

Abstract
Aggressive B-cell lymphomas are diverse group of neoplasms that arise at different stages of B-cell development and by various mechanisms of neoplastic transformation. The aggressive B-cell lymphomas include many types, subtypes and variants of diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL), mantle cell lymphoma and its blastoid variant, and B lymphoblastic lymphoma. Differences in histology, cytogenetic and molecular abnormalities, as well as the relationship with the tumor microenvironment, help define characteristic signatures for these neoplasms, and in turn dictate potential therapeutic targets. Rather than survey the entire spectrum of aggressive B-cell lymphomas, this report aims to identify and characterize important clinically aggressive subtypes of DLBCL, and explore the relationship of DLBCL to BL and the gray zone between them (B-cell lymphoma unclassifiable with features intermediate between DLBCL and BL).
Keywords: aggressive B-cell lymphoma, Burkitt, double hit, high grade, unclassifiable
Introduction
Aggressive B-cell lymphomas (BCL) include both precursor lymphoid neoplasms (B lymphoblastic leukemia/lymphoma NOS and B lymphoblastic leukemia lymphoma with recurrent genetic abnormalities) and numerous mature B-cell neoplasms, including mantle cell lymphoma (MCL), primary effusion lymphoma (PEL), Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL) and its many types, subtypes and variants, and B-cell lymphoma unclassifiable with features intermediate between DLBCL and Burkitt lymphoma. DLBCL includes a large number of disparate entities with marked differences in morphology, phenotype, molecular pathogenesis and clinical behavior (Table 1). There is an ongoing effort to tailor therapy based on specific subtypes of DLBCL, and prognostic markers are becoming increasingly important. Rather than a discussion of all types of aggressive BCL, this report attempts to identify those entities within the spectrum DLBCL that have distinctive pathologic and clinical significance, and the differential with Burkitt lymphoma, and high-grade unclassifiable B-cell lymphoma.
Table 1.
WHO 2008 DLBCL variants, subgroups and subtypes/entities63
| Diffuse large B-cell lymphoma NOS |
| Common morphologic variants |
| Centroblastic |
| Immunoblastic |
| Anaplastic and other rare morphologic variants |
| Molecular subgroups |
| GCB |
| Activated B-cell-like (non-GCB) |
| Immunohistochemical subgroups |
| CD5 + DLBCL |
| Primary DLBCL of the CNS |
| Primary cutaneous DLBCL, leg type |
| EBV + DLBCL of the elderly |
| Diffuse large B-cell lymphoma subtypes |
| T-cell/histiocyte-rich large B-cell lymphoma |
| Primary DLBCL of the CNS |
| Primary cutaneous DLBCL, leg type |
| EBV-positive DLBCL of the elderly |
| Other lymphomas of large B cells |
| Primary mediastinal (thymic) LBCL |
| Intravascular large B-cell lymphoma |
| DLBCL associated with chronic inflammation |
| Lymphomatoid granulomatosis |
| ALK-positive LBCL |
| Plasmablastic lymphoma |
| Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease |
| Primary effusion lymphoma |
| Borderline cases |
| B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt’s lymphoma |
| B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma |
Abbreviations: CNS, central nervous system; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; GCB, germinal center B cell.
Diffuse large B-cell lymphoma
DLBCL is characterized by diffuse nodal architectural effacement or extranodal infiltration by sheets of large cells of B-cell phenotype. Small T-lymphocytes/histiocytes are usually present unlike Burkitt lymphomas that are more homogeneous and uniform. In some subtypes, such as T-cell/histiocyte rich large B-cell lymphoma, the background small lymphocytes and histiocytes may outnumber the large B cells. Sclerosis is variable but may be prominent, and mitotic figures are easily identified. Of clinical importance is classification of diffuse large cell lymphoma in patients with grade 3 follicular lymphoma. If there are diffuse areas containing greater than 15 centroblasts per high power field, a separate diagnosis of DLBCL should be rendered, and this should precede the diagnosis of follicular lymphoma. Staining for follicular dendritic cells with antibodies to CD21 or CD23 may be helpful in highlighting areas of follicular proliferation in a partially diffuse infiltrate (and vice versa).
The concept of separating the lymphomas into ‘clinical grades’ dates back to the mid-1970s and the working formulation, and was based on actuarial survival curves from patients in an international study group. The working formulation divided the DLBCL into large cell and immunoblastic, with the immunoblastic in the high-grade category often requiring central nervous system (CNS) prophylaxis. Since then DLBCL has become increasingly complex (Table 1), and it has been termed the ‘waste basket’ of lymphoma classification.1 Although the addition of rituximab has improved the outcome for DLBCL, up to 40% of cases become refractory to chemotherapy or relapse with poor outcome. Appropriate therapy requires stratification of different subtypes of DLBCL in a schema that is clinically meaningful. In addition to histology, immunohistochemistry, molecular and cytogenetic characteristics, prognosis may be influenced by diverse factors such as the ‘stromal signature’ of the background2 and host factors such as serum levels of vitamin D.3 Increasingly newer tools including gene expression profiling (GEP), gene sequencing and micro RNA (miRNA) profiling4 have provided prognostically relevant information. Newer treatment modalities include the use of proteosome inhibitors that target the NF-kB pathway required by activated B-cell (ABC) type DLBCL, small molecule inhibitors of signal transduction pathways and agents like lenalidomide, which modulate the cytokines and tumor microenvironment.5–7 Pathologic features will be important in determining which patients may benefit from these agents.8
Clinical features
DLBCL is the most common histologic subtype of NHL and comprises 30% of adult NHL in the west and greater than 40% in Asia. Although the median age at presentation is in the 7th decade at age 64, DLBCL occur at any age. Cases of DLBCL account for about 20% of non-Hodgkin lymphomas in children under 14, and up to 37% of non-Hodgkin lymphomas in adolescents aged 15–19 years.9 It is slightly more common in males. About 40% of DLBCL are extranodal stage I or II, or patients may present with a nodal rapidly enlarging mass or masses. The most common sites of extranodal involvement are the stomach, ileocecal region, bone, testis, spleen, Waldeyer’s ring, salivary gland, thyroid, liver, kidney and adrenal. The outcome in DLBCL has been improved with rituximab therapy, first used in elderly patients with DLBCL. However, a significant percentage of patients (20–40%) become refractory to treatment, and the prognosis for relapsed DLBCL is dismal.10 Unfavorable factors in DLBCL include older age >60 years, poor performance status (ECOG>2), advanced stage (III–IV), extranodal involvement >2 sites and high serum LDH.
Pathogenesis
DLBCL can arise de novo or as a progression or transformation event in a patient with low-grade lymphoma, including CLL/SLL, follicular lymphoma and marginal zone lymphoma. DLBCL may be a final common pathway in many patients who are longterm survivors of indolent lymphoma.11 Transformation of CLL/SLL to DLBCL is termed Richter’s syndrome. Even in the rituximab era, high-grade transformation remains an important challenge in the management of patients with low-grade lymphoma.12 Immunodeficiency is a risk factor for DLBCL, and about 10% of DLBCL are EBV +. The incidence of EBV + DLBCL increases with increasing age and may relate to senescence of the immune system.13 As the presence of EBV may adversely affect prognosis, the question arises if DLBCL’s from older patients should be screened for EBV, particularly if there are aggressive clinical features or atypical nodal, mucosal or cutaneous sites of involvement.13
Differential diagnosis
Although the diagnosis of DLBCL is usually not difficult, there are situations in which it is problematic. DLBCL may form cohesive sheets or involve sinusoids, mimicking carcinoma, melanoma etc. In particular, nasopharyngeal carcinomas, neuroendocrine carcinomas, germ cell tumors (seminomas) and granulocytic sarcomas (chloromas) can be mistaken for DLBCL and should be considered in the differential diagnosis. These can usually be identified using a standard panel of immunohistochemical markers including a keratin antibody such as AE1/3 or CAM5.2, neuroendocrine markers such as NSE, synaptophysin, chromogranin, germ cell tumor markers such as PLAP and OCT3/4, and melanoma markers such as S100, HMB45 and Mart1. In some cases, there may be aberrant phenotypic characteristics that make DLBCL difficult to identify (for example, plasmablastic lymphoma (PBL) may lack CD20, as may ALK + DLBCL). DLBCL may also share histologic or phenotypic features with other lymphomas, probably due to common pathogenic pathways such as the ‘gray zone’ lymphoma discussed below. Some DLBCL may contain Hodgkin and Reed Sternberg (H–RS)-like cells. This is particularly common in those that are associated with immunodeficiency and infection with EBV, so that differential with classical Hodgkin lymphoma can be problematic.
