given the documented ability of BFA to promote autophagy (Ding et al, 2007), which is also evident in the immunoblotting experiments that we performed (Fig4D). These results prompted us to investigate whether the structural or functional integrity of the Golgi apparatus is affected by the administration of FAs. The abundance of several markers of the Golgi apparatus including LMAN1, golgin A2 (GOLGA2) and golgi brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1) did not change in U2OS cells maintained in conditions in which oleate and palmitate efficiently promoted p62 degradation (Supplementary Fig S1B). Moreover, while BFA compromised protein glycosylation and inhibited the exocytic/endocytic functions of U2OS cells, palmitate and oleate failed to do so (Supplementary Fig S1C–E).
Figure 4.

- A–C Impact of brefeldin A on the colocation of autophagic markers with the Golgi apparatus induced by oleate. GFP-GALT/RFP-LC3-expressing (A), GFP-LC3-expressing (B) or GFP-GALT-expressing (C) U2OS cells were cultured in control conditions (Co) or treated with 500 μM oleate (OL), alone or combined with 10 μg/ml brefeldin A (BFA), for 6 h. Thereafter, cells were processed to assess the colocalization of autophagic markers (GFP-LC3+ dots and p62) and GFP-GALT+ structures. Data are normalized means ± SEM of at least 3 independent experiments (**P < 0.01, ***P < 0.001 versus untreated cells; #P < 0.05, ##P < 0.01 versus OL-treated cells). Scale bars, 10 μm.
- D,E Inhibition of oleate-induced autophagy by Golgi apparatus-disrupting agents. Wild-type U2OS (D) or HeLa (E) cells were maintained in control or nutrient-free (NF) conditions or treated with 500 μM palmitate (PA) or 500 μM OL, alone or combined with 10 μg/ml BFA, for 6 h. Thereafter, cells were processed to assess LC3 lipidation and p62 degradation by immunoblotting (D) or long-lived protein degradation (E). In (D), β-actin levels were monitored to ensure equal loading of lanes. In (E), data are normalized means ± SEM of four replicate assessments from one representative experiment out of three performed (n.s., non-significant, **P < 0.01 versus residual degradation upon BFA administration to cells maintained in control conditions).
Taken together, these observations suggest that oleate has the unique capacity to stimulate an autophagic response that relies on, but does not compromise, the structural and functional integrity of the Golgi apparatus.
Induction of non-canonical autophagy by oleate
The siRNA-mediated depletion of AMPK, PKR and JNK1 (Supplementary Fig S2A) inhibited the ability of palmitate, but not oleate, to induce the accumulation of GFP-LC3+ dots in cultured U2OS cells (Fig5A). This may relate to the fact that SFAs (such as palmitate), but not UFAs (such as oleate), promote the activation of JNK1 in vitro (Holzer et al, 2011). Thus, it seems that palmitate and oleate trigger autophagy via distinct biochemical mechanisms. In line with this notion, the knockdown of carnitine palmitoyltransferase 1A (CPT1A) (Supplementary Fig S2A), which is required for the import of long-chain FAs destined to β oxidation into the mitochondrial matrix (Galluzzi et al, 2013), inhibited the formation of GFP-LC3+ triggered by palmitate, but not by oleate (Fig5B).
Figure 5.

- A,B Implication of AMPK, PKR, JNK1 and CPT1A in palmitate-induced, but not oleate-induced, autophagy. GFP-LC3-expressing U2OS cells were transfected with a control siRNA (siUNR) or with siRNAs targeting AMPK (siAMPK), PKR (siPKR), JNK1 (siJNK1) or CPT1A (siCPT1A) for 48 h and either maintained in control conditions (Co) or treated with 500 μM palmitate (PA) or 500 μM oleate (OL). Six hours later, GFP-LC3+ dots were quantified by automated fluorescence microscopy. Data are means ± SEM of at least three independent experiments (*P < 0.05, **P < 0.01 versus untreated siUNR-transfected cells; #P < 0.05, ##P < 0.01, ###P < 0.001 versus siUNR-transfected cells treated with PA; n.s., non-significant versus siUNR-transfected cells treated with OL). Scale bars, 10 μm.
- C,D Upstream regulators of palmitate-induced, but not oleate-induced, autophagy. Wild-type (WT, D) and GFP-LC3-expressing (C) U2OS cells were transfected with siUNR or with siRNAs specific to BECN1 (siBECN1), PIK3C3 (siPIK3C3) and ULK1 (siULK1) for 48 h and either kept in control conditions or treated with 500 μM PA or 500 μM OL for additional 6 h. Thereafter, cells were processed for the quantification of GFP-LC3+ dots by automated fluorescence microscopy, or for the assessment of LC3 lipidation and p62 degradation by immunoblotting. In (C), data are means ± SEM of at least three independent experiments (**P < 0.01 versus untreated siUNR-transfected cells; #P < 0.05 versus siUNR-transfected cells treated with PA; n.s., non-significant versus siUNR-transfected cells treated with OL). Scale bars = 10 μm. In (D), β-actin levels were monitored to ensure equal loading of lanes, and densitometry was employed to quantify the abundance of lipidated LC3 (LC3-II) and p62 (both normalized to β-actin levels). Data are means ± SD of at least 3 independent experiments (*P < 0.05, **P < 0.01 versus untreated siUNR-transfected cells; #P < 0.05 versus siUNR-transfected cells treated with PA; n.s., non-significant versus siUNR-transfected cells treated with OL).
- E–H Effects of PIK3C3 inhibitors on palmitate and oleate-induced autophagy. GFP-LC3-expressing U2OS cells (E, F) and WT HeLa cells (G, H) were cultured in control conditions, maintained in nutrient-free (NF) medium or treated with the indicated concentrations of PA (500 μM where not specified) and OL (500 μM where not specified), alone or in the presence of 5 mM 3-methyladenine (3MA) or 200 nM wortmannin (Wort). Six hours later, cells were processed for the quantification of GFP-LC3+ dots by automated fluorescence microscopy (E, F), for the evaluation of LC3A, LC3B and GABARAPL2 lipidation and p62 degradation by immunoblotting (G), or for the assessment of long-lived protein degradation (H). In (E, F), data are means ± SEM of at least two independent experiments (**P < 0.01 versus untreated cells, #P < 0.05, ##P < 0.01, ###P < 0.001 versus cells treated with PA; n.s., non-significant versus cells treated with OL). Scale bars, 10 μm. In (G), β-actin levels were assessed to monitor lane loading and densitometry was employed to quantify the abundance of lipidated LC3-like proteins (LC3A-II, LC3B-II and GATE16-II) and p62 (both normalized to β-actin levels). In (H), data are normalized means ± SEM of four replicate assessments from one representative experiment out of three performed (n.s., non-significant, **P < 0.01 versus residual degradation upon 3MA administration to cells maintained in control conditions).
The knockdown of ULK1, BECN1 and PIK3C3 with specific siRNAs affected several manifestations of the autophagic response of U2OS cells to palmitate, including the accumulation of GFP-LC3+ dots (Fig5C), the lipidation of endogenous LC3 and the degradation of p62 (Fig5D), but failed to interfere with oleate-induced autophagy. Accordingly, two distinct pharmacological inhibitors of PIK3C3, namely, 3-methyladenine and wortmannin, limited the ability of palmitate, but not that of oleate, to promote the formation of GFP-LC3+ puncta (Fig5E and F) and the lipidation of endogenous LC3, LC3A and GABARAP-like 2 (GABARAPL2, best known as GATE16) in U2OS cells, as well as p62 degradation in HeLa cells (Fig5G). Moreover, 3-methyladenine suppressed the degradation of long-lived proteins as promoted in HeLa cells by nutrient deprivation and palmitate, but not oleate (Fig5H). The siRNA-mediated downregulation of BECN1 and PIK3C3 actually inhibited the accumulation of GFP-LC3+ dots triggered by a panel of SFAs, but not by UFAs (Supplementary Fig S2B). Accordingly, SFA-induced autophagy, but not its UFA-induced counterpart, proceeded along with the generation of phosphatidylinositol 3-phosphate [PtdIns(3)P] (Supplementary Fig S2C), as determined by a RFP-tagged short peptide (FYVE) that specifically binds to PtdIns(3)P-containing membranes (Zhang et al, 2007). As an internal control, the accumulation of RFP-FYVE+ dots triggered by SFAs was abolished by the depletion of BECN1 and PIK3C3 (Supplementary Fig S2C).
In an attempt to obtain further insights into the molecular mechanisms that are responsible for the autophagic response to oleate, we individually downregulated several proteins that have also been implicated in non-canonical variants of autophagy, including autophagy/beclin-1 regulator 1 (AMBRA1), ATG14, high-mobility group box 1 (HMGB1), KIAA0226 (best known as RUBICON), PTEN-induced putative kinase 1 (PINK1), SH3-domain GRB2-like endophilin B1 (SH3GLB1, best known as BIF1), UV radiation resistance associated (UVRAG) and vacuole membrane protein 1 (VMP1) (Codogno et al, 2012). None of these interventions, however, abolished the ability of oleate to promote the accumulation of GFP-LC3+ dots in U2OS cells (Supplementary Fig S3A). We therefore investigated whether oleate-induced autophagy is influenced by MTORCI signaling. To this aim, we took advantage of mouse embryonic fibroblasts (MEFs) lacking tuberous sclerosis 2 (TSC2), a key component of the upstream MTORCI-inhibiting TSC complex (Inoki et al, 2002). Similar to nutrient and growth factor deprivation, both palmitate and oleate inhibited the catalytic activity of MTORCI in WT MEFs, as assessed by the phosphorylation state of the MTORCI substrate ribosomal protein S6 kinase, 70 kDa, polypeptide 1 (RPS6KB1, best known as p70S6K) (Fig6). Tsc2−/− MEFs manifested a reduced autophagic response to palmitate and oleate, as monitored by p62 degradation and LC3 lipidation, correlating with an increased phosphorylation of p70S6K (Fig6). These results were corroborated in U2OS cells subjected to the siRNA-mediated downregulation of TSC2 or phosphatase and tensin homolog (PTEN), another upstream inhibitor of MTORCI (Supplementary Fig S3B). Oleate is known to modulate the enzymatic activity of both phospholipase D1, phosphatidylcholine specific (PLD1), and phospholipase D2 (PLD2) (Kim et al, 1999), and these enzymes have previously been shown to influence autophagic responses (Dall'Armi et al, 2010; Liu et al, 2013). We therefore tested whether PLDs would contribute to oleate-induced autophagy. We found that VU0155069 (a selective inhibitor of PLD1), BML-279 (a mixed PLD1/PLD2 inhibitor) and BML-280 (a selective inhibitor of PLD2) all fail to inhibit LC3 lipidation as induced by oleate in U2OS cells (Supplementary Fig S4A). Rather, these pharmacological inhibitors of PLDs per se promoted LC3 lipidation, at least to some extent. Along similar lines, the partial depletion of PLD1 by RNA interference failed to interfere with oleate-induced LC3 lipidation in U2OS cells (Supplementary Fig S4B).
Figure 6.
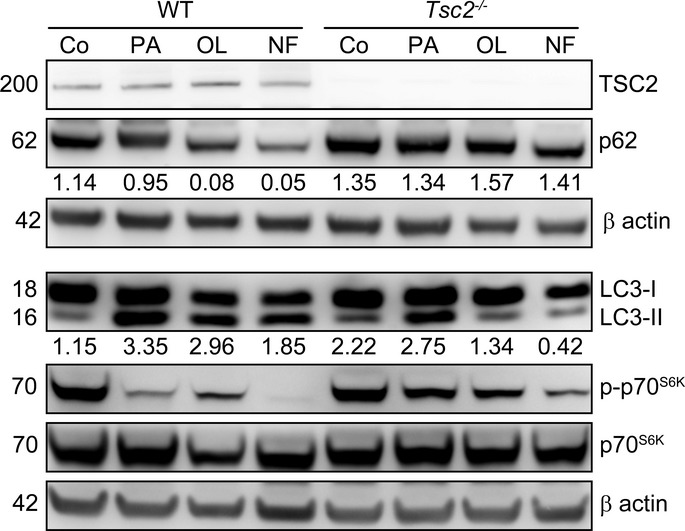
Mechanistic insights into oleate-induced autophagy
Sensitivity of oleate-induced autophagy to MTORCI signaling. Wild-type (WT) or Tsc2−/− MEFs were cultured in control conditions, maintained in nutrient-free (NF) medium or exposed to 500 μM palmitate (PA) or 500 μM oleate (OL) for 6 h, followed by the assessment of LC3 lipidation, p62 degradation and p70S6K kinase phosphorylation. TSC2 and β-actin levels were monitored as a genotype control and to ensure equal loading of lanes, respectively, and densitometry was employed to quantify the abundance of lipidated LC3 (LC3-II) and p62 (both normalized to β-actin levels).
Altogether, these data indicate that SFAs promote canonical autophagy while UFAs can induce a non-canonical autophagic response that does not rely on the class III phosphatidylinositol-3 kinase activity of the BECN1/PIK3C3 complex, but proceeds along with the inhibition of MTORCI and is responsive to MTORCI hyperactivation.
BECN1-independent autophagic responses to oleate in vivo
To test the pathophysiological relevance of our results, we administered palmitate and oleate to WT or Becn1+/− C57BL/6 mice, followed by the assessment of autophagic markers in multiple organs. Oleate promoted the lipidation of LC3 and the degradation of p62, as determined by immunoblotting, in both WT and Becn1+/− livers, while palmitate did so only in the former (Fig7A). Similar results were obtained in the kidneys (Supplementary Fig S5). The lipidation of LC3 induced by oleate in Becn1+/− livers was exacerbated by the co-administration of leupeptin, an inhibitor of lysosomal proteases (Ezaki et al, 2011) (Fig7B), indicating that oleate is capable of upregulating the autophagic flux in vivo, even in the context of Becn1 haploinsufficiency.
Figure 7.
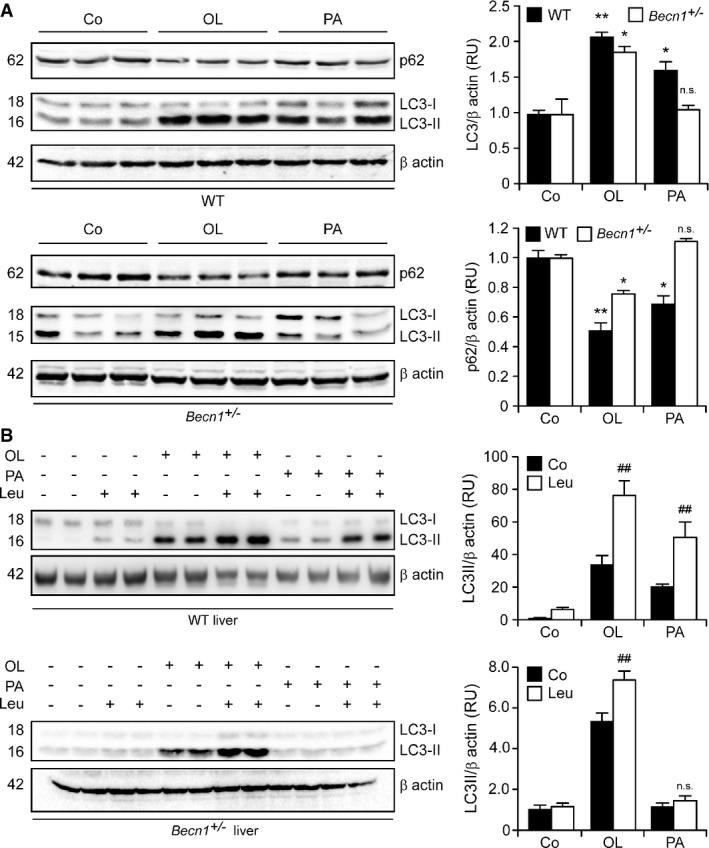
- A,B Becn1-independent induction of the autophagic flux by oleate in mice. Wild-type (WT) or Becn1+/− mice were injected intraperitoneally with vehicle, 100 mg/kg palmitate (PA) or 100 mg/kg oleate (OL). Four hours later, mice received vehicle or 15 mg/kg leupeptin (Leu) later. After 2 h, animals were euthanatized, followed by the immunoblotting-assisted assessment of LC3 lipidation and p62 degradation in the liver. β-actin levels were monitored to ensure equal loading of lanes, and densitometry was employed to quantify the relative abundance of lipidated LC3 (LC3-II) and p62, both normalized to β-actin levels. Results are means ± SD of 2 or 3 mice (n.s., non-significant, *P < 0.05, **P < 0.01 versus untreated mice of the same genotype; ##P < 0.01 versus mice of the same genotype treated with the same fatty acid).
Moreover, a Saccharomyces cerevisiae strain lacking atg6 (coding for the yeast ortholog of mammalian BECN1) was impaired in its ability to target palmitate-derived, but not oleate-derived, lipid droplets to VPH1+ lysosomes via autophagy (Fig8A). Along similar lines, the knockdown of BEC-1, the Caenorhabditis elegans ortholog of mammalian BECN1, inhibited the accumulation of LGG-1+ puncta in worms exposed to palmitate, but not in nematodes treated with oleate (Fig8B).
Figure 8.
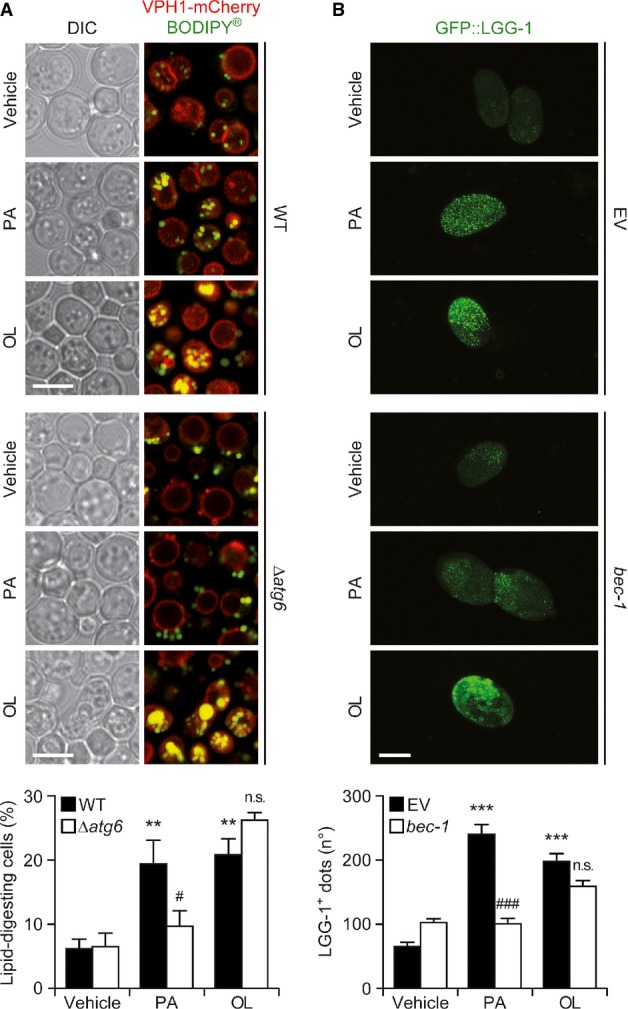
- Atg6-independent induction of autophagy by oleate in yeast. Exponentially growing WT or Δatg6 S. cerevisiae cells were exposed to vehicle, 1.8 mM palmitate (PA) or 1.8 mM oleate (OL) for 72 h, followed by the immunofluorescence microscopy-assisted quantification of cells exhibiting BODIPY®+ lipid droplets within VPH1+ vacuoles (lipid-digesting cells). Scale bars, 5 μm. Data are means ± SEM of 4 independent experiments (**P < 0.01 versus WT cells treated with vehicle; n.s., not significant, #P < 0.05 versus WT cells treated with the same fatty acid). DIC, differential interference contrast.
- BEC-1-independent induction of autophagy by oleate in nematodes. A C. elegans strain expressing GFP::LGG-1 was reared from the L4 stage on plates containing vehicle only, 500 μM PA or 500 μM OL and fed with E. coli transformed with either an empty vector (EV) or with a construct for the downregulation of BEC-1. Gravid animals were treated with sodium hypochlorite, and the number of GFP::LGG-1+ dots was quantified in released embryos by fluorescence microscopy. Scale bars, 30 μm. Data are means ± SEM of 2 independent experiments (***P < 0.001 versus worms fed with the EV and treated with vehicle; n.s., not significant, ###P < 0.001 versus worms fed with the EV and treated with the same fatty acid).
These experiments confirm that UFAs induce a non-canonical, BECN1-independent autophagic program in mice, yeast cells and nematodes.
Discussion
A number of stimuli including several chemotherapeutics and toxins have been shown to promote non-canonical, BECN1-independent autophagic responses, generally in cultured cells (Dupont & Codogno, 2013). In most of these settings, non-canonical autophagy accompanies cell death, representing an unsuccessful attempt of adaptation to stress (Galluzzi et al, 2015). It has been proposed that BECN1-independent autophagy may have evolved along with the ability of several viruses to suppress cell-intrinsic adaptive responses to infection by inhibiting BECN1 (Munz, 2011). In this scenario, an autophagic response promoting the demise of cells that cannot clear infection by autophagy would indeed be beneficial for the whole organism. The pathophysiological significance of non-canonical autophagic responses to oleate remains to be elucidated. To this aim, it will be interesting to determine whether and how the absence of BECN1 affects oleate-induced cell death, as well as the implication of the Golgi apparatus in this process. Intriguingly, oleate triggers non-canonical autophagy in organisms as evolutionary distant as yeast, nematodes and mammals. It is therefore tempting to speculate that BECN1-independent autophagic responses arose relatively early during evolution, perhaps as an adaptation to metabolic, rather than viral, cues.
Several genetic and pharmacological interventions that disrupt the Golgi apparatus have been shown to induce autophagy in human cell lines. This applies to the knockdown of GBF1 and N-ethylmaleimide-sensitive factor attachment protein α (NAPA), as well as to the administration of BFA, which inhibits GBF1, ADP-ribosylation factor guanine nucleotide exchange factor 1 (ARFGEF1) and ARFGEF2, or golgicide A, which selectively targets GBF1. Among these Golgi apparatus-disrupting interventions, the depletion of NAPA is the only one that reportedly triggers BECN1-independent autophagy (Naydenov et al, 2012). This said, oleate failed to dismantle the Golgi apparatus in our experiments (Supplementary Fig S3), implying that this is not the mechanism whereby oleate promotes non-canonical autophagy. Rather, disrupting the Golgi apparatus with BFA interfered with the ability of oleate to promote (i) the colocalization of LC3+ autophagosomes with the autophagic substrate p62, (ii) LC3 lipidation, (iii) p62 degradation and (iv) the catabolism of long-lived proteins (Fig4), suggesting that oleate-induced autophagy obligatorily relies on an intact Golgi apparatus. Intriguingly, oleate-induced autophagy is not influenced by the downregulation of AMPK (Fig5), but proceeds along with the inhibition of MTORCI (Fig6) and can be partially suppressed by MTORCI hyperactivation. Further experiments are required to clarify in detail the molecular pathways through which oleate triggers non-canonical autophagy.
In summary, our findings demonstrated that unsaturated FAs can induce non-canonical, BECN1-independent autophagy in vitro and in vivo through a phylogenetically conserved mechanism that requires an intact Golgi apparatus.
Materials and Methods
Chemicals, cell lines and culture conditions
Unless otherwise specified, chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA), culture media and supplements for cell culture from Gibco®-Life Technologies™ (Carlsbad, CA, USA) and plasticware from Corning Inc. (Corning, NY, USA). Golgicide A and VU0155069 were from Tocris Bioscience (Bristol, UK). BML-279 and BML-280 were from Enzo Life Sciences Inc. (Farmingdale, NY, US). Human cervical carcinoma HeLa cells as well as WT human osteosarcoma U2OS cells and their RFP-FYVE-, GFP-GALT-, GFP-GALT/RFP-LC3- and GFP-LC3-expressing derivatives were maintained at 37°C under 5% CO2, in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 mg/l sodium pyruvate, 10 mM HEPES buffer, 100 units/ml penicillin G sodium and 100 μg/ml streptomycin sulfate. WT and Tsc2−/− MEFs were maintained in the same medium further supplemented with non-essential amino acids. Cells were seeded in 6- or 12-well culture plates or in 10 or 15 Ø cm culture dishes and allowed to adapt for 24 h before experimental interventions. Serum and nutrient deprivation was achieved by culturing cells in serum-free Earle's balanced salt solution (EBSS).
RNA interference
Cells at 50% confluence were transfected with a custom-made, non-targeting siRNA (siUNR, 5′-GCCGGUAUGCCGGUUAAGUdTdT-3′) (Galluzzi et al, 2010) or with siRNAs specific to AMBRA1 (#217068 and #25229, from Ambion®-Life Technologies™), AMPK (#HA02727114, from Sigma-Aldrich), ATG5 (5′-UUUCUUCUUAGGCCAAAGGdTdT-3′), ATG7 (5′-UUGAGAUUUAGAUCCACUGAdTdT-3′), ATG14 (5′-GACGAUCCCAUAUAUUAGAdTdT-3′ and 5′-GCAUACCCUCAGGAAUCUAdTdT-3′), BECN1 (5′-UUCCGUAAGGAACAAGUCGGdTdT-3′), CPT1A (#SASI_Hs01 00231321, from Sigma-Aldrich), EIF2AK2 (5′-UAAUAACGUAAGUCUUUCCGdTdT-3′), HMGB1 (#106695 and #106696, from Ambion®-Life Technologies™), KIAA0226 (#272287 and #272288, from Ambion®-Life Technologies™), MAPK8 (from Cell Signaling Technology, Danvers, MA, USA), PIK3C3 (5′-ACGGTGATGAATCATCTCCdTdT-3′), PINK1 (#SI00287931 and #SI00287924, from Qiagen, Hilden, Germany), PLD1 (5′-CUGGAAGAUUACUUGACAAdTdT-3′), PTEN (5′-GUAUAGAGCGUGCAGAUAAdTdT-3′), SH3GLB1 (#134763 and #134764, from Ambion®-Life Technologies™), ULK1 (#SI02223270, from Qiagen), TSC2 (#5269468, Invitrogen®-Life Technologies™), UVRAG (5′-CGUGGCGAAGUCUCGAUUUdTdT-3′ and 5′-GCUGUUGAUUGAAUGGAAAdTdT-3′), or VMP1 (5′-UAUACGUUGCACAUACUGUUGAUGCdTdT-3′ and 5′-GCAUCAACAGUAUGUGCAACGUAUAdTdT-3′) by means of the HiPerfect Transfection Reagent (Qiagen), as previously described (Galluzzi et al, 2012). Transfection efficiency was monitored 48 h later by immunoblotting (see below).
Immunofluorescence microscopy
Immunofluorescence microscopy was performed according to standardized procedures (Hoffmann et al, 2008; Vitale et al, 2010). In brief, cells grown on coverslips were fixed with 4% paraformaldehyde (PFA, w:v in H2O) for 15 min at room temperature and permeabilized with 0.1% Triton X-100 for 10 min, followed by the blocking of non-specific binding sites by incubation in 5% bovine serum albumin (BSA, w:v in PBS) for 15 min. Thereafter, cells were incubated overnight at 4°C with antibodies specific to the detection of LMAN1 (#ALX-804-602-C100, clone G1/93, Enzo Life Sciences Inc.), RAB7A (#9367, clone D95F2, Cell Signaling Technology), SQSTM1 (#H00008878-M01, clone 2C11, Abnova, Taipei, Taiwan) and TGOLN2 (#AHP500G, AbD Serotec, Raleigh, NC, USA). Primary antibodies were detected with appropriate AlexaFluor™ conjugates (Molecular Probes®-Life Technologies™). Nuclear counterstaining was invariably achieved by staining cells with 10 μg/ml Hoechst 33342 conjugates (Molecular Probes®-Life Technologies™). Epifluorescence and confocal fluorescence microscopy assessments (40–63×) were performed on a DM IRE2 HC FLUO microscope (Leica Microsystems, Wetzlar, Germany) equipped with a CoolSNAP EZ CCD camera (Photometrics, Tucson, AZ, USA), on an LSM 510 microscope equipped with an ApoTome.2 system (both from Carl Zeiss, Jena, Germany) or a TCS SPE confocal microscope (Leica Microsystems).
Automated fluorescence microscopy
U2OS cells stably expressing GFP-GALT, GFP-LC3, RFP-FYVE or GFP-GALT/RFP-LC3 were seeded in 96-well BD High-Content Imaging Plates (BD Falcon, Franklyn Lanes, NJ, USA) and allowed to adapt for 24 h. Upon treatment, cells were fixed with 4% PFA for 15 min at room temperature and counterstained with 10 μg/ml Hoechst 33342. Images were acquired using a Pathway™ 855 High-Content Imager (BD Imaging Systems, San José, CA, USA) equipped with a 40× objective (Olympus, Center Valley, CA, USA) coupled to a robotized Twister II plate handler (Caliper Life Sciences, Hopkinton, MA, USA). Images were analyzed for the presence of GFP-LC3+, RFP-LC3+ and RFP-FYVE+ cytoplasmic dots and/or for GFP-GALT+ subcellular compartments by means of the Attovision v.1.7.1 software package (BD Imaging Systems). To this aim, region of interests (ROIs) corresponding to cells were segmented and subdivided into a cytoplasmic and nuclear area based on Hoechst 33342 staining, according to the proprietary procedures. 2 × 2 Rolling Ball (RB) and Marr-Hildreth or 5 × 5 RB algorithms were used to recognize cytoplasmic GFP-LC3+, RFP-FYVE+, RFP-LC3+ or GFP-GALT+ regions. Co-localization studies were performed based on M1 and M2 co-localization coefficients, estimating the contribution of one channel to the area co-localized with the other channel, independently of signal intensity. Statistical analyses were implemented on the R software package (http://www.r-project.org/).
Degradation of long-lived proteins
The autophagic degradation of long-lived proteins was performed as previously described (Dupont et al, 2014b). Briefly, cells were incubated with 0.2 μCi/ml [14C]-valine for 18 h at 37°C. Unincorporated radioisotope was removed by three washes in PBS (pH 7.4). Cells were then maintained in the presence of 0.1% BSA and 10 mM unlabeled valine for 1 h. Thereafter, the culture medium was discarded, and cells were placed in control conditions or pre-incubated for 1 h with 10 μg/ml brefeldin A, 5 mM 3-methyladenine or 10 μg/ml E-64d plus 10 μg/ml pepstatin. Cells were then placed in control conditions, subjected to serum and nutrient deprivation or treated with 500 μM oleate or palmitate for additional 4 h. Finally, radiolabeled proteins from the 4-h chase medium were precipitated in 10% trichloroacetic acid (v/v in H2O) at 4°C. Precipitated proteins were separated from soluble radioactivity by centrifugation at 600 g for 10 min and dissolved in 250 μl Soluene® 350 (Perkin Elmer, Waltham, MA, USA). The rate of protein degradation was calculated starting from the acid-soluble radioactivity recovered from both cells and the culture medium.
Immunoblotting
Cells were washed with cold PBS and lysed in a buffer containing 1% NP-40, 20 mM HEPES (pH 7.9), 10 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM orthovanadate, 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 mM dithiothreitol, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 10 μg/ml pepstatin, as previously described (Galluzzi et al, 2012). Lysates were separated (25 μg proteins per lane) on pre-cast 4–12% polyacrylamide NuPAGE® Novex® Bis-Tris gels (Invitrogen®-Life Technologies™) or 12% Tris-Glycine Mini-PROTEAN® TGX™ gels (Bio-Rad, Hercules, CA, USA) and electrotransferred to Immobilon™ membranes (Millipore Corporation, Billerica, MA, USA). Membranes were incubated in 0.05% Tween-20 (v:v in TBS) supplemented with 5% non-fat powdered milk (w:v in TBS) for 1 h to saturate unspecific binding sites and sliced in parts to allow for the simultaneous detection of proteins with different MW in the same experiment. Thereafter, membranes were incubated overnight at 4°C with primary antibodies specific to P-AMPK (#2525, clone 40H9, Cell Signaling Technologies), AMPK (#2603, clone 23A3, Cell Signaling Technologies), ATG5 (#A2859, clone ATG5-18, Sigma-Aldrich), ATG7 (#A2856, Sigma-Aldrich), BECN1 (#sc-11427, clone H-300, Santa Cruz Biotechnology Inc., Santa Crus, CA, USA), CPT1A (#12252, clone D3B3, Cell Signaling Technologies), GABARAPL2 (#PM038, MBL International Corp., Woburn, MA, USA), GBF1 (#612116, BD Biosciences, San José, CA, USA), GOLGA2 (#ab52649, clone EP892Y, Abcam, Cambridge, UK), EIF2AK2 (#3072, Cell Signaling Technologies), LMAN1 (#ALX-804-602-C100, clone G1/93, Enzo Life Sciences Inc.), MAP1LC3A (#12135-1-AP, ProteinTech, Chicago, IL, US), MAP1LC3B (#2775, Cell Signaling Technologies), MAPK8 (#9258, clone 56G8, Cell Signaling Technologies), PIK3C3 (#5149, clone D9A5, Cell Signaling Technologies), PLD1 (#3832, Cell Signaling Technologies), PTEN (#9552, Cell Signaling Technologies), P-RPS6KB1 (#9205, Cell Signaling Technologies), RPS6KB1 (#2708, clone 49D7, Cell Signaling Technologies), SQSTM1 (#H00008878-M01, clone 2C11, Abnova), TSC2 (#3612, Cell Signaling Technologies) and ULK1 (#8054, clone D8H5, Cell Signaling Technologies). Antibodies specific to glyceraldehyde-3-phosphate dehydrogenase (GAPDH, #ab9484, Abcam) or β-actin (#ab6276, clone AC-15, Abcam) were employed to monitor the equal loading of lanes. Primary antibodies were detected with appropriate horseradish peroxidase (HRP)-labeled secondary antibodies (Southern Biotech, Birmingham, AL USA) and the SuperSignal West Pico chemiluminescent substrate (Thermo Fischer Scientific, Waltham, MA, USA). Chemiluminiscence images were acquired on a ImageQuant LAS 4000 digital imaging system (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Transmission electron microscopy
For ultrastructural studies, cultured cells were fixed in 1.6% glutaraldehyde (v:v in 0.1 M phosphate buffer) for 1 h, scraped off the plastic dish, centrifuged and post-fixed as a cell pellet in 1% osmium tetroxide (w:v in 0.1 M phosphate buffer) for 2 h. Following dehydration through a graded ethanol series, cells were embedded in Epon™ 812. Ultrathin sections were stained with standard uranyl acetate and lead citrate. For immunogold studies, cells were fixed with either 4% formaldehyde or 1.6% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) for 1 h at 4°C. Cell pellets were dehydrated in methanol and embedded in Lowicryl® K4M at −20°C in a AFS2 Freeze Substitution Processor apparatus (Leica Microsystems). Polymerization under UV light was carried out for 2 days at −20°C, followed by 2 days at 20°C. Ultrathin sections were incubated with primary antibodies specific to GFP (#ab290, Abcam) and SQSTM1 (#610832, BD Biosciences), for 1 h at room temperature, and then with secondary antibodies conjugated to 10- or 15-nm gold particles (BBI International, Cardiff, UK), as appropriate. Images were acquired with a Tecnai 12 electron microscope (FEI, Eindhoven, the Netherlands). The distance between gold particles was measured by means of the ImageJ software (http://rsb.info.nih.gov/ij/).
Assessment of Golgi apparatus functions
Protein glycosylation was measured by means of the Pierce™ Glycoprotein Staining Kit (#24562, Thermo Fischer Scientific), as per manufacturer's instructions. Densitometry was employed to obtain quantitative assessments, and data were normalized with respect to control conditions (U2OS cells maintained in normal medium for the duration of the assay). The transport of proteins to the Golgi apparatus was monitored in HeLa cells transiently expressing a thermosensitive, EGFP-tagged variant of the vesicular stomatitis virus G protein (plasmid #11912, from Addgene, Cambridge, MA, US), as previously described (Hirschberg et al, 1998). Endocytosis was evaluated in HeLa cells with pHrodo™ Red Dextran (#P10361, from Molecular Probes®-Life Technologies™), according to the manufacturer's recommendations.
Mouse experiments
WT C57BL/6 mice (Janvier Labs, Le Genest Saint Isle, France) and Becn1+/− C57BL/6 mice (a kind gift of Dr. Beth Levine, UT Southwestern Medical Center, Dallas, TX, USA) (Qu et al, 2003) were bred and maintained in compliance with the FELASA guidelines, the European Community regulations for animal experiments (2010/63UE), the Local Ethics Committee for Animal Welfare (CE n. 26: 2012-65, 2012-67; Val de Marne, France) and/or the regulations of the Italian Ministry of Health (D.Lgs 116/92; Rome, Italy). Mice were housed in a temperature-controlled environment with 12 h light/dark cycles and received food and water ad libitum. All mice used in this study were 6-week-old female animals. Animals received a single intraperitoneal injection of 100 mg/kg palmitate or 100 mg/kg oleate in 10% bovine serum albumin (BSA, w:v in PBS) or an equivalent volume of vehicle, in combination with an intraperitoneal injection of 15 mg/kg leupeptin (in 150 mM NaCl) or an equivalent volume of vehicle, and were sacrificed 1 or 2 h later. Then, organs were collected and processed for the immunoblotting-assisted detection of LC3 lipidation and p62 degradation, as previously described (Marino et al, 2014).
Yeast studies
WT BY4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0) S. cerevisiae cells expressing VPH1-mCherry, which marks vacuolar membranes (Ruckenstuhl et al, 2014), were employed to generate Δatg6 mutants by means of the URA3 gene disruption cassette (Guldener et al, 1996). WT and Δatg6 BY4741 cells were grown to stationary growth phase and then diluted in synthetic complete medium as previously described (Ruckenstuhl et al, 2014). According to experimental needs, the medium was further supplemented with 1.8 mM palmitate (in Tergitol™, final concentration = 0.5%), 1.8 mM oleate or an equivalent volume of solvent. Seventy-two hours later, cells were collected, stained with BODIPY® 493/503 (Molecular Probes®-Life Technologies™) as per manufacturer recommendations and imaged on a SP5 (Leica Microsystems) or LSM510 (Carl Zeiss) confocal microscope equipped with high numerical aperture (NA 1.4) oil immersion objectives. Image deconvolution was performed with Huygens Pro 4.0 (Scientific Volume Imaging, Hilversum, the Netherlands). Images were adjusted for brightness/contrast and assembled with the ImageJ software. Finally, z-stacks were interactively inspected to calculate the percentage of cells that unambiguously harbored lipid droplets within vacuolar structures (but not cells in which lipid droplets were localized in the close proximity of the vacuolar membrane).
C. elegans studies
DA2123 worms (adIs2122[plgg-1GFP::LGG-1; rol-6(df)], expressing a GFP::LGG-1 chimera) (Kang et al, 2007) were maintained according to standard procedures (Brenner, 1974). Gravid adults were treated with sodium hypochlorite to obtain tightly synchronized embryos that were allowed to develop into the L4 developmental stage. Thereafter, L4 larvae were transferred to fresh RNAi plates containing 0.5 mM oleate (Larodan, Fine Chemicals AB, Limhamn, Sweden), 0.5 mM palmitate or an equal volume of ethanol and fed with HT115 (DE3) Escherichia coli transformed with either the pL4440 empty vector (control conditions) or with a pL4440-based construct for the downregulation of BEC-1 (Tavernarakis et al, 2008). To improve the efficiency of RNAi, worms were reared for at least 2 generations on bec-1 dsRNA-producing bacteria. Gravid adults were treated with sodium hypochlorite, and released embryos were imaged with a Axio Imager Z2 Plus epifluorescence microscope equipped with a 40× objective (both from Carl Zeiss), using a 480 ± 10 nm band-pass excitation filter and a 515 ± 15 nm band-pass emission filter. Images were acquired under the same exposure. The number of GFP+ dots was analyzed using the ImageJ software. For each condition, we processed at least 15 images obtained across 2 independent experiments.
Statistical analyses
Unless otherwise mentioned, data are reported as mean ± SD of triplicate determinations and experiments were repeated at least twice yielding similar results. Data were analyzed using Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) or Excel (Microsoft Co., Redmond, WA, USA), and statistical significance was assessed by means of two-tailed Student's t-test or ANOVA tests, as appropriate.
Acknowledgments
We are indebted to Dr. Beth Levine (UT Southwestern Medical Center, Dallas, TX, USA) for the kind gift of Becn1+ /− mice and Klara Hellauer (University of Graz, Graz, Austria) for technical assistance. This work was supported by grants to GK from the Ligue Nationale contre le Cancer (Equipes labellisée), Agence Nationale pour la Recherche (ANR), Projets blancs, ANR under the frame of E-Rare-2 (the ERA-Net for Research on Rare Diseases), the Longevity Research Chair of the AXA Foundation, Association pour la Recherche sur le Cancer, European Commission (ArtForce), European Research Council (Advanced Investigator Award), Fondation pour la Recherche Médicale, Institut National du Cancer, Cancéropôle Ile-de-France, Fondation Bettencourt-Schueller, the LabEx Immuno-Oncology and the Paris Alliance of Cancer Research Institutes; to PC from Institut National du Cancer (INCa); to FC from AIRC (IG2010 and 2013), FISM (2009 and 2013), the Telethon Foundation (GGP10225), the Italian Ministry of University and Research (PRIN 2009 and FIRB Accordi di Programma 2011) and the Italian Ministry of Health (RF 2009); and to RT and SL from the Comision Nacional de Investigacion Cientifica y Tecnologica de Chile (FONDAP 15130011, FONDECYT 11130285 and 1120212). MN-S and ND are funded by Fondation Recherche Medicale (FRM). SAM received a grant from the Higher Education Commission (HEC) of Pakistan. VC is funded by the Lundbeck Foundation. FM is grateful to the Austrian Science Fund FWF (Austria) for grants LIPOTOX, P23490-B12, P24381-B20 and F3005-B12, as well as to the University of Graz for grant “Unconventional Research”. FC receives funding from the Danish Cancer Society (KBVU R72-A4408 and KBVU R72-A4647), The Bjarne Saxhof Foundation, the Lundbeck Foundation (n. R167-2013-16100) and NovoNordisk (n. 7559).
Author contributions
MCM, LG and GK designed the study. MN-S, SAM, FP, JMB-S, GM, ABY, RT, VS, VI, OK, PR and HW performed in vitro experiments. SAM, FP and VC performed in vivo experiments. KC performed immunohistochemical determinations. NT and MM worked on C. elegans. CB and ND performed protein degradation assays. FH and GP performed electron microscopy-based determinations. MN-S, FM, SL, PC, NT and FC analyzed data. MN-S, LG and GK wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure Legends
Review Process File
References
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Cascio G, Schiera G, Di Liegro I. Dietary fatty acids in metabolic syndrome, diabetes and cardiovascular diseases. Curr Diabetes Rev. 2012;8:2–17. doi: 10.2174/157339912798829241. [DOI] [PubMed] [Google Scholar]
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- Dall'Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, Duff K, Frohman MA, Wenk MR, Yamamoto A, Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nat Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- Dupont N, Codogno P. Non-canonical autophagy: facts and prospects. Curr Pathobiol Rep. 2013;1:263–271. [Google Scholar]
- Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014a;24:609–620. doi: 10.1016/j.cub.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Orhon I, Bauvy C, Codogno P. Autophagy and autophagic flux in tumor cells. Methods Enzymol. 2014b;543:73–88. doi: 10.1016/B978-0-12-801329-8.00004-0. [DOI] [PubMed] [Google Scholar]
- Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, Iwata J, Tanida I, Furuya N, Zheng DM, Tada N, Tanaka K, Kominami E, Ueno T. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fediuc S, Gaidhu MP, Ceddia RB. Regulation of AMP-activated protein kinase and acetyl-CoA carboxylase phosphorylation by palmitate in skeletal muscle cells. J Lipid Res. 2006;47:412–420. doi: 10.1194/jlr.M500438-JLR200. [DOI] [PubMed] [Google Scholar]
- Fukui M, Kang KS, Okada K, Zhu BT. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: role of ROS accumulation, caspase-8 activation, and autophagy induction. J Cell Biochem. 2013;114:192–203. doi: 10.1002/jcb.24354. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, Servant N, Paccard C, Hupe P, Robert T, Ripoche H, Lazar V, Harel-Bellan A, Dessen P, Barillot E, Kroemer G. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–1803. doi: 10.1158/0008-5472.CAN-09-3112. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, Goubar A, Martins I, Michels J, Kratassiouk G, Carmona-Gutierrez D, Scoazec M, Vacchelli E, Schlemmer F, Kepp O, Shen S, Tailler M, Niso-Santano M, Morselli E, Criollo A, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep. 2012;2:257–269. doi: 10.1016/j.celrep.2012.06.017. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD, White E. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, Skuballa W, Vivet S, Lichtner RB, Vicencio JM, Panaretakis T, Siemeister G, Lage H, Nanty L, Hammer S, Mittelstaedt K, Winsel S, Eschenbrenner J, Castedo M, Demarche C, et al. Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res. 2008;68:5301–5308. doi: 10.1158/0008-5472.CAN-08-0237. [DOI] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21:2161–2171. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, Hallstrom S, Rockenfeller P, Konya V, Heinemann A, Madeo F, Graier WF, Malli R. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem. 2012;287:21110–21120. doi: 10.1074/jbc.M111.319129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim Y, Lee SD, Lopez I, Arnold RS, Lambeth JD, Suh PG, Ryu SH. Selective activation of phospholipase D2 by unsaturated fatty acid. FEBS Lett. 1999;454:42–46. doi: 10.1016/s0014-5793(99)00745-0. [DOI] [PubMed] [Google Scholar]
- Kim HS, Montana V, Jang HJ, Parpura V, Kim JA. Epigallocatechin gallate (EGCG) stimulates autophagy in vascular endothelial cells: a potential role for reducing lipid accumulation. J Biol Chem. 2013;288:22693–22705. doi: 10.1074/jbc.M113.477505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–42544. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kach A, Ziegler U, Ong AC, Wallace DP, Arcaro A, Serra AL. The role of phospholipase D in modulating the MTOR signaling pathway in polycystic kidney disease. PLoS ONE. 2013;8:e73173. doi: 10.1371/journal.pone.0073173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernandez AF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, et al. Regulation of autophagy by cytosolic acetyl-coenzyme a. Mol Cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, Masiello P, Marchetti P, De Tata V. Palmitate activates autophagy in INS-1E beta-cells and in isolated rat and human pancreatic islets. PLoS ONE. 2012;7:e36188. doi: 10.1371/journal.pone.0036188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz C. Beclin-1 targeting for viral immune escape. Viruses. 2011;3:1166–1178. doi: 10.3390/v3071166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov NG, Harris G, Morales V, Ivanov AI. Loss of a membrane trafficking protein alphaSNAP induces non-canonical autophagy in human epithelia. Cell Cycle. 2012;11:4613–4625. doi: 10.4161/cc.22885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke EJ, Kuballa P, Xavier R, Ruvkun G. omega-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 2013;27:429–440. doi: 10.1101/gad.205294.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauloin A, Chat S, Pechoux C, Hue-Beauvais C, Droineau S, Galio L, Devinoy E, Chanat E. Oleate and linoleate stimulate degradation of beta-casein in prolactin-treated HC11 mouse mammary epithelial cells. Cell Tissue Res. 2010;340:91–102. doi: 10.1007/s00441-009-0926-3. [DOI] [PubMed] [Google Scholar]
- Peng G, Li L, Liu Y, Pu J, Zhang S, Yu J, Zhao J, Liu P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology. 2011;152:2206–2218. doi: 10.1210/en.2010-1369. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–R34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Ruckenstuhl C, Netzberger C, Entfellner I, Carmona-Gutierrez D, Kickenweiz T, Stekovic S, Gleixner C, Schmid C, Klug L, Sorgo AG, Eisenberg T, Buttner S, Marino G, Koziel R, Jansen-Durr P, Frohlich KU, Kroemer G, Madeo F. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014;10:e1004347. doi: 10.1371/journal.pgen.1004347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Kepp O, Michaud M, Martins I, Minoux H, Metivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene. 2011;30:4544–4556. doi: 10.1038/onc.2011.168. [DOI] [PubMed] [Google Scholar]
- Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L, Galluzzi L, Kepp O, Pierron G, Maiuri MC, Hikita H, Kroemer R, Kroemer G. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–680. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ren M, Gao GQ, Gong B, Xin W, Guo H, Zhang XJ, Gao L, Zhao JJ. Chronic palmitate exposure inhibits AMPKalpha and decreases glucose-stimulated insulin secretion from beta-cells: modulation by fenofibrate. Acta Pharmacol Sin. 2008;29:443–450. doi: 10.1111/j.1745-7254.2008.00717.x. [DOI] [PubMed] [Google Scholar]
- Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) J Biol Chem. 2012;287:14364–14376. doi: 10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy. 2008;4:870–873. doi: 10.4161/auto.6730. [DOI] [PubMed] [Google Scholar]
- Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, Metivier D, Vivet S, Tajeddine N, Joza N, Valent A, Castedo M, Kroemer G. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010;29:1272–1284. doi: 10.1038/emboj.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- Wijendran V, Hayes KC. Dietary n-6 and n-3 fatty acid balance and cardiovascular health. Annu Rev Nutr. 2004;24:597–615. doi: 10.1146/annurev.nutr.24.012003.132106. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure Legends
Review Process File