

Abstract
The H3.3 histone variant has been a subject of increasing interest in the field of chromatin studies due to its two distinguishing features. First, its incorporation into chromatin is replication independent unlike the replication-coupled deposition of its canonical counterparts H3.1/2. Second, H3.3 has been consistently associated with an active state of chromatin. In accordance, this histone variant should be expected to be causally involved in the regulation of gene expression, or more generally, its incorporation should have downstream consequences for the structure and function of chromatin. This, however, leads to an apparent paradox: In cells that slowly replicate in the organism, H3.3 will accumulate with time, opening the way to aberrant effects on heterochromatin. Here, we review the indications that H3.3 is expected both to be incorporated in the heterochromatin of slowly replicating cells and to retain its functional downstream effects. Implications for organismal aging are discussed.
Keywords: aberrant repair, aneuploidy, chromatin, epigenetic information, Hayflick limit, somatic stem cells
Organismal vs. replicative aging and epigenetic vs. genetic information
Many mechanistic studies of aging performed at both the molecular and cellular levels focus on the understanding of cellular (or replicative) senescence, that is, the loss of proliferative potential in long-term cultures of primary cells. This phenomenon was first discovered when cells were passed in culture until, after approximately 50 cumulative population doublings (CPDs), they could not be further stimulated to proliferate and reached the so-called Hayflick limit (Hayflick, 1985). Premature induction of irreversible growth arrest and other marks of cellular senescence [including specific cell morphology, senescence-associated beta-galactosidase (SABG), and senescence-associated heterochromatin foci (SAHFs)] (Bayreuther et al., 1988; Dimri et al., 1995; Campisi & d'Adda di Fagagna, 2007) can be provoked by various stress inducers (Brack et al., 2000), including oncogenic stress (Serrano et al., 1997), oxidative stress (Horikoshi et al., 1986; Ogryzko et al., 1996), and even inhibitors of histone deacetylases (HDAC) (Ogryzko et al., 1996).
For clarification, cellular senescence should be distinguished from the true subject of gerontology – organismal senescence – ‘a progressive deterioration of physiological function, an intrinsic age-related process of loss of viability and increase in vulnerability’ (de Magalhaes, 2004; Campisi, 2005; Jeyapalan & Sedivy, 2008). On the one hand, the contribution of cellular senescence to organismal aging appears rather straightforward, as the loss of proliferative potential can be directly linked to diminished capacity for tissue regeneration, decreased immune response (Effros, 1996), and a deteriorating endocrine system (Gosden, 1996). On the other hand, the existence of aging-related diseases such as Alzheimer's disease and age-related macular degeneration, the increased incidence and morbidity of cardiovascular, autoimmune, and oncological pathologies, as well as the accumulation of birth defects in the progeny of aging individuals together illustrate that not all aspects of aging can be easily explained solely on the basis of a loss of cellular proliferative potential.
In this respect, we propose to focus on cells that either do not replicate in adults or accomplish very few divisions during the lifespan of an organism – that is, far less than set by the Hayflick limit. For the purpose of this review, we will term these cells below Hayflick limit (BHL) cells. Below Hayflick limit cells include postmitotic cells such as terminally differentiated neurons and muscle cells, and female ova, which are formed during embryonic development and remain in a nonproliferating state for decades (Macklon & Fauser, 1999). Adult stem cells in their dormant stage can also be included since for decades before initiating proliferation, they might not enter into division (Orford & Scadden, 2008; Sottocornola & Lo Celso, 2012), as well as cells (e.g., some liver, kidney, and stomach cells) that enter the G0 phase semi-permanently after differentiation. Below Hayflick limit cells are interesting for the following reason: On the one hand, they are far from entering the replicative senescence state; on the other hand, due to the constant molecular turnover and active metabolism in these cells (even in the absence of replication), the lifespan of an adult organism should lead to accumulation of irreversible changes, which could contribute to organismal aging.
A second important clarification concerns the nature of the molecular clock that counts the age of a cell (or an organism). In accordance with the genocentric view, which has dominated the biology field until the end of the twentieth century and which considers DNA as the only source of stable information determining cell phenotype, it was natural to expect that the ticks and tocks of the clock are ultimately of a genetic nature; that is, they will somehow reshape the genetic molecular code script through real changes in the genome, for example, telomere shortening (Olovnikov, 1973; Wright & Shay, 2001) or mutations (Vijg & Dolle, 2002), or at least through accumulating DNA lesions due, for example, to oxidative damage (Gensler & Bernstein, 1981; Hoeijmakers, 2009). With the recent surge in epigenetic research, a greater interest has emerged in cellular changes that (i) have a nongenetic nature and (ii) are sufficiently stable to irreversibly accumulate in cells and contribute to the phenomenon of aging.
Epigenetics focuses on the mechanisms of processing of epigenetic information – defined here as information that is both (i) necessary to determine the state of an organism in addition to its DNA sequence (i.e., genetic information) and (ii) relatively stable compared to the characteristic times of metabolic changes and cellular lifespan (Russo et al., 1996; Bird, 2007). These mechanisms play a role in the maintenance of differentiated phenotypes in cell lineages during embryonic development and in adult ages, although their primary evolutionary role might have been to protect genetic information, for example, via suppression of parasitic genetic elements (Saade & Ogryzko, 2014).
One principal carrier of epigenetic information is chromatin – a hierarchically organized complex of DNA, histones, and nonhistone proteins (Bernstein et al., 2007). Not surprisingly, the recent interest in ‘all things epigenetic’ begat new ideas on the role of chromatin in aging. It has been known since the 60s that DNA methylation is progressively lost with aging (Pogribny & Vanyushin, 2010). With the more recent works of Guarante in yeast (Kaeberlein et al., 1999; Guarente, 2000; Lin et al., 2000), the discovery of the role of sirtuins and the effects of reservatrol (Howitz et al., 2003; Kaeberlein et al., 2005), the role of chromatin in the aging process has become a more fashionable field of research (Chatterjee & Williams, 1962; Dimauro & David, 2009; Pegoraro & Misteli, 2009; Feser & Tyler, 2011; McCord et al., 2013). So far, however, most of the mechanistic studies have focused on cellular senescence; that is, they have been concerned with how irreversible changes in chromatin could account for the loss of cellular proliferative potential. For example, the group of Bruce Howard suggested the existence of cellular checkpoint mechanisms that monitor the proper maintenance of heterochromatin domains during cell proliferation, and further proposed that defects in their maintenance could contribute to the phenomenon of replicative senescence (Howard, 1996; Ogryzko et al., 1996). This early idea is consistent with a more recent observation of the large-scale unraveling of peri/centromeric satellite chromatin (senescence-associated distension of satellites, or SADS), which could manifest the loss of proper maintenance of heterochromatin in aging cells (Cruickshanks et al., 2013; De Cecco et al., 2013a,b; Swanson et al., 2013).
Now, could epigenetic changes also irreversibly accumulate with time in BHL cells thus contributing to organismal, but not replicative, senescence? At first, one might shy away from the idea as, by current definition, epigenetic changes have to be heritable. Accordingly, one should not even formulate the question as the notion of heritability cannot apply to nonproliferating (postmitotic) cells. To deal with this terminological obstacle, the more general term of ‘epigenetic stability’ (i.e., preservation of stable traits regardless of whether cells proliferate or not) can be used to take into account that epigenetic mechanisms (either chromatin based or other) are most likely also involved in long-term preservation of phenotypic traits in nonreplicating cells (Ogryzko, 2008). With this slight adjustment in the scope of epigenetics, we can legitimately ask whether epigenetic factors and/or changes can affect the properties of nonproliferating cells.
In this minireview, we discuss the possibility that in nonreplicating cells, epigenetic modifications, and more specifically very particular changes in chromatin structure – the gradual replacement of canonical histones H3.1/H3.2 with variant histone H3.3 – could contribute to organismal aging by inducing aberrations in gene regulation and other functions in BHL cells. Although this hypothesis has not been directly supported by a plethora of experimental data as yet, the aggregation of existing claims and accumulating evidence leads almost inevitably to paradoxical conclusions about the role of H3.3 in BHL cells with tempting implications with regard to the aging process. The ‘H3.3 dilemma’, as we term this situation in the field, is both sufficiently intriguing and convincing to be worth-raising, in the hope that it will trigger new directions and efforts for research.
Alternative histones in general and H3.3 in particular
Alternative histone variants (replacement histones) are the latest addition to the growing list of potential epigenetic marks carried by chromatin, which also include DNA methylation and histone post-translational modifications. Although discovered a long time ago, these variants have attracted renewed interest in the last 10 years with the recognition of their various roles in genome function (Luger et al., 2012). Thus, their presence was found to correlate with particular functional states of chromatin (Table1); for example, histone macroH2A is enriched in silenced chromatin, whereas H2A.BBD is associated with euchromatin and splicing/RNA processing (Costanzi & Pehrson, 1998; Chadwick & Willard, 2001a,b).
Table 1.