Immunohistochemistry
DLBCL characteristically express pan B-cell markers including CD19, CD20, CD22, CD79a and PAX5. Surface/cytoplasmic immunoglobulin (IgM>IgG>IgA) is present in 50–75% of cases. CD30 is variable, positive in the anaplastic variant of DLBCL. Unlike Hodgkin lymphoma, DLBCL usually express a complete B-cell program, including expression of transcription factors such as OCT-2 and BOB.1. CD3 is negative, although a CD3 + variant of DLBCL has been described.14 Other markers commonly present in DLBCL include CD10 (40%), BCL-6 (60%), BCL2 (50%), CD43 (20%), CD5 (<10%), CD30 (10%), IRF4/MUM1 (40%) and p53 (30%). Usually there are 30–90% Ki67 positive cells. Monoclonal antibodies allow nuclear localization of MYC protein to identify MYC+ DLBCL (Figure 1). MYC immunohistochemistry is strongly positive in DLBCL with MYC translocations, but overexpression can also occur by other mechanisms such as amplification.15 The antibody may serve to screen DLBCL for MYC overexpression, and such cases can then be probed with MYC FISH using a break apart probe or other methods to identify cases with translocations. Regardless of translocations, expression of MYC and BCL2 in DLBCL appears to independently predict inferior overall and progression-free survival in DLBCL.16,17
Figure 1.
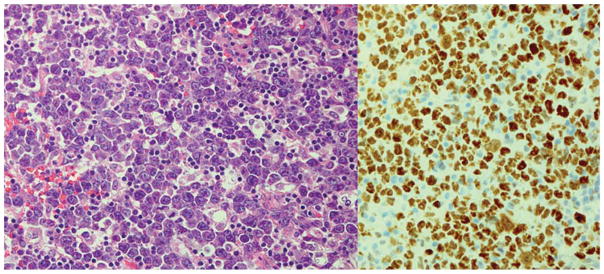
Diffuse large B-cell lymphoma MYC positive. The cells are large with one or more nucleoli and there is infiltration by small lymphocytes and histiocytes. Immunostains for MYC is strongly positive in the nuclei of almost all the malignant cells and FISH detected a MYC translocation with the break apart probe. Left: Hematoxylin and eosin. Right: Immunoperoxidase stain for MYC protein, hematoxylin counterstain. Original × 400.
Aberrant phenotypes are not uncommon in DLBCL, and may be responsible for confusion in the diagnosis. DLBCL may lack one or more B-cell markers such as CD19, CD20, CD22 and CD79a. DBCL may co-express IRF4/MUM1 and BCL6 (unlike normal germinal center cells). DLBCL may be positive for CD5 (discussed below) or rarely express cyclin D1. Immunohistochemistry may also help identify other unique variants such as PBLs, which have a plasma cell phenotype and are often negative for PAX5 and positive for CD138 (see below).
Cyclin D1+ DLBCL
Cyclin D1-positive CD5 negative cases of DLBCL lack the characteristic t(11;14) translocation of MCL, but may have additional copies of CCND1/chromosome 11.18 Also unlike most cases of MCL, cyclin D1+ DLBCL cases are negative for SOX11.19 One study revealed deletion in genes involved in the AKT/glycogen synthase kinase-3 β cascade controlling nuclear levels of cyclin D1, suggesting that posttranslational events regulating cyclin D1 may occur in a subset of DLBCL’s.20 Most cases of cyclin D1+ DLBCL have centroblastic morphology and a post germinal center phenotype positive for IRF4/MUM121 (Figure 2).
Figure 2.

Diffuse large B-cell lymphoma with GCB phenotype positive for cyclin D1. The cells resemble centroblasts with oval nuclei and one or more nucleoli mostly at the nuclear membrane (top). The cyclin D1 stain reveals nuclear staining. FISH was negative for t(11;14). Top: Hematoxylin and eosin, bottom: immunoperoxidase stain for cyclin D1 hematoxylin counterstain. Original × 240.
CD5+DLBCL
Expression of CD5 is also associated with a worse prognosis even in the rituximab era.22 It is thought that this subtype may arise from the same progenitor cell as the mutated variant of CD5+ SLL/CLL cell, but these cases arise de novo without prior history of CLL. Clinical features include a higher age distribution (mean 66 years), female predominance, advanced stage at diagnosis, ‘B’ symptoms and a more aggressive clinical course. A subset of patients with intravascular large B-cell lymphoma also expresses CD5.
Bone marrow involvement in DLBCL
Marrow involvement occurs in about 16% of DLBCL. Two main patterns of bone marrow involvement occur. Bone marrow involvement may be concordant in which the cells infiltrating the marrow are large (large cell lymphoma). This variant is generally associated with poorer overall and progression-free survival.23 In other cases, the lymphoma cells infiltrating the marrow are small and characteristic of low-grade lymphoma (so-called discordant histology). This pattern of marrow infiltration is more common and has been associated with more frequent relapse but a similar 5-year survival to cases without bone marrow involvement. In up to 30% of patients with bone marrow disease circulating cells in the peripheral blood can be identified with flow cytometry.
The role of the microenvironment in DLBCL
There is increasing evidence that not only the malignant cells but the microenvironment and the inflammatory response may also provide clues to the pathogenesis and behavior of DLBCL. Lenz et al2 identified two patterns of stromal signature in DLBCL. The stromal microenvironment associated with a favorable prognosis included genes associated with the extracellular matrix such as fibronectin and collagens, as well as anti-angiogenic factors, monocytes and thrombospondin. The adverse stromal signature was associated with angiogenesis and included endothelial cell markers such as CD31, and angiogenesis factors such as VEGF. This may have therapeutic implications and clinical trials are in progress using drugs such as lenalidomide that have a potential effect on stromal cells, angiogenesis and the microenvironment. 24 Genes expressed by the immune microenvironment such as TNFRSF9 have been shown to predict overall survival in patients with DLBCL.25 Immunostains for stromal markers such as SPARC (secreted protein, acidic and rich in cysteine) may have prognostic significance in a subset of DLBCL.26
DLBCL, NOS
DLBCL, NOS is commonly centroblastic or immunoblastic in appearance. In the centroblastic variant (Figure 2), which is the most common morphologic variant, the cells resemble large centroblasts. The cells are intermediate to large with round or oval nuclei, vesicular chromatin and 2–4 nucleoli often at the nuclear membrane. There is moderate amphophilic to basophilic cytoplasm. There may be a monomorphic proliferation of centroblasts or there may be an admixed component of immunoblasts. Occasionally, the nuclei may be multilobated especially in extranodal locations such as primary bone DLBCL. In the immunoblastic variant, >90% of the cells have an immunoblastic appearance with a large centrally located nucleolus, and basophilic or amphophilic cytoplasm (Figure 3), and this histology is usually associated with the non-germinal center type of DLBCL. Immunoblastic morphology may be an independent adverse prognostic factor in DLBCL.27 The anaplastic variant is characterized by large round oval or polygonal cells with bizarre pleomorphic nuclei. These cells may resemble Hodgkin or RS cells. In some cases, the cells can resemble those seen in anaplastic large cell lymphoma, and show sinusoidal and/or cohesive growth pattern. However, these should not be confused with anaplastic large cell lymphoma that is of T-cell origin or ALK positive large B-cell lymphoma. These cases must also be differentiated from metastatic undifferentiated carcinoma. Other rare morphologic variants of DLBCL include cases with microvillous, spindle cell or signet ring cells in which the cytoplasm contains large amounts of protein displacing the nucleus (Figure 4).
Figure 3.

Diffuse large B-cell lymphoma (DLBCL) with non-germinal center B-cell (GCB) phenotype and an immunoblastic appearance. The cells are large and somewhat pleomorphic, many with large central nucleoli and amphophilic cytoplasm. Hematoxylin and eosin, original × 400.
Figure 4.
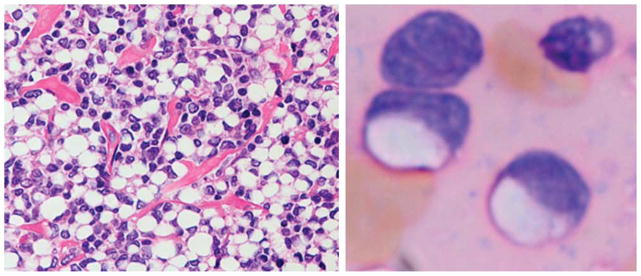
Diffuse large B-cell lymphoma (DLBCL) with signet ring cells mimicking carcinoma. This germinal center B-cell (GCB) phenotype DLBCL has cells with central cytoplasmic clear zones in histologic preparations and touch imprints. The cytoplasm forms a ring about a central clear vacuole. Hematoxylin and eosin, original × 240 (left) and × 400 (right).
Molecular and genetic features
DLBCL is characterized by clonal rearrangements of immunoglobulin heavy and light chain genes. There are somatic hypermutations in the variable regions in most cases. Based on GEP, DLBCL can be divided into two main subtypes: germinal center B-cell (GCB) with a GCB signature (50% cases) and an ABC group, which has a gene profile similar to that induced by in vitro activation of peripheral blood lymphocytes.28 The non-GCB group has been associated with an adverse prognosis even in the rituximab era.29 Not all cases of DLBCL can be characterized as ABC or GCB, and there is a third less well-characterized group that does not clearly fit the two main molecular subtypes. The proportion of ABC subtype DLBCL increases with age, and patients 70 or older are more likely to have non-GCB lymphomas.30,31 Immunohistochemical markers for the ABC group include IRF4/MUM1 and CD138. ABC lymphomas exhibit higher expression levels of NFκB, and constitutive NFκB signaling is required for survival of the malignant cells.28,32 The most common genetic findings in DLBCL, NOS are abnormalities in 3q27 involving BCL6 in 30–40% of cases (more in the ABC type of DLBCL), abnormalities of BCL2 t(14;18) in 20–40% and MYC rearrangement involving 8q24 in 10% of cases. BCL2 and MYC translocations are usually associated with germinal center-related DLBCL. BCL2 is the most highly mutated gene in GCB DLBCL.33 MYC rearrangements may be associated with an adverse prognosis either as a ‘simple’ hit involving the MYC gene alone34 or additional hits involving MYC/BCL2, MYC/BCL6 or MYC/BCL2/BCL6 (discussed below).35 Tumors with extra MYC signals plus IgH@BCL2 or MYC rearrangement plus extra BCL2 signals also appear to have a poor prognosis.36
Immunohistochemical subgroups as surrogates for GEP
A number of investigators have attempted to recapitulate the molecular subgroups described above using a limited panel of immunohistochemical reagents to identify GCB and non-GCB lymphomas.29 The Hans classifier of DLBCL37 has been widely used in clinical trials. According to this scheme, DLBCL that are CD10+ in more than 30% of the cells are in the GCB group. Cases that are CD10 negative and BCL6+, IRF4/MUM1 negative are also in the GCB group. Cases that are BCL6−, CD10− or BCL6+ and IRF4/MUM1+ are non-GCB. There are a number of newer algorithms attempting to improve on the Hans schema. These include the use of FOXP1 and GCET1 by Choi et al38 and the use of LMO2 by Natkunam et al.39 One study comparing different algorithms on a large patient cohort showed an 87% concordance with GEP for the Choi method and 86% for Hans.29 In a multi-center study, performed by the cancer and leukemia group B using dose-adjusted EPOCH-R for previously untreated DLBCL, significantly improved time to progression, event-free survival and overall survival was noted in the GCB group as defined by the Hans method.40 The efficacy of DA-EPOCH-R on GCB-DLBCL may relate to its effect on BCL6.40 A report from the international DLBCL rituximab-CHOP consortium program study used an algorithm based on expression of CD10, FOXP1 and BCL6, which had a 92.6% concordance with GEP.41 Although most studies find that immunohistochemical algorithms correlate with prognosis in DLBCL, there are contrary reports arguing that these methods are not reliable as surrogates for molecular testing.10,27,42
DLBCL based on an anatomic site
Some subtypes of DLBCL are peculiarly related to their sites of presentation. Examples include primary DLBCL of the CNS, primary cutaneous DLBCL leg type and primary mediastinal (thymic) large B-cell lymphoma.
Primary DLBCL of the CNS (PCNSL)
This subtype of DLBCL is rare (4% of primary brain tumors and less than 1% of non-Hodgkin lymphomas), and defined as primary intracerebral or intraocular lymphoma. Lymphomas of the dura, intravascular lymphoma, lymphomas secondarily involving the CNS and immunodeficiency lymphomas are excluded from this group. Primary CNS lymphoma is most common in older males and has a poor outcome in those over 60.43 They may present as single space occupying lesions in the brain, although multiple lesions occur in about 30% of patients. The particular microenvironment in the CNS sanctuary site likely contributes to the adverse prognosis.44 Intraocular DLBL are also in this category, and patients with intraocular disease have a high incidence of spread to the CNS. Studies with GEP suggest that there is a characteristic gene profile associated with primary CNS lymphomas.45,46 There is no consistent pattern of ABC vs GCB signature, but the histogenesis may relate to a germinal center exit B cell.
Pathology of primary DLBCL of the CNS
Tumor cells mostly resemble large centroblasts or immunoblasts, and characteristically involve perivascular spaces (Figure 5). The malignant cells may occur in sheets or be intermixed with reactive small lymphocytes, macrophages, active microglial cells and reactive astrocytes. Necrosis is frequently present. They are positive for pan B-cell markers, and frequently express BCL6 and IRF4/MUM1. Major issues that need to be resolved in PCNSL include the origin of the tumor cells, transforming events and role of the microenvironment of the CNS.47
Figure 5.
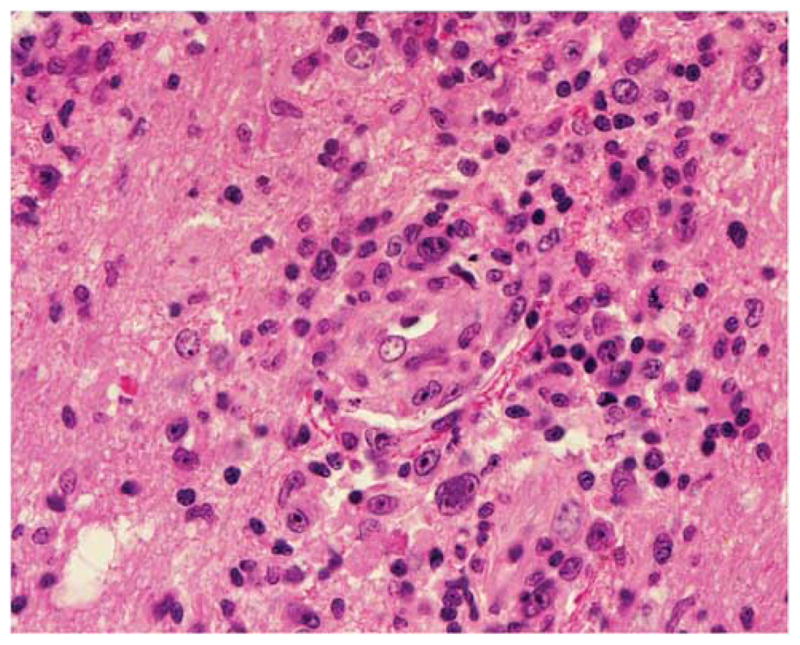
Diffuse large B-cell lymphoma of the central nervous system. Malignant cells are large and pleomorphic with prominent nucleoli. They are seen ringing a blood vessel and admixed with small lymphocytes and glial cells. Hematoxylin and eosin × 400.
Primary cutaneous B-cell lymphoma, leg type
This subtype of DLBCL occurs mostly in elderly females, presenting with rapidly growing tumors on one or both lower legs. It is characterized morphologically by sheets of centroblasts and immunoblasts and frequent mitoses (Figure 6). In addition to pan B-cell markers, these lymphomas characteristically express BCL-2 (unlike primary cutaneous follicle center lymphoma—PCFCL), BCL6 and IRF4/MUM1+, but not CD10. This is an aggressive form of DLBCL with only a 50% 5-year survival. Analogous to other nodal and extranodal LBCL, but unlike PCFCL, immunohistochemical expression of IgM is characteristically present and may be related to an ABC-like phenotype. As DLBCL leg type has genetic similarities with DLBCL’s at other sites, it is not entirely clear that the leg type of DLBCL is a distinct entity. However, characteristic molecular findings have been identified, and include defects of the CDKN2A suppressor pathway, phosphorylation of Rb protein and expression of p53.48
Figure 6.
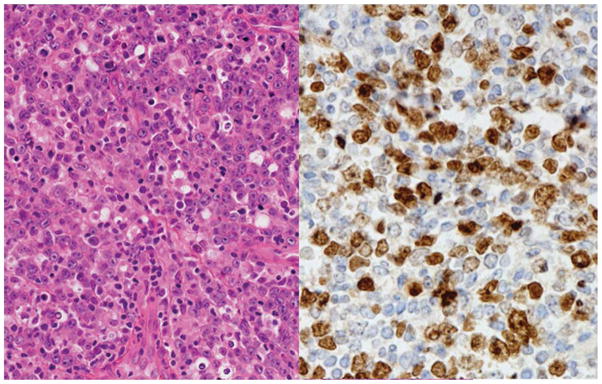
Primary cutaneous diffuse large B-cell lymphoma, ‘leg type’. The section is from a large ulcerating mass on the lower limb. There is a diffuse infiltrate of large lymphoid cells with oval nuclei. Immunoperoxidase stain for IRF4/MUM1 is positive. Left: Hematoxylin and eosin, original × 400, Right immunoperoxidase stain for IRF4/MUM1, hematoxylin counterstain.
DLBCL associated with immune dysfunction or infectious agents
Aggressive B-cell lymphomas associated with infectious agents include the spectrum of HIV-and Kaposi’s sarcoma-associated herpes virus (KSHV)-related lymphomas. Discussion in this review will be centered on lymphomas associated with EBV and senescence of the immune system.
EBV-positive DLBCL of the elderly
This is an aggressive form of DLBCL included as a provisional subtype in WHO 2008. It is defined as a clonal EBV+ large cell lymphoma in a patient >50 years old with no known cause for immunodeficiency or prior lymphoma. The median age is 70 years. The incidence appears higher in Asia and some Latin American countries.49 It can occur in younger individuals, but one must rigorously exclude the possibility of an underlying immunodeficiency. It is critical to exclude other more specific types of EBV-related lymphoproliferative disorders such as lymphomatoid granulomatosis, infectious mononucleosis and PEL. EBV should be localized in the malignant cells not bystander lymphocytes, and the characteristic pattern of viral latency in the large malignant cells is EBER+, LMP1+ and EBNA2+/−. DLBCL of the elderly must be also be distinguished from EBV-positive mucocutaneous ulcer, which may have a similar polymorphous or lymphomatous appearance, as well as clonal gene rearrangements, yet pursues an indolent and often self limited clinical course (Figure 7).13
Figure 7.

EBV+ Mucocutaneous ulcer. The biopsy is from an elderly patient present with an oral mucosal ulcer. The top panel is a whole mount showing ulcerated oral mucosa and a deep lymphoid infiltrate. The section lower left shows a pleomorphic infiltrate including numerous large lymphoid cells that resemble a large cell lymphoma. The immunostain for EBV LMP1 is positive in the cytoplasm of the large cells (right). This patient was treated conservatively with resolution of the lesion. Hematoxylin and eosin, original × 40 (top) and × 240 (lower left). Immunostain for EBV LMP1 (right) with hematoxylin counterstain × 300.
Clinical features
Cases of EBV+ DLBCL of the elderly may be nodal or extranodal in presentation. Many have a high IPI and prognosis is inferior to EBV-negative DLBL even if adjusted for age. Most are refractory to initial therapy with poor overall and progression-free survival.50
Pathologic features
Common to many EBV-related lymphoproliferative disorders, the histology may vary over a spectrum from polymorphic to monomorphic. Geographical necrosis is common (Figure 8). Particularly, the polymorphic cases can resemble post-transplantation lymphoproliferative disorders. H–RS cells may be present, and classical Hodgkin lymphoma in the elderly must be considered in the differential diagnosis. Most cases have a non-GCB phenotype (CD10−, IRF4/MUM1+), and are characterized by prominent classical and alternative NF-kB pathway activation.50 Most cases have monoclonal immunoglobulin gene rearrangements and in addition may have clonal T-cell populations.50
Figure 8.
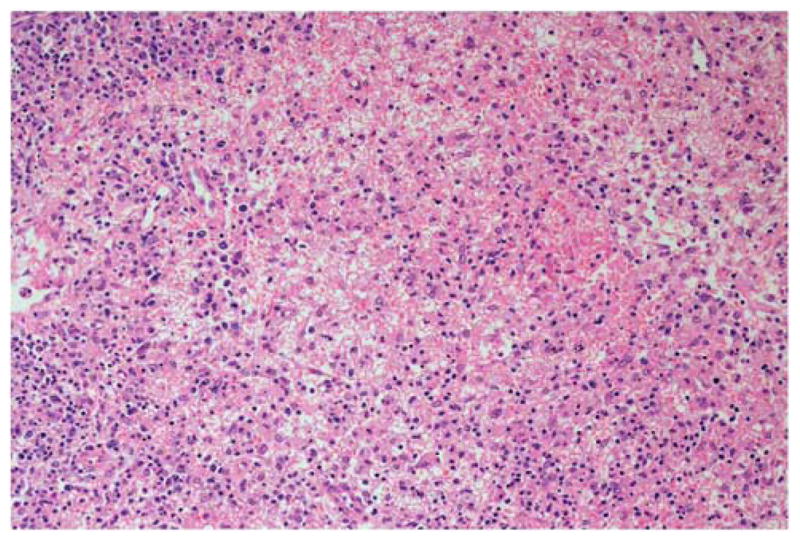
EBV + DLBCL of the elderly. There is marked geographic necrosis and a pleomorphic infiltrate that includes large lymphoid cells with irregular nuclei and prominent nucleoli. Hematoxylin and eosin, original × 200.
Immunphenotypic features
Immunophenotypic studies are helpful in the differential as most cases are CD20+, CD79a+, IRF4/MUM1+, CD10− and BCL6−. The H–RS-like cells are characteristically EBV+, including expression of EBV LMP1, CD20 and CD30 in 75% of cases, but are CD15 negative. However, the H–RS cells may have decreased expression of CD20 and there may be expression of CD15. EBV should be present in the majority of the tumor cells.51 EBNA2 is found in about 30% of cases, and is an indicator of an impaired immune system.51 This viral latency pattern differs from that seen in PBL and may be helpful with this differential diagnosis (see below). Cases of EBV+ DLBCL of the elderly have clonal immunoglobulin gene rearrangements, which may be helpful in differentiating atypical reactive processes and infectious mononucleosis of the elderly.
Plasmablastic lymphoma
PBL is defined as a diffuse proliferation of large cells, which resemble B immunoblasts, but in which all of the neoplastic cells have the phenotype of plasma cells. Other lymphomas with plasmablastic features should be considered in the differential including anaplastic lymphoma kinase-positive large B-cell lymphoma, and the spectrum of KSHV-associated lymphomas (PEL), extracavitary PEL and large B-cell lymphomas arising in KSHV+ multicentric Castleman disease. KSHV-positive lymphomas are not included within the spectrum of PBLs.
PBL of the oral cavity type
PBL of the oral cavity type is an aggressive large B-cell lymphoma generally associated with HIV infection, and most common in men with a median age of 40. There is a predilection for the oral cavity, nasal and paranasal areas, but it may occur at other extranodal sites including the skin, soft tissue and gastrointestinal tract.52 Neoplastic cells are usually uniform and cohesive, and there is a morphologic spectrum with many cases resembling B immunoblasts or centroblasts, whereas in other cases there is more obvious plasmacytic differentiation (Figure 9). The phenotype reveals evidence of strong terminal differentiation with weak or absent CD20 and CD45, variable BCL6, and staining for CD138 and IRF4/MUM1. PBLs generally do not express PAX5, which is associated with early and mature stages of B-cell differentiation, and are positive for XBP1 and BLIMP 1, which are thought to drive plasmacytic differentiation.53 Montes-Moreno et al53 describe a variant phenotype of PBL that has low or absent expression of CD138. CD79a may be positive, and many cases express cytoplasmic immunoglobulin, usually IgG. EBV-EBER is positive in most cases. EBV LMP1 and EBNA2 are not expressed (EBV viral latency pattern I). This differs from the viral latency pattern seen in EBV+ DLBCL of the elderly, which more closely resembles post transplant lymphomas. Rearrangement of the MYC gene is seen in up to 60% of cases, and MYC may be a common genetic mechanism that imparts plasmablastic morphology and aggressive clinical course.54,55 MYC rearrangements are more common in EBV+ PBLs and may be associated with gains in multiple chromosomal loci.56 Complex karyotypes are frequently encountered, and abnormalities include increased copy number and amplification of CCND1.54 Although it has been suggested that oral and extraoral PBL’s have distinct clinicopathologic features,57 this is not always the case (Table 2).
Figure 9.

Plasmablastic lymphoma of the oral cavity type. The cells have a blastic appearance, but plasmacytic differentiation is not marked. The lymphoma is negative for B-cell markers including CD79a, but positive for plasma cell marker CD138 and positive for MYC protein. Hematoxylin and eosin (top left), immunoperoxidase stains, hematoxylin counterstain × 240.
Table 2.
Plasmablastic lymphoma
| PBL of the oral mucosa type | PBL with plasmacytic differentiation | |
|---|---|---|
| Histology | May have minimal plasmacytic differentiation | Plasmacytic differentiation marked |
| Clinical associations | Usually associated with HIV | May be associated with transplants, autoimmune disease, advanced age, rarely occurs in immunocompetent individuals |
| Site | Usually extranodal Often involves the oral cavity, nasal and paranasal sinuses |
May involve lymph nodes Uncommon in the oral cavity |
| EBV-EBER | EBV positive in almost all cases | EBV positive in 60–75% of the cases |
Abbreviations: DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; PBL, plasmablastic lymphoma.
PBL not associated with HIV
In addition to oral cavity type PBL associated with HIV, similar lymphomas are seen in patients with non-HIV-related immunosuppression, and occasionally in immunocompetent patients.53,57,58 This form of PBL may present in lymph nodes, and usually does not involve the oral mucosa. The lymphoma cells have a markedly plasmacytic appearance. Clinical information may be essential in making the differential between plasmablastic myeloma and lymphoma, as in the case illustrated in Figure 10, which was a PBL in the bone marrow. Although EBV-EBER is more often associated with PBL than myeloma, plasmablastic myeloma in immunocompetent patients is occasionally positive for EBER.59
Figure 10.

Plasmablastic lymphoma involving the bone marrow. The cells are blastic with abundant cytoplasm and paranuclear clearing or ‘hof’. Differential from blastic myeloma may be difficult without adequate clinical information. Hematoxylin and eosin, original × 400.
Burkitt lymphoma
Clinical features
Burkitt lymphoma (BL) is the most common non-Hodgkin lymphoma (NHL) in children and adolescents accounting for over 40% of NHL’s in those under the age of 20.9 However, because of the relatively low incidence of NHL in childhood, about 30% of cases of BL occur in adults over 60. It is essential to differentiate these cases from DLBCL as BL may be cured with intensive chemotherapy and CNS prophylaxis, as well as adequate supportive care that may include prevention of tumor lysis syndrome.60 Bone marrow involvement at presentation is common particularly in HIV-associated BL. Although BL is a single pathologic entity, there are different clinical variants. Endemic BL usually occurs in equatorial Africa and is associated with EBV in greater than 90% of cases. Children with endemic BL present with tumor masses in the jaws, gonads and kidneys. Sporadic Burkitt lymphoma is seen in the west in immunocompetent patients, and usually involves the abdomen, terminal ileum and Waldeyer’s ring. Both sporadic and HIV-related BL are only associated with EBV in about 40% of cases. Nodal involvement is more common in adults, as is bulky disease.60 BL associated with HIV occurs in patients with relatively high CD4 T-cell counts, greater than 50/μl,61 and there is frequent lymph node, gastrointestinal tract and bone marrow localization. Despite this clinical diversity endemic, sporadic and HIV-related BL have similar pathologic features and almost identical miRNA profiles. MYC and NFkB pathway-associated miRNA’s can also be used to differentiate between BL and DLBCL.62
Pathologic features
BL is characterized by a monomorphic proliferation cohesive of medium sized B cells with numerous mitoses and frequently a ‘starry sky’ pattern caused by the presence of phagocytic or tingible body macrophages. The neoplastic cells resemble the small non-cleaved cells (centroblasts) seen in the germinal center, with round nuclei, dispersed chromatin, basophilic cytoplasm and squared off cell borders, cytoplasmic vacuoles, medium-sized paracentral nucleoli. There are few small lymphocytes and histiocytes in the background. BL’s are typically described as uniform in their cellular makeup, but there may be some variation in size and shape particularly following therapy. In AIDS-related Burkitt lymphomas, the neoplastic cells typically have a more plasmacytoid cytoplasmic appearance (Figure 11). The 2008 WHO classification appropriately recognizes the histologic range seen in cases of BL and eliminates the category of atypical BL or Burkitt-like lymphoma.63 Cases that resemble BL in all aspects but exhibit greater variation in size and shape should still be classified as BL, and this finding does not justify a ‘gray zone’ diagnosis.
Figure 11.

Burkitt lymphoma in a patient with HIV. The cells are intermediate in size and exhibit some variation in size, with many having a plasmacytoid appearance. Hematoxylin and eosin, original × 400.
Immunophenotype
The neoplastic cells have a germinal center phenotype positive for CD10 and BCL6. The immunoglobulin genes are hypermutated but the cells are positive for IgM and there is no class switch. BCL2 and TdTare characteristically negative, and the Ki67 proliferation rate approaches 100%. Although the diagnosis of BL is usually not problematic, questions arise in cases that are not entirely typical, either because of morphologic or immunophenotypic overlap with other lymphomas. For example, BL may be negative for BCL6, and although BL is characteristically negative for BCL2, a subset may be weakly positive.
Molecular/cytogenetic characteristics
The translocation in Burkitt lymphoma involves the MYC gene on chromosome 8, plus the heavy chain gene on chromosome 14, the kappa chain gene on chromosome 2, the lambda light chain gene on chromosome 22, or rarely other translocation partners. In endemic BL, the MYC gene is typically translocated to the IgH joining region, whereas in sporadic BL, the MYC gene is juxtaposed to the IgH MU switch region. The MYC translocation is characteristic of BL but can be seen in other non-Hodgkin lymphomas, including DLBCL and so-called double-hit lymphomas that have both BCL2 and MYC translocations (see below). A MYC break apart probe is usually used in interphase FISH to identify MYC translocations as it is not dependent on the specific translocation partner, but not all cases of BL are positive and a negative FISH result does not exclude the diagnosis. BL is characteristically ‘MYC simple’ in that translocation involving MYC is the primary event with few other karyotypic abnormalities. It is important to realize that translocation involving MYC cannot always be documented in BL, and no single parameter is the gold standard for diagnosis.
GEP has revealed distinct molecular signatures for BL and DLBCL.64,65 BL is distinguished by the high level of expression of MYC, the expression of a subgroup of germinal center genes and low level of NFκB. Studies using GEP as the ‘gold standard’ to differentiate BL from DLBCL indicate that a significant percentage of cases may be misclassified by expert pathologists using standard criteria.64–66 Using different platforms (Affymetrix U133 GeneChips in the case of Hummel et al65 and a custom oligonucleotide microarray with 2524 unique genes in the case of Dave et al64), GEP identified an intermediate group of cases with signature intermediate between DLBCL and BL. These included cases that were diagnosed by pathologists as atypical Burkitt lymphoma that lacked a characteristic molecular BL signature, and likely would fall into the unclassifiable category using current criteria (see below). Table 3 contrasts the clinical and pathologic features of DLBCL, BL and B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma (BLU).
Table 3.
Differential between DLBCL, BL and B-cell lymphoma unclassifiable, with features intermediate between DLBCL and BL (BLU)
| DLBCL | BL | BLU | |
|---|---|---|---|
| Age at presentation | Usually older but can occur at any age | Children, young adults | Older adults |
| Pathogenesis | May be related to the germinal center (GCB), activated B cell or other pathway | GCB derived | GCB derived |
| Growth rate | Rapid | Extremely rapid, Ki67 approaching 100% | Extremely rapid but usually less than 100% |
| Stage | Even distribution, 50% stage 1 or 2 | Usually high stage | Usually advanced III/IV |
| Bone marrow involvement | Uncommon, often terminal | Common | Common |
| CNS involvement | Unusual | Leptomeningeal disease common at presentation in children and adults60 | Common |
| EBV | Uncommon in the absence of immunodeficiency or age-related senescence | >90% in endemic BL 40% in sporadic and HIV-related BL |
Negative |
| MYC translocation | Uncommon, usually a secondary event associated with a complex karyotype | Almost always present as initiating event and single abnormality (MYC simple) | Often double hits with translocations involving MYC, plus BCL2 and/or sometimes BCL6 |
Abbreviations: BL, Burkitt lymphoma; CNS, central nervous system; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; GCB, germinal center B cell.
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma (BLU)
B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma (BLU) is a heterogeneous category that is not considered a distinct entity by the WHO, but may be used as a working classification for cases that may have morphological and genetic features of both DLBCL and BL but do not fulfill diagnostic criteria for either entity. Criteria for applying the BLU category are still evolving, and are expected to be refined as additional case series are reported. Some cases more closely resemble BL but have an atypical phenotype such as expression of BCL2, or a complex karyotype that includes BCL2, as well as MYC translocations and other abnormalities. Morphology is variable, but there is a uniform diffuse proliferation of intermediate to large lymphoid cells without significant numbers of infiltrating lymphocytes. Proliferation rate is high (80% or more), but often less than the 100%, which characterizes BL. Other cases resemble DLBCL with centroblastic morphology and many of these are ‘double’ or ‘triple hit’ lymphomas with translocations involving the BCL2 gene, the MYC gene and sometimes the BCL6 gene as well (Figure 12). About 2/3 of cases with BCL6 and MYC translocations also have BCL2 translocations.67 The term BLU should be used with discretion and does not apply to DLBCL cases simply because of a high growth faction, starry sky macrophages or MYC translocations. Similarly the term is not valid in cases of BL cases because of atypical cytology. Blastoid lymphomas with cyclin D1 and MYC rearrangements are considered blastoid MCLs. Transformed follicular lymphomas may also be considered separately even if they show complex genotypes that include MYC translocations.
Figure 12.

B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma (BLU). There is a diffuse proliferation of intermediate to large lymphoid cells with small nucleoli. Few infiltrating small lymphocytes or histiocytes are apparent. The lower panel shows immunostain for MYC protein in a paraffin section. FISH studies revealed translocations in MYC and BCL2 (double hit lymphoma). Top: Hematoxylin and eosin × 400; bottom: Immunoperoxidase stain for MYC, hematoxylin counterstain × 240.
Immunophenotype
Phenotypically the cells are positive for pan-B cell markers although expression of CD20 and immunoglobulin light chains may be decreased.68 They usually have a germinal center signature and are usually CD10+, BCL6+, IRF4/MUM1 negative. Unlike Burkitt lymphoma, most are positive for BCL2.67,69 High protein expression of MYC may be seen even in cases that lack the translocation, and may relate to amplification or extra copies of the gene.70,71 Most cases with MYC translocations have high levels of nuclear staining for MYC with immunohistochemical techniques (greater than 70–80% of the malignant cells positive), and this may be used as a screening test to determine which cases may require further genetic testing.
Clinical features
BLU occurs in older adults most of whom present at advanced stage (III or IV), with generalized lymphadenopathy.71 Patients frequently have high levels of LDH, high IPI, bone marrow involvement and CNS involvement, although the frequency of CNS disease either at presentation or relapse varies widely in different studies (9–50%).67 The disease is aggressive with a poor clinical outcome and is not curable with conventional chemotherapy.
Cytogenetic and molecular studies
Cytogenetic studies usually reveal a complex karyo-type unlike Burkitt lymphoma. Many cases are ‘double-hit’ or ‘triple hit’ lymphomas with translocations involving MYC in addition to BCL2 and sometimes BCL6 as well. Only about 60% of cases with MYC translocations involve the heavy chain gene t(8;14), and in 40% of the cases there are other translocation partners.72 The significance of the translocation partner is unknown, but one study suggests a better outcome if there is a non-Ig/MYC translocation.72 BCL6+/MYC+ double hit lymphomas are less common and most represent BCL2+/MYC+/BCL6+ triple hits.67 From a genetic perspective, the gray zone between Burkitt lymphoma and DLBCL may be a temporary repository for different biologic entities.73 The pathogenetic mechanism and aggressive behavior may relate to synergism between the proliferative activity of MYC and the anti-apoptotic effect of BCL2.
Conclusions
Refinements in diagnostic hematopathology incorporating newer immunohistochemical and molecular techniques, as well as attention to clinical parameters, have resulted in a long list of aggressive B-cell lymphomas that need to be recognized to provide appropriate therapy. In addition to describing new variants and subtypes of DLBCL, such as the primary cutaneous leg type of DLBCL, the WHO classification now recognizes areas of acceptable uncertainty such as BLU, and age and EBV infection as a defining factor in DLBCL. GEP, miRNA profiling, signaling pathway analysis, whole genome sequencing and other techniques have ushered in a new era in the characterization of aggressive B-cell lymphomas, 74 and it is anticipated that these advances will result in the identification of rational targeted therapeutic targets. In addition, there is growing appreciation of the role of the tumor microenvironment, and the importance of the immune background of the patient. There are a significant percentage of patients with DLBCL who fail conventional therapy, and the number of ongoing clinical trials attest to the search for novel-targeted agents tailored towards specific molecules or pathways.75 One example is the promising use of PIM2 inhibitors, which act on cytokine pathways (IL2, IL6) and the JAK-STAT pathway.6 Finding the ‘Achilles heel’ of DLBCL will require case selection based on gene expression, cytokine and signal transduction pathways among others. Clearly, the classification of aggressive B-cell lymphomas is changing and the entities that emerge must have clinical relevance that translates to effective therapy.
Acknowledgments
I am grateful to Drs Elias Campo and Steven Swerdlow for their editorial review and helpful suggestions.
Footnotes
Disclosure/conflict of interest
The author declares no conflict of interest.
References
- 1.Said J. Diffuse aggressive B-cell lymphomas. Adv Anat Pathol. 2009;16:216–235. doi: 10.1097/PAP.0b013e3181a9d5d2. [DOI] [PubMed] [Google Scholar]
- 2.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drake MT, Maurer MJ, Link BK, et al. Vitamin D insufficiency and prognosis in non-Hodgkin’s lymphoma. J Clin Oncol. 2010;28:4191–4198. doi: 10.1200/JCO.2010.28.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alencar AJ, Malumbres R, Kozloski GA, et al. Micro-RNAs are independent predictors of outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Clin Cancer Res. 2011;17:4125–4135. doi: 10.1158/1078-0432.CCR-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chanan-Khan AA, Cheson BD. Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol. 2008;26:1544–1552. doi: 10.1200/JCO.2007.14.5367. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Abad C, Pisonero H, Blanco-Aparicio C, et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood. 2011;118:5517–5527. doi: 10.1182/blood-2011-03-344374. [DOI] [PubMed] [Google Scholar]
- 7.Pfreundschuh M. How I treat elderly patients with diffuse large B-cell lymphoma. Blood. 2010;116:5103–5110. doi: 10.1182/blood-2010-07-259333. [DOI] [PubMed] [Google Scholar]
- 8.de Jong D, Balague Ponz O. The molecular background of aggressive B cell lymphomas as a basis for targeted therapy. J Pathol. 2011;223:274–282. doi: 10.1002/path.2807. [DOI] [PubMed] [Google Scholar]
- 9.Miles RR, Arnold S, Cairo MS. Risk factors and treatment of childhood and adolescent Burkitt lymphoma/leukaemia. Br J Haematol. 2012;156:730–743. doi: 10.1111/j.1365-2141.2011.09024.x. [DOI] [PubMed] [Google Scholar]
- 10.Gutierrez-Garcia G, Cardesa-Salzmann T, Climent F, et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood. 2011;117:4836–4843. doi: 10.1182/blood-2010-12-322362. [DOI] [PubMed] [Google Scholar]
- 11.Said J. Transformation to aggressive B-cell lymphoma: morphology, immunophenotype, and molecular characteristics. Appl Immunohistochem Mol Morphol. 2003;11:199–205. [PubMed] [Google Scholar]
- 12.Montoto S, Fitzgibbon J. Transformation of indolent B-cell lymphomas. J Clin Oncol. 2011;29:1827–1834. doi: 10.1200/JCO.2010.32.7577. [DOI] [PubMed] [Google Scholar]
- 13.Dojcinov SD, Venkataraman G, Pittaluga S, et al. Age-related EBV-associated lymphoproliferative disorders in the Western population: a spectrum of reactive lymphoid hyperplasia and lymphoma. Blood. 2011;117:4726–4735. doi: 10.1182/blood-2010-12-323238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Chen C, Lau S, et al. CD3-positive large B-cell lymphoma. Am J Surg Pathol. 2009;33:505–512. doi: 10.1097/PAS.0b013e318185d231. [DOI] [PubMed] [Google Scholar]
- 15.Ruzinova MB, Caron T, Rodig SJ. Altered subcellular localization of c-Myc protein identifies aggressive B-cell lymphomas harboring a c-MYC translocation. Am J Surg Pathol. 2010;34:882–891. doi: 10.1097/PAS.0b013e3181db83af. [DOI] [PubMed] [Google Scholar]
- 16.Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, Doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3460–3467. doi: 10.1200/JCO.2011.41.4342. [DOI] [PubMed] [Google Scholar]
- 17.Slack GW, Tan K, Scott DW, et al. Co-expression of MYC and BCL2 protein in R-CHOP treated de novo diffuse large B-cell lymphoma predits poor outcome. Modern Pathol. 2012;25(Supplement 2):370A. [Google Scholar]
- 18.Izquierdo F, Suarez D. CD5(−) diffuse large B-cell lymphoma with peculiar cyclin D1+ phenotype Pathologic and molecular characterization of a single case. Hum Pathol. 2012;43:1344–1345. doi: 10.1016/j.humpath.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 19.Hsiao SC, Cortada IR, Colomo L, et al. SOX11 is useful in differentiating cyclin D1-positive diffuse large B-cell lymphoma from mantle cell lymphoma. Histopathology. doi: 10.1111/j.1365-2559.2012.04260.x. e-pub ahead of print 29 May 2012. [DOI] [PubMed] [Google Scholar]
- 20.Lucioni M, Novara F, Riboni R, et al. CD5(−) diffuse large B-cell lymphoma with peculiar cyclin D1+ phenotype. Pathologic and molecular characterization of a single case. Hum Pathol. 2011;42:1204–1208. doi: 10.1016/j.humpath.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Vela-Chavez T, Adam P, Kremer M, et al. Cyclin D1 positive diffuse large B-cell lymphoma is a post-germinal center-type lymphoma without alterations in the CCND1 gene locus. Leuk Lymphoma. 2011;52:458–466. doi: 10.3109/10428194.2010.540361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salles G, de Jong D, Xie W, et al. Prognostic significance of immunohistochemical biomarkers in diffuse large B-cell lymphoma: a study from the Lunenburg Lymphoma Biomarker Consortium. Blood. 2011;117:7070–7078. doi: 10.1182/blood-2011-04-345256. [DOI] [PubMed] [Google Scholar]
- 23.Chigrinova E, Mian M, Scandurra M, et al. Diffuse large B-cell lymphoma with concordant bone marrow involvement has peculiar genomic profile and poor clinical outcome. Hematol Oncol. 2011;29:38–41. doi: 10.1002/hon.953. [DOI] [PubMed] [Google Scholar]
- 24.Zinzani PL, Pellegrini C, Gandolfi L, et al. Combination of lenalidomide and rituximab in elderly patients with relapsed or refractory diffuse large B-cell lymphoma: a phase 2 trial. Clin Lymphoma Myeloma Leuk. 2011;11:462–466. doi: 10.1016/j.clml.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Alizadeh AA, Gentles AJ, Alencar AJ, et al. Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood. 2011;118:1350–1358. doi: 10.1182/blood-2011-03-345272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer PN, Fu K, Greiner T, et al. The stromal cell marker SPARC predicts for survival in patients with diffuse large B-cell lymphoma treated with rituximab. Am J Clin Pathol. 2011;135:54–61. doi: 10.1309/AJCPJX4BJV9NLQHY. [DOI] [PubMed] [Google Scholar]
- 27.Ott G, Ziepert M, Klapper W, et al. Immunoblastic morphology but not the immunohistochemical GCB/nonGCB classifier predicts outcome in diffuse large B-cell lymphoma in the RICOVER-60 trial of the DSHNHL. Blood. 2010;116:4916–4925. doi: 10.1182/blood-2010-03-276766. [DOI] [PubMed] [Google Scholar]
- 28.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362:1417–1429. doi: 10.1056/NEJMra0807082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer PN, Fu K, Greiner TC, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol. 2011;29:200–207. doi: 10.1200/JCO.2010.30.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mareschal S, Lanic H, Ruminy P, et al. The proportion of activated B-cell like subtype among de novo diffuse large B-cell lymphoma increases with age. Haematologica. 2011;96:1888–1890. doi: 10.3324/haematol.2011.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thunberg U, Enblad G, Berglund M. Classification of diffuse large B-cell lymphoma by immunohistochemistry demonstrates that elderly patients are more common in the non-GC subgroup and younger patients in the GC subgroup. Haematologica. 2012;97:e3. doi: 10.3324/haematol.2011.057240. author reply e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bavi P, Uddin S, Bu R, et al. The biological and clinical impact of inhibition of NF-kappaB-initiated apoptosis in diffuse large B cell lymphoma (DLBCL) J Pathol. 2011;224:355–366. doi: 10.1002/path.2864. [DOI] [PubMed] [Google Scholar]
- 33.Schuetz JM, Johnson NA, Morin RD, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–1390. doi: 10.1038/leu.2011.378. [DOI] [PubMed] [Google Scholar]
- 34.Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol. 2010;28:3360–3365. doi: 10.1200/JCO.2009.26.3947. [DOI] [PubMed] [Google Scholar]
- 35.Cuccuini W, Briere J, Mounier N, et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood. 2012;119:4619–4624. doi: 10.1182/blood-2012-01-406033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, Lin P, Fayad LE, et al. B-cell lymphomas with MYC/8q24 rearrangements and IGH@BCL2/t(14;18) (q32;q21): an aggressive disease with heterogeneous histology, germinal center B-cell immunophenotype and poor outcome. Mod Pathol. 2012;25:145–156. doi: 10.1038/modpathol.2011.147. [DOI] [PubMed] [Google Scholar]
- 37.Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–282. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- 38.Choi WW, Weisenburger DD, Greiner TC, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res. 2009;15:5494–5502. doi: 10.1158/1078-0432.CCR-09-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Natkunam Y, Farinha P, Hsi ED, et al. LMO2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with anthracycline-based chemotherapy with and without rituximab. J Clin Oncol. 2008;26:447–454. doi: 10.1200/JCO.2007.13.0690. [DOI] [PubMed] [Google Scholar]
- 40.Wilson WH, Jung SH, Porcu P, et al. A Cancer and Leukemia Group B multi-center study of DA-EPOCH-rituximab in untreated diffuse large B-cell lymphoma with analysis of outcome by molecular subtype. Haematologica. 2012;97:758–765. doi: 10.3324/haematol.2011.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visco C, Li Y, Xu-Monette ZY, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia. 2012;26:2103–2113. doi: 10.1038/leu.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castillo JJ, Beltran BE, Song MK, et al. The Hans algorithm is not prognostic in patients with diffuse large B-cell lymphoma treated with R-CHOP. Leuk Res. 2012;36:413–417. doi: 10.1016/j.leukres.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Pels H, Schmidt-Wolf IG, Glasmacher A, et al. Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol. 2003;21:4489–4495. doi: 10.1200/JCO.2003.04.056. [DOI] [PubMed] [Google Scholar]
- 44.Ferreri AJ. How I treat primary CNS lymphoma. Blood. 2011;118:510–522. doi: 10.1182/blood-2011-03-321349. [DOI] [PubMed] [Google Scholar]
- 45.Sung CO, Kim SC, Karnan S, et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood. 2011;117:1291–1300. doi: 10.1182/blood-2010-07-297861. [DOI] [PubMed] [Google Scholar]
- 46.Tun HW, Personett D, Baskerville KA, et al. Pathway analysis of primary central nervous system lymphoma. Blood. 2008;111:3200–3210. doi: 10.1182/blood-2007-10-119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montesinos-Rongen M, Siebert R, Deckert M. Primary lymphoma of the central nervous system: just DLBCL or not? Blood. 2009;113:7–10. doi: 10.1182/blood-2008-04-149005. [DOI] [PubMed] [Google Scholar]
- 48.Kaune KM, Neumann C, Hallermann C, et al. Simultaneous aberrations of single CDKN2A network components and a high Rb phosphorylation status can differentiate subgroups of primary cutaneous B-cell lymphomas. Exp Dermatol. 2011;20:331–335. doi: 10.1111/j.1600-0625.2010.01226.x. [DOI] [PubMed] [Google Scholar]
- 49.Hofscheier A, Ponciano A, Bonzheim I, et al. Geographic variation in the prevalence of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: a comparative analysis of a Mexican and a German population. Mod Pathol. 2011;24:1046–1054. doi: 10.1038/modpathol.2011.62. [DOI] [PubMed] [Google Scholar]
- 50.Montes-Moreno S, Odqvist L, Diaz-Perez JA, et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol. 2012;25:968–982. doi: 10.1038/modpathol.2012.52. [DOI] [PubMed] [Google Scholar]
- 51.Adam P, Bonzheim I, Fend F, et al. Epstein-Barr virus-positive diffuse large B-cell lymphomas of the elderly. Adv Anat Pathol. 2011;18:349–355. doi: 10.1097/PAP.0b013e318229bf08. [DOI] [PubMed] [Google Scholar]
- 52.Qing X, Sun N, French SW, et al. Oral and extraoral plasmablastic lymphoma: similarities and differences in clinicopathologic characteristics. Am J Clin Pathol. 2011;135:977–978. doi: 10.1309/AJCPOEOXRYIXQZ0F. author reply 8–9. [DOI] [PubMed] [Google Scholar]
- 53.Montes-Moreno S, Gonzalez-Medina AR, Rodriguez-Pinilla SM, et al. Aggressive large B-cell lymphoma with plasma cell differentiation: immunohistochemical characterization of plasmablastic lymphoma and diffuse large B-cell lymphoma with partial plasmablastic phenotype. Haematologica. 2010;95:1342–1349. doi: 10.3324/haematol.2009.016113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boy SC, van Heerden MB, Babb C, et al. Dominant genetic aberrations and coexistent EBV infection in HIV-related oral plasmablastic lymphomas. Oral Oncol. 2011;47:883–887. doi: 10.1016/j.oraloncology.2011.06.506. [DOI] [PubMed] [Google Scholar]
- 55.Bogusz AM, Seegmiller AC, Garcia R, et al. Plasmablastic lymphomas with MYC/IgH rearrangement: report of three cases and review of the literature. Am J Clin Pathol. 2009;132:597–605. doi: 10.1309/AJCPFUR1BK0UODTS. [DOI] [PubMed] [Google Scholar]
- 56.Valera A, Balague O, Colomo L, et al. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol. 2010;34:1686–1694. doi: 10.1097/PAS.0b013e3181f3e29f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansra D, Montague N, Stefanovic A, et al. Oral and extraoral plasmablastic lymphoma: similarities and differences in clinicopathologic characteristics. Am J Clin Pathol. 2010;134:710–719. doi: 10.1309/AJCPJH6KEUSECQLU. [DOI] [PubMed] [Google Scholar]
- 58.Hsi ED, Lorsbach RB, Fend F, et al. Plasmablastic lymphoma and related disorders. Am J Clin Pathol. 2011;136:183–194. doi: 10.1309/AJCPV1I2QWKZKNJH. [DOI] [PubMed] [Google Scholar]
- 59.Chang ST, Liao YL, Lu CL, et al. Plasmablastic cytomorphologic features in plasma cell neoplasms in immunocompetent patients are significantly associated with EBV. Am J Clin Pathol. 2007;128:339–344. doi: 10.1309/27H8XJH31F3GUNAT. [DOI] [PubMed] [Google Scholar]
- 60.Linch DC. Burkitt lymphoma in adults. Br J Haematol. 2012;156:693–703. doi: 10.1111/j.1365-2141.2011.08877.x. [DOI] [PubMed] [Google Scholar]
- 61.Smith SM. AIDS-related BL and CD4 count: a clue? Blood. 2010;116:5435–5436. doi: 10.1182/blood-2010-09-306407. [DOI] [PubMed] [Google Scholar]
- 62.Lenze D, Leoncini L, Hummel M, et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia. 2011;25:1869–1876. doi: 10.1038/leu.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. WHO Press; Geneva, Switzerland: 2008. [Google Scholar]
- 64.Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431–2442. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 65.Hummel M, Bentink S, Berger H, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–2430. doi: 10.1056/NEJMoa055351. [DOI] [PubMed] [Google Scholar]
- 66.Harris NL, Horning SJ. Burkitt’s lymphoma—the message from microarrays. N Engl J Med. 2006;354:2495–2498. doi: 10.1056/NEJMe068075. [DOI] [PubMed] [Google Scholar]
- 67.Aukema SM, Siebert R, Schuuring E, et al. Double-hit B-cell lymphomas. Blood. 2011;117:2319–2331. doi: 10.1182/blood-2010-09-297879. [DOI] [PubMed] [Google Scholar]
- 68.Wu D, Wood BL, Dorer R, et al. ‘Double-Hit’ mature B-cell lymphomas show a common immunophenotype by flow cytometry that includes decreased CD20 expression. Am J Clin Pathol. 2010;134:258–265. doi: 10.1309/AJCP7YLDTJPLCE5F. [DOI] [PubMed] [Google Scholar]
- 69.Carbone A, Gloghini A, Aiello A, et al. B-cell lymphomas with features intermediate between distinct pathologic entities. From pathogenesis to pathology. Hum Pathol. 2010;41:621–631. doi: 10.1016/j.humpath.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 70.Perry A, Dave B, Crockett D, et al. High-grade B-cell lymphoma with featues intermediate between Burkitt lymphoma and diffuse large B-cell lymphoma (grey zone lymphoma): A clinicopathologic analysis of 39 cases. Modern Pathol. 2012;25(Supplement 2):361A. [Google Scholar]
- 71.Lin P, Dickason TJ, Fayad LE, et al. Prognostic value of MYC rearrangement in cases of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. Cancer. 2012;118:1566–1573. doi: 10.1002/cncr.26433. [DOI] [PubMed] [Google Scholar]
- 72.Johnson NA, Savage KJ, Ludkovski O, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114:2273–2279. doi: 10.1182/blood-2009-03-212191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salaverria I, Siebert R. The gray zone between Burkitt’s lymphoma and diffuse large B-cell lymphoma from a genetics perspective. J Clin Oncol. 2011;29:1835–1843. doi: 10.1200/JCO.2010.32.8385. [DOI] [PubMed] [Google Scholar]
- 74.Nogai H, Dorken B, Lenz G. Pathogenesis of non-Hodgkin’s lymphoma. J Clin Oncol. 2011;29:1803–1811. doi: 10.1200/JCO.2010.33.3252. [DOI] [PubMed] [Google Scholar]
- 75.Janakiram M, Thirukonda VK, Sullivan M, Petrich AM. Emerging therapeutic targets in diffuse large B-cell lymphoma. Curr Treat Options Oncol. doi: 10.1007/s11864-011-0178-9. e-pub ahead of print 2 February 2012. [DOI] [PubMed] [Google Scholar]