Core histone variants, their functions, and features
| Histone | Biological function and features | Conservation | References |
|---|---|---|---|
| H3 variants | |||
| H3.3 | Gene activation, silencing, and chromosome segregation. Can be deposited in replication-independent way | Yes, but in yeast, it is the only noncentromeric H3 variant | Ahmad & Henikoff (2002), Elsaesser et al. (2010), Filipescu et al. (2013) |
| CenpA | Epigenetic marker of centromere | Present in most of eukaryotes, but less conserved than other H3 histones | Palmer et al. (1991), Cleveland et al. (2003), Bailey et al. (2013) |
| H3.X | Euchromatin in primates | Primate specific | Wiedemann et al. (2010) |
| H3.4/H3t | Sperm genome and nucleolus of somatic cells | Mammalian specific | Tachiwana et al. (2010) |
| H3.Y | Euchromatin in primates | Primate specific | Wiedemann et al. (2010) |
| H3.5/H3.3c | Euchromatin in hominid testis | Hominid specific | Schenk et al. (2011) |
| H2A variants | |||
| H2AZ | Poising genes for activation. Gene activation, gene silencing, and chromosome segregation | Present in most of eukaryotes | Faast et al. (2001), Creyghton et al. (2008), Eirin-Lopez et al. (2009), Hu et al. (2013) |
| macroH2A | Association with repressed/silenced chromatin, large size due to an additional C-terminal domain | Vertebrate specific | Costanzi & Pehrson (1998), Buschbeck et al. (2009), Gamble et al. (2010) |
| H2A.BBD | Splicing, replication Active transcription | Mammalian specific | Ioudinkova et al. (2012), Tolstorukov et al. (2012) |
| H2AX | Double-strand break repair/meiotic remodeling of sex chromosomes and genome integrity. The function is mediated by the phosphorylated form γH2A.X | Present in most of eukaryotes | Fernandez-Capetillo et al. (2003), Sedelnikova et al. (2003) |
| H2B variants | |||
| TH2B | Chromatin to nucleoprotamine transition | Gineitis et al. (2000), Li et al. (2005), Govin et al. (2007), Montellier et al. (2013) | |
| H2BFWT | Sperm telomere binding | Primate specific | Gineitis et al. (2000), Churikov et al. (2004) |
| H2BE | Transcription regulation in olfactory neurons | Santoro & Dulac (2012) | |
A striking example for an epigenetic role of replacement histones is that of CenpA, an H3 variant that has been shown to serve as a self-perpetuating mark on chromatin, important in the maintenance and reproduction of centromere chromatin regardless of its underlying DNA sequence (Cleveland et al., 2003; Bailey et al., 2013). As another distinct feature, the sequence of CenpA varies significantly between homologs in different species, which might reflect the role of improper chromosome segregation (and resulting meiotic incompatibility) in speciation (Henikoff et al., 2004; Probst et al., 2009).
Our main focus here concentrates on another H3 variant – the alternative histone H3.3. Unlike CenpA, H3.3 is conserved through a wide range of species and differs by only few amino acids from its canonical counterparts H3.1 and H3.2 (4 and 5 replacements, respectively, mostly in the ‘AAIG’ vs. ‘SAVM’ patch at aa 87–90 in the histone sequence) (Elsaesser et al., 2010; Filipescu et al., 2013). Despite their relative modesty, these changes have two dramatic consequences.
First, unlike the replication-coupled (RC) deposition of its canonical counterparts into chromatin, the deposition of H3.3 is replication independent (RI) (Ahmad & Henikoff, 2002). Whereas H3.1/2 copurify with the histone chaperone CAF-1, which is responsible for their RC deposition, there are different RI pathways for H3.3 incorporation into chromatin. HIRA (Lamour et al., 1995) is the chaperone complex that is responsible for H3.3 deposition at actively transcribed regions (Tagami et al., 2004; Goldberg et al., 2010), whereas the DAXX–ATRX complex (Drane et al., 2010; Goldberg et al., 2010) and DEK (Sawatsubashi et al., 2010) can deposit H3.3 into heterochromatin and regulatory regions, including that of intermediate response genes in neurons (Michod et al., 2012). Interestingly, changing any one of the residues specific to H3.1/2 to those present in H3.3 relieves the block to RI assembly and allows histone H3 deposition outside of S phase (Ahmad & Henikoff, 2002), suggesting that RI deposition is the default pathway and that H3.1/2 are actively recognized and blocked from RI deposition. That in the yeast Saccharomyces cerevisiae, the only H3 histone involved in regular chromatin structure is similar to H3.3, reinforces the notion that H3.3 assembly is the default H3 deposition pathway. Consistently, it was shown that unlike H3.3, H3.1 colocalizes with replication sites (Ray-Gallet et al., 2011). The same study showed that H3.3 can be deposited at replication sites when H3.1 deposition is impaired, but that the opposite is not true: H3.1 cannot replace H3.3 when the incorporation of the latter is affected.
As another remarkable feature, H3.3 is associated with actively transcribed chromatin. Several lines of evidence established this correlation. In 1984, H3S, an H3.3-like histone, was found in ciliates only in the active macronucleus, whereas the canonical H3-like histone H3F was found in the transcriptionally inactive micronucleus (Allis & Wiggins, 1984). Two decades later, H3.3 deposition in Drosophila was shown to localize to active rDNA arrays and euchromatin but not heterochromatin (Ahmad & Henikoff, 2002). Working on human cells, Janicki et al. (2004) showed that H3.3 deposition took place on a transgene array whose transcription was activated.
The association of H3.3 with active chromatin is supported by the analysis of histone post-translational modifications (PTMs). Mass spectrometry analysis of Drosophila H3.3 shows that it is enriched in active chromatin marks (methylation at K4 and K79 and acetylation at K9, K14, and K18 + K23) and that it has lost PTMs’ characteristic of repressed chromatin such as dimethyl lysine 9 (McKittrick et al., 2004; Mito et al., 2005). In mammalian cells, the majority of modifications detected on H3.3 are also marks of active chromatin, including methylation of K4 and K79 and acetylation of K14, K18, and K23 (Hake & Allis, 2006).
Finally, using chromatin immunoprecipitation (ChIP) technology, H3.3 was shown specifically incorporated throughout the gene body of transcribed genes and highly enriched in promoter regions in both Drosophila and mammalian cells, its presence correlating with that of bound RNA polymerase II (Janicki et al., 2004; Chow et al., 2005; Wirbelauer et al., 2005; Daury et al., 2006; Mito et al., 2007; Nakayama et al., 2007; Jin et al., 2009; Sutcliffe et al., 2009; Tamura et al., 2009).
H3.3 – a transcription player or a ‘placeholder dummy’? The ‘H3.3 dilemma’
The simplest possibility (the ‘zero hypothesis’) to account for the association of H3.3 with active chromatin derives from the facts that (i) active chromatin is more open, therefore more dynamic and more prone to molecular turnover and (ii) the deposition of canonical H3.1/2 histone is strictly replication-coupled. Let us designate τa the characteristic time of H3 histone turnover at active chromatin sites, τi the analogous time for inactive chromatin, and τr the characteristic time of cell replication. Photobleaching experiments indicate that τi ≥ τr for typical cells in culture (Kimura & Cook, 2001), whereas τa could be significantly shorter than τr. Accordingly, for active states of chromatin, the deposition of canonical H3 forms is too slow (∼τr) to catch up with the molecular turnover, the only alternative being in the deposition of H3.3 leading to its accumulation at corresponding sites. Thus, in the framework of the ‘zero hypothesis’, H3.3 is only a ‘placeholder dummy’ that replaces H3.1/2 in a nonreplicative context. Its association with active genes is therefore nothing more than a downstream consequence of an open state of chromatin (Fig.1a).
Fig 1.
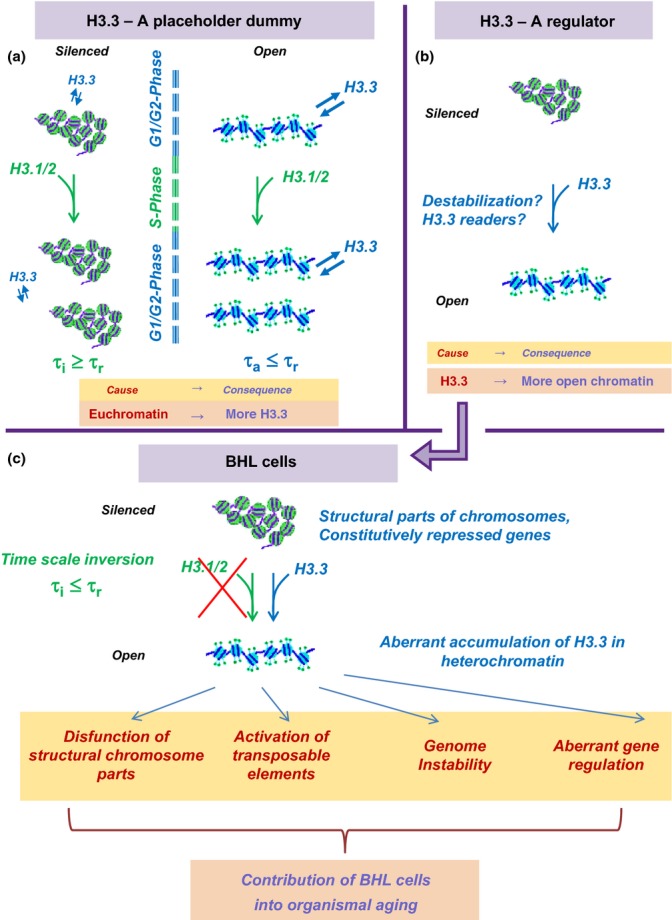
H3.3 Dilemma. Top. Alternative explanations for association of H3.3 with active chromatin. (a) A placeholder dummy. Euchromatin (right) is more open and more prone to histone damage and exchange than heterochromatin (left). Time of turnover of H3 histones in active chromatin τa is shorter than time of cell replication τr. Accordingly, replication-coupled deposition of canonical H3.1/2 cannot be responsible for the turnover of all H3 histones, which is expected to lead to a preferential accumulation of H3.3 in open chromatin. To the contrary, the turnover rate of H3 in heterochromatin τi is slower than τr, and the replication-coupled H3.1/2 deposition can be sufficient for H3 replacement. (b) A regulator. H3.3-containing chromatin is ‘special’ in some respect, and H3.3 replacement of the canonical H3.1/2 leads to chromatin opening or other consequences for its structure and function. Down. Consequences of the ‘regulator’ model in the case of below Hayflick limit (BHL) cells. The replication time of BHL cells is expected to be slower than the rate of H3 exchange in heterochromatin, which should lead to an accumulation of H3.3 histones in heterochromatin of BHL cells with potentially negative consequences in terms of structure and function.
A more attractive possibility has it that the H3.3 variant is causally involved in the establishment of an active/open chromatin state (Fig.1b). There are many ways whereby the replacement of H3.1/2 by H3.3 could affect chromatin, including changes in nucleosome stability or creation of docking sites for regulatory proteins (i.e., ‘H3.3 readers’), etc. Understandably, the idea that H3.3 is a player in gene regulation is a more stimulating hypothesis. The notion that H3.3-containing nucleosomes are in some way ‘special’ serves as a direct motivation for the hypothesis of their semiconservative replication (Nakatani et al., 2004; Jin et al., 2009). It is also more consistent with the mechanistic roles of other histone replacement variants such as CenpA, macroH2A, and H2AZ, in the establishment of particular functional chromatin states (Bonisch & Hake, 2012; Skene & Henikoff, 2013). However, the differences between these histone variants and their canonical counterparts are more significant than the 4(5) aminoacid difference between H3.3 and H3.1/2. This calls for more rigor in establishing the possible downstream effects of H3.3 presence. In addition, the fact that H3.1/2 deposition on chromatin requires replication sets a constraint on the plausible properties of H3.3 histone, which could be formulated as a dilemma that emerges when considering two features of H3.3 deposition in BHL cells (Fig.1c).
Feature 1. τr ≥ τi in BHL cells
In an adult organism, BHL cells are expected to accumulate H3.3 in their heterochromatin. Indeed, due to their slow replication time, they cannot incorporate the replication-coupled canonical H3.1/2 histones at a speed sufficient to compensate for H3 molecular turnover in all chromatin types.
There are many causes for molecular turnover of histones, including thermal fluctuations, oxidation, proteolysis (Adams-Cioaba et al., 2011) and active chromatin remodeling due to DNA repair, all events expected to regularly necessitate the incorporation of new histone molecules in chromatin. Whereas in actively replicating cells τi ≥ τr, that is, the rate of canonical H3 deposition should be sufficient to compensate for the loss of H3 in inactive chromatin, BHL cells live for decades with, at best, little replication. The decades-long time scales are not comparable with the rate of molecular turnover of proteins in a living cell. This is, essentially, the same problem that motivated Francis Crick to propose his ‘epigenetic templating’ model of long-term synaptic potentiation in neurobiological memory (Crick, 1984; Ogryzko, 2008).
Exacerbating the problem, H3 is the only core histone with cysteine residues and thus should be more sensitive to oxidation than other histones. Moreover, without timely replacement of a histone molecule, this oxidation will progress irreversibly: Unlike disulfide forms, RS-SR which can be reduced back to thiol groups RSH, sulfenic (RSO−), sulfinic (RSO2−), and sulfonic (RSO3−) forms of cysteine cannot be restored.
With the inverted relation τr ≥ τi in BHL cells, H3 would eventually require a replacement at all sites in the genome, thus including repressed chromatin. Unlike the replication-coupled H3.1/2 deposition, the H3.3 RI pathway remains available at all times in BHL cells; hence, one should expect a gradual substitution of H3.1/2 by H3.3 in repressed chromatin.
Feature 2. Aberrant effects on repressed chromatin
Now, let us consider that the association of H3.3 with active chromatin is due to its role in establishing an active and open chromatin state. In this case, one should expect that its incorporation into heterochromatin could affect BHL cells in an undesirable way. Heterochromatin-based silencing is an essential mechanism employed to restrict gene expression to housekeeping and lineage-specific genes, as well as to suppress parasitic selfish elements (e.g., transposons). Repetitive sequences with a structural role (such as satellite DNA) need also be transcriptionally silenced. One can see how the opening of otherwise silenced chromatin in inappropriate contexts would lead to unwanted transcriptional activation (or else competition with other genomic sites for binding of available transcription factors) with negative consequences due to perturbed epigenetic programs and induction of genome instability (via activation of transposable elements and/or affecting structural parts of the chromosomes).
Accordingly (unless we are prepared to consider the consequences for organismal aging discussed below), we are faced with ‘the H3.3 dilemma’: (i) either H3.3 is a mundane ‘placeholder dummy’; that is, H3.3-containing nucleosomes are not ‘special’ and are tolerable in any amounts at any place in the genome, (ii) or H3.3 is not incorporated in BHL cells at the most inappropriate genome locations, perhaps because in these cells, chromatin deposition of H3.1/2 is not as strictly replication-coupled as imagined and thus occurs at a low albeit sufficient rate.
At this point, it is worth-noting that the DAXX-ATRX chaperone system has been shown to facilitate H3.3 deposition in many nongenic repeat regions of the genome (Filipescu et al., 2013). Consistent with this fact, recent studies indicate that the notion of H3.3 associated only with gene activation is a clear oversimplification. Instead, the emerging view implicates H3.3 in the establishment of a chromatin landscape which would allow proper gene expression upon cell differentiation, for example the bivalent chromatin landscape in embryonic stem cells (Banaszynski et al., 2013). Still, despite its nuances, this view remains consistent with the notion of H3.3 incorporation affecting chromatin properties and function. Thus, the problem persists of the downstream effects of a H3.3 presence at inappropriate sites of the genome in BHL cells.
Unlike H3 histones, the H2A/H2B histones and their variants are subject to a relatively fast exchange (at a time scale less than the replication time of a cell) and their deposition is not coupled to DNA replication. Two distinct features pertain to H3 histones: (i) the difference between the RC and RI pathways and (ii) their relatively slow rate of exchange; this is what is responsible for the H3.3-specific dilemma.
In the next section, we will review how both horns of the dilemma fare with regard to the experimental evidence.
Horns of the dilemma – experimental observations
H3 protein is mostly represented by the H3.3 variant in terminally differentiated and quiescent cells
Despite H3 being the only core histone containing cysteine and thus more prone to oxidation, no age-related accumulation of oxidized histone H3 (Carter & Chae, 1975) has been detected, indicating that cells have a way to replace oxidized H3 histones.
In this respect, a recent proteome-wide study that measured protein molecular turnover using stable isotope chase combined with mass spectrometry (Toyama et al., 2013) must be discussed. In mammals, most proteins have an average half-life of 1–2 days. Some, however, (e.g., crystallins, nucleoporins) exhibit exceptionally long half-lives up to several months (as judged by the significant presence of heavy isotope-labeled versions of corresponding peptides in 6- or 12-month-old tissues). Strikingly, histone H3.1 has the slowest turnover with only 10% of the protein replaced in 6 months in rat brain tissue. The anomalously high stability of H3.1 would appear to invalidate the main premise of the H3.3 dilemma. However, the steady state assumption used to justify the half-life estimations of protein stability is not valid for the canonical H3 histone. The case of H3.1 molecular turnover is special because in nondividing cells, it is replaced by a different molecule – H3.3. It is thus not surprising that most of the H3.1 present in nonreplicating cells is represented by molecules deposited at a young age, because even if the total levels of H3.1 fall dramatically in aged tissue, the replacement comes not in the form of fresh H3.1 molecules, but as H3.3 histone.
Indeed, H3.3 has been shown to progressively replace most H3.1/2 in terminally differentiated cells in vertebrates. In quiescent human T lymphocytes, for instance, H3.3 is the only H3 variant synthesized and is the major variant by mass (73%) (Wu et al., 1983). It also becomes the predominant form in chicken liver and kidney and also represents up to 90% of H3 molecules in terminally differentiated rat neurons (Urban & Zweidler, 1983; Pina & Suau, 1987).
The latter studies have all been performed in model organisms with a typical lifespan of several years. Due to the molecular protein turnover, it should take no more than a year to substitute H3.1/2 with H3.3. In this regard, the predominance of H3.3 should not come as a surprise, but this begs the question of why so much of canonical (replication-coupled) H3 still remains in chromatin in terminally differentiated cells. The same question is even more acute when considering humans who live much longer.
A partial explanation for the remaining canonical H3 is, of course, DNA repair. Virtually every pathway of DNA repair requires DNA synthesis and involves the PCNA molecule which can recruit the H3.1/2 chaperone CAF1 for deposition of H3.1/2 in the absence of replication (e.g., according to the ‘access-repair-restore’ model (Smerdon, 1991)). However, it has recently been reported that for DNA repair, H3.3 can also be deposited by the HIRA chaperone at sites of DNA damage, important for recovery of transcriptional activity (Adam et al., 2013) as well as for progression of the DNA replication fork after UV damage (Frey et al., 2014), suggesting that even in the case of repair, not all newly deposited H3 histones are canonical H3.1/2. Furthermore, common wisdom (Goodarzi & Jeggo, 2012; Lemaitre & Soutoglou, 2014) has it that heterochromatin represents a barrier for repair machinery. Accordingly, it is an open question of how much the repair-coupled H3.1/2 deposition can contribute to the maintenance of canonical H3.1/2 at heterochromatin loci in BHL cells. It is possible that some alternative yet to be discovered mechanisms of H3.1/2 deposition at heterochromatin loci (whether linked to slow background DNA synthesis or else DNA synthesis independent) take place in terminally differentiated and other BHL cells. Although it would be useful to confirm these data with modern techniques (such as mass spectrometry and Western blotting with H3.3-specific antibodies), it is indeed very likely that the vast majority of H3 histones in these cells are represented by H3.3, with the consequence that in their heterochromatin (the largest part in the genome), H3.1/2 should be substituted by H3.3.
Experimental evidence for a special nature of the H3.3 nucleosome
The biological role of H3.3 has been the subject of intensive recent research. Adding more urgency to this effort, increasing evidence implicates H3.3 and its chaperones in cancer (Schwartzentruber et al., 2012; Behjati et al., 2013; Fontebasso et al., 2013; Aihara et al., 2014; Venneti et al., 2014). Most remarkable is the tumor type specificity of the H3.3 mutations that have been detected - whereas K27 and G34 of H3.3 are affected in 31% of childhood brain tumors (Schwartzentruber et al., 2012), 95% of chondroblastomas exhibit K36M alterations, and 92% of giant cell tumors of bone have K27 mutated in this protein (Behjati et al., 2013). These facts are hard to reconcile with H3.3 being a simple placeholder for canonical H3. Other recent data provide additional support for the notion that replacement of canonical H3.1/2 by H3.3 has downstream effects on chromatin properties and function.
Incidentally, functional knockouts of the H3.3 gene reveal partial lethality in adult Drosophila males (Sakai et al., 2009) and misregulation of gene activation in mammals (Placek et al., 2009; Sakai et al., 2009; Tamura et al., 2009; Banaszynski et al., 2013; Bush et al., 2013). This, however, cannot serve as direct evidence for a special nature of H3.3-containing nucleosomes. Instead, it could be argued that the observed gene expression effects could be simply due to nucleosome depletion in the absence of a functional replication-independent histone deposition pathway; in addition, nucleosome depletion could compromise genome stability. Moreover, H3.3 is the only noncentromeric H3 histone in yeast, and it is also not essential for transcription or viability in Tetrahymena (Cui et al., 2006). This suggests that, in any case, the search for a special role of H3.3-containing nucleosomes would be most productive in higher eukaryotic systems.
Another class of evidence based on the functional knockouts of the H3.3 chaperones HIRA or DAXX (Yang et al., 2011; Pchelintsev et al., 2013; Soni et al., 2014) and on the interaction of these proteins with known transcriptional regulators, such as BRG1 or HP1γ (Kim et al., 2011; Pchelintsev et al., 2013), also cannot be interpreted straightforwardly in support of the special nature of H3.3 nucleosomes. It could be that these proteins perform additional and independent roles in gene regulation, which still would be consistent with a placeholder role for H3.3. For example, HIRA binds many genomic sites in the absence of UBN1 and ASF1a, its usual partners in H3.3 deposition, and these ‘HIRA-only’ sites are also not enriched in H3.3 (Pchelintsev et al., 2013). Concerning DAXX, its nonchaperone functions have been reviewed (Lindsay et al., 2008; Salomoni, 2013); in addition to depositing H3.3, it has also been shown recently to be involved in the deposition of CenpA in aberrant locations in the genome (Lacoste et al., 2014). Nevertheless, given the mechanistic association of the HIRA chaperone with gene activation (Yang et al., 2011; Pchelintsev et al., 2013), it remains tempting to speculate that the role of the DAXX in H3.3 deposition in pericentromeric and other nongenic repeat chromatin domains (Morozov et al., 2012; Corpet et al., 2014) could be to avoid any adverse consequences of the involvement of HIRA chaperone in H3.3 deposition in the case of heterochromatin and other nongenic repeat sequences (Banaszynski et al., 2013).
Biophysical studies are more direct in addressing the issue of the special nature of the H3.3-containing chromatin. Albeit somewhat controversial, they indicate subtle effects on nucleosome stability and positioning (Thakar et al., 2009) and increased sensitivity of a H3.3-containing nucleosome to salt-dependent disruption, exacerbated in the presence of a H2AZ histone variant within the same nucleosome (Jin et al., 2009). On the other hand, the hybrid CenpA/H3.3 nucleosome is unusually stable, an observation that was linked to CenpA mislocalization and resulting chromosome aberrations in cancer (Arimura et al., 2014). Another study (Chen et al., 2013) points to the higher-order folding of chromatin as the level of chromatin organization where the effects of H3.3 are mostly manifest.
The most convincing evidence for a special nature and/or role of H3.3-containing chromatin would be whether proteins or protein domains were found specialized in distinguishing between H3.1/2 and H3.3 (i.e., ‘H3.3 readers’, consistent with the influential concept of the ‘histone code’ (Hake & Allis, 2006)). Remarkably, three recent papers claim to accomplish Just that, both pointing at the crucial role of aminoacid A-S(T)31 replacement. First, the potential tumor-suppressor protein ZMYND11 has been shown to specifically recognize H3.3, trimethylated at K36, and to curtail RNA polymerase II-driven RNA elongation (Wen et al., 2014). Importantly, in addition to K36me3 bound by the ZMYND11 PWWP domain, the H3.3-specific S31 (replaced by alanine in other H3 histones) also contributes to this interaction by being lodged into the bromo-ZnF-PWWP ‘valley’, greatly augmenting the affinity between the two proteins. More recently, a related study revealed the importance of this recognition for regulation of RNA splicing (i.e., intron retention) (Guo et al., 2014).
Second, ATXR5/6, a specific histone methyltransferase from Arabidopsis, has been shown to do the opposite, specifically targeting the H3.1 variant, ‘reading’ the alanine 31 which is replaced in the H3.3 histone (Jacob et al., 2014). The authors proposed a model whereby a specific heterochromatin mark (H3K27me1) is maintained during DNA replication. The important implication is that this mark cannot be maintained after H3.1/2 has been replaced by H3.3 (due to the inability of ATXR5/6 to methylate H3.3), which could contribute to the opening of a previously inactive chromatin, and be responsible for, for example, the induction of transposable elements.
H3.3 dilemma and aging – organismal and replicative
Let us now return to the H3.3 dilemma. The experimental evidence strongly suggests that H3.3 nucleosomes are both (i) ‘special’ and (ii) do eventually replace H3.1/2 in the heterochromatin of BHL cells. Accordingly, as far as BHL cells are concerned, it cannot be ‘business as usual’ and there are obvious implications for organismal aging.
We are far from proposing that H3.3 accumulation in BHL cells would provide a unifying theory of organismal aging, a multifaceted phenomenon that cannot be reduced to one universal cause. The question we ask, however, is whether some aspects of organismal aging could be due to the eventual accumulation of H3.3 in heterochromatin of BHL cells leading to aberrations in gene expression and genome instability. Various experimental models can be examined in this respect, pertaining to diverse aspects of an aging organism.
Aging females
With age, fertility decreases, the number of miscarriages increases as well as the frequency of congenital birth defects in the newborns (Gosden, 1985; Hassold & Chiu, 1985; Stein, 1985; Gindoff & Jewelewicz, 1986; Piette et al., 1990) (Fig.2a). The most prominent factor is aneuploidy (e.g., strikingly increasing trisomy 21), indicating that ova quality decreases with age. Given that germline proliferation in the ovary terminates during fetal development, women's eggs have to remain in a nonproliferative state for decades and thus should qualify as bona fide BHL cells subject to protein turnover and H3.3 accumulation in heterochromatin. Intriguingly, although the causes of age-related aneuploidy (Angell, 1997; Lamb et al., 1997; Wolstenholme & Angell, 2000) are still under debate, the loss of cohesion between homologous chromosomes or chromatids which produce segregation errors appears to be an important mechanism (Wolstenholme & Angell, 2000; Schramm et al., 2002; Pellestor, 2004; Pellestor et al., 2006). Investigations in mice suggest that a loss of cohesin complex could be responsible (Chiang et al., 2010; Lister et al., 2010), but in humans (Garcia-Cruz et al., 2010) no differences are observed in the levels of meiotic cohesins in oocytes of different ages, indicating that loss of cohesin cannot be the only cause for chromosome nondisjunction in the eggs of aging females. It is obviously tempting to speculate that an improper accumulation of H3.3 in specific structural parts of chromosomes (such as centromeric and pericentromeric chromatin, which are heterochromatic) could affect cohesion and other properties, thereby contributing to meiotic defects and aneuploidy.
Fig 2.
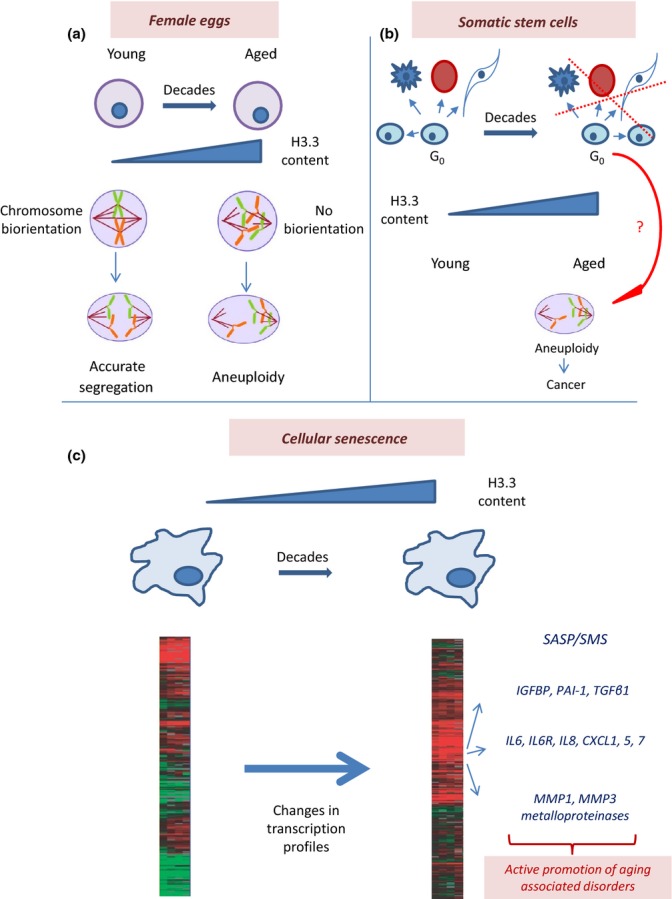
Possible relationship between the H3.3 dilemma and organismal aging. (a) Aging eggs might accumulate H3.3 in structural parts of their chromosomes, leading to negative consequences in chromosome/chromatid cohesion, resulting in increased aneuploidy. (b) Somatic stem cells might accumulate H3.3 in their heterochromatin, which could result in epigenetic reprogramming and negative consequences in terms of differentiation and self-renewal properties. Also, aneuploidy might contribute to increased cancer frequency in aged individuals. (c) In senescent cells, a Misincorporation of H3.3 at inappropriate genome locations could lead to changes in transcription profiles responsible for the specific (e.g., proinflammatory) properties of senescent cells (SASP/SMS phenomenon), which could actively contribute to organismal aging.
Adult/somatic stem cells
Another particular class of BHL cells is represented by adult (somatic) stem cells (SC) (Fig.2b). An important and relevant feature is their ability to stay in a quiescent state for decades (Orford & Scadden, 2008; Li & Bhatia, 2011; Sottocornola & Lo Celso, 2012) before being induced to proliferate and/or differentiate. This property has been linked to their long-term reconstituting capacity (Reya et al., 2001). Defects in regulation of quiescence can lead to premature exhaustion of the SC pool causing failure in tissue regeneration (Cheshier et al., 1999; Arai et al., 2004) needed, for example, following myelotoxic insults (Cheshier et al., 1999). Thus, the H3.3 dilemma considerations do pertain to the biology of adult SC, especially during their nonreplicating stage, even though they are little affected by the Hayflick limit. Despite many technical challenges in isolating and working with dormant adult SC, future research should shed light on how the accumulation of H3.3 in heterochromatin could affect their main characteristics: capacity to self-renew and differentiate, which is directly relevant to organismal aging. Furthermore, chromosome cohesion defects and the resulting aneuploidy upon exiting a long-term dormant stage could have oncological implications. An additional open question – which could be seen as an offshoot of the H3.3 dilemma when applied to adult SC – is whether a high H3.3 content could serve as a marker for dormant somatic SC.
Cellular senescence (Fig.2c)
Up till now, we have restricted our discussion to the possible role of BHL cells in organismal aging, intentionally excluding the phenomenon of cellular senescence. In fact, H3.3 has been previously discussed in such a context (Rai & Adams, 2012), and more recent data suggest that this variant histone and its proteolytically processed form could drive cellular senescence (Duarte et al., 2014), most likely through stress-induced mechanisms.
This section, however, focuses on how H3.3 can be relevant for a different aspect of the biology of senescent cells – that is, for understanding the postmitotic stage of cellular senescence and its contribution to organismal aging. Indeed, given that cells can be growth-arrested for decades, the logic behind molecular turnover and H3.3 accumulation should apply to the cellular senescent state as well. One can ask whether some distinguishing characteristics of senescent cells could relate to the accumulation of H3.3 and consequent aberrations in chromatin function. For example, the above-mentioned SADS phenomenon (Cruickshanks et al., 2013; De Cecco et al., 2013a,b; Swanson et al., 2013) could be a manifestation of improper incorporation of H3.3 in the absence of replication – that is, it might be not the upstream cause of growth arrest (the suggestion consistent with the Howard hypothesis (Howard, 1996; Ogryzko et al., 1996)), but rather a downstream consequence thereof.
More importantly, changes in the gene expression profiles due to H3.3 misincorporation can help to explain how cellular senescence contribute to organismal aging – in ways that come in addition to the mere loss of proliferation potential and limited tissue regeneration. One of the marks of cellular senescence is senescence-associated secretory phenotype (SASP), which results in the secretion of various growth factors, cytokines, and proteases (called summarily senescence messaging secretome (SMS) (Kuilman & Peeper, 2009)), leading to age-related tissue dysfunction and disruption (Coppe et al., 2008; Rodier & Campisi, 2011). The SMS includes IGFBP, PAI-1, TGFβ1 (Tremain et al., 2000; Kortlever et al., 2006; Wajapeyee et al., 2008), and immune regulators such as IL6, IL6R, IL8, CXCL1, 5, and 7 (Shelton et al., 1999; de Magalhaes et al., 2004; Xue et al., 2007; Acosta et al., 2008; Kuilman et al., 2008), and secretion of proinflammatory proteins by senescent cells may be involved in positive feedback loops inducing senescence in neighbor cells (Acosta et al., 2008; Kuilman et al., 2008; Freund et al., 2010). Also, the activity of the metalloproteinases MMP1 and MMP3 which degrade the extracellular matrix is increased in senescent cells (Krizhanovsky et al., 2008). Most importantly, it has been shown that clearance of senescent cells delays aging-associated disorders in mice (Baker et al., 2011), strongly supporting the notion that the presence of senescent cells actively promotes (presumably, via SASP/SMS) these disorders, cancer included (Krtolica et al., 2001). A crucial question is how changes occur in the transcriptional program during cellular senescence. A tempting, although still speculative, explanation is provided by the H3.3 dilemma – misincorporation of H3.3 into the otherwise suppressed chromatin of senescent cells could lead to their reprogramming and aberrant gene expression, with SASP an eventual (although not necessarily direct) result of such changes. Consistent with this idea, a recent study from the Adams group demonstrates that the HIRA chaperone plays an important role in chromatin dynamics in senescent cells and is responsible for the changes in gene expression profiles (Rai et al., 2014).
Conclusion. ‘Aberrant Chromatin repair’?
Natural wear and tear of chromatin is unavoidable during the decades-long lifespan of BHL cells. With chromatin considered the principal carrier of epigenetic information, an important question arises as to the way its histone components are reconstituted. H3 presents a special challenge in this regard. On the one hand, it is buried deep inside the nucleosome and cannot be readily replaced by passive off-and-on molecular exchanges. On the other hand, the presence of cysteine residues makes it more prone to oxidative stress. In most eukaryotes, evolution has chosen a way, somewhat similar to two alternative strategies of dealing with DNA damage: Whereas replication-coupled deposition of H3.1/2 corresponds to passive removal of a ‘lesion’ via its dilution among the exponentially growing number of normal copies, replication-independent H3.3 deposition is an active replacement, corresponding to, for example, the DNA nucleotide excision repair or base excision repair pathways. Consistent with the notion that epigenetic information can be damaged and thus needs be repaired, RI H3.3 deposition could be considered a specialized ‘epigenetic repair pathway’.
Recent evidence, however, indicates that H3.3 is not a simple doppelganger of H3.1/2, but, at least in higher eukaryotes, has its own personality, a unique voice in the epigenetic cellular orchestra. If this is not a placeholder dummy and the information content of chromatin is changed after H3.1/2 replacement, a more apt analogy would be an aberrant DNA repair (Kim et al., 2014; Talhaoui et al., 2014). Here, we have considered some constraints that the properties of H3.1/2 and H3.3 set on plausible models of H3.3 function and we have discussed the potential implications of ‘aberrant chromatin repair’ for organismal aging, with some tackling of the properties of senescent cells. We believe that these considerations, while admittedly speculative at the moment, have a potential to trigger further research.
We see another angle where DNA repair could be relevant in the context of the H3.3 dilemma. DNA lesions are repaired less efficiently in heterochromatin, in part because the effects of such lesions are not expected to manifest themselves in silenced genes, repair being thus less necessary (Goodarzi & Jeggo, 2012; Lorat et al., 2012). Improper H3.3 accumulation in heterochromatin could activate otherwise silenced genes and synergize with the negative effect of unrepaired DNA lesions which accumulate therein. This is another facet of the H3.3 dilemma that awaits further investigation.
We have provided a number of arguments to suggest that the H3.3 replacement histone could play an important role in organismal and cellular aging via improper incorporation into the heterochromatin of BHL cells. Accordingly, the ability to manipulate H3.3 deposition and/or its downstream effects might open a new way for epigenetic treatment and prophylaxis of aging.
Acknowledgments
We thank Drs. Murat Saparbaev and Alexander Ishchenko for the discussion and Dr. Nikolay Ogryzko for critical reading of the manuscript.
Funding
This work was supported by Association pour la Recherche sur le Cancer (subvention no. SFI20121205936 to VO and PDF20130606592 to VO and RA).
Conflict of interest
None declared.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Adam S, Polo SE, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Adams-Cioaba MA, Krupa JC, Xu C, Mort JS, Min J. Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat. Commun. 2011;2:197. doi: 10.1038/ncomms1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell. 2002;9:1191–1200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S, Tatsuno K, Yamamoto S, Takayanagi S, Narita Y, Shibui S, Aburatani H, Saito N. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol. 2014;16:140–146. doi: 10.1093/neuonc/not144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Wiggins JC. Proteolytic processing of micronuclear H3 and histone phosphorylation during conjugation in Tetrahymena thermophila. Exp. Cell Res. 1984;153:287–298. doi: 10.1016/0014-4827(84)90601-3. [DOI] [PubMed] [Google Scholar]
- Angell R. First-meiotic-division nondisjunction in human oocytes. Am. J. Hum. Genet. 1997;61:23–32. doi: 10.1086/513890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Arimura Y, Shirayama K, Horikoshi N, Fujita R, Taguchi H, Kagawa W, Fukagawa T, Almouzni G, Kurumizaka H. Crystal structure and stable property of the cancer-associated heterotypic nucleosome containing CENP-A and H3.3. Sci. Rep. 2014;4:7115. doi: 10.1038/srep07115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai PJ, Bai DL, Russell DH, Macara IG, Shabanowitz J, Hunt DF, Black BE, Foltz DR. Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proc. Natl Acad. Sci. USA. 2013;110:11827–11832. doi: 10.1073/pnas.1300325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, Elsasser SJ, Chapgier A, Goldberg AD, Canaani E, Rafii S, Zheng D, Allis CD. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc. Natl Acad. Sci. USA. 1988;85:5112–5116. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goody V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ, Flanagan AM. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- Bonisch C, Hake SB. Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res. 2012;40:10719–10741. doi: 10.1093/nar/gks865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack C, Lithgow G, Osiewacz H, Toussaint O. EMBO WORKSHOP REPORT: molecular and cellular gerontology Serpiano, Switzerland, September 18–22, 1999. EMBO J. 2000;19:1929–1934. doi: 10.1093/emboj/19.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Uribesalgo I, Wibowo I, Rue P, Martin D, Gutierrez A, Morey L, Guigo R, Lopez-Schier H, Di Croce L, et al. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 2009;16:1074–1079. doi: 10.1038/nsmb.1665. [DOI] [PubMed] [Google Scholar]
- Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O'Geen H, Cotterman RF, Knoepfler PS. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin. 2013;6:7. doi: 10.1186/1756-8935-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Carter DB, Chae CB. Composition of liver histones in aging rat and mouse. J. Gerontol. 1975;30:28–32. doi: 10.1093/geronj/30.1.28. [DOI] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF. Histone H2A variants and the inactive X chromosome: identification of a second macroH2A variant. Hum. Mol. Genet. 2001a;10:1101–1113. doi: 10.1093/hmg/10.10.1101. [DOI] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J. Cell Biol. 2001b;152:375–384. doi: 10.1083/jcb.152.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee BR, Williams RP. Cytological changes in aging bacterial cultures. J. Bacteriol. 1962;84:340–344. doi: 10.1128/jb.84.2.340-344.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhao J, Wang Y, Wang M, Long H, Liang D, Huang L, Wen Z, Li W, Li X, Feng H, Zhao H, Zhu P, Li M, Wang QF, Li G. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27:2109–2124. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl Acad. Sci. USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr. Biol. 2010;20:1522–1528. doi: 10.1016/j.cub.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CM, Georgiou A, Szutorisz H, Maia e Silva A, Pombo A, Barahona I, Dargelos E, Canzonetta C, Dillon N. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 2005;6:354–360. doi: 10.1038/sj.embor.7400366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churikov D, Siino J, Svetlova M, Zhang K, Gineitis A, Morton Bradbury E, Zalensky A. Novel human testis-specific histone H2B encoded by the interrupted gene on the X chromosome. Genomics. 2004;84:745–756. doi: 10.1016/j.ygeno.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, Olbrich T, Gwerder M, Fink D, Stucki M. Dynamics of histone H3.3 deposition in proliferating and senescent cells reveals a DAXX-dependent targeting to PML-NBs important for pericentromeric heterochromatin organization. Cell Cycle. 2014;13:249–267. doi: 10.4161/cc.26988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature. 1998;393:599–601. doi: 10.1038/31275. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008;135:649–661. doi: 10.1016/j.cell.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, Adams PD. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013;15:1495–1506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Liu Y, Gorovsky MA. Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol. Cell. Biol. 2006;26:7719–7730. doi: 10.1128/MCB.01139-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daury L, Chailleux C, Bonvallet J, Trouche D. Histone H3.3 deposition at E2F-regulated genes is linked to transcription. EMBO Rep. 2006;7:66–71. doi: 10.1038/sj.embor.7400561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013a;12:247–256. doi: 10.1111/acel.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peterson AL, Neretti N, Sedivy JM, Kreiling JA. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging. 2013b;5:867–883. doi: 10.18632/aging.100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimauro T, David G. Chromatin modifications: the driving force of senescence and aging? Aging. 2009;1:182–190. doi: 10.18632/aging.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte LF, Young AR, Wang Z, Wu HA, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma'ayan A, Narita M, Bernstein E. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014;5:5210. doi: 10.1038/ncomms6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effros RB. Insights on immunological aging derived from the T lymphocyte cellular senescence model. Exp. Gerontol. 1996;31:21–27. doi: 10.1016/0531-5565(95)00017-8. [DOI] [PubMed] [Google Scholar]
- Eirin-Lopez JM, Gonzalez-Romero R, Dryhurst D, Ishibashi T, Ausio J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009;9:31. doi: 10.1186/1471-2148-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser SJ, Goldberg AD, Allis CD. New functions for an old variant: no substitute for histone H3.3. Curr. Opin. Genet. Dev. 2010;20:110–117. doi: 10.1016/j.gde.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JR, Taylor H, Matthaei K, Rathjen PD, Tremethick DJ, Lyons I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001;11:1183–1187. doi: 10.1016/s0960-9822(01)00329-3. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Feser J, Tyler J. Chromatin structure as a mediator of aging. FEBS Lett. 2011;585:2041–2048. doi: 10.1016/j.febslet.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipescu D, Szenker E, Almouzni G. Developmental roles of histone H3 variants and their chaperones. Trends Genet. 2013;29:630–640. doi: 10.1016/j.tig.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Fontebasso AM, Liu XY, Sturm D, Jabado N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol. 2013;23:210–216. doi: 10.1111/bpa.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A, Listovsky T, Guilbaud G, Sarkies P, Sale JE. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014;24:2195–2201. doi: 10.1016/j.cub.2014.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, Kraus WL. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010;24:21–32. doi: 10.1101/gad.1876110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cruz R, Brieno MA, Roig I, Grossmann M, Velilla E, Pujol A, Cabero L, Pessarrodona A, Barbero JL, Garcia Caldes M. Dynamics of cohesin proteins REC8, STAG3, SMC1 beta and SMC3 are consistent with a role in sister chromatid cohesion during meiosis in human oocytes. Hum. Reprod. 2010;25:2316–2327. doi: 10.1093/humrep/deq180. [DOI] [PubMed] [Google Scholar]
- Gensler HL, Bernstein H. DNA damage as the primary cause of aging. Q. Rev. Biol. 1981;56:279–303. doi: 10.1086/412317. [DOI] [PubMed] [Google Scholar]
- Gindoff PR, Jewelewicz R. Reproductive potential in the older woman. Fertil. Steril. 1986;46:989–1001. doi: 10.1016/s0015-0282(16)49869-9. [DOI] [PubMed] [Google Scholar]
- Gineitis AA, Zalenskaya IA, Yau PM, Bradbury EM, Zalensky AO. Human sperm telomere-binding complex involves histone H2B and secures telomere membrane attachment. J. Cell Biol. 2000;151:1591–1598. doi: 10.1083/jcb.151.7.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Jeggo PA. The heterochromatic barrier to DNA double strand break repair: how to get the entry visa. Int. J. Mol. Sci. 2012;13:11844–11860. doi: 10.3390/ijms130911844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosden RG. Maternal age: a major factor affecting the prospects and outcome of pregnancy. Ann. N. Y. Acad. Sci. 1985;442:45–57. doi: 10.1111/j.1749-6632.1985.tb37504.x. [DOI] [PubMed] [Google Scholar]
- Gosden R. Cheating Time. New York: W. H. Freeman & Company; 1996. [Google Scholar]
- Govin J, Escoffier E, Rousseaux S, Kuhn L, Ferro M, Thevenon J, Catena R, Davidson I, Garin J, Khochbin S, Caron C. Pericentric heterochromatin reprogramming by new histone variants during mouse spermiogenesis. J. Cell Biol. 2007;176:283–294. doi: 10.1083/jcb.200604141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, Wang Z, Yang P, Wu F, Hui J, Fu X, Shi X, Shi YG, Xing Y, Lan F, Shi Y. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell. 2014;56:298–310. doi: 10.1016/j.molcel.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc. Natl Acad. Sci. USA. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Chiu D. Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum. Genet. 1985;70:11–17. doi: 10.1007/BF00389450. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The cell biology of aging. Clin. Geriatr. Med. 1985;1:15–27. [PubMed] [Google Scholar]
- Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 2004;20:320–326. doi: 10.1016/j.tig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Horikoshi T, Balin AK, Carter DM. Effect of oxygen on the growth of human epidermal keratinocytes. J. Invest. Dermatol. 1986;86:424–427. doi: 10.1111/1523-1747.ep12285695. [DOI] [PubMed] [Google Scholar]
- Howard BH. Replicative senescence: considerations relating to the stability of heterochromatin domains. Exp. Gerontol. 1996;31:281–293. doi: 10.1016/0531-5565(95)00022-4. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, Ge K, Levens D, Crane-Robinson C, Zhao K. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013;12:180–192. doi: 10.1016/j.stem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioudinkova ES, Barat A, Pichugin A, Markova E, Sklyar I, Pirozhkova I, Robin C, Lipinski M, Ogryzko V, Vassetzky YS, Razin SV. Distinct distribution of ectopically expressed histone variants H2A.Bbd and MacroH2A in open and closed chromatin domains. PLoS One. 2012;7:e47157. doi: 10.1371/journal.pone.0047157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Y, Bergamin E, Donoghue MT, Mongeon V, LeBlanc C, Voigt P, Underwood CJ, Brunzelle JS, Michaels SD, Reinberg D, Couture JF, Martienssen RA. Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science. 2014;343:1249–1253. doi: 10.1126/science.1248357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, Spector DL. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, Kennedy BK. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Kim H, Heo K, Choi J, Kim K, An W. Histone variant H3.3 stimulates HSP70 transcription through cooperation with HP1gamma. Nucleic Acids Res. 2011;39:8329–8341. doi: 10.1093/nar/gkr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Dejsuphong D, Adelmant G, Ceccaldi R, Yang K, Marto JA, D'Andrea AD. Transcriptional repressor ZBTB1 promotes chromatin remodeling and translesion DNA synthesis. Mol. Cell. 2014;54:107–118. doi: 10.1016/j.molcel.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Lacoste N, Woolfe A, Tachiwana H, Garea AV, Barth T, Cantaloube S, Kurumizaka H, Imhof A, Almouzni G. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell. 2014;53:631–644. doi: 10.1016/j.molcel.2014.01.018. [DOI] [PubMed] [Google Scholar]
- Lamb NE, Feingold E, Savage A, Avramopoulos D, Freeman S, Gu Y, Hallberg A, Hersey J, Karadima G, Pettay D, Saker D, Shen J, Taft L, Mikkelsen M, Petersen MB, Hassold T, Sherman SL. Characterization of susceptible chiasma configurations that increase the risk for maternal nondisjunction of chromosome 21. Hum. Mol. Genet. 1997;6:1391–1399. doi: 10.1093/hmg/6.9.1391. [DOI] [PubMed] [Google Scholar]
- Lamour V, Lecluse Y, Desmaze C, Spector M, Bodescot M, Aurias A, Osley MA, Lipinski M. A human homolog of the S. cerevisiae HIR1 and HIR2 transcriptional repressors cloned from the DiGeorge syndrome critical region. Hum. Mol. Genet. 1995;4:791–799. doi: 10.1093/hmg/4.5.791. [DOI] [PubMed] [Google Scholar]
- Lemaitre C, Soutoglou E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair (Amst.) 2014;19:163–168. doi: 10.1016/j.dnarep.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia R. Stem cell quiescence. Clin. Cancer Res. 2011;17:4936–4941. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Maffey AH, Abbott WD, Conde e Silva N, Prunell A, Siino J, Churikov D, Zalensky AO, Ausio J. Characterization of nucleosomes consisting of the human testis/sperm-specific histone H2B variant (hTSH2B) Biochemistry. 2005;44:2529–2535. doi: 10.1021/bi048061n. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Lindsay CR, Morozov VM, Ishov AM. PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front Biosci. 2008;13:7132–7142. doi: 10.2741/3216. [DOI] [PubMed] [Google Scholar]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y, Jessberger R, Kirkwood TB, Hoog C, Herbert M. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 2010;20:1511–1521. doi: 10.1016/j.cub.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Lorat Y, Schanz S, Schuler N, Wennemuth G, Rube C, Rube CE. Beyond repair foci: DNA double-strand break repair in euchromatic and heterochromatic compartments analyzed by transmission electron microscopy. PLoS One. 2012;7:e38165. doi: 10.1371/journal.pone.0038165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012;13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklon NS, Fauser BC. Aspects of ovarian follicle development throughout life. Horm. Res. 1999;52:161–170. doi: 10.1159/000023456. [DOI] [PubMed] [Google Scholar]
- de Magalhaes JP. From cells to ageing: a review of models and mechanisms of cellular senescence and their impact on human ageing. Exp. Cell Res. 2004;300:1–10. doi: 10.1016/j.yexcr.2004.07.006. [DOI] [PubMed] [Google Scholar]
- de Magalhaes JP, Chainiaux F, de Longueville F, Mainfroid V, Migeot V, Marcq L, Remacle J, Salmon M, Toussaint O. Gene expression and regulation in H2O2-induced premature senescence of human foreskin fibroblasts expressing or not telomerase. Exp. Gerontol. 2004;39:1379–1389. doi: 10.1016/j.exger.2004.06.004. [DOI] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKittrick E, Gafken PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl Acad. Sci. USA. 2004;101:1525–1530. doi: 10.1073/pnas.0308092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michod D, Bartesaghi S, Khelifi A, Bellodi C, Berliocchi L, Nicotera P, Salomoni P. Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron. 2012;74:122–135. doi: 10.1016/j.neuron.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. Histone replacement marks the boundaries of cis-regulatory domains. Science. 2007;315:1408–1411. doi: 10.1126/science.1134004. [DOI] [PubMed] [Google Scholar]
- Montellier E, Boussouar F, Rousseaux S, Zhang K, Buchou T, Fenaille F, Shiota H, Debernardi A, Hery P, Curtet S, Jamshidikia M, Barral S, Holota H, Bergon A, Lopez F, Guardiola P, Pernet K, Imbert J, Petosa C, Tan M, Zhao Y, Gerard M, Khochbin S. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev. 2013;27:1680–1692. doi: 10.1101/gad.220095.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov VM, Gavrilova EV, Ogryzko VV, Ishov AM. Dualistic function of Daxx at centromeric and pericentromeric heterochromatin in normal and stress conditions. Nucleus. 2012;3:276–285. doi: 10.4161/nucl.20180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Ray-Gallet D, Quivy JP, Tagami H, Almouzni G. Two distinct nucleosome assembly pathways: dependent or independent of DNA synthesis promoted by histone H3.1 and H3.3 complexes. Cold Spring Harb. Symp. Quant. Biol. 2004;69:273–280. doi: 10.1101/sqb.2004.69.273. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Nishioka K, Dong YX, Shimojima T, Hirose S. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev. 2007;21:552–561. doi: 10.1101/gad.1503407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV. Erwin Schroedinger, Francis Crick and epigenetic stability. Biol. Direct. 2008;3:15. doi: 10.1186/1745-6150-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol. Cell. Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973;41:181–190. doi: 10.1016/0022-5193(73)90198-7. [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Palmer DK, O'Day K, Trong HL, Charbonneau H, Margolis RL. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc. Natl Acad. Sci. USA. 1991;88:3734–3738. doi: 10.1073/pnas.88.9.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchelintsev NA, McBryan T, Rai TS, van Tuyn J, Ray-Gallet D, Almouzni G, Adams PD. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013;3:1012–1019. doi: 10.1016/j.celrep.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegoraro G, Misteli T. The central role of chromatin maintenance in aging. Aging. 2009;1:1017–1022. doi: 10.18632/aging.100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellestor F. Maternal age and chromosomal abnormalities in human oocytes. Med. Sci. (Paris) 2004;20:691–696. doi: 10.1051/medsci/2004206-7691. [DOI] [PubMed] [Google Scholar]
- Pellestor F, Andreo B, Anahory T, Hamamah S. The occurrence of aneuploidy in human: lessons from the cytogenetic studies of human oocytes. Eur. J. Med. Genet. 2006;49:103–116. doi: 10.1016/j.ejmg.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Piette C, de Mouzon J, Bachelot A, Spira A. In-vitro fertilization: influence of women's age on pregnancy rates. Hum. Reprod. 1990;5:56–59. doi: 10.1093/oxfordjournals.humrep.a137041. [DOI] [PubMed] [Google Scholar]
- Pina B, Suau P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev. Biol. 1987;123:51–58. doi: 10.1016/0012-1606(87)90426-x. [DOI] [PubMed] [Google Scholar]
- Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009;83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribny AP, Vanyushin BF. Age-related genomic hypomethylation. In: Tollefsbol TO, editor. Epigenetics of Aging. New York: Springer; 2010. pp. 11–28. [Google Scholar]
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Rai TS, Adams PD. Lessons from senescence: chromatin maintenance in non-proliferating cells. Biochim. Biophys. Acta. 2012;1819:322–331. doi: 10.1016/j.bbagrm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014;28:2712–2725. doi: 10.1101/gad.247528.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LE, Almouzni G. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rodier F, Campisi J. Four faces of cellular senescence. J. Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo VEA, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Woodbury: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- Saade E, Ogryzko V. Epigenetics: what it is about? Biopolym. Cell. 2014;30:3–9. [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr. Biol. 2009;19:1816–1820. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomoni P. The PML-interacting protein DAXX: histone loading gets into the picture. Front. Oncol. 2013;3:152. doi: 10.3389/fonc.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro SW, Dulac C. The activity-dependent histone variant H2BE modulates the life span of olfactory neurons. Elife. 2012;1:e00070. doi: 10.7554/eLife.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E, Tanabe M, Zhao Y, Kimura S, Fujiyama S, Ueda T, Umetsu D, Ito T, Takeyama K, Kato S. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 2010;24:159–170. doi: 10.1101/gad.1857410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk R, Jenke A, Zilbauer M, Wirth S, Postberg J. H3.5 is a novel hominid-specific histone H3 variant that is specifically expressed in the seminiferous tubules of human testes. Chromosoma. 2011;120:275–285. doi: 10.1007/s00412-011-0310-4. [DOI] [PubMed] [Google Scholar]
- Schramm RD, Paprocki AM, Bavister BD. Features associated with reproductive ageing in female rhesus monkeys. Hum. Reprod. 2002;17:1597–1603. doi: 10.1093/humrep/17.6.1597. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol. Ther. 2003;2:233–235. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr. Biol. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. Histone variants in pluripotency and disease. Development. 2013;140:2513–2524. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- Smerdon MJ. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991;3:422–428. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- Soni S, Pchelintsev N, Adams PD, Bieker JJ. Transcription factor EKLF (KLF1) recruitment of the histone chaperone HIRA is essential for beta-globin gene expression. Proc. Natl Acad. Sci. USA. 2014;111:13337–13342. doi: 10.1073/pnas.1405422111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottocornola R, Lo Celso C. Dormancy in the stem cell niche. Stem Cell Res. Ther. 2012;3:10. doi: 10.1186/scrt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein ZA. A woman's age: childbearing and child rearing. Am. J. Epidemiol. 1985;121:327–342. doi: 10.1093/oxfordjournals.aje.a114004. [DOI] [PubMed] [Google Scholar]
- Sutcliffe EL, Parish IA, He YQ, Juelich T, Tierney ML, Rangasamy D, Milburn PJ, Parish CR, Tremethick DJ, Rao S. Dynamic histone variant exchange accompanies gene induction in T cells. Mol. Cell. Biol. 2009;29:1972–1986. doi: 10.1128/MCB.01590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013;203:929–942. doi: 10.1083/jcb.201306073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Osakabe A, Kawaguchi K, Shiga T, Hayashi-Takanaka Y, Kimura H, Kurumizaka H. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc. Natl Acad. Sci. USA. 2010;107:10454–10459. doi: 10.1073/pnas.1003064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- Talhaoui I, Couve S, Gros L, Ishchenko AA, Matkarimov B, Saparbaev MK. Aberrant repair initiated by mismatch-specific thymine-DNA glycosylases provides a mechanism for the mutational bias observed in CpG islands. Nucleic Acids Res. 2014;42:6300–6313. doi: 10.1093/nar/gku246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Smith M, Kanno T, Dasenbrock H, Nishiyama A, Ozato K. Inducible deposition of the histone variant H3.3 in interferon-stimulated genes. J. Biol. Chem. 2009;284:12217–12225. doi: 10.1074/jbc.M805651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakar A, Gupta P, Ishibashi T, Finn R, Silva-Moreno B, Uchiyama S, Fukui K, Tomschik M, Ausio J, Zlatanova J. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009;48:10852–10857. doi: 10.1021/bi901129e. [DOI] [PubMed] [Google Scholar]
- Tolstorukov MY, Goldman JA, Gilbert C, Ogryzko V, Kingston RE, Park PJ. Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells. Mol. Cell. 2012;47:596–607. doi: 10.1016/j.molcel.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, 3rd, Hetzer MW. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene. 2000;19:1698–1709. doi: 10.1038/sj.onc.1203471. [DOI] [PubMed] [Google Scholar]
- Urban MK, Zweidler A. Changes in nucleosomal core histone variants during chicken development and maturation. Dev. Biol. 1983;95:421–428. doi: 10.1016/0012-1606(83)90043-x. [DOI] [PubMed] [Google Scholar]
- Venneti S, Santi M, Felicella MM, Yarilin D, Phillips JJ, Sullivan LM, Martinez D, Perry A, Lewis PW, Thompson CB, Judkins AR. A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol. 2014;128:743–753. doi: 10.1007/s00401-014-1338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Dolle ME. Large genome rearrangements as a primary cause of aging. Mech. Ageing Dev. 2002;123:907–915. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, Li B, Barton MC, Li W, Li H, Shi X. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann SM, Mildner SN, Bonisch C, Israel L, Maiser A, Matheisl S, Straub T, Merkl R, Leonhardt H, Kremmer E, Schermelleh L, Hake SB. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010;190:777–791. doi: 10.1083/jcb.201002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbelauer C, Bell O, Schubeler D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 2005;19:1761–1766. doi: 10.1101/gad.347705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme J, Angell RR. Maternal age and trisomy–a unifying mechanism of formation. Chromosoma. 2000;109:435–438. doi: 10.1007/s004120000088. [DOI] [PubMed] [Google Scholar]
- Wright WE, Shay JW. Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr. Opin. Genet. Dev. 2001;11:98–103. doi: 10.1016/s0959-437x(00)00163-5. [DOI] [PubMed] [Google Scholar]
- Wu RS, Tsai S, Bonner WM. Changes in histone H3 composition and synthesis pattern during lymphocyte activation. Biochemistry. 1983;22:3868–3873. doi: 10.1021/bi00285a023. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Song Y, Seol JH, Park JY, Yang YJ, Han JW, Youn HD, Cho EJ. Myogenic transcriptional activation of MyoD mediated by replication-independent histone deposition. Proc. Natl Acad. Sci. USA. 2011;108:85–90. doi: 10.1073/pnas.1009830108. [DOI] [PMC free article] [PubMed] [Google Scholar